

Abstract
Targeted therapy has become a valuable approach in adenocarcinoma of the lung. The number of actionable mutations has been continuously increasing with significant acceleration from discovery to clinical application. Herein, we present a case of innovative treatment using targeted therapy of a 75 year-old female with two primary adenocarcinomas of the lung. The first tumor was found to carry an activating mutation in the exon 19 of the epidermal growth receptor (EGFR), and responded favorably to treatment with erlotinib. The second primary was found to carry an isolated amplification of the c-met gene, but no EGFR mutation. Off-label use of crizotinib, a potent inhibitor of c-met, was prescribed. Within four weeks of treatment initiation, the tumor and dependent lymphadenopathy responded with rapid shrinkage. This observation stresses the need for re-biopsy of tumors upon progression or change of biological behavior for selection of appropriate targeted therapy.
Keywords: c-met, epidermal growth factor receptor, adenocarcinoma, lung, crizotinib
The discovery of new “driver mutations” has augmented the therapeutic armamentarium for the treatment of lung cancer1. We herein describe an interesting case of rapid response to crizotinib in a de novo c-met amplified adenocarcinoma in a patient with a pre-existing adenocarcinoma of the lung carrying an activating mutation in exon 19 of the epidermal growth factor receptor (EGFR). A left lower lobe lung (LLL) mass, several indeterminate pulmonary nodules and a lytic spinal lesion was discovered in a 75 year old female on annual surveillance in follow-up for early stage breast cancer that had been surgically treated seven years ago. The patient had a 40 pack year smoking history and an excellent performance status (ECOG 0-1). Past medical history was significant for bilateral breast cancer (2002 and 2004) and a well differentiated papillary carcinoma of the thyroid (2008), all of which were treated surgically and were in remission. The patient had no significant active medical conditions. The LLL mass was biopsied and found to be morphologically compatible with a moderately-to-well differentiated adenocarcinoma. Immunohistochemical analysis revealed diffuse positivity for TTF-1 and Napsin A. The tumor was negative for mammaglobin, estrogen receptor and progesterone receptor expression. This immunoprofile supported the diagnosis of an adenocarcinoma of pulmonary origin. The molecular profile revealed an activating mutation in exon 19 of the EGFR with deletion of amino acids 746 through 750. PCR and Sanger DNA sequencing were utilized to detect these mutations. Erlotinib was initiated with good partial radiographic response based on RECIST 1.1 criteria (Fig. 1, upper panel). After three months of treatment, an enlarging right upper lobe (RUL) lesion and adjacent lymphadenopathy were discovered on follow-up CT scans of the chest (Fig. 1, lower panel). The other tumor lesions remained stable. This enlarging RUL lesion was biopsied and confirmed morphologically to be a poorly differentiated carcinoma with solid growth pattern. Immunohistochemical studies demonstrated tumor cells with diffusely positive staining for CK7, and patchy positive pattern for TTF-1 staining, while CK20, Napsin A, p63, CK5/6, estrogen receptor, progesterone receptor, PAX8, Gata-3, and CDX2 were not detected. Taken together, the morphologic features and immunoprofile are consistent with poorly differentiated adenocarcinoma of lung primary (Fig. 2).
Figure 1.
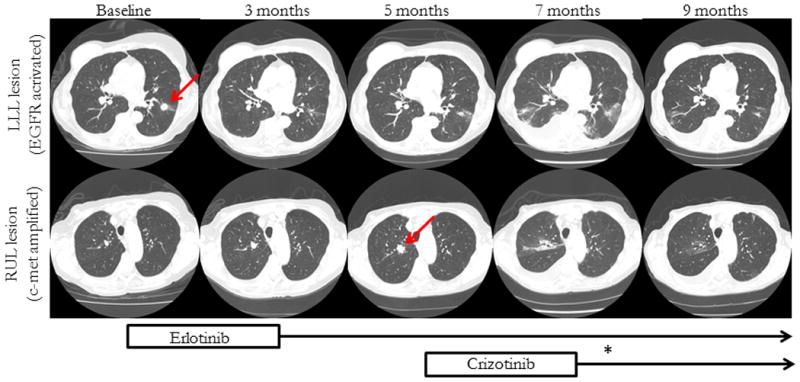
Radiographic evaluation of LLL (upper series) and RUL (lower series). The biopsied lesions are marked by arrows. The timeline refers to time since initial diagnosis of LLL lesion. The upper series demonstrates continuous shrinkage after initiation of erlotinib. The patchy infiltrates at 7 months are biopsy proven to be bronchiolitis obliterans organizing pneumonia. The lower series demonstrates a RUL lesion with interval growth between baseline and 5 months, followed by significant shrinkage after initiation of crizotinib. (*) indicates the 2 week time frame of drug discontinuation due to transaminitis
Figure 2.
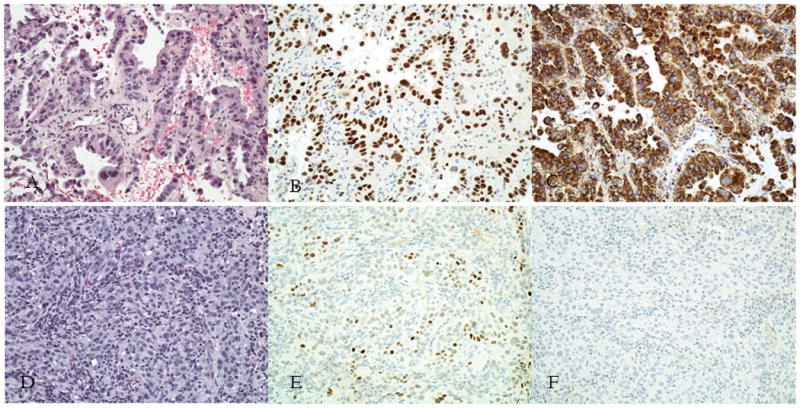
Morphologic features and immunohistochemical stains for LLL (A-C) and RUL (D-F) tumors. H&E stains for LLL (A) and RUL (D). Positive TTF-1 stains in LLL (B) and RUL (E). Positive Napsin A staining in LLL (C) and negative staining in RUL (F).
Molecular examination of the erlotinib-refractory RUL mass for resistance markers to EGFR inhibition was negative for any EGFR mutation, including the previously detected exon 19 or a T790M mutation; however, high levels of c-met amplification were discovered. C-met amplification is a well-established mechanism of resistance to treatment with EGFR inhibitors2 and part of our institutional reflex panel in setting of resistance to EGFR inhibitor. The amplification of c-met, semi-quantitated by fluorescence in situ hybridization (FISH), revealed a c-met/CEP7 ratio of 6.5 in the background of a diploid chromosome 7, therefore suggesting true amplification at a high level. DNA sequencing of EGFR exons 18 through 21 revealed a wild-type genotype. Since the EGFR mutation was felt to be the driver mutation in the LLL lesion, and no mutation of EGFR was found in the RUL lesion, the RUL tumor was felt to be a distinct second primary adenocarcinoma of the lung and propagated by a different molecular pathway. Of note, anaplastic lymphoma kinase (ALK) fusion was not detected in either biopsy specimen3. After presenting this case our institutional tumor board and discussion with the patient, off-label use of crizotinib in this setting was pursued based on early phase I trial data presented at this year's ASCO meeting4.
Crizotinib is a potent multikinase inhibitor with IC50 of c-met inhibition in the low nanomolar range. The phase I study suggested a good correlation between level of c-met amplification and response to crizotinib4. Therefore, crizotinib was initiated in addition to continuation of erlotinib, which was given the first primary adenocarcinoma carrying an activating EGFR mutation, which she had previously tolerated well.
After four weeks of treatment, the c-met amplified tumor had shrunk by over fifty percent in maximum dimension, consistent with a good partial response. Dependent hilar lymphadenopathy equally responded with significant size reduction. On therapy with combined crizotinib and erlotinib, the patient also experienced a significant increase in toxicity, including a mild case of biopsy-proven bronchiolitis obliterans organizing pneumonia (BOOP) without significant clinical pulmonary symptoms and grade 3 transaminitis. The patient was seen by our pulmonary services and was felt to be appropriate for observation and continuation of drug therapy. Her transaminitis resolved with discontinuation within 2 weeks and remained without recurrence on subsequent dose reduction (both erlotinib (75 mg per day) and crizotinib (250 mg per day) were adjusted by 50%). She has since continued both medications for two months with lower extremity edema being the only noticeable adverse event. The increase in drug-related toxicity may be related to synergistic effects of multi-kinase pathway inhibition on combination therapy, or alternatively through increased plasma levels as a result of competitive inhibition of the cytochrome p450 3A4 system, a mechanism through which both drugs are metabolized. Theoretically, this could lead to higher plasma levels of both crizotinib and erlotinib. It is of note that the EGFR mutated LLL lesion, which initially responded to treatment with a good partial response and then stabilized its radiographic appearance, demonstrated further improvement in radiographic appearance after initiation of crizotinib (Fig. 1). It remains speculative whether increasing plasma levels of erlotinib, additional pathway inhibition through crizotinib or a combination of both is responsible for this observation. The c-met pathway also intersects with multiple other signaling pathways, including EGFR. In pre-clinical models, concurrent EGFR/c-met inhibition showed increased anti-tumor activity, enhanced erlotinib sensitivity and applied negative selection pressure against clones with c-met amplification upon continued stimulation with hepatocyte growth factor (HGF). It is also conceivable that the LLL lesion may have carried a secondary alteration within the c-met pathway which could explain further shrinkage, however this testing was not done in the absence of a clinical indication with ongoing response to erlotinib.
In summary, this case provides further evidence of c-met as a primary driver mutation in adenocarcinomas of the lung. It further underlines the efficacy of crizotinib in c-met driven tumors, especially those with high levels of amplification that was previously reported by Camidge et al.4 Moreover, this case stresses the importance of molecular re-evaluation by repeat biopsy in the context of disease progression and change in clinical behavior. As in our case, the possibility of a synchronous multiple primary lung cancer (SMPLC) with a second driver mutation should be considered as it may provide additional therapeutic opportunities. This approach will require further verification the detection of early pulmonary carcinomas is expected to rise with increased screening efforts and improved survival.
Acknowledgments
Martin Frederik Dietrich received support from the NIH T32 institutional grant.
Abbreviations
- EGFR
Epidermal growth factor receptor
- c-met
c-met proto-oncogene
- HGF
hepatocyte growth factor
- ASCO
American Society of Clinical Oncology
- LLL
left lower lobe
- RUL
right upper lobe
- SMPLC
synchronous multiple primary lung cancers
References
- 1.Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nature medicine. 2012;18:349–51. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 3.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. The New England journal of medicine. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camidge D Ross, S-HIO, Shapiro Geoffrey, Otterson Gregory Alan, et al. J Clin Oncol. 2014;32:5s. suppl; abstr. 8001. [Google Scholar]