

Abstract
Decreased insulin sensitivity, also referred to as insulin resistance (IR), is a fundamental abnormality in patients with type 2 diabetes and a risk factor for cardiovascular disease. While IR predisposition is heritable, the genetic basis remains largely unknown. The GENEticS of Insulin Sensitivity consortium conducted a genome-wide association study (GWAS) for direct measures of insulin sensitivity, such as euglycemic clamp or insulin suppression test, in 2,764 European individuals, with replication in an additional 2,860 individuals. The presence of a nonsynonymous variant of N-acetyltransferase 2 (NAT2) [rs1208 (803A>G, K268R)] was strongly associated with decreased insulin sensitivity that was independent of BMI. The rs1208 “A” allele was nominally associated with IR-related traits, including increased fasting glucose, hemoglobin A1C, total and LDL cholesterol, triglycerides, and coronary artery disease. NAT2 acetylates arylamine and hydrazine drugs and carcinogens, but predicted acetylator NAT2 phenotypes were not associated with insulin sensitivity. In a murine adipocyte cell line, silencing of NAT2 ortholog Nat1 decreased insulin-mediated glucose uptake, increased basal and isoproterenol-stimulated lipolysis, and decreased adipocyte differentiation, while Nat1 overexpression produced opposite effects. Nat1-deficient mice had elevations in fasting blood glucose, insulin, and triglycerides and decreased insulin sensitivity, as measured by glucose and insulin tolerance tests, with intermediate effects in Nat1 heterozygote mice. Our results support a role for NAT2 in insulin sensitivity.
Keywords: Cardiology, Endocrinology, Genetics, Metabolism
Introduction
Insulin resistance (IR) is present in the vast majority of patients who eventually develop type 2 diabetes and seems to be a necessary, but not sufficient, abnormality leading to hyperglycemia in these individuals. Insulin-resistant persons who continue to secrete enough insulin to prevent gross decompensation of glucose homeostasis are still at increased risk to develop cardiovascular disease. It is estimated that 25% to 33% of the US population is sufficiently insulin resistant to be at risk for adverse clinical consequences (1, 2), and the worldwide prevalence of IR is increasing as a consequence of the obesity epidemic (3). A greater understanding of the genetic basis of insulin sensitivity could lead to better diagnostic and therapeutic options.
Insulin-mediated glucose uptake is dictated primarily by skeletal muscle and adipose tissue and varies by as much as 6-fold in apparently healthy individuals (1, 4, 5). Direct measures of insulin sensitivity include the euglycemic-hyperinsulinemic clamp (6, 7) and the insulin suppression test (IST) (8), which are highly negatively correlated (r < –0.9) (9).
The heritability of insulin sensitivity is approximately 40% to 50%, both prior to and after adjusting for an estimate of adiposity, such as BMI or waist circumference (10, 11). However, very large GWAS of surrogate measures of IR or consequences of IR (such as diabetes or fasting insulin) have identified few novel loci that appear to influence insulin sensitivity. Of the 65 type 2 diabetes variants, the vast majority appear to affect insulin synthesis, processing and secretion, and/or pancreatic development, with only a few loci having been associated consistently with surrogate measures of IR (e.g., PPARG, IRS1) (12–17). Surrogate measures of IR are only modestly correlated with direct measures (with correlation coefficients of r ~ 0.35–0.75 for measures based on fasting insulin and/or glucose or measures based on oral glucose tolerance test [GTT]) (5, 13, 18), supporting a partly overlapping genetic structure between surrogates and quantitative measures of IR.
To promote investigation into the genetic basis of insulin sensitivity, we formed the GENESIS (GENEticS of Insulin Sensitivity) consortium. Here, we report what we believe to be a novel insulin sensitivity locus (N-acetyltransferase 2 [NAT2]) identified through a combined approach, incorporating a GWAS meta-analysis of 2,764 individuals and replication in 2,860 individuals with direct, reference measures of insulin sensitivity, followed by in vitro and in vivo functional validation. To our knowledge, this study includes the vast majority of individuals in the world who have had both a reference measure of insulin sensitivity and have DNA available for genetic studies.
Results
GWAS single-marker association testing for insulin sensitivity.
We analyzed GWAS data from nondiabetic participants of European ancestry from 4 studies who provided DNA for genome-wide genotyping and underwent a direct measure of insulin sensitivity (Table 1), including either euglycemic-hyperinsulinemic clamp or IST. In total, 2,764 European subjects passed quality control procedures and entered the GWAS analysis. In the initial meta-analyses (adjusted for age, gender, and ethnicity, with or without adjustment for BMI), no SNPs reached GWAS significance levels of P < 5 × 10–8. We took forward variants representing 4 of the top signals into follow-up studies (Figure 1 and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI74692DS1).
Table 1. Summary details of relevant characteristics of GWAS (RISC, ULSAM, EUGENE2, and Stanford IST) cohorts.
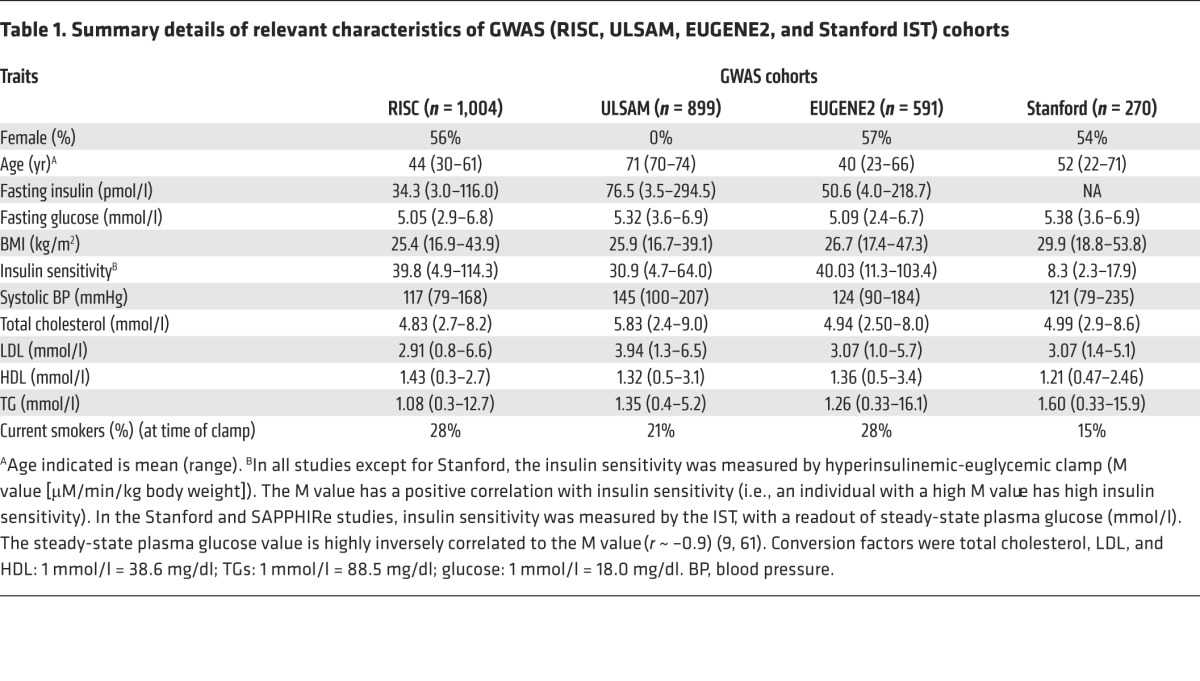
Figure 1. Manhattan plot for the age-, gender-, and BMI-adjusted GWAS analyses (genomic positions from Hg19).

Insulin sensitivity measures were fitted in a linear regression model with age, gender, center, and BMI, with the first two principal components for race/ethnicity included as predictors.
In silico replication and de novo genotyping in independent cohorts.
We performed an in silico follow-up of 5 SNPs from the 4 top loci most strongly associated with insulin sensitivity in 1,601 Hispanic subjects from 3 cohorts within the GUARDIAN consortium (19) who had previously undergone hyperinsulinemic-euglycemic clamp and GWAS genotyping (Supplemental Table 1). These SNPs were chosen because they had initial GWAS P values of less than 6 × 10–6, with evidence of multiple supporting SNPs (Table 2 and Figure 1). The most strongly associated SNPs at one of these loci, NAT2, included two common, nonsynonymous coding SNPs [rs1208 (803A>G, K268R) and rs1801280 (341T>C, I114T)], which are in modest pairwise linkage disequilibrium; r2 ~ 0.75) (Figure 2).
Table 2. Top GWAS signals for insulin sensitivity.
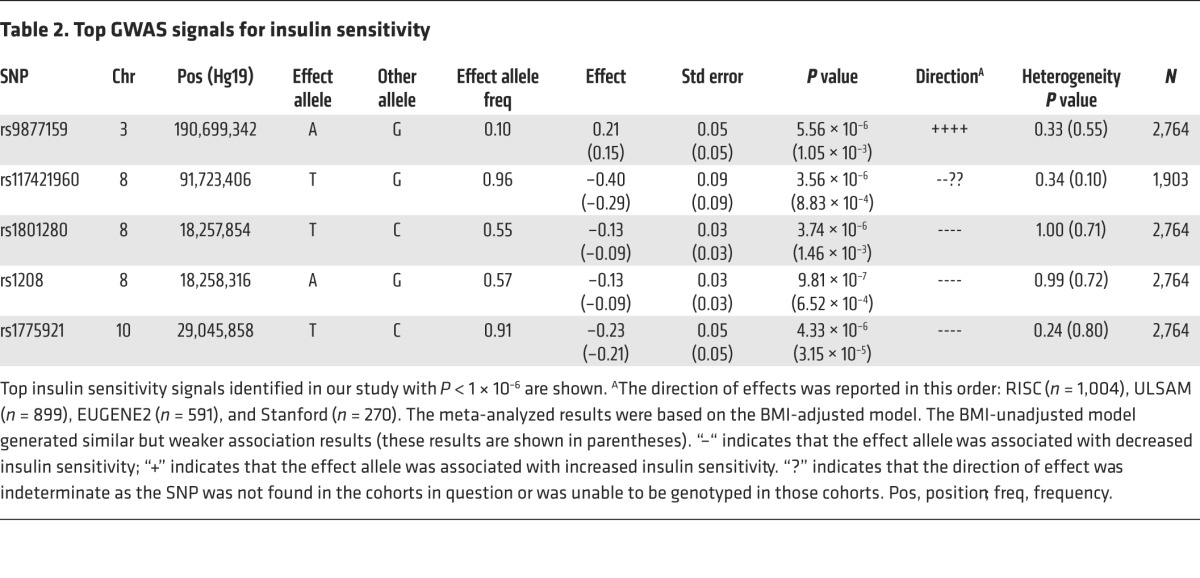
Figure 2. Regional association plots for NAT2.

Regional association plots for NAT2 (A) in age- and gender-adjusted analyses and (B) age-, gender-, and BMI-adjusted analyses. The –log10 of P values of the imputed SNPs are plotted on the y axis against genomic position (Hg19) on the x axis. Purple diamonds represent the top signal. Estimated recombination rates (taken from HapMap) are plotted to reflect the local linkage disequilibrium structure around the associated SNPs and their correlated proxies (according to a blue-to-red scale from r2 = 0 to r2 = 1, based on pairwise r2 values from Hg19/1000 Genomes).
In the GUARDIAN analysis, only SNPs in the NAT2 locus were directionally consistent with the initial GWAS findings for insulin sensitivity (P = 0.09 for rs1801280 and P = 0.10 for rs7832071, which was selected as proxy for rs1208 with r2 = 0.97) (Figure 3 and Supplemental Table 2). We also genotyped the rs1208 and rs1801280 SNPs in adult nondiabetic European subjects from Minnesota (n = 930) and Scandinavia (n = 329). As shown in Figure 3, the effect size and directionality of the association were consistent across cohorts.
Figure 3. Forest plot and association statistics for the lead SNP in NAT2, rs1208 (effect allele “A”, frequency 0.57), in all cohorts in analyses adjusted for age, gender, and BMI.

We performed an inverse variance–weighted fixed-effects meta-analysis of the combined discovery and replication cohorts (with the analyses adjusted for age, gender, and BMI). The vertical black line indicates the null or “0” effect. The dashed vertical line indicates the overall effect size (ES) after the meta-analysis. Rectangles are proportional to the sample size of the referenced cohort. Horizontal lines through the rectangles indicate the standard error. Diamonds represent the overall effect size for each subtotal and for the overall analysis. The width of the diamond spans the standard error. “Subtotal” refers to either the subtotal of the discovery phase GWAS or the subtotal of the replication. *, Scandinavian replication cohort; **, part of GUARDIAN consortium, Hispanic replication cohort; ***, Minnesota replication cohort.
Finally, we tested the association of rs1208 and rs1801280 in 455 East Asian samples that had previously undergone GWAS genotyping, and the direction of effect was consistent for these SNPs. However, the minor allele frequencies of these SNPs in East Asian individuals is much lower than those in European individuals, at only approximately 4%.
Meta-analysis.
Following the in silico and de novo analyses in the GUARDIAN, Scandinavian, and Minnesota studies, we performed an inverse variance–weighted, fixed-effects meta-analysis of the combined discovery and replication cohorts (with the analyses adjusted for age, gender, BMI) (n = 5,624). This meta-analysis showed that the ancestral alleles at both rs1208 (the “A” allele, frequency 0.57) and rs1801280 (“T” allele, frequency 0.55) were associated with a greater degree of IR (P = 2.8 × 10–6 and P = 5.7 × 10–5, respectively). There was evidence of heterogeneity in the data (P for heterogeneity of 0.05) that was no longer evident after the exclusion of the smallest of the cohorts from Minnesota (NaKs cohort, n = 115). With the exclusion of this cohort, the association for rs1208 and rs1801280 became stronger (P = 6.4 × 10–7 and P = 1.1 × 10–5, respectively) (Figure 3, Supplemental Table 2, and Supplemental Figure 2).
For each “A” allele at rs1208, the effect on measures of insulin sensitivity was modest, explaining 0.5%–0.8% of the trait variance for M value derived from euglycemic clamp or steady-state plasma glucose derived from the IST.
Predicted NAT2 acetylator phenotype is not associated with insulin sensitivity.
It is well established that variation in NAT2 has effects on metabolism of certain drugs and toxins (20, 21). NAT2 is highly polymorphic, with over 65 known allelic variants (http://louisville.edu/medicine/departments/pharmacology/news-information/nat). However, humans can be classified into “rapid,” “intermediate,” or “slow” acetylators of isoniazid or sulfamethazine (the canonical substrates) on the basis of the haplotype structure that can be inferred with great accuracy in European populations from a subset of the 7 more common coding SNPs (22, 23). To assess whether the association that we observed with insulin sensitivity was being driven by predicted acetylator status, we used a 6-SNP model to predict acetylator phenotype (http://nat2pred.rit.albany.edu/). There was no significant association of predicted acetylator phenotype with insulin sensitivity (P = 0.23 for BMI-adjusted insulin sensitivity and P = 0.83 for the unadjusted model).
NAT2 SNPs are not associated with insulin clearance or insulin secretion.
Given the role of NAT2 as a drug-metabolizing gene, we wanted to exclude an effect on insulin clearance, which can be calculated by the insulin infusion rate divided by the steady-state plasma insulin level during the euglycemic clamp. In analyses analogous to those performed for insulin sensitivity, we determined that rs1208 and rs1801280 are not associated with insulin clearance (P > 0.05).
We also queried publically available data (24) and ascertained that rs1208 is not associated with indices of glucose-stimulated insulin secretion during oral GTT, such as the disposition index (P = 0.34, n = 10,500).
NAT2 SNPs are associated with other cardiometabolic traits in large-scale GWAS.
We hypothesized that SNPs that affect insulin sensitivity might also affect cardiometabolic risk. To assess whether NAT2 SNPs are also associated with other glycemic traits and lipid levels, we used publicly available GWAS data from MAGIC (Meta-Analyses of Glucose and Insulin-related traits Consortium) (17, 25) and GLGC (Global Lipids Genetics consortium) (26). The rs1208 and rs1801280 SNPs were nominally associated with hemoglobin A1C (HbA1C) and fasting glucose in approximately 133,000 individuals from the MAGIC study and with triglycerides (TGs) and total cholesterol in approximately 100,000 individuals (26). The effects are all in the expected direction (e.g., the effect allele/ancestral allele “A” of rs1208, which is associated with impaired insulin sensitivity, is also associated with unfavorable changes of lipids levels) (Table 3). These SNPs were not associated with HDL cholesterol levels. The NAT2 SNPs have a borderline association (P = 0.06) in the expected direction with type 2 diabetes in the DIAGRAM consortium (27). The rs1208 SNP is also nominally associated with an increase in coronary artery disease risk (P = 0.02) in data from the CARDIoGRAM (Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis study) consortium (28).
Table 3. Top insulin sensitivity signals in our meta-analysis and associations to other glycemic and cardiovascular traits in public GWAS data.
Perturbations in Nat1 affect insulin sensitivity and adipogenesis in vitro.
To further explore the possible role of human NAT2 (or its mouse ortholog, Nat1; ref. 29) in insulin sensitivity, we conducted in vitro studies related to cellular insulin response. Mouse 3T3-L1 adipocytes are a commonly used model in the study of IR. In these cells, stimulation with insulin decreased the mRNA levels of mouse Nat1 by approximately 50%, without an effect on mouse Nat2 (the ortholog to human NAT1) (ref. 21 and Supplemental Figure 3). siRNAs directed against mouse Nat1 resulted in a 65% decrease in Nat1 levels, without affecting mouse Nat2 levels (Supplemental Figure 4).
Nat1 knockdown decreased insulin-stimulated glucose uptake by 34% (P < 0.001) and also inhibited insulin-stimulated glucose uptake by 25% in the presence of the competitive glucose transport inhibitors apigenin and cytochalasin B (P < 0.001) (Figure 4). In contrast to the effects seen with suppression of Nat1 levels, when Nat1 was overexpressed in 3T3-L1 cells, there was an increase in insulin-stimulated glucose uptake (31% vs. control, P < 0.001) (Figure 4 and Supplemental Figure 5). As expected, the total amount of glucose uptake was suppressed by the presence of cytochalasin b or apigenin, but overexpression of Nat1 was still associated with an increase in glucose uptake compared with that in control cells. In addition, overexpression of mouse Nat1 in mouse C2C12 myotubes increased insulin-mediated glucose uptake (Supplemental Figure 6).
Figure 4. Effect of Nat1 knockdown and overexpression on glucose uptake.

(A) Nat1 knockdown decreased basal and insulin-stimulated glucose uptake in 3T3-L1 adipocytes. 3T3-L1 adipocytes transfected with scrambled siRNA (scr siRNA) or with siRNA against Nat1 (si Nat1). Cells were incubated in the presence or absence of insulin and the GLUT4 inhibitor apigenin (data for apigenin are shown, though similar results were obtained with cytochalasin B) in glucose and serum-free culture medium. 2-NDBG glucose uptake was measured using a plate reader. Values indicate mean ± SD of 3 separate experiments, with 3 to 5 wells per experiment. **P ≤ 0.01, ***P ≤ 0.001 relative to basal scrambled controls or the indicated comparison bar. (B) Nat1 increased glucose uptake in 3T3-L1 adipocytes. 3T3-L1 adipocytes transfected with pCMV control and the overexpression plasmid for Nat1 (pNat1). Cells were incubated in the presence or absence of insulin and/or inhibitor in glucose and serum-free culture medium. 2-NBD glucose uptake was measured using a plate reader. Values indicate mean ± SD of 3 separate experiments, with 3 to 5 wells per experiment. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, relative to basal pCMV controls or the indicated comparison bar, ANOVA.
Nat1 silencing increased basal and isoproterenol-stimulated lipolysis, while decreasing insulin-mediated suppression of lipolysis, in 3T3-L1 cells. Basal lipolysis was increased by 2.3-fold by Nat1 siRNA, and isoproterenol-stimulated lipolysis was significantly augmented from the 6.3-fold increase in control cells (receiving scrambled siRNA) to 8.4-fold in the presence of Nat1 siRNA (P < 0.001). In the presence of siRNA to Nat1, insulin was not able to fully suppress isoproterenol-induced lipolysis (Figure 5). Conversely, Nat1 overexpression decreased isoproterenol-stimulated lipolysis by 66% (P < 0.001) and augmented the insulin-mediated suppression of lipolysis by 69% (P < 0.001) (Figure 5) compared with cells transfected with an empty vector.
Figure 5. Effect of Nat1 knockdown and overexpression on lipolysis.

(A) Knockdown of Nat1 increased both basal and stimulated lipolysis. 3T3-L1 adipocytes were transfected with scrambled siRNA or with siRNA against Nat1. Cells were serum starved for overnight and stimulated with 1 μM isoproterenol (ISO) or 100 nM insulin for 1 hour. FFA release in the media was measured and normalized with total protein. Data represent the mean ± SD of 3 separate experiments, with 3 to 5 wells per experiment. ***P ≤ 0.001, relative to basal scrambled control or the indicated comparison bar. (B) Nat1 overexpression decreased basal and isoproterenol-induced lipolysis. 3T3-L1 adipocytes were transfected with pCMV control plasmid or pNat1. Cells were serum starved for overnight and stimulated with 1 μM isoproterenol or 100 nM insulin for 1 hour. FFA release in the media was measured and normalized with total protein. Data represent the mean ± SD of 3 separate experiments, with 3 to 5 wells per experiment. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, relative to basal pCMV controls or the indicated comparison bar, ANOVA.
Finally, Nat1 silencing also decreased 3T3-L1 adipocyte differentiation by 32% (P < 0.001) compared with control, as judged by the accumulation of cytoplasmic lipid droplets and 29% decreased TG content (P < 0.01). This effect was accompanied by a decrease in the levels of adipogenic genes, such as Pparg, Cebpa, and Lep (Figure 6).
Figure 6. Silencing Nat1 decreased adipogenesis.

3T3-L1 preadipocyte cells were transfected with scrambled or Nat1 siRNA and left untreated or treated with a mixture of methylisobutylxanthine, dexamethasone, and insulin (differentiation media). (A) Ten days after the addition of differentiation media, the cells were photographed. Original magnification, ×10. (B) Cell differentiation was quantified using FACS and expressed relative to scrambled control. Results represent mean ± SD from 5 separate experiments, with 3 to 5 wells per experiment. ***P ≤ 0.001, 2-tailed unpaired Student’s t test and ANOVA. (C) Total TGs were measured using a plate reader. Results represent mean ± SD from 3 separate experiments, with 3 to 5 wells per experiment. **P ≤ 0.01, 2-tailed unpaired Student’s t test and ANOVA. (D) 3T3-L1 preadipocytes were treated as above, and the mRNA levels of the indicated genes were determined by real-time quantitative PCR, normalized to cyclophilin, and expressed relative to scrambled control. Results represent mean ± SEM from at least 3 separate experiments, with 3 to 5 wells per experiment. *P = 0.05, **P = 0.01, ***P = 0.001, 2-tailed unpaired Student’s t test and ANOVA.
Knockout of Nat1 through gene targeting leads to decreased insulin sensitivity in vivo.
Through homologous recombination, we generated mice that were wild-type (Nat1WT), heterozygous (Nat1Het), or homozygous (Nat1KO) for a global knockout of Nat1. Fasting plasma glucose, insulin, and TG levels were markedly elevated in Nat1KO mice compared with those in Nat1WT mice (136%, 150%, 154%, respectively), and heterozygote mice also had significant elevations in fasting blood glucose and TGs, with a (nonsignificant) increase in fasting plasma insulin compared with that in Nat1WT mice.
Nat1KO mice had significantly elevated plasma glucose levels during intraperitoneal GTTs and a decreased response to insulin with higher plasma glucose levels during intraperitoneal insulin tolerance test (ITTs) compared with Nat1WT mice (AUC increased by 144% for GTT, P < 0.001, and 124% for ITT, P < 0.01). For both the GTT and ITT, plasma glucose levels were significantly elevated by 30 minutes and remained elevated at 120 minutes. Importantly, in both the GTT and ITT, there was a gene dosage effect, with Nat1Het mice having an intermediate phenotype between Nat1WT and Nat1KO (Figure 7). There was no difference in body weight in the gene-targeted mice compared with that in wild-type mice (Supplemental Figure 7).
Figure 7. Nat1 deficiency impaired insulin sensitivity in vivo.

(A and B) Intraperitoneal GTTs and ITTs were performed on fasted male mice (n = 13–21 per genotype). The area under the curve was measured and plotted. Het, heterozygote. (C) Blood glucose (n = 13–20 per genotype), (D) plasma insulin (n = 13–20 per genotype), and (E) TG (n = 13–16 per genotype) concentrations were determined in mice fasted overnight. Values represent mean ± SEM. *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001, ANOVA.
Discussion
Our study, which coupled genetic data from 5,624 individuals with direct measures of IR with detailed in vitro and in vivo functional characterization provides strong evidence for the role of the NAT2 gene in insulin sensitivity.
This study is, to our knowledge, the first association study arising from a GWAS meta-analysis with these phenotypes. Although our association statistics did not reach formal levels of genome-wide significance, several supporting strands of evidence present a compelling case for NAT2. First, in our GWAS, we observed the strongest association for a nonsynonymous SNP in NAT2 [rs1208 (803A>G, K268R)], with the ancestral “A” allele (frequency 0.57) associated with a greater degree of IR in analyses adjusted for age, gender, and BMI (P = 2.8 × 10–6). Importantly, the same ancestral “A” allele at rs1208 was also nominally associated with various cardiometabolic traits, including higher fasting glucose, HbA1C, TG levels, total and LDL cholesterol, and coronary artery disease in the expected direction. In a lookup of the publically available data from the DIAGRAM consortium, rs1208 has a borderline nominal association with type 2 diabetes (P = 0.06) in the expected direction. Finally, in another recent study, SNP rs7825609 in NAT2 was associated with a combined phenotype of cardiovascular disease and type 2 diabetes (30). As IR is a common risk factor for both cardiovascular disease and type 2 diabetes, the association of this NAT2 SNP with the combined phenotype is in line with a role of NAT2 in IR.
Second, the results of our in vitro functional experiments support direct effects of NAT2 perturbations on glucose uptake, lipolysis, and adipocyte differentiation. In mouse 3T3-L1 adipocytes, silencing of Nat1 (the mouse ortholog to NAT2) decreased insulin-mediated glucose uptake. Furthermore, Nat1 silencing also decreased 3T3-L1 adipocyte differentiation and increased basal and isoproterenol-stimulated lipolysis, while decreasing insulin-mediated suppression of lipolysis. Importantly, opposite effects were seen with Nat1 overexpression both in mouse 3T3-L1 adipocytes as well as mouse C2C12 myoblasts.
Finally, our in vivo studies in mice deficient or heterozygous for mouse Nat1 (the mouse ortholog of human NAT2) (21, 31) affirm that decreased Nat1 levels increased fasting glucose, insulin, and TG levels. Even more compelling are the data from the GTTs and ITTs, showing that loss of Nat1 results in an IR phenotype. Mice lacking either/both Nat1 and Nat2 have not previously been described to have an overt phenotype (32, 33) other than a reduction in arylamine metabolism (34). However, prior to this report, little was known about the effects on metabolic parameters or glucose metabolism (D.W. Hein, personal communication).
NAT2 and the related NAT1 comprise a unique family of cytosolic enzymes that catalyze acetyl-CoA–dependent N- and O-acetylation reactions and have distinct substrate specificity and tissue distribution despite a high degree of homology (~80% amino acid identity) (35–37). Human NAT2 has a single 870-bp coding exon encoding the 290–amino acid, 34-kDa enzyme (38), which is highly expressed in liver and intestine, with low levels of expression in other tissues (Genotype Tissue Expression project; http://www.gtexportal.org/home/gene/NAT2). Over 50 years ago, variation in NAT2 causing altered acetylation activity was shown to be responsible for isoniazid-induced toxicity (39). NAT2 plays a role in the biotransformation and detoxification of many commonly used hydrazine and arylamine drugs as well as xenobiotics and carcinogens, resulting in the well-described relationship between the “slow” acetylation status of NAT2 and increased risk of industrial bladder cancer caused by exposure to arylamine dyes and cigarette smoke (38, 40, 41). Common SNPs in NAT2 are also associated strongly with other endophenotypes, such as various metabolites in blood and urine (42, 43). More recently, another variant in NAT2 (rs1495741), which has modest linkage disequilibrium with rs1208 (r2 = 0.23) but tags the NAT2 acetylator phenotype, has been associated with skin fluorescence, a noninvasive marker of advanced glycation endproducts (44). Interestingly, the rapid acetylator genotype was associated with lower skin fluorescence but higher fasting glucose and HbA1C values. While the crystal structure of NAT2 has been resolved in complex with CoA, neither K268R (rs1208) nor I114T (rs1801280) are known to play a direct role in these interactions, but several of the coding SNPs, including rs1801280, are thought to increase protein degradation (31, 45, 46), thereby affecting the acetylator phenotype.
The search for an explanation for the presence of multiple common coding SNPs in NAT2 has been the source of intense investigation, and the prevalence of NAT2 haplotypes has been determined for a large number of human populations (38, 45); the “rapid” acetylator NAT2*4 haplotype is considered ancestral and predominates in Southern Africa and East Asia, but higher frequencies of the most common “slow” acetylator haplotypes NAT2*5 (defined by the presence of rs1801280) and NAT2*6 (defined by the presence of rs1799930) predominate in Europe, Northern Africa, and the Middle East (47). Importantly, our analysis based on predicted acetylator status did not support a stronger association with insulin sensitivity for “slow” versus “rapid” acetylators, which suggests that the mechanism by which NAT2 may affect insulin sensitivity might be independent of the ability to acetylate the canonical drug substrates. In this regard, it is interesting that, although NATs were among the first examples of pharmacogenetic genes to be described, the endogenous substrate for NAT2 is unknown (21, 48).
The strengths of our study include the rigorous, quantitative measurement of insulin-mediated glucose uptake in the various cohorts, which in fact represent the majority of subjects in the world with both “clamp” data and DNA available for genotyping. We have also performed detailed in vitro and in vivo characterization to buttress support for NAT2 as an IR gene. The weaknesses of the study are the relative paucity of samples with this quantitative phenotype and the inability to easily collect more subjects with these measurements. It is plausible that a larger sample size would uncover additional variants, as has been seen in most other quantitative phenotypes, such as height, BMI, and lipid levels (26, 49, 50).
Even with this report, there remain only a handful of variants associated with IR, which may be at least partly explained by the complex pathophysiology and difficulty of measuring IR. The manifestation of IR entails intricate compensations across multiple parallel pathways, which might mask the effects of genetic variants, whereas β cell dysfunction mainly involves one organ and manifests as one easily measurable outcome (i.e., plasma concentration of insulin). We have analyzed the largest sample size so far, with insulin sensitivity measured by gold standard methods. These quantitative measures of insulin sensitivity are invasive, labor-intensive, and time-consuming procedures. However, these measures are considered the best reflection of peripheral insulin action. Our study provides an example of how new information from more detailed measures of insulin sensitivity can contribute to the understanding of IR pathophysiology. Further investigation of NAT2 is warranted, given these unexpected findings that potentially indicate a new role for this very well-known pharmacogenetic locus.
Methods
GWAS populations and phenotyping
The four studies in the GWAS included the RISC consortium (n = 1,004), ULSAM (Uppsala Longitudinal Study of Adult Men; n = 899), the EUGENE2 consortium (n = 591), and the Stanford IST cohort (n = 270). Descriptive characteristics of the participants are shown in Table 1. In brief, we excluded individuals with established heart disease or diabetes (or those on diabetic medications), pregnancy, fasting plasma glucose ≥7.0 mmol/l; 2-h plasma glucose (on a 75-g oral GTT) ≥11.0 mmol/l. Additional details are available in the Supplemental Methods.
Replication cohorts
The GUARDIAN cohort is comprised of nondiabetic Mexican-American subjects from 7 independent cohorts, but euglycemic clamps (using an insulin infusion rate of 60 mU/m2/mi for 2 hours) were performed in the following three studies: the Mexican-American Coronary Artery Disease study, the Hypertension-IR study, and the NIDDM-Atherosclerosis study. A total of 1,601 subjects from these cohorts passed quality control and were included in the analyses.
Following the lookup in the GUARDIAN samples, the strongest evidence for association was observed for nonsynonymous coding SNPs in NAT2 (rs1208 and rs1801280). Therefore, we performed de novo genotyping in additional European, nondiabetics from several studies in Minnesota (IR Study, PHPBC, NaKs) and Scandinavia (BOTNIA, MALMO-SIB).
A limited number of Chinese or Japanese subjects (461 siblings from 202 families) that had undergone an IST as part of the SAPPHIRe Cohort study were also used to lookup top association SNPs in NAT2.
Additional descriptions of the replication cohorts are shown in the Supplemental Methods.
GWAS genotyping, quality control, and imputation of individual studies
The RISC and Stanford samples were genotyped using the Affymetrix 6.0 microarray platform. The ULSAM samples were genotyped using the Illumina Omni2.5M platform. The EUGENE2 samples were genotyped using the Illumina 550K platform. For all studies, we used standard quality control criteria, including excluding samples that were duplicated or those with gender discordance or non-European ancestry. We excluded SNPs with genotyping call rates <95%, Hardy-Weinberg equilibrium P values of less than 0.0001 and MAF <1%. Haplotypes were phased using MACH (51) (RISC, EUGENE2, and Stanford). Missing genotypes were imputed on the 1000 Genomes Project data (interim 20101123 phase 1) against all-population reference panel using either MiniMac (52) (RISC, EUGENE2, Stanford) or IMPUTE (ULSAM). Additional details are available in the Supplemental Methods.
In silico lookup and de novo replication genotyping
Based on the initial GWAS, we carried forward 5 SNPs from 4 loci that had been implicated through the initial “discovery” analysis (of the 4 GWAS cohorts) for examination within the GUARDIAN cohort. These loci were chosen because they had P values of approximately 10–6 in the discovery analysis and also had multiple supporting SNPs within the locus (Table 2). Two SNPs (rs1208 and rs1801280, r2 ~ 0.75) from the NAT2 locus were included because they were both nonsynonymous coding SNPs. Additional details on these SNPs are in the Supplemental Methods. Following the in silico lookup in GUARDIAN, rs1208 and rs1801280 were genotyped in the Scandinavian and Minnesota samples using TaqMan.
Finally, we examined whether the NAT2 SNPs were associated with the metabolic clearance rate of insulin, which could be calculated as the insulin infusion rate divided by the steady-state plasma insulin level during the euglycemic clamp or IST. Insulin clearance data were available from GWAS cohorts, excluding the Stanford cohort.
NAT2 predicted acetylator phenotype analysis
NAT2 haplotypes have been shown to be predictive of NAT2 acetylator status (22). To determine whether the insulin sensitivity association would be stronger when comparing “rapid,” “intermediate,” and “slow” predicted acetylator phenotypes with one another based on haplotypes, we performed an analysis using a 6-SNP model that has been shown to accurately predict the rapid, intermediate, and slow acetylator phenotype (as defined by the ability of NAT2 to acetylate the canonical substrate isoniazid). The acetylator phenotype of each individual was predicted using NAT2PRED (http://nat2pred.rit.albany.edu/) (23).
The associations between insulin sensitivity SNPs and glycemic traits, lipids levels, and cardiovascular risk
To assess the potential overlap among the SNPs associated with insulin sensitivity, glycemic traits, and lipid levels, we downloaded the publicly available GWAS data for fasting insulin, fasting glucose, HbA1C, homeostasis model assessment-IR, homeostasis model assessment–β cell function, BMI, TGs, total cholesterol, HDL cholesterol, and LDL cholesterol from MAGIC (17, 25) and GLGC (26). We pulled out independent SNPs (pruned based on a 500-kb window) with P < 1 × 10–3 (n = 185 SNPs) from our meta-analyzed GWAS data (insulin sensitivity adjusted for age, gender, and BMI) and cross-referenced the top signals of insulin sensitivity with other glycemic traits and lipids in large-scale publicly available GWAS data from the MAGIC and GLGC consortia. We investigated whether the SNPs with low P values in our study showed associations with these traits more than expected by chance (Supplemental Figure 8).
Furthermore, we investigated associations for the fasting insulin– and fasting glucose–associated SNPs reported by Scott et al. (17) and the 65 type 2 diabetes SNPs reported by Morris et al. (53) with insulin sensitivity in our study. We ran queries to examine whether these known glycemic-related and diabetes-related SNPs showed an enrichment of low P values for insulin sensitivity associations and whether the direction of effects was consistent with what we would expect (Supplemental Tables 3 and 4 and Supplemental Figure 9).
For the NAT2 SNPs, we also accessed information on coronary artery disease from the CARDIoGRAMplusC4D consortium (28).
Cell culture
The murine preadipocyte line 3T3-L1 was obtained from ATCC and cultured according to the manufacturer’s instructions. Differentiation was as described previously (54) (see the Supplemental Methods for additional details). Mouse C2C12 myoblasts obtained from ATCC were cultured in DMEM supplemented with 10% FBS. To induce differentiation, media were replaced with DMEM containing 2% horse serum. Experiments were performed in differentiated C2C12 myotubes.
RNA extraction, reverse transcription, and real-time PCR
Total RNA from cells and mouse tissues were isolated, and quantitative PCR was performed as described previously (55) using primers shown in Supplemental Table 5. The data were analyzed by the public domain program relative expression software tool (REST).
Transient transfection of 3T3-L1 cells
For knockdown experiments, differentiated 3T3-L1 adipocytes were transfected with 50 nM synthetic predesigned siRNA targeting Nat1 (mouse homolog to NAT2) or nonsilencing siRNA (Origene) using Lipofectamine 2000 transfection reagent (Life Technologies) following the manufacturer’s recommended protocol. For overexpression studies, cells were transiently transfected with expression plasmids for Nat1 and pCMV (control) (500 ng per well). After 48 hours of transfection, the media were changed and cells were exposed to insulin (100 nM) or vehicle. After 6 hours, cell extracts were prepared for RNA and protein.
Lipolysis and glucose uptake assays in 3T3-L1 cells
Lipolysis and glucose uptake assays were performed as previously described in fully differentiated 3T3-L1 adipocytes (54, 56). See the Supplemental Methods for additional details.
Adipogenesis of 3T3-L1 preadipocytes
To determine the impact of Nat1 expression (overexpression or knockdown) on the normal differentiation of 3T3-L1 preadipocytes into mature adipocytes, transient transfection in 3T3-L1 preadipocytes was used on day 0 and day 4 of a standard differentiation protocol (see the Supplemental Methods for additional details). For knockdown experiments, 3T3-L1 adipocytes were transfected with 20 nM synthetic predesigned siRNA targeting Nat1 or nonsilencing siRNA. On day 10, pictures of the cells were taken and cell lysates were collected for RNA isolation or protein quantitative analysis. At days 11 and 12, when the control adipocytes reached maturity, adipocyte differentiation was quantitated by “side scatter” (scatter of laser light caused by fat droplets in adipocytes) assessed by flow cytometry. Total TGs were measured using plate reader after dissolving the lipids in isopropanol.
Nat1-targeted mice
Nat1-targeted mice (Nat1KO mice) were produced by homologous recombination at the Knockout Mouse Project Repository and the Mouse Biology Program (http://www.mousebiology.org) at the University of California, Davis, using a C57BL/6N ES cell clone (11571A-A3). Though we attempted to breed 5 chimeric mice to achieve germline transmission, all mice in these studies arose from the successful mating of a single 70% chimeric male mouse with C57BL/6 female mice, transmitting the Nat1-targeted allele to a single female heterozygote mouse.
Mice used in experiments were generated through breeding with C57BL/6 mice or heterozygote crosses, and age-matched wild-type littermate mice were used as controls. Mice were housed in a pathogen-free barrier facility with a 12-hour light/dark cycle and fed standard rodent chow diet and water ad libitum. Body weight was monitored weekly for 12 weeks. Male mice, between 10 and 14 weeks old, were used in all the experiments. For genotyping, PCR analysis was used, using the following primers: Nat1-wt F: CCTGCCATCTTCCTTTTGACAGAGG; Nat1-wt R: TGTGCTTACAAACACAGATGCTGGC; Neo F: GCAGCCTCTGTTCCACATACACTTCA; Nat1-R: GGCTTGAGTTCTGTTTTGAGGACTGG.
Blood glucose, plasma insulin, and plasma lipid composition assessment
Overnight fasting blood glucose levels from the tail vein were measured with a glucometer (TRUEbalance, Nipro Diagnostics Inc.). Blood was collected by retro-orbital bleeding from anesthetized mice, and plasma TGs levels were determined in mice fasted overnight using enzymatic kits (L-Type TG M, Wako Diagnostics) (57). Plasma insulin levels were determined using the Ultra-Sensitive Mouse Insulin ELISA Kit (Crystal Chem Inc.).
GTTs and ITTs
GTTs and ITTs were performed on 10- to 12-week-old mice, as previously reported (58). For GTTs, mice were injected i.p. after overnight fasting with 2 g glucose/kg body weight. For ITTs, mice were injected i.p after 6-hour fasting with insulin at 1.0 U/kg body weight. See the Supplemental Methods for additional details.
Statistics
GWAS analyses and study-specific statistical analyses.
We performed GWAS of insulin sensitivity in a total sample size of 2,764 individuals of European descent. In the primary analyses, insulin sensitivity measures were fitted in a linear regression model with age, gender, center (for RISC and EUGENE2, which were conducted at multiple centers), BMI, and the first two principal components for race/ethnicity (derived from EIGENSTRAT; ref. 59) included as predictors. We also ran the analysis without adjustment for BMI in an attempt to identify loci associated with insulin sensitivity via adiposity. The standardized residuals from the linear regression analysis were normalized by inverse-normal transformation prior to GWAS to create a normally distributed trait for GWAS analysis. To account for imputation uncertainty, we used the 1000 Genomes imputation allele dosage in linear models. We carried out GWAS separately within each cohort using an additive genetic model. Additional details are in the Supplemental Methods.
Meta-analysis.
Following the in silico lookup and de novo genotyping efforts in the GUARDIAN, Scandinavian, and Minnesota samples, we performed an inverse variance–weighted fixed-effects meta-analysis of the combined discovery and replication cohorts (with the analyses adjusted for age, gender, BMI) for the NAT2 SNPs rs1208 and rs1801280. We used the METAL program (60) to meta-analyse individual studies by combining the study-specific regression coefficient estimates. In total, 12,106,458 SNPs passed quality control (minor allele count [MAC] >5 in individual cohorts; imputation quality >0.3 in MACH or >0.4 in IMPUTE) and were included in the meta-analysis. Meta-analyses were performed after reversing the signs of the effect sizes in the Stanford cohort (as the correlation of IST with M value is inverse).
During the meta-analysis, significant heterogeneity was demonstrated for the smallest cohort that was part of the genotyping effort in Minnesota (n = 115, P for heterogeneity = 0.05); hence, additional analyses were performed excluding this cohort.
Statistical analysis of acetylator phenotype.
Association analysis was performed in STATA to see whether the predicted acetylator phenotype was associated with insulin sensitivity, with acetylator phenotypes coded as follows: 0 = S (slow), 1 = I (intermediate), and 2 = R (rapid).
Statistical analyses of in vitro data.
Statistical analyses of the experiments were performed with GraphPad Prism 5.0. Two-tailed unpaired Student’s t test and ANOVA were applied to determine statistical significance.
Study approval
All human genetic studies were conducted according to the principles of the Declaration of Helsinki. The IRBs at Stanford University, Uppsala University (ULSAM), Lund University (Scandinavian cohorts), and University of Minnesota (Minnesota studies) as well as all of the institutions represented in the RISC, EUGENE2, GUARDIAN, and SAPPHIRe studies approved these studies. The institutions represented are referenced in the supplemental material. Written informed consent was received from participants prior to inclusion in the study. All animal protocols were approved by the Administrative Panel on Laboratory Animal Care at Stanford University and were performed in accordance with the guidelines of the American Association for the Accreditation of Laboratory Animal Care.
Supplementary Material
Acknowledgments
See the supplemental material for the RISC consortium, the EUGENE2 study, the GUARDIAN consortium, the SAPPHIRe study, ULSAM, Stanford, Minnesota, and Scandinavian consortia/study details. The GENESIS consortium would not have been possible without the collaborative efforts of many collaborating cohorts/consortia. The initial genotyping of the Stanford, SAPPHIRe, and RISC samples was funded by Merck & Co Inc. The work with the Stanford cohort was funded in part by grants from the Stanford Cardiovascular Institute and the Fondation Leducq. Joshua W. Knowles was supported by an American Heart Association Fellow-to-Faculty Transition Award (10FTF3360005). Joshua W. Knowles and Thomas Quertermous were authorized to act on behalf of the Stanford group for this manuscript. The RISC study was supported by European Union grant QLG1-CT-2001-01252 and AstraZeneca. Timothy M. Frayling is supported by European Research Council grant (SZ-50371-GLUCOSEGENES-FP7-IDEAS-ERC: 323195). Mark Walker was authorized to act on behalf of the RISC group for this manuscript. EUGENE2 was supported by European Community FP6 EUGENE2 no. LSHM-CT-2004-512013. Markku Laakso was authorized to act on behalf of the EUGENE2 group for this manuscript. For ULSAM, Erik Ingelsson was supported by grants from the Swedish Research Council, the Swedish Heart-Lung Foundation, and the Swedish Foundation for Strategic Research, while working on this project. Andrew P. Morris is a Wellcome Trust Senior Fellow in Basic Biomedical Science (grant no. WT098017 and WT064890). Erik Ingelsson was authorized to act on behalf of the ULSAM group for this manuscript. GUARDIAN was supported by the National Institutes of Health (GUARDIAN study DK085175). Mark O. Goodarzi and Jerome I. Rotter were authorized to act on behalf of the GUARDIAN group for this manuscript. The work by SAPPHIRe was supported in part by grants from the National Health Research Institutes, Taiwan (PH-099-PP-03, PH-100-PP-03, PH-101-PP-03). Chao A. Hsiung was authorized to act on behalf of the SAPPHIRe group for this manuscript. The Minnesota studies were supported by grants HL34659, HL52851, DK072124, and M01RR00400 from the National Institutes of Health. Alan Sinaiko and James Pankow were authorized to act on behalf of the Minnesota group for this manuscript. Leif Groop was authorized to act on behalf of the Scandinavian cohorts for this manuscript.
Footnotes
Conflict of interest: Danielle M. Greenawalt is currently an employee of and has income from AstraZeneca. Cliona Molony is an employee of and has income from Merck. Pek Lum was an employee of Merck at the time that some of the work herein was performed and is now an employee of Capella Biosciences.
Reference information:J Clin Invest. 2015;125(4):1739–1751. doi:10.1172/JCI74692.
References
- 1.Facchini FS, Hua N, Abbasi F, Reaven GM. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86(8):3574–3578. doi: 10.1210/jcem.86.8.7763. [DOI] [PubMed] [Google Scholar]
- 2.Yip J, Facchini FS, Reaven GM. Resistance to insulin-mediated glucose disposal as a predictor of cardiovascular disease. J Clin Endocrinol Metab. 1998;83(8):2773–2776. doi: 10.1210/jcem.83.8.5005. [DOI] [PubMed] [Google Scholar]
- 3.Einhorn D, et al. American College of Endocrinology position statement on the insulin resistance syndrome. Endocr Pract. 2003;9(3):237–252. [PubMed] [Google Scholar]
- 4.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 5.Yeni-Komshian H, Carantoni M, Abbasi F, Reaven GM. Relationship between several surrogate estimates of insulin resistance and quantification of insulin-mediated glucose disposal in 490 healthy nondiabetic volunteers. Diabetes Care. 2000;23(2):171–175. doi: 10.2337/diacare.23.2.171. [DOI] [PubMed] [Google Scholar]
- 6.Ferrannini E, Mari A. How to measure insulin sensitivity. J Hypertens. 1998;16(7):895–906. doi: 10.1097/00004872-199816070-00001. [DOI] [PubMed] [Google Scholar]
- 7.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237(3):E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 8.Pei D, Jones CN, Bhargava R, Chen YD, Reaven GM. Evaluation of octreotide to assess insulin-mediated glucose disposal by the insulin suppression test. Diabetologia. 1994;37(8):843–845. doi: 10.1007/BF00404344. [DOI] [PubMed] [Google Scholar]
- 9.Knowles JW, et al. Measurement of insulin-mediated glucose uptake: direct comparison of the modified insulin suppression test and the euglycemic, hyperinsulinemic clamp. Metabolism. 2013;62(4):548–553. doi: 10.1016/j.metabol.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosy-Westphal A, et al. Common familial influences on clustering of metabolic syndrome traits with central obesity and insulin resistance: the Kiel obesity prevention study. Int J Obes (Lond). 2007;31(5):784–790. doi: 10.1038/sj.ijo.0803481. [DOI] [PubMed] [Google Scholar]
- 11.Mills GW, et al. Heritability estimates for beta cell function and features of the insulin resistance syndrome in UK families with an increased susceptibility to type 2 diabetes. Diabetologia. 2004;47(4):732–738. doi: 10.1007/s00125-004-1338-2. [DOI] [PubMed] [Google Scholar]
- 12.Altshuler D, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26(1):76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 13.Ingelsson E, et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes. 2010;59(5):1266–1275. doi: 10.2337/db09-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voight BF, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42(7):579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dupuis J, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rung J, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet. 2009;41(10):1110–1115. doi: 10.1038/ng.443. [DOI] [PubMed] [Google Scholar]
- 17.Scott RA, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44(9):991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen-Torvik LJ, et al. Heritability and genetic correlations of insulin sensitivity measured by the euglycaemic clamp. Diabet Med. 2007;24(11):1286–1289. doi: 10.1111/j.1464-5491.2007.02271.x. [DOI] [PubMed] [Google Scholar]
- 19.Goodarzi MO, et al. Insulin sensitivity and insulin clearance are heritable and have strong genetic correlation in Mexican Americans. Obesity. 2014;22(4):1157–1164. doi: 10.1002/oby.20639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walraven JM, Zang Y, Trent JO, Hein DW. Structure/function evaluations of single nucleotide polymorphisms in human N-acetyltransferase 2. Curr Drug Metab. 2008;9(6):471–486. doi: 10.2174/138920008784892065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sim E, Walters K, Boukouvala S. Arylamine N-acetyltransferases: from structure to function. Drug Metab Rev. 2008;40(3):479–510. doi: 10.1080/03602530802186603. [DOI] [PubMed] [Google Scholar]
- 22.Hein DW, Doll MA. Accuracy of various human NAT2 SNP genotyping panels to infer rapid, intermediate and slow acetylator phenotypes. Pharmacogenomics. 2012;13(1):31–41. doi: 10.2217/pgs.11.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuznetsov IB, McDuffie M, Moslehi R. A web server for inferring the human N-acetyltransferase-2 (NAT2) enzymatic phenotype from NAT2 genotype. Bioinformatics. 2009;25(9):1185–1186. doi: 10.1093/bioinformatics/btp121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prokopenko I, et al. A central role for GRB10 in regulation of islet function in man. PLoS Genet. 2014;10(4):e1004235. doi: 10.1371/journal.pgen.1004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manning AK, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44(6):659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voight BF, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42(7):579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deloukas P, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hein DW, Boukouvala S, Grant DM, Minchin RF, Sim E. Changes in consensus arylamine N-acetyltransferase gene nomenclature. Pharmacogenet Genomics. 2008;18(4):367–368. doi: 10.1097/FPC.0b013e3282f60db0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan KH, et al. Shared molecular pathways and gene networks for cardiovascular disease and type 2 diabetes mellitus in women across diverse ethnicities. Circ Cardiovasc Genet. 2014;7(6):911–919. doi: 10.1161/CIRCGENETICS.114.000676. [DOI] [PubMed] [Google Scholar]
- 31.Sim E, Westwood I, Fullam E. Arylamine N-acetyltransferases. Expert Opin Drug Metab Toxicol. 2007;3(2):169–184. doi: 10.1517/17425255.3.2.169. [DOI] [PubMed] [Google Scholar]
- 32.Cornish VA, et al. Generation and analysis of mice with a targeted disruption of the arylamine N-acetyltransferase type 2 gene. Pharmacogenomics J. 2003;3(3):169–177. doi: 10.1038/sj.tpj.6500170. [DOI] [PubMed] [Google Scholar]
- 33.Sugamori KS, et al. Generation and functional characterization of arylamine N-acetyltransferase Nat1/Nat2 double-knockout mice. Mol Pharmacol. 2003;64(1):170–179. doi: 10.1124/mol.64.1.170. [DOI] [PubMed] [Google Scholar]
- 34.Sugamori KS, et al. Reduced 4-aminobiphenyl-induced liver tumorigenicity but not DNA damage in arylamine N-acetyltransferase null mice. Cancer Lett. 2012;318(2):206–213. doi: 10.1016/j.canlet.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blum M, Grant DM, McBride W, Heim M, Meyer UA. Human arylamine N-acetyltransferase genes: isolation, chromosomal localization, and functional expression. DNA Cell Biol. 1990;9(3):193–203. doi: 10.1089/dna.1990.9.193. [DOI] [PubMed] [Google Scholar]
- 36.Grant DM, Blum M, Beer M, Meyer UA. Monomorphic and polymorphic human arylamine N-acetyltransferases: a comparison of liver isozymes and expressed products of two cloned genes. Mol Pharmacol. 1991;39(2):184–191. [PubMed] [Google Scholar]
- 37.Kawamura A, et al. Eukaryotic arylamine N-acetyltransferase. Investigation of substrate specificity by high-throughput screening. Biochem Pharmacol. 2005;69(2):347–359. doi: 10.1016/j.bcp.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 38.Hein DW, et al. Molecular genetics and epidemiology of the NAT1 and NAT2 acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev. 2000;9(1):29–42. [PubMed] [Google Scholar]
- 39.Evans DA, Manley KA, McKusick VA. Genetic control of isoniazid metabolism in man. Br Med J. 1960;2(5197):485–491. doi: 10.1136/bmj.2.5197.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cartwright RA, et al. Role of N-acetyltransferase phenotypes in bladder carcinogenesis: a pharmacogenetic epidemiological approach to bladder cancer. Lancet. 1982;2(8303):842–845. doi: 10.1016/s0140-6736(82)90810-8. [DOI] [PubMed] [Google Scholar]
- 41.Rothman N, et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat Genet. 2010;42(11):978–984. doi: 10.1038/ng.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suhre K, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477(7362):54–60. doi: 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suhre K, et al. A genome-wide association study of metabolic traits in human urine. Nat Genet. 2011;43(6):565–569. doi: 10.1038/ng.837. [DOI] [PubMed] [Google Scholar]
- 44.Eny KM, et al. GWAS identifies an NAT2 acetylator status tag single nucleotide polymorphism to be a major locus for skin fluorescence. Diabetologia. 2014;57(8):1623–1634. doi: 10.1007/s00125-014-3286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grant DM, et al. Human acetyltransferase polymorphisms. Mutat Res. 1997;376(1):61–70. doi: 10.1016/s0027-5107(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 46.Zang Y, Doll MA, Zhao S, States JC, Hein DW. Functional characterization of single-nucleotide polymorphisms and haplotypes of human N-acetyltransferase 2. Carcinogenesis. 2007;28(8):1665–1671. doi: 10.1093/carcin/bgm085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luca F, et al. Multiple advantageous amino acid variants in the NAT2 gene in human populations. PLoS One. 2008;3(9):e3136. doi: 10.1371/journal.pone.0003136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalow W. Ethnic differences in drug metabolism. Clin Pharmacokinet. 1982;7(5):373–400. doi: 10.2165/00003088-198207050-00001. [DOI] [PubMed] [Google Scholar]
- 49.Lango Allen H, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467(7317):832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yue P, et al. Apelin decreases lipolysis via G(q), G(i), and AMPK-dependent mechanisms. Endocrinology. 2011;152(1):59–68. doi: 10.1210/en.2010-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chennamsetty I, et al. Farnesoid X receptor represses hepatic human APOA gene expression. J Clin Invest. 2011;121(9):3724–3734. doi: 10.1172/JCI45277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blodgett AB, Kothinti RK, Kamyshko I, Petering DH, Kumar S, Tabatabai NM. A fluorescence method for measurement of glucose transport in kidney cells. Diabetes Technol Ther. 2011;13(7):743–751. doi: 10.1089/dia.2011.0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chennamsetty I, et al. Nicotinic acid inhibits hepatic APOA gene expression: studies in humans and in transgenic mice. J Lipid Res. 2012;53(11):2405–2412. doi: 10.1194/jlr.M029769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yue P, et al. Apelin is necessary for the maintenance of insulin sensitivity. American journal of physiology. Am J Physiol Endocrinol Metab. 2010;298(1):E59–E67. doi: 10.1152/ajpendo.00385.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 60.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greenfield MS, Doberne L, Kraemer F, Tobey T, Reaven G. Assessment of insulin resistance with the insulin suppression test and the euglycemic clamp. Diabetes. 1981;30(5):387–392. doi: 10.2337/diab.30.5.387. [DOI] [PubMed] [Google Scholar]
- 62.Soranzo N, et al. Common variants at 10 genomic loci influence hemoglobin A(1) (C) levels via glycemic and nonglycemic pathways. Diabetes. 2010;59(12):3229–3239. doi: 10.2337/db10-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.