

Abstract
In response to adenosine 5′-diphosphate, the P2Y1 receptor (P2Y1R) facilitates platelet aggregation, and thus serves as an important antithrombotic drug target. Here we report the crystal structures of the human P2Y1R in complex with a nucleotide antagonist MRS2500 at 2.7Å resolution, and with a non-nucleotide antagonist BPTU at 2.2Å resolution. The structures reveal two distinct ligand binding sites, providing atomic details of P2Y1R’s unique ligand binding modes. MRS2500 recognizes a binding site within the seven transmembrane bundle of P2Y1R, which, however, is different in shape and location from the nucleotide binding site in previously determined P2Y12R structure. BPTU binds to an allosteric pocket on the external receptor interface with the lipid bilayer, making it the first structurally characterized selective G protein-coupled receptor (GPCR) ligand located entirely outside of the helical bundle. These high-resolution insights into P2Y1R should enable discovery of new orthosteric and allosteric antithrombotic drugs with reduced adverse effects.
Human purinergic GPCRs are divided into two subfamilies, Gq-coupled P2Y1R-like receptors and Gi-coupled P2Y12R-like receptors1. Both P2Y1R and P2Y12R are activated by adenosine 5′-diphosphate (ADP) to induce platelet activation, which plays a pivotal role in thrombosis formation2,3. The blockade of either receptor significantly decreases ADP-induced platelet aggregation4. While most of the available antithrombotic drugs act on P2Y12R, P2Y1R has been suggested as a new promising target, which may offer a safety advantage over P2Y12R inhibitors in terms of reduced bleeding liabilities2. Besides platelet aggregation, P2Y1R is also involved in many other physiological processes, such as vascular inflammation, and Ca2+ wave propagation and activation of extracellular signal-regulated kinase in astrocytes5–7.
(1′R,2′S,4′S,5′S)-4-(2-Iodo-6-methylaminopurin-9-yl)-1-[(phosphato)methyl]-2(phosphato)bicycle[3.1.0]-hexane (MRS2500) is a potent P2Y1R antagonist that completely blocks ADP-induced platelet aggregation and effectively reduces arterial thrombosis with only a moderate prolongation of the bleeding time, which makes it an attractive candidate as an antithrombotic agent8,9. Another ligand, 1-(2-(2-(tert-butyl)phenoxy)pyridin-3-yl)-3-(4-(trifluoromethoxy)phenyl)urea (BPTU), was recently discovered by Bristol-Myers Squibb as a novel P2Y1R antagonist that substantially reduces platelet aggregation with a minimal effect on bleeding, for the treatment of thrombosis10. To understand how these anti-thrombotic ligands recognize their purinoceptor target and to enable new drug discovery, we solved X-ray crystal structures of the human P2Y1R receptor bound to MRS2500 and BPTU (Extended Data Table 1), and performed a structure-guided mutagenesis study (Extended Data Table 2).
Overall architecture of P2Y1R
The P2Y1R structures share a canonical seven-transmembrane (7TM) helical bundle architecture with other known GPCR structures (Fig. 1a, 1b and Extended Data Fig. 1). There are two disulfide bonds, connecting the N-terminus to helix VII, and helix III to the second extracellular loop (ECL2), stabilizing the conformations of the ECLs. The ECL2 of P2Y1R exhibits a hairpin structure, which was previously observed in all the known peptide-bound GPCR structures, such as PAR1, NTSR1, chemokine and opioid receptors11–15. The conserved D[E]R3.50Y motif in class A GPCR family is replaced by an HR3.50Y motif in P2Y1R, making the crystal structure of P2Y1R the first GPCR structure with a basic histidine residue at position 3.49 (Ballesteros-Weinstein nomenclature16) (Extended Data Fig. 2a). Distinct from many other class A GPCRs, which contain a salt bridge between D[E]3.49 and R3.50, H1483.49 in P2Y1R repels R1493.50, resulting in a more extended side chain conformation of this residue. Consequently, R1493.50 forms a hydrogen bond with the main chain of A3277.56 at the intracellular tip of helix VII and stabilizes the C-terminus in a different conformation compared to many other known class A GPCR structures. Like PAR1 and some other GPCRs, the P2Y1R structures lack helix VIII, and the C-terminal region beyond R338 appears disordered.
Figure 1. Structures of the P2Y1R-MRS2500 and P2Y1R-BPTU complexes.

a and b, Side view of the P2Y1R-MRS2500 (a) and P2Y1R-BPTU (b) structures. The receptor is shown in blue (a) and orange (b) cartoon representation. The ligands MRS2500 and BPTU are shown in sphere representation with magenta and green carbons, respectively. The disulfide bonds are shown as yellow (a) and red (b) sticks. The membrane boundaries (brown) are adapted from the OPM database31 with P2Y12R (PDB ID: 4NTJ) as a model. c and d, Structural comparison of the helical bundles between P2Y1R and P2Y12R. (c) Top view of the extracellular side; (d) bottom view of the intracellular side. The receptors are in cartoon representation. The P2Y1R-MRS2500, P2Y1R-BPTU, P2Y12R-AZD1283 (PDB ID: 4NTJ) and P2Y12R-2MeSADP (PDB ID: 4PXZ) structures are colored blue, orange, grey and green, respectively.
The two P2Y1R structures are structurally similar (Cα RMSD within the entire receptor is 0.9 Å), except for subtle differences at the extracellular ends of helices I and II. Additionally, in the P2Y1R-MRS2500 structure, a salt bridge between R195 in ECL2 and MRS2500 shifts the β-hairpin tip of ECL2 by 2.6 Å toward the central axis of the helical bundle compared to the P2Y1R-BPTU structure (Extended Data Fig. 2b). Compared with the recently solved structure of P2Y12R17,18 belonging to a separate Gi-coupled P2YR subfamily, P2Y1R is structurally distinct from either the agonist-bound or the antagonist-bound P2Y12R, with Cα RMSD within the helical bundle of 2.2 and 2.6 Å, respectively (Fig. 1c and 1d). The extracellular tip of P2Y1R’s helix VI has a position intermediate between the agonist and antagonist-bound P2Y12R structures; while helix VII in P2Y1R is in a relatively similar conformation to the antagonist-bound P2Y12R structure. Unlike P2Y12R, P2Y1R has a highly conserved in class A GPCR residue P2295.50, which leads to a helical kink and displaces the extracellular end of helix V by over 4 Å compared to the P2Y12R structures. Another substantial difference between the helical bundles of the two purinergic receptors is that the extracellular end of helix III shifts away from the axis of the 7TM helical bundle by over 5 Å in P2Y1R compared to the P2Y12R structures. This apparent shift is likely due to the different conformations that the ECL2 adopts in P2Y1R and P2Y12R. The intracellular halves of the two P2Y receptors, however, are very similar with all helices overlaying each other relatively well.
Ligand binding mode of P2Y1R-MRS2500
In the P2Y1R-MRS2500 structure, the ligand occupies a pocket defined by residues mainly from N-terminus, ECL2 and helices VI and VII (Fig. 2 and Extended Data Fig. 3a). The adenine ring of MRS2500 inserts into a binding crevice with R2876.62 and L44 on either side, and its N6H and N7 are coordinated by two hydrogen bonds with the N2836.58 side chain, similar to adenine recognition in A2A adenosine receptor (N6.55) and P2Y12R (N5.40) but at a different location. The 2-iodo group precisely fits into a small sub-pocket shaped by the P2Y1R’s N-terminus, and interacts with the main chain carbonyl of C42. This substituent has been shown to be critical for a high ligand binding affinity to P2Y1R. Derivatives containing 2-bromo, chloro and fluoro substitutions exhibit much lower affinities than MRS25008. The N6-methyl group extends into another sub-pocket between helices VI and VII, forming hydrophobic interactions with A2866.61 and N2997.28. The corresponding 6-amino analogue is 16-fold less potent, and any alkyl substitution larger than ethyl abolishes binding to P2Y1R8. The (N)-methanocarba ring makes a hydrophobic contact with the phenyl group of Y203 in ECL2. This is in agreement with the observation that the Y203F mutation, but not Y203A, retains the ability to bind the nucleotide antagonist MRS217919 (Extended Data Fig. 4). Both phosphate groups of nucleotide-like antagonists have been proven to be important for the high affinity interactions with P2Y1R20. In the P2Y1R-MRS2500 structure, each terminal oxygen of the two phosphates forms at least one contact with the receptor. The 3′-phosphate makes hydrogen bonds with Y1102.63 and Y3037.32 and is engaged in two salt-bridge interactions with K46 at the N-terminus and R195 in ECL2. The 5′-phosphate forms a salt-bridge with R3107.39 and makes hydrogen bonds with T205 in ECL2 and Y3067.35.
Figure 2. P2Y1R ligand binding pocket for MRS2500.

a, Key residues in P2Y1R for MRS2500 binding. MRS2500 (magenta carbons) and P2Y1R residues (cyan carbons) involved in ligand binding are shown in stick representation. The receptor is shown in blue cartoon representation. Other elements are colored as follows: oxygen, red; nitrogen, dark blue; sulfur, yellow; phosphorus, orange; iodine, purple. Salt bridges are displayed as red dashed lines and hydrogen bonds as blue dashed lines. b, Schematic representation of interactions between P2Y1R and MRS2500. Hydrophobic interactions are indicated as green dashed lines.
Most of the above key residues of P2Y1R have not been previously tested for their involvement in either agonist or antagonist binding. Our mutagenesis studies have further confirmed the critical roles of these residues in ligand binding of P2Y1R, showing that the L44A, Y1102.63F, Y203A, T205A and N2836.58A mutants had greatly reduced binding affinity for [3H]2-methylthio-adenosine 5′-diphosphate (2MeSADP), while the K46A, R195A, Y3037.32F mutants, surrounding the 3′-phosphate that is absent in 2MeSADP, selectively decreased the binding affinity of MRS2500 without affecting the binding of 2MeSADP and BPTU (Extended Data Table 2 and Extended Data Fig. 5). Removing the hydroxyl group coordinating the 5′-phosphate that is present in MRS2500 and 2MeSADP, Y3067.35F mutant displayed greatly reduced affinity of both nucleotides, consistent with a common binding site for both ligands. The requirement for T205 and N2836.58 for 2MeSADP recognition is also consistent with a similar orientation of the both nucleotide ligands.
Previous mutagenesis of some residues in the helical bundle, such as H1323.33, Y1363.37, T2225.43, F2265.47, K2806.55, etc. reduced P2Y1R binding affinity of some agonist and antagonist ligands20,21. All of these residues are located much deeper than the MRS2500 binding site, but overlap well with the corresponding 2MeSADP binding site in the P2Y12R structure. This observation raises the possibility of a second potential nucleotide binding site in the P2Y1R receptor. Indeed, the P2Y1R-MRS2500 structure has a deeper cavity that partially overlaps with the position of the adenine ring of 2MeSADP bound to P2Y12R (Extended Data Fig. 6a). In the current P2Y1R structure, however, the cavity is too small and the ECL2 extends deep into the ligand binding pocket, blocking access to this potential binding site. Rearrangement of ECL2, as well as the helical bundle, would be required for a nucleotide to enter to this site. Nevertheless, it should be noted that the current data do not rule out the possibility that mutations of those residues may affect binding by changing the receptor conformation rather than by direct contact with a ligand.
The binding site of MRS2500 in P2Y1R locates much closer to the extracellular surface than the small-molecule ligand binding sites in the other known GPCR structures (Extended Data Fig. 6b). Comparing the antagonist binding sites in the three solved δ group class A GPCR structures, the binding sites of AZD1283 in P2Y12R and vorapaxar in PAR1 are closer to helices IV and V than the MRS2500 binding site in P2Y1R (Extended Data Fig. 6c). Although recognized by the same endogenous ligand ADP, P2Y1R and P2Y12R structures reveal very different features in binding their nucleotide-like ligands (Fig. 3). The ligand binding sites for MRS2500 in P2Y1R and 2MeSADP in P2Y12R are spatially distinct with only a minor overlap of phosphate binding regions near the residues at position 7.35. The adenine groups of the two ligands are in different orientations. In the P2Y1R-MRS2500 structure, the adenine ring is adjacent to P2Y1R’s helices VI and VII, while the adenine group of 2MeSADP reaches deep into the binding pocket to form hydrophobic interactions with helices III and IV in the P2Y12R structure.
Figure 3. Comparison of the ligand-binding modes between P2Y1R-MRS2500 and P2Y12R-2MeSADP.

a and b, Side view (a) and top view (b) of the comparison of the ligand-binding sites between P2Y1R-MRS2500 and P2Y12R-2MeSADP. MRS2500 and 2MeSADP are shown in stick representation with magenta and yellow carbons, respectively. Only P2Y1R represented as blue cartoon is shown. c and d, Comparison of the ligand-binding pockets between P2Y1R-MRS2500 (c) and P2Y12R-2MeSADP (d). The P2Y1R and P2Y12R structures are shown in cartoon and molecular surface representations, and colored in blue and green, respectively.
A unique binding site for BPTU
The non-nucleotide ligand BPTU and other diarylurea P2Y1R antagonists have been recently introduced as novel antiplatelet agents10,22,23. Surprisingly, the P2Y1R-BPTU crystal structure reveals that instead of interacting within the 7TM helical bundle, BPTU binds to P2Y1R on the lipidic interface of the TM domain. The relatively shallow ligand binding pocket, formed by aromatic and hydrophobic residues of helices I, II and III and ECL1, accommodates BPTU predominantly through hydrophobic interactions (Fig. 4 and Extended Data Fig. 3b). The only polar interactions are represented by two hydrogen bonds between the nitrogen atoms of BPTU’s urea group and the mainchain carbonyl of L1022.55. This carbonyl is available for bidentate coordination of this selective P2Y1R antagonist because the residue P1052.58 above precludes intrahelical hydrogen-bonding. Previous structure-activity relationship (SAR) studies demonstrated that replacing the urea linker of BPTU with other two to four atom linkers greatly reduced potency24. This could be explained by the importance of the two hydrogen-bond interactions for retaining P2Y1R binding affinity of this chemical series. The pyridyl group forms hydrophobic interactions with A1062.59 and F119, while its nitrogen atom is not involved in any interaction. However, if the pyridyl is substituted by a phenyl group, an extra hydrophobic contact with M1233.24 may be introduced. This is supported by the fact that a corresponding phenyl derivative showed higher ligand binding affinity and antiplatelet activity22. A1062.59 is unique to P2Y1 among P2YRs; other subtypes have larger side chains at this position, which could sterically hinder binding of BPTU’s phenyl ring to this site, consistent with its P2Y1R selectivity. Significantly, the sterically hindered A1062.59W/F/L mutants lost the ability to bind BPTU, while retaining recognition of nucleotide agonist 2MeSADP and antagonist MRS2500 (Extended Data Table 2 and Extended Data Fig. 5). The benzene ring within the phenoxy group of BPTU wedges into a cavity between helices II and III, interacting with T1032.56, M1233.24, L1263.27 and Q1273.28. The hydrophobic nature of this sub-pocket is consistent with previous studies that have shown that the lipophilicity of this aryl group of the ligand is important for binding affinity and in vitro functional activity22. Similar to the A1062.59W mutant, the T1032.56W mutation abolished the binding affinity of BPTU to P2Y1R, but did not affect the binding of 2MeSADP and MRS2500 (Extended Data Table 2). The tert-butyl substituent of the phenoxy ring forms a hydrophobic contact with L1022.55 in the P2Y1R-BPTU structure. SAR studies have shown that a lipophilic substitution of the phenoxy ring at either the ortho or meta position is preferred10, consistent with the fact that a para substitution may cause spatial clashes with receptor’s helix III based on the P2Y1R-BPTU structure. At the other end of the ligand, the ureido phenyl ring forms two aromatic edge-to-face interactions with F621.43 and F661.47. This aligns with the SAR requirement of aromatic monosubstitution of the urea N distal to the pyridine ring; replacing this aromatic ring with aliphatic substituents or inserting one or two methylene groups between a phenyl ring and the urea group decreased P2Y1R binding affinity dramatically10. The above consistency between the P2Y1R structure and the SAR studies has been validated also with docking simulations of several related, potent urea derivatives (Extended Data Figure 7) that support the observed BPTU ligand binding mode.
Figure 4. P2Y1R ligand binding pocket for BPTU.

a, Key residues in P2Y1R for BPTU binding. BPTU (green carbons) and P2Y1R residues (brown carbons) involved in ligand binding are shown in stick representation. The receptor is shown in orange cartoon representation. Lipid molecules in close contacts with BPTU are shown in white line representation. Hydrogen bonds are blue dashed lines. b, Schematic representation of interactions between P2Y1R and BPTU. Hydrophobic interactions are green dashed lines. The BPTU binding site remains intact in the P2Y1R-MRS2500 complex. c and d, Side view (c) and zoom-in view (d) of the BPTU ligand binding site in P2Y1R. The receptor is shown in molecular surface representation and colored by binding property (Yellow: hydrophobic surface; red: hydrogen bond acceptor potential; blue: hydrogen bond donor potential; white: neutral surface). The figure was prepared using ICM software (http://www.molsoft.com).
Previous efforts to reduce the lipophilicity of BPTU (HPLC logP = 5.7) have shown that adding polar or basic amine groups to this ligand decreases binding affinity and antiplatelet activity; in fact, the binding affinity in this chemical series correlates directly with the HPLC logP value24. These data agree with our structure where the ligand BPTU binds in a highly hydrophobic environment. The structural features of the ligand binding site and the high lipophilicity of BPTU suggest that this ligand most likely enters the ligand binding pocket via the lipid bilayer, rather than through the highly charged nucleotide approach route that leads to the P2Y1R orthosteric site. The approach routes of hydrophobic GPCR ligands through the lipid bilayer have been proposed for several different receptors, rhodopsin, S1P1 and GPR40 etc.25–27. However, the ligands bound to these receptors at least partially occupy the conventional GPCR ligand binding pocket, and to our knowledge, BPTU is the first structurally characterized selective and high affinity GPCR ligand that binds entirely outside of the helical bundle of GPCRs. The location of this ligand binding site indicates that BPTU acts as an allosteric modulator of P2Y1R. The allosteric regulation of P2Y1R by BPTU was studied by investigating its ability to influence the dissociation of [3H]2MeSADP from the receptor. The data indicate that BPTU can substantially accelerate the dissociation of 2MeSADP, while the mutation A1062.59W abolishes the allosteric effect on the receptor by BPTU (Extended Data Fig. 8).
Structural insights into inhibition mechanisms of MRS2500 and BPTU
Although MRS2500 and BPTU bind to distinct sites in P2Y1R, these two ligands stabilize the receptor in similar inactive conformations. In comparing the active state structures of several different GPCRs with their inactive structures, a relatively stable bundle of helices I to IV, and a more mobile module consisting of helices V, VI and VII have been observed, demonstrating that the rearrangements of helices V, VI and VII play important roles in the process of GPCR activation28–30. In P2Y1R, the antagonist MRS2500 potentially prevents such movements and stabilizes the receptor in an inactive state by interacting with helices VI and VII. In addition, it also bridges to the less mobile module through its 3′-phosphate, a requirement for antagonism in this chemical series. However, in the BPTU-bound P2Y1R structure, the ligand makes no contacts with receptor’s helices V, VI and VII, implying that BPTU inhibits receptor function in a different way. Besides the movements of helices V to VII, rotation of helix III was also reported to be involved in the transformation from the inactive conformation to the active conformation of some GPCRs, although it is more subtle compared to the conformational changes of helices V to VII17,28–30. The P2Y1R binding mode of BPTU suggests that this ligand most likely inhibits agonist-induced receptor activation by blocking relative movement of helices II and III, possibly a rotation of helix III. The above findings suggest that the movement of this less mobile module is equally critical for GPCR activation, in addition to the conformational changes of helices V to VII. Hindrance of the conformational plasticity of either domain may result in a severe loss of receptor function.
Conclusions
The P2Y1R structures reveal atomic details of two completely distinct ligand binding sites having chemically and structurally contrasting characteristics: a hydrophilic and charged site for the nucleotide-like antagonist MRS2500 with numerous anchor points on different receptor domains and a shallow site on the outer, lipid-exposed surface for the non-nucleotide ligand BPTU stabilized by a single carbonyl polar interaction. A comparison between P2Y1R and P2Y12R shows that these representative structures of two different P2YR subfamilies interact with their nucleotide ligands in disparate binding modes, which deepen our understanding of the diversity of signal recognition mechanisms in GPCRs. The external, hydrophobic binding site of BPTU suggests the entry of this antagonist occurs through the lipid membrane, and opens new opportunities to broaden the scope of future GPCR ligand discovery to target novel allosteric sites outside of the canonical ligand binding pocket. The P2Y1R structures also provide insights into the inhibition mechanisms of receptor activation by MRS2500 and BPTU through interactions with different domains of the receptor.
Methods
Cloning and expression of engineered P2Y1R proteins
The wild-type (WT) human P2Y1R DNA was synthesized by Genewiz and then cloned into a modified pFastBac1 vector (Invitrogen) containing an expression cassette with an HA signal sequence followed by a FLAG tag at the N-terminus and a PreScission protease site followed by a 10xHis tag at the C-terminus. An engineered construct (construct 1) was generated by overlap extension PCR to insert M1-E54 of rubredoxin32 between K247 and P253 in the intracellular loop 3 (ICL3) of P2Y1R. The P2Y1R gene was further modified by introducing the D3207.49N mutation to improve protein yield and stability. Another P2Y1R construct (construct 2) was made by adding A23-L128 of a thermostabilized BRIL (PDB ID: 1M6T) prior to the residue A8 of the receptor sequence in construct 1. High-titer recombinant baculovirus (>108 viral particles per ml) was obtained using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Spodoptera frugiperda (Sf9) cells at cell density of 2–3×106 cells/ml were infected with virus at MOI (multiplicity of infection) of 5. Cells were harvested by centrifugation at 48 h post infection and stored at -80 °C until use.
Purification of Sf9-expressed P2Y1R proteins for crystallization
Insect cell membranes were disrupted by thawing frozen cell pellets in a hypotonic buffer containing 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl and EDTA-free complete protease inhibitor cocktail (Roche). Cell membranes were disrupted by repeated dounce homogenization. Extensive washing of the membranes was performed by centrifugation in the same hypotonic buffer (one more time), followed by a high osmotic buffer containing 1 M NaCl, 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl (three times), thereby removing soluble and membrane associated proteins from the suspension of membranes, and then the hypotonic buffer (one more time) to remove the high concentration of NaCl. Purified membranes were resuspended in 10 mM HEPES, pH 7.5, 30% (v/v) glycerol, 10 mM MgCl2, 20 mM KCl and EDTA-free complete protease inhibitor cocktail, flash-frozen with liquid nitrogen, and stored at −80 °C until further use.
Prior to solubilization, the purified membranes of construct 1-expressed materials were thawed on ice in the presence of 1 mM ATP (Sigma), 2 mg/mL iodoacetamide (Sigma) and EDTA-free protease inhibitor cocktail (Roche). After incubating at 4°C for 1h, the membranes were then solubilized in 50 mM HEPES, pH 7.5, 300 mM NaCl, 0.5% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace), 0.1% (w/v) cholesterol hemisuccinate (CHS) (Sigma), and 500 μM ATP for 3h at 4°C. The supernatant was isolated by centrifugation at 160,000 × g for 30 min, supplemented with 30 mM imidazole, pH 7.5, and incubated with TALON IMAC resin (Clontech) overnight at 4°C. The resin was washed with ten column volumes of 25 mM HEPES, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, 40 mM imidazole, 0.05% (w/v) DDM, 0.01% (w/v) CHS and 1 mM ATP, followed by ten column volumes of 25 mM HEPES, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10 mM MgCl2, 5 mM ATP, and fifteen column volumes of 25 mM HEPES, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS and 50 μM MRS2500. The protein was eluted by 25 mM HEPES, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, 300 mM imidazole, 0.05% (w/v) DDM, 0.01% (w/v) CHS and 100 μM MRS2500 in five column volumes. PD MiniTrap G-25 column (GE healthcare) was used to remove imidazole and increase the compound concentration to 1 mM. The protein was then treated overnight with His-tagged PreScission protease (home-made) and His-tagged PNGase F (home-made) to remove the C-terminal His-tag and de-glycosylate the receptor. PreScission protease, PNGaseF and the cleaved His tag were removed by Ni-NTA superflow resin (Qiagen) incubation at 4 °C for 1 hour. The His-tag cleaved protein was collected in the Ni-NTA column flow through, and then concentrated to 40–50 mg/ml with a 100 kDa molecular weight cut-off Vivaspin concentrator (Sartorius Stedim Biotech). Receptor purity, monodispersity and concentration were estimated using SDS-PAGE and analytical size-exclusion chromatography (aSEC).
The P2Y1R-BPTU complex protein was purified following a protocol similar to the above procedure. The membranes of construct 2-expressed material were incubated in 50 μM BPTU, 2 mg/mL iodoacetamide (Sigma), and EDTA-free protease inhibitor cocktail (Roche) at 4 °C for 1 hour, and then solubilized in 50 mM HEPES, pH 7.5, 300 mM NaCl, 0.5% (w/v) DDM, 0.1% (w/v) CHS and 25 μM BPTU for 3h at 4°C. After overnight binding to the TALON IMAC resin, the resin was washed with thirty column volumes of 25 mM HEPES, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, 40 mM imidazole, 0.05% (w/v) DDM, 0.01% (w/v) CHS and 25 μM BPTU, and then eluted by five column volumes of 25 mM HEPES, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, 300 mM imidazole, 0.05% (w/v) DDM, 0.01% (w/v) CHS and 50 μM BPTU. Imidazole was removed using the PD MiniTrap G-25 column. The protein was further purified by the treatment with His-tagged PreScission protease and PNGase F.
Lipidic cubic phase crystallization of P2Y1R-MRS2500 and P2Y1R-BPTU complexes
Purified protein samples of P2Y1R were reconstituted into lipidic cubic phase (LCP) by mixing with molten lipid in a mechanical syringe mixer33. The protein solution was mixed with molten monoolein/cholesterol (10:1 by mass) lipids at weight ratio of 1:1.5 (protein: lipid). After formation of a transparent lipidic cubic phase, the mixture was dispensed onto 96-well glass sandwich plates (Shanghai FAstal BioTech, Inc.) in 40–50 nl drops and overlaid with 800 nl precipitant solution using a Mosquito LCP robot (TTP Labtech). Protein reconstitution in LCP and crystallization trials were performed at room temperature (19–22 °C). Plates were incubated and imaged at 20 °C using an automated incubator/imager (RockImager, Formulatrix). The crystals of the P2Y1R-MRS2500 complex grew to their full size (70–150 μm) within two weeks in 20–30% PEG400 (v/v), 50–100 mM sodium citrate, 50 μM MRS2500, and 0.1 M HEPES, pH 7.0 or 0.1 M Tris-HCl, pH 8.0. The P2Y1R-BPTU complex was crystallized in 100–300 mM ammonium phosphate dibasic, 0–10% PEG2000 MME, 50 μM BPTU, and 0.1 M sodium citrate, pH 6.5, and the crystals reached their maximum size (100–130 μm) within two weeks. The P2Y1R crystals were collected directly from LCP using 50–100 μm micromounts (M2-L19–50/100, MiTeGen) and flash frozen in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data were collected at the SPring-8 beam line 41XU, Hyogo, Japan, using a Pilatus3 6M detector (X-ray wavelength 1.0000 Å). The crystals were exposed with a 10 μm mini-beam for 1 second and 1° oscillation per frame. XDS34 was used for integrating and scaling data from 36 best-diffracting crystals of the P2Y1R–MRS2500 complex and 12 crystals of the P2Y1R–BPTU complex. Initial phase information of the P2Y1R-MRS2500 complex was obtained by molecular replacement (MR) with Phaser35 using the receptor portion of PAR1 (PDB ID: 3VW7), converted to polyalanines, and rubredoxin structure (PDB ID: 1IRO) as search models. The correct MR solution contained two P2Y1R-rubredoxin molecules packed antiparallel in the asymmetric unit. Refinement was performed with REFMAC536 and BUSTER37 followed by manual examination and rebuilding of the refined coordinates in the program COOT38 using both |2Fo| - |Fc| and |Fo| - |Fc| maps. The final model includes 296 residues (38–247 and 253–338) of the 345 residues of P2Y1R and residues 1 to 54 of rubredoxin. The remaining N- and C-terminal residues are disordered and were not refined. The P2Y1R–BPTU complex structure was solved using P2Y1R in the P2Y1R–MRS2500 complex and rubredoxin as starting models and refined under the same procedure. The final model of the P2Y1R–BPTU complex contains 291 residues (38–156, 158–247 and 253–334) of P2Y1R and the 54 residues of rubredoxin. Without clear electronic density, the N-terminal fused BRIL was not traced. The crystal packing shows that there is no room to fit BRIL, indicating that it most likely degraded.
Ligand-binding assays
Materials: 2MeSADP and MRS2500 were from Tocris (Minneapolis, MN, USA). [3H]2MeSADP (3.5 Ci/mmol) was purchased from Moravek, Brea, CA, USA. BPTU was synthesized as reported using a 2-aryloxy-3-isothiocyanatopyridine as intermediate10.
Membrane preparations from Sf9 cells and COS-7 cells expressing WT and mutant human P2Y1Rs were used for all the ligand-binding assays. Protein concentrations were measured using Bio-Rad protein assay reagents. For saturation experiments, 50 μl [3H]2MeSADP (from 5 to 200 nM) was incubated with 100 μl WT and mutant P2Y1R membrane preparations (5 μg/tube) in a total assay volume of 200 μl Tris-HCl buffer containing 10 mM MgCl2. MRS2500 or 2MeSADP (10 μM) was used to determine the non-specific binding. For displacement experiments using the membrane preparations from Sf9 cells, increasing concentrations of MRS2500 or BPTU were incubated with WT or mutant P2Y1R membrane preparations (5–10 μg) and 25 nM [3H]2MeSADP at 25 °C for 30 min. Using the membrane preparations from COS-7 cells, increasing concentrations of MRS2500 or BPTU were incubated with WT or mutant P2Y1R membrane preparations (20 μg) and 2 nM [3H]2MeSADP at 25 °C for 30 min.
For dissociation experiments, WT or mutant P2Y1R membrane preparations from Sf9 cells (5 μg) were first pre-equilibrated with 25 nM [3H]2MeSADP at 4°C for 30 min. Using the membrane preparations from COS-7 cells, membranes containing 20 μg of protein were pre-equilibrated with 2 nM [3H]2MeSADP at 4°C for 30 min. Then the dissociation was initiated at 4°C by mixing with 10 μM MRS2500 in the absence or presence of 10 μM BPTU (note that the dissociation at 25°C was too fast to be measured). The reaction was terminated by harvesting with a 24-channnel Brandel cell harvester (Brandel, Gaithersburg, MD, USA) and followed by washing twice with 5 ml cold Tris-HCl buffer containing 10 mM MgCl2. Radioactivity was measured using a scintillation counter (Tri-Carb 2810TR). Data were analyzed using Prism 6 (GraphPad, San Diego, CA, USA).
Docking simulations of BPTU derivatives
The P2Y1R-BPTU structure was prepared using the Protein Preparation Wizard tool implemented in the Schrödinger suite, adding all the hydrogen atoms and the missing side chains of residues whose backbone coordinates were observed in the structure. The orientation of polar hydrogens was optimized, the protein protonation states were adjusted and the overall structure was minimized with harmonic restraints on the heavy atoms, to remove strain. Then, all the hetero groups and water molecules were deleted.
The SiteMap tool of the Schrödinger suite was used to identify potential binding sites in the structure. In addition to the canonical orthosteric binding site within the TM bundle, a shallow pocket was identified on external receptor interface with the lipid bilayer in correspondence of the BPTU crystallographic pose and was selected as the docking site. Molecular docking of several BPTU derivatives at the P2Y1R structure was performed by means of the Glide package from the Schrödinger suite. In particular, a Glide Grid was centered on the centroid of residues located within 5 Å from the previously identified cavity. The Glide Grid was built using an inner box (ligand diameter midpoint box) of 14 Å × 14 Å × 14 Å and an outer box (within which all the ligand atoms must be contained) that extended 20 Å in each direction from the inner one. Docking of ligands was performed in the rigid binding site using the SP (standard precision) procedure. The top scoring docking conformations accurately reproduced the binding mode observed for BPTU in the crystal and were in agreement with previous SAR findings for this class of compounds.
Extended Data
Extended Data Figure 1. Crystal packing of P2Y1R–MRS2500 and P2Y1R–BPTU complexes.

a, Overall structure of the P2Y1R–MRS2500 complex. P2Y1R and rubredoxin are colored in blue and grey, respectively. MRS2500 is shown in magenta stick representation. b and c, Crystal packing of the P2Y1R–MRS2500 complex shown in two different views. d, Overall structure of the P2Y1R–BPTU complex. P2Y1R and rubredoxin are colored in orange and grey, respectively. BPTU is shown in green stick representation. e and f, Crystal packing of the P2Y1R–BPTU complex shown in two different views. Lipid molecules are shown in white line representation.
Extended Data Figure 2.
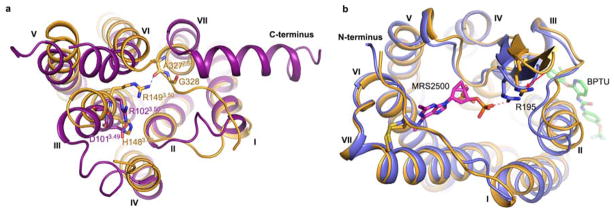
a, Comparison of the HR3.50Y motif in P2Y1R and the DR3.50Y motif in A2A adenosine receptor (A2AAR, PDB ID: 3EML). P2Y1R and A2AAR are shown in cartoon representation, and colored in orange and purple, respectively. Residues at positions 3.49 and 3.50 are shown as sticks. The hydrogen bond formed by R1493.50 and A3277.56 in P2Y1R is displayed as a blue dashed line. b, Comparison of the P2Y1R–MRS2500 complex and the P2Y1R–BPTU complex structures. The P2Y1R–MRS2500 structure is colored in blue, and the P2Y1R–BPTU structure is orange. The ligands MRS2500 and BPTU are shown in stick representation with magenta and green carbons, respectively. The residues R195 in both structures are shown as sticks. The salt-bridge between MRS2500 and R195 in the P2Y1R–MRS2500 structure is displayed as a red dashed line. The red arrow indicates the shift at the β-hairpin tip of ECL2 toward the center axis of the helical bundle in the P2Y1R-MRS2500 structure compared to the P2Y1R-BPTU structure.
Extended Data Figure 3. Electron density of MRS2500 (a) and BPTU (b).
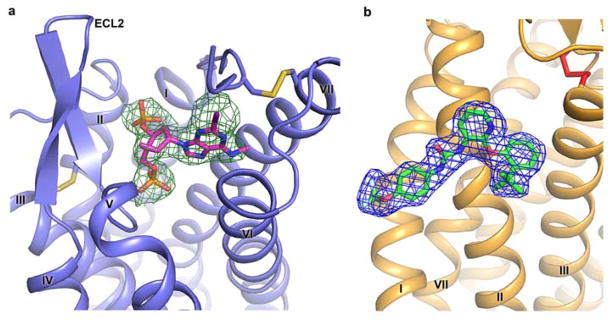
a, MRS2500 is shown in stick representation with magenta carbons. The receptor is shown in blue cartoon representation. Electron density is contoured at 1.0σ from an |2Fo| - |Fc| map, and colored in green. The disulfide bonds are shown as yellow sticks. b, BPTU is shown in stick representation with green carbons. The receptor is shown in orange cartoon representation. Electron density is contoured at 1.0σ from an |2Fo| - |Fc| map, and colored in blue. The disulfide bond is shown as a red stick.
Extended Data Figure 4. Chemical structures of different P2Y1R ligands that are discussed in the paper.

a, N6-methyl-2′-deoxyadenosine-3′,5′-bisphosphate (MRS2179); b, 2MeSADP.
Extended Data Figure 5. Inhibition of [3H]2MeSADP binding by MRS2500 and BPTU.

Data shown are from a representative experiment performed in duplicate. a–d, Inhibition of [3H]2MeSADP binding to membrane preparations from Sf9 cells expressing mutant P2Y1Rs by MRS2500 (a and b) and BPTU (c and d). Construct 1 is the P2Y1R construct used for MRS2500 co-crystallization. Construct 2 is the P2Y1R construct used for BPTU co-crystallization. The Ki values for MRS2500 or BPTU from at least three independent experiments are listed in Extended Data Table 2. The affinity of MRS2500 at Construct 1 is 18.4 ± 1.5 nM, which is significantly different from its affinity at the mutant Y303F (2880 ± 854 nM) (P<0.05, unpaired t-test). The affinity of BPTU for construct 1 is 161 ± 47 nM, which is not significantly different from its affinity for Y303F (309 ± 36 nM) (P>0.05, unpaired t-test). e and f, Inhibition of [3H]2MeSADP binding to membrane preparations from COS-7 cells expressing WT and mutant P2Y1Rs by MRS2500 (e) and BPTU (f).
Extended Data Figure 6. Structural comparison of the ligand binding sites between P2Y1R and other class A GPCRs.

a, Comparison of the ligand binding sites between P2Y1R-MRS2500 and P2Y12R-2MeSADP. Only P2Y1R (blue) is shown in cartoon and surface representations. The ligands MRS2500 and 2MeSADP are shown as sticks with magenta and yellow carbons, respectively. The red arrow points to the small cavity that partially overlaps with the position of the adenine ring of 2MeSADP bound to P2Y12R in the P2Y1R-MRS2500 structure. b, Comparison of the small-molecule ligand binding sites of P2Y1R, β2 adrenergic receptor (β2AR, PDB ID: 2RH1), A2AAR (PDB ID: 3EML), CXCR4 (PDB ID: 3ODU), protease-activated receptor 1 (PAR1, PDB ID: 3VW7), δ-opioid receptor (dOR, PDB ID: 4EJ4) and P2Y12R (PDB ID: 4NTJ). Only P2Y1R helices are shown (blue). The ligands MRS2500 (for P2Y1R, magenta), carazolol (for β2AR, orange), ZM241385 (for A2AAR, red), ID1t (for CXCR4, wheat), vorapaxar (for PAR1, yellow), naltrindole (for dOR, green) and AZD1283 (for P2Y12R, cyan) are shown in stick representation. c, Top view of the antagonist binding sites of P2Y1R (blue), P2Y12R (cyan) and PAR1 (yellow). The antagonists, MRS2500, AZD1283 and vorapaxar are shown in stick representation and colored as b.
Extended Data Figure 7. Docking poses of selected BPTU derivatives.

Superposition of the crystallographic pose of BPTU (green carbons) with the docking poses obtained for: BPTU (yellow carbons), compound 3l from ref. 22 (pink carbons) and compound 2d from ref. 23 (cyan carbons). The P2Y1R–BPTU structure is shown in cartoon representation and colored in orange. P2Y1R residues (grey carbons) involved in ligand binding are shown in stick representation. Hydrogen bonds are shown as red dashed lines. All the compounds overlay very well with the BPTU crystal pose, showing hydrogen bonds with the mainchain carbonyl of L1022.55. An additional hydrogen bond with Q1273.28 helps to stabilize the binding mode of compound 2d.
Extended Data Figure 8. The allosteric effects of BPTU on [3H]2MeSADP dissociation.
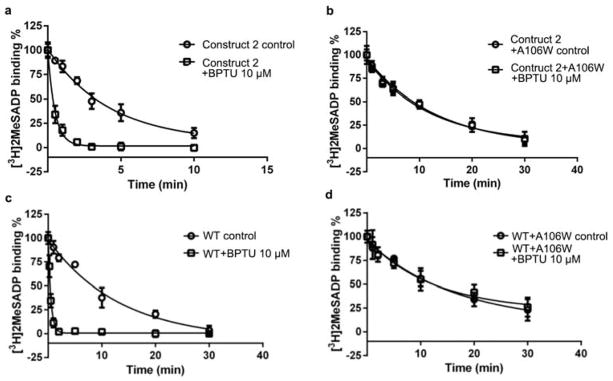
a and b, The allosteric effects of BPTU on [3H]2MeSADP dissociation from the membrane preparations from Sf9 cells expressing construct 2 (a, the P2Y1R construct used for BPTU co-crystallization) and the A1062.59W mutant P2Y1R (b). The dissociation of [3H]2MeSADP was tested in the absence (○) or presence (□) of 10 μM BPTU. c and d, The allosteric effects of BPTU on [3H]2MeSADP dissociation from the membrane preparations from COS-7 cells expressing WT (c) and the A1062.59W mutant P2Y1R (d).
Extended Data Table 1.
Data collection and structure refinement statistics.
| Data Collection | |||
|---|---|---|---|
| P2Y1R-BPTU | P2Y1R-MRS2500 | ||
| Number of crystals used | 12 | 36 | |
| Space group | R3:H | P1 | |
| Cell dimensions | |||
| a, b, c (Å) | 66.3, 66.3, 239.1 | 40.6, 65.3, 76.8 | |
| α,β,γ (°) | 90.0, 90.0, 120.0 | 101.9, 95.1, 101.4 | |
| Number of reflections processed | 143,276 | 100,372 | |
| Number of unique reflections | 19,881 | 20,471 | |
| Resolution (Å) | 50.0-2.20 (2.30-2.20) * | 50.0-2.70 (2.80-2.70) * | |
| Rmerge (%) | 10.1 (97.9) | 16.1 (83.2) | |
| CC1/2 | 0.998(0.619) | 0.972 (0.608) | |
| Mean I/σ(I) | 12.1 (1.8) | 18.9 (2.3) | |
| Completeness (%) | 99.9 (99.7) | 98.2 (96.2) | |
| Redundancy | 7.2 (4.6) | 4.9 (3.6) | |
| Refinement | |||
| Resolution (Å) | 50.0-2.20 | 50.0-2.70 | |
| Number of reflections (test set) | 19,881 (1,017) | 19,283 (1,027) | |
| Rwork/Rfree (%) | 20.7/23.0 | 21.8/26.7 | |
| Number of atoms | A | B | |
| Protein | 2,713 | 2,769 | 2,800 |
| Ligand | 32 | 29 | 29 |
| Zn | 1 | 1 | 1 |
| Cholesterol and CHS | 133 | n/a | n/a |
| Lipids, PEG and waters | 235 | 6 | 4 |
| Overall B values (Å2) | A | B | |
| P2Y1R | 53.2 | 47.1 | 46.4 |
| Rubredoxin | 129.3 | 47.2 | 78.6 |
| Ligand | 51.4 | 54.7 | 57.2 |
| Zn | 87.0 | 42.0 | 76.4 |
| Cholesterol and CHS | 52.1 | n/a | n/a |
| Lipids and waters | 69.8 | 32.5 | 46.4 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.010 | 0.009 | |
| Bond angles (°) | 1.04 | 1.33 | |
| Ramachandran plot statistics (%) † | |||
| Favored regions | 96.7 | 94.6 | |
| Allowed regions | 3.3 | 5.4 | |
| Disallowed regions | 0.0 | 0.0 | |
Values in parentheses are for highest-resolution shell.
As defined in MolProbity.
Extended Data Table 2.
Binding of [3H]2MeSADP to membrane preparations from Sf9 cells (a) and COS-7 cells (b) expressing WT and mutant P2Y1Rs and inhibition by MRS2500 or BPTU.*
| a. Ligand binding to membrane preparations from Sf9 cells
| |||
|---|---|---|---|
| WT/mutants | Inhibition of binding | Saturation binding | |
|
| |||
| Ki (nM), MRS2500 | Ki (nM), BPTU | Kd (nM), 2MeSADP | |
| WT | 16.8±6.6 | 173±45 | 53.6±6.1 |
| Construct 1† | 18.4±1.5 | 161±47 | 33.7±12.2 |
| Construct 1+L44A mutation | ND | ND | NS |
| Construct 1+K46A mutation | 425±156§ | 100±19§ | 68.3±10.0 |
| Construct 1+Y1102.63F mutation | ND | ND | NS |
| Construct 1+R195A mutation | 157±40§ | 119±39§ | 59.8±11.6 |
| Construct 1+Y203A mutation | ND | ND | NS |
| Construct 1+T205A mutation | ND | ND | NS |
| Construct 1+N2836.58A mutation | ND | ND | NS |
| Construct 1+A2866.61W mutation | 825±522§ | 146±59|| | 47.4±10.5 |
| Construct 1+Y3037.32F mutation | 2880±854§ | 309±36|| | 41.9±11.0 |
| Construct 1+Y3067.35F mutation | 2370±650§ | 184±143|| | >300 |
| Construct 2‡ | 22.5±7.9 | 226±57 | 36.7±7.6 |
| Construct 2+T1032.56W mutation | 35.0±15.2|| | >10,000 | 45.0±11.6 |
| Construct 2+A1062.59W mutation | 44.8±15.8|| | >10,000 | 23.4±5.8 |
| Construct 2+A1062.59F mutation | 30.5±16.9|| | >10,000 | 57.4±21.8 |
| Construct 2+A1062.59L mutation | 15.2±1.8|| | 4350±522§ | 41.9±4.1 |
| b. Ligand binding to membrane preparations from COS-7 cells
| |||
|---|---|---|---|
| WT/mutants | Inhibition of binding | Saturation binding | |
|
| |||
| Ki (nM), MR2500 | Ki (nM), BPTU | Kd (nM), 2MeSADP | |
| WT | 9.89±0.81 | 120±8 | 1.88±0.59 |
| WT+Y3037.32F mutation | 635±158§ | 171±48|| | 1.78±0.16 |
| WT+A1062.59W mutation | 11.5±3.7|| | >10,000 | 1.96±0.11 |
Data represent mean ± SEM from 3–5 separate experiments performed in duplicate.
The P2Y1R construct used for MRS2500 co-crystallization.
The P2Y1R construct used for BPTU co-crystallization.
Significant difference from the affinity for the control constructs (P<0.05, One-way ANOVA or unpaired t-test where appropriate).
No significant difference from the affinity for the control constructs (P>0.05, One-way ANOVA). NS, negligible specific binding for [3H]2MeSADP. ND, not determined.
Acknowledgments
This work was supported by the “National Basic Research Program of China” grants 2012CB518000, 2014CB910400 and 2012CB910400 (Q.Z., B.W.), CAS Strategic Priority Research Program XDB08020300 (B.W.), the National Science Foundation of China grants 31422017 (B.W.), 31370729 (Q.Z.) and 91313000 (H.J.), the National Science and Technology Major Project 2013ZX09507001 (H.J., Q.Z., B.W.), NIDDK, NIH Intramural Research Program grant Z01 DK031116-26 (K.A.J.), and the National Institutes of Health grant U54 GM094618 (V.C., V.K., R.C.S.). The authors thank A. Walker for assistance with manuscript preparation and S.M. Moss for technical assistance.
Footnotes
Author information: Atomic coordinates and structure factors for the P2Y1R-MRS2500 and P2Y1R-BPTU structures have been deposited in the Protein Data Bank with indentification codes 4XNW and 4XNV.
Author Contributions: D.Z. optimized the construct, developed the purification procedure and purified the P2Y1R proteins for crystallization, performed crystallization trials and optimized crystallization conditions. Z.G.G. designed, performed and analysed ligand binding and competition assays of wild-type and mutant P2Y1R. K.Z. helped in construct and crystal opimization, and collected diffraction data. E.K. and J.W. helped in ligand synthesis of P2Y1R. S.C. performed and analysed ligand binding assays. S.P. performed and analysed docking assays. C.Y. and L.M. expressed the P2Y1R proteins. W.Z. developed the initial expression and purification protocol for P2Y1R. G.W.H. helped to analyse the structures. H.L. oversaw ligand synthesis of P2Y1R. V.C. and V.K. helped to analyse the structures and assisted with manuscript preparation. H.J. and R.C.S. oversaw structure analysis/interpretation of P2Y1R. K.A.J. oversaw, designed and analysed ligand binding assays, oversaw ligand synthesis, and assisted with manuscript preparation. Q.Z. and B.W. initiated the project, planned and analysed experiments, solved the structures, supervised the research and wrote the manuscript.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Abbracchio MP, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacological reviews. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gachet C. P2 receptors, platelet function and pharmacological implications. Thrombosis and haemostasis. 2008;99:466–472. doi: 10.1160/TH07-11-0673. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson KA, Deflorian F, Mishra S, Costanzi S. Pharmacochemistry of the platelet purinergic receptors. Purinergic signalling. 2011;7:305–324. doi: 10.1007/s11302-011-9216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin J, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:8070–8074. doi: 10.1073/pnas.95.14.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zerr M, et al. Major contribution of the P2Y1 receptor in purinergic regulation of TNFalpha-induced vascular inflammation. Circulation. 2011;123:2404–2413. doi: 10.1161/CIRCULATIONAHA.110.002139. [DOI] [PubMed] [Google Scholar]
- 6.Fam SR, Gallagher CJ, Salter MW. P2Y1 purinoceptor-mediated Ca2+ signaling and Ca2+ wave propagation in dorsal spinal cord astrocytes. The Journal of neuroscience. 2000;20:2800–2808. doi: 10.1523/JNEUROSCI.20-08-02800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neary JT, Kang Y, Willoughby KA, Ellis EF. Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. The Journal of neuroscience. 2003;23:2348–2356. doi: 10.1523/JNEUROSCI.23-06-02348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HS, et al. 2-Substitution of adenine nucleotide analogues containing a bicyclo[3.1.0]hexane ring system locked in a northern conformation: enhanced potency as P2Y1 receptor antagonists. Journal of medicinal chemistry. 2003;46:4974–4987. doi: 10.1021/jm030127+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hechler B, et al. MRS2500 [2-iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate], a potent, selective, and stable antagonist of the platelet P2Y1 receptor with strong antithrombotic activity in mice. The Journal of pharmacology and experimental therapeutics. 2006;316:556–563. doi: 10.1124/jpet.105.094037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chao H, et al. Discovery of 2-(phenoxypyridine)-3-phenylureas as small molecule P2Y1 antagonists. Journal of medicinal chemistry. 2013;56:1704–1714. doi: 10.1021/jm301708u. [DOI] [PubMed] [Google Scholar]
- 11.Zhang C, et al. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White JF, et al. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu B, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan Q, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H, et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of strcture-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 17.Zhang J, et al. Agonist-bound structure of the human P2Y12 receptor. Nature. 2014;509:119–122. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K, et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014;509:115–118. doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. Journal of medicinal chemistry. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moro S, et al. Human P2Y1 receptor: molecular modeling and site-directed mutagenesis as tools to identify agonist and antagonist recognition sites. Journal of medicinal chemistry. 1998;41:1456–1466. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo D, von Kugelgen I, Moro S, Kim YC, Jacobson KA. Evidence for the Recognition of Non-Nucleotide Antagonists Within the Transmembrane Domains of the Human P2Y1 Receptor. Drug development research. 2002;57:173–181. doi: 10.1002/ddr.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiao JX, et al. Conformationally constrained ortho-anilino diaryl ureas: discovery of 1-(2-(1′-neopentylspiro[indoline-3,4′-piperidine]-1-yl)phenyl)-3-(4-(trifluoromethoxy)phenyl)urea, a potent, selective, and bioavailable P2Y1 antagonist. Journal of medicinal chemistry. 2013;56:9275–9295. doi: 10.1021/jm4013906. [DOI] [PubMed] [Google Scholar]
- 23.Yang W, et al. Discovery of 4-aryl-7-hydroxyindoline-based P2Y1 antagonists as novel antiplatelet agents. Journal of medicinal chemistry. 2014;57:6150–6164. doi: 10.1021/jm5006226. [DOI] [PubMed] [Google Scholar]
- 24.Wang TC, et al. Discovery of diarylurea P2Y1 antagonists with improved aqueous solubility. Bioorganic & medicinal chemistry letters. 2013;23:3239–3243. doi: 10.1016/j.bmcl.2013.03.125. [DOI] [PubMed] [Google Scholar]
- 25.Hildebrand PW, et al. A Ligand Channel through the G Protein Coupled Receptor Opsin. Plos One. 2009;4 doi: 10.1371/journal.pone.0004382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanson MA, et al. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava A, et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 2014;513:124–127. doi: 10.1038/nature13494. [DOI] [PubMed] [Google Scholar]
- 28.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 29.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 32.Chun E, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.Smart OS, et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]