

Abstract
1. Establishing the genetic and molecular basis underlying adaptive traits is one of the major goals of evolutionary geneticists in order to understand the connection between genotype and phenotype and elucidate the mechanisms of evolutionary change. Despite considerable effort to address this question, there remain relatively few systems in which the genes shaping adaptations have been identified.
2. Here, we review the experimental tools that have been applied to document the molecular basis underlying evolution in several natural systems, in order to highlight their benefits, limitations and suitability. In most cases, a combination of DNA, RNA and functional methodologies with field experiments will be needed to uncover the genes and mechanisms shaping adaptation in nature.
Keywords: adaptation, candidate genes, DNA mapping, expression, functional analysis, QTL, resequencing
Understanding how diversity arises and is maintained in nature has been a major question in evolutionary biology since ancient times (Lewontin 1974; Mayr 1982). Until recently, addressing this question was methodologically limited, but the recent and increasing development of molecular and genomic tools has now equipped us better to face the challenge. Consequently, much of modern research in evolution is devoted to identifying the genes shaping adaptive phenotypes (Hoekstra & Coyne 2007; Stern & Orgogozo 2009; Nadeau & Jiggins 2010; Jones et al. 2012; Kunte et al. 2014). This has and will continue to contribute to answering important questions about how evolution proceeds: Do adaptations arise gradually through many small mutations, via large leaps of major effect or both? Is the evolution of similar traits in different lineages the product of mutations in the same genes? Are particular kinds of adaptive mutations more likely than others, such as gene regulatory or protein-coding mutations? Here, we will focus on the methods used to address these questions.
The narrowing of the genomic regions controlling adaptations has employed mainly three different approaches termed forward genetics, reverse genetics and candidate gene (Stinchcombe & Hoekstra 2007; Nadeau & Jiggins 2010). Forward genetics approaches seek to identify genes underlying a known adaptive trait (Stinchcombe & Hoekstra 2007; Nadeau & Jiggins 2010; Stapley et al. 2010), while reverse genetics approaches refer to the detection of selection signatures across the genome without necessarily have a prior knowledge of the associated phenotype (Stinchcombe & Hoekstra 2007; Bonin 2008; Ellegren & Sheldon 2008; Stapley et al. 2010). Both forward and reverse genetics approaches are currently benefiting from the recent advances in sequencing technology, although its application still faces several challenges related to data storage, data analysis and cost that, although decreasing, can be challenging especially for organisms with large genomes (Wang, Gerstein & Snyder 2009).
Alternatively, the candidate gene approach relies on existing knowledge about the genes participating in the formation of the adaptive phenotype under investigation in other organisms; a correlation between trait variation and allelic polymorphism suggests the use of the candidate gene in shaping the adaptive trait (Luikart et al. 2003; Haag et al. 2005; Hoekstra et al. 2006; Mundy 2009). Any of the above approaches, alone or often combined, can narrow a strong set of candidate genes underlying adaptations. A further validation of these candidates can benefit from the exploration of the gene expression patterns and the application of functional assays such as knockouts, knockdowns and/or transgenics. However, a relation between a candidate gene and a presumptive adaptive phenotype does not constitute the unequivocal detection of the ‘loci of evolution’. The definite connection between gene function and adaptation also requires the implementation of selection experiments that test the adaptive consequences of the individual alleles of these candidates (Fig.1). Unfortunately, this has been rarely achieved, perhaps due to the difficulty of replicating evolutionary processes under controlled conditions (Colosimo et al. 2005).
Fig 1.
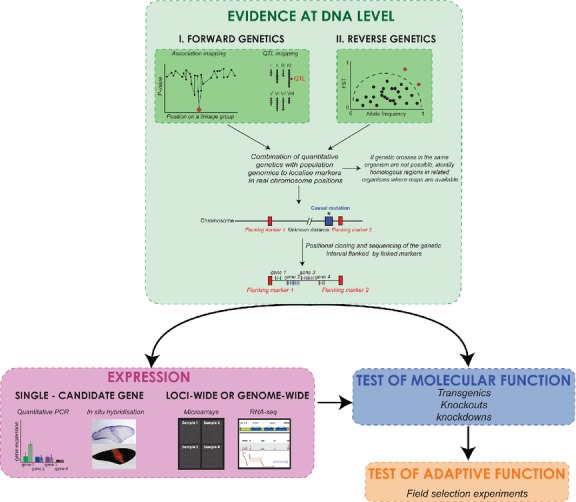
Methodological processes useful to identify the loci underlying adaptation. Ideally, phenotype–genotype association studies, followed by the profiling of gene expression, functional tests and selection tests should be combined to identify a gene(s) as involved in shaping an adaptive trait. Evidence at the DNA level was adapted and modified from (Stinchcombe & Hoekstra 2007; Barrett & Hoekstra 2011). In situ hybridization shows expression of the gene optix in wings of Heliconius melpomene (Photo: Bob Reed) (Reed et al. 2011). Photographs of microarray and RNA-seq by Carolina Pardo-Diaz.
Narrowing down adaptations at the DNA level
Forward genetics approaches
Forward genetics methods work towards finding the genetics controlling a known adaptive trait (i.e. one that is known to increase organismal fitness and/or reproduction in a particular context and where coefficients of selection have been measured and/or the response to changes in the selective agent has been quantified; see section IV: assays of allele effects on fitness with field studies). Forward genetics methods consist mainly of two different approaches: (i) association mapping using sexual populations that recombine in nature and (ii) quantitative trait loci (QTL) mapping using pedigrees, both of which survey a large number of molecular markers across the genome of individuals segregating for the adaptive trait of interest in order to identify gene regions as well as genes that are responsible for its variation (Stinchcombe & Hoekstra 2007; Nadeau & Jiggins 2010; Slate et al. 2010; Stapley et al. 2010; Martin & Jiggins 2013; Savolainen, Lascoux & Merila 2013; Wray 2013; Dittmar et al. 2014; Zuellig, Kenney & Sweigart 2014). Crucially in both cases, the methods involve identifying associations between the phenotypes of individuals with genetic variants. Thus, forward genetics is only useful where there is variation in a population for a particular trait or where such a variable population can be generated in the laboratory via crosses. This approach, however, is not useful to map differences in species that are completely isolated reproductively.
The association mapping approach, termed a ‘genome-wide association study’ (GWAS) when applied across the whole genome, takes advantage of the historical recombination in wild populations in order to detect non-random associations between genomic markers scattered across the genome and the adaptive trait of interest (Shimizu & Purugganan 2005; Stinchcombe & Hoekstra 2007; Hunter, Wright & Bomblies 2013). Using historical recombination increases the resolution in the detection of the locus (or loci) controlling the adaptive trait under study as the segregation would be much larger than that of a progeny of a experimental biparental population (Hwang et al. 2014). Thus, GWAS works by identifying non-random association of alleles between a locus with the adaptive trait (i.e. linkage disequilibrium, LD), as consequence of the action of natural selection (Long & Langley 1999; Shimizu & Purugganan 2005; Stranger, Stahl & Raj 2011). Until recently, GWAS applied genetic markers such as AFLPs, microsatellites and single copy gene markers to sample genetic variation (Table 1) (Holliday, Ritland & Aitken 2010; Holliday, Wang & Aitken 2012), but more recently, high-throughput sequencing approaches are used [such as reduced representation sequencing (Altshuler et al. 2000; Hohenlohe et al. 2010), low-coverage genotyping (Andolfatto et al. 2011; Elshire et al. 2011) and genome resequencing (Nielsen et al. 2011)]. Coupled with advances in bioinformatics, the use of GWAS to investigate the genetics of adaptation is increasingly popular (Hunter, Wright & Bomblies 2013). In lodgepole pines, for example, the use of restriction-site associated DNA sequencing (RAD-seq) generated markers for a GWAS of variation in serotiny (adaptation of cones to remain closed and retain seeds until a forest fire opens them) and revealed that at least 11 loci are involved in the natural variation of this trait (Parchman et al. 2012). Similarly, in Arabidopsis thaliana, a GWAS identified a cis-regulatory polymorphism at the AtHKT1;1 locus as the factor controlling adaptive variation in leaf Na+ accumulation capacity (Baxter et al. 2010a). Additionally, several GWASs have identified loci with signals of human adaptations including genes involved in immunity, cancer, infection, reproduction, healing and height (Hancock et al. 2011b; Jarvis et al. 2012; Lachance et al. 2012; Scheinfeldt & Tishkoff 2013). The power of association-based methods to detect a true association between a SNP and an adaptive trait largely depends on the phenotypic variance of the population explained by the SNP. Such phenotypic variance is determined by how strongly the alternative allelic variants differ in their phenotypic effects (effect size) and their frequency in the sample (Korte & Farlow 2013). Association-based methods are therefore biased towards detecting large-effect loci, although this is probably true of all methods described here (Rockman 2011; Martin & Jiggins 2013). This bias can be reduced by using extremely large sample sizes to maximize the genetic variance within the sample (Bodmer & Bonilla 2008; Korte & Farlow 2013). However, in some cases GWAS in humans using large sample sizes have shown that a single phenotype may be controlled by many minor effect loci that may explain only a small proportion of the trait heritability, which has limited the identification of causal variants (Rockman 2011). This limitation of GWAS is highly relevant to the study of the genetics of adaptive traits with polygenic inheritance, which may be the majority of adaptive traits (Rockman 2011; Turchin et al. 2012).
Table 1.
Examples of natural adaptations investigated at the molecular level, the methodological approaches followed and the genes identified (if they have)
| Organism | Adaptive trait | DNA evidence | Expression Profiling | Functional test | Genes identified/suggested | References |
|---|---|---|---|---|---|---|
| Arabidopsis thaliana | Flowering time | Forward genetics approaches – association mapping and QTL mapping | Microarray | Complementation test | FRI and FLC | Aranzana et al. (2005), Lempe et al. (2005), Bergelson & Roux (2010), Salomé et al. (2011) |
| Arabidopsis thaliana | Pathogen resistance | Forward genetics approaches – association mapping and QTL mapping | RNA blot analysis | Mutagenesis | Rpm1 | Grant et al. (1995), Tornero et al. (2002), de Torres et al. (2003), Aranzana et al. (2005) |
| Gasterosteus aculeatus (threespine stickleback) | Armour plate patterning | Forward genetics approaches – association mapping and QTL mapping | qRT-PCR | Transgenics | EDA | Colosimo et al. (2005), Jones et al. (2012) |
| Gasterosteus aculeatus (threespine stickleback) | Loss of pelvic spines | Forward genetics approaches – QTL mapping | qRT-PCR and in situ hybridization | Transgenics | Pitx1 | Shapiro et al. (2004), Shapiro, Bell & Kingsley (2006), Chan et al. (2010) |
| Peromyscus sp. | Coat colour | Forward genetics approaches – QTL mapping and reverse genetics approaches | qRT-PCR, in situ hybridization and antibodies | Transgenics | Agouti | Manceau et al. (2011), Linnen et al. (2013) |
| Tetrapoda | Land colonization | Reverse genetics approaches – comparative genomics | RNA-seq, in situ hybridization | Transgenics | Several genes | Amemiya et al. (2013) |
| Drosophila sp. | Male wing pigmentation involved in courtship | Reverse genetics approaches – genotyping of candidate genes | Immunochemistry | Transgenics | Yellow | Gompel et al. (2005), Prud'homme et al. (2006) |
| Spatial regulation of pigmentation genes involved in wing spot formation | NA | In situ hybridization, Western blot, immunochemistry, microarray, qRT-PCR | RNAi screen, mutants, transgenics | Dll | Arnoult et al. (2013) | |
| Thlaspi caerulescens | Zinc accumulation | Forward genetics approaches – association mapping and QTL mapping | Microarray and Northern blot | Yeast complementation analysis | MT2 and MT3 | Assunção et al. (2006), Hassinen et al. (2007) |
| Picea sitchensis (sitka spruce conifer) | Wood physical attributes | Forward genetics approaches – association mapping | Microarray | NA | β-expansin, Tubulin 3B, Galactosyl-transferase | Holliday, Ritland & Aitken (2010), Beaulieu et al. (2011) |
| Gasterosteus aculeatus (threespine stickleback) | Gill pigmentation | Forward genetics approaches – QTL mapping | In situ hybridization and allele-specific expression | NA | Kitlg | Miller et al. (2007) |
| Heliconius melpomene and Heliconis erato | Wing colour pattern | Forward genetics approaches – QTL mapping | Microarray, in situ hybridization, qRT-PCR | NA | Optix, WntA | Jiggins et al. (2005), Baxter et al. (2008, 2010b), Counterman et al. (2010), Reed et al. (2011), Hines et al. (2012), Martin et al. (2012) |
| Papilio polytes | Wing mimicry | Forward genetics approaches – association mapping | Immunochemistry, qRT-PCR, RNA-seq | NA | doublesex | Kunte et al. (2014) |
| Rattus norvegicus | Warfarin resistance | Reverse genetics approaches – microsatellite | Semi-quantitative RT-PCR | NA | Vitamin K epoxide reductase (VKOR) | Kohn, Pelz & Wayne (2003), Lasseur et al. (2006) |
| Cichlids | Visual pigment diversification | Reverse genetics approaches – genotyping of candidate genes | qRT-PCR on candidate genes | NA | Opsin genes | Hofmann et al. (2009) |
| Ovis aries (Soay sheep) | Coat colour | Forward genetics approaches – association mapping | qRT-PCR | NA | Tyrp1 | Beraldi et al. (2006), Gratten et al. (2007) |
| Vertebrates | Pigmentation | Reverse genetics approaches – genotyping of candidate genes | qRT-PCR | NA | Mc1r | Hoekstra & Nachman (2003), Nachman, Hoekstra & D’ Agostino (2003), Mundy et al. (2004), Rosenblum, Hoekstra & Nachman (2004), Hoekstra et al. (2006), Hubbard et al. (2010) |
| Chrysomela aeneicollis (montane beetle) | Adaptation to local thermal conditions | Reverse genetics approaches – genome-wide SNP typing | Western blot | NA | Pgi – Hsp70 | Rank (1992), Dahlhoff & Rank (2000) |
| Chaenocephalus aceratus and Pleuragramma antarcticum (Antarctic notothenioid fishes) | Craniofacial skeletal morphology | Reverse genetics approaches – genotyping of candidate genes | In situ hybridization | NA | col1a1, col2a1b and col10a1 | Albertson et al. (2010) |
| Pseudopodoces humilis (ground tit) | High altitude adaptations | Reverse genetics approaches – whole-genome sequencing | NA | NA | Adrenaline response and hormone biosynthesis genes | Cai et al. (2013) |
| Chrysemys picta bellii (western painted turtle) | Extreme anoxia and tissue freezing | Reverse genetics approaches – whole-genome sequencing | RNA-seq | NA | Tumour suppression genes, glucose transport genes and the miR-29b micro RNA | Bradley Shaffer et al. (2013) |
| Tapeworms | Parasitism adaptations | Reverse genetics approaches – whole-genome sequencing | RNA-seq | NA | Apomucin gene family, antigen B gene family, Hsp70 gene family | Tsai et al. (2013) |
| Falco peregrinus (peregrine falcon) and Falco cherrug (saker falcon) | Predation adaptations | Reverse genetics approaches – whole-genome sequencing | RNA-seq | NA | Olfactory receptor genes, beak development genes | Zhan et al. (2013) |
| Pteropus alecto (fruit bat) and Myotis davidii (insectivorous bat) | Flight and immune adaptations | Reverse genetics approaches – whole-genome sequencing | NA | NA | Repair of genetic damage genes, skin elasticity genes, muscle contraction genes, innate immunity genes | Zhang et al. (2013) |
| Ambystoma mexicanum (Mexican axolotl) | Paedomorphosis | Forward genetics approaches – QTL mapping | Microarray | NA | Thyroid hormone-response genes | Voss & Shaffer (1997), Page et al. (2008, 2009, 2010), Huggins et al. (2012) |
| Cichlid fish | Colour pattern | Forward genetics approaches – association mapping and QTL mapping | qRT-PCR | NA | Pax7 | Streelman, Albertson & Kocher (2003), Roberts, Ser & Kocher (2009) |
| Bicyclus anynana | Eyespots | Genotyping of candidate genes | Immunohistochemistry | NA | Distal-less | Beldade, Brakefield & Long (2002) |
| Rana chensinensis and R. kukunoris (ranid frogs) | Adaptation to high elevation | NA | Transcriptomic analysis | NA | 125 protein-coding genes | Yang et al. (2012) |
| Ipomoea sp. | Floral colour | Reverse genetics approaches – genotyping of candidate genes | In situ hybridization and Northern blot | Complementation test and enzyme assay | Genes in flavonoid biosynthesis | Durbin et al. (2003), Zufall & Rausher (2003, 2004) |
| Coregonus spp. (lake whitefish) | Growth, swimming activity, gill rakers and condition factor | Reverse genetics approaches – genome-wide SNP typing Forward genetics approaches – QTL mapping | Microarray | NA | Several genes | Rogers & Bernatchez (2005), Derome et al. (2008), Renaut et al. (2011) |
| Littorina saxatilis | Local adaptation | Reverse genetics approaches – AFLPs | ESTs genomic scan | NA | Two loci | Wood et al. (2008), Galindo, Grahame & Butlin (2010), Westram et al. (2014) |
| Melitaea cinxia (Glanville fritillary butterfly) | Dispersal rate and flight metabolism | Reverse genetics approaches – genotyping of candidate genes | NA | NA | Pgi | Haag et al. (2005) |
| Anser indicus and Chloephaga melanoptera (geese) | High altitude adaptation | Reverse genetics approaches – candidate genes | NA | Protein assay with same mutation in heterologous human protein | Haemoglobins | Jessen et al. (1991) |
| Chicken and Japanese Quail | Plumage colour | Reverse genetics approaches – candidate genes Forward genetics approaches – association mapping | Quantification of mRNA decay in mutant variant | NA | SLC45A2 | Gunnarsson et al. (2007) |
| Lake Victoria cichlids | Light spectrum sensitivity – visual system | Reverse genetics approaches – candidate genes | NA | Protein assay | LWS | Terai et al. (2006) |
| Lakes Tanganyika and Malawi cichlids | Visual adaptation to deep-water habitats | Reverse genetics approaches–- genotyping of candidate genes | NA | Protein assay | RH1 | Sugawara et al. (2005) |
| Astyanax fasciatus (Mexican cave tetra) | Albinism | Forward genetics approaches – QTL mapping | NA | Cell-based functional assay | Oca2 | Protas et al. (2006) |
| Ostrinia nubilalis (European corn borer) | Sexual isolation pheromones | Forward genetics approaches–- association mapping | NA | NA | Pher and Resp | Dopman, Robbins & Seaman (2010) |
| Cervus elaphus (red deer) | Birth weight | Forward genetics approaches – association mapping and QTL mapping | NA | NA | One major QTL | Slate et al. (2002) |
| Peromyscus mice | Behavioural differences – burrow architecture | Forward genetics approaches – association mapping and QTL mapping | NA | NA | Four QTL | Weber, Peterson & Hoekstra (2013) |
| Rana temporaria (common frog) | Adaptation to altitude | Reverse genetics approaches – AFLPs | NA | NA | Eight outlier loci | Bonin et al. (2006) |
| Pinus contorta (lodgepole pine) | Cone serotiny | Reverse genetics approaches – genome-wide SNP typing | NA | NA | Eleven candidate loci | Parchman et al. (2012) |
| Triticum sp. (wheat) | Drought adaptation | Forward genetics approaches – association mapping | NA | NA | Several loci | Maccaferri et al. (2011) |
| Phoxinus phoxinus (cyprinid fish European minnow) | Body shape variation | Reverse genetics approaches – AFLPs | NA | NA | Several loci | Collin & Fumagalli (2011) |
| Mimulus sp. | Floral morphology | Forward genetics approaches – QTL mapping | NA | NA | Several loci | Bradshaw et al. (1998) |
| Floral pigmentation | Reverse genetics approaches – candidate genes | qRT-PCR | Virus-induced gene silencing (VIGS) | R2R3-MYB | Streisfeld & Rausher (2009), Streisfeld, Young & Sobel (2013) | |
| Leptosiphon sp. | Floral characters associated with the mating system | Forward genetics approaches – association mapping and QTL mapping | NA | NA | Several loci | Goodwillie, Ritland & Ritland (2006) |
| Fundulus heteroclitus | Adaptation to local water temperature | NA | In vivo reporter gene expression of candidate gene | Protein activity | Lactate dehydrogenase-B gene (Ldh-B) | Schulte et al. (2000) |
| Drosophila yakuba and Drosophila santomea | Body colouration | NA | Expression constructs with mutant versions (in situ hybridization) | Transgenics | Tan and Yellow | Jeong et al. (2008) |
It is worth-noting that the suitability of a GWAS approach in discovering adaptation genes depends on additional factors that are highly dependent on the study system. For instance, the extent of genomic LD can introduce bias in a GWAS, so it is recommended to use high-density genotyping platforms that generate enough SNPs to examine the background level of LD in each population in order to differentiate LD outliers from genomic background (Alhaddad et al. 2013; Porto-Neto, Kijas & Reverter 2014). Also, obtaining the minimum number of markers necessary for a successful GWAS relies on factors such as genome size, number of individuals and number of groups or populations included, which all influence the genotyping effort required to get enough coverage and overlap between samples (Davey et al. 2011). Technical replicates with sufficient coverage may also be needed in order to assess the reproducibility of the genotyping technique and the quality of SNP calling, both of which can affect the proper calling of heterozygous sites and the reliability of GWAS (Hong et al. 2012; Nielsen et al. 2012).
QTL mapping is an alternative approach to understand the genetic basis of a known adaptive trait. It relies on the generation of mapping crosses to create a genetically variable and recombinant population in the laboratory and uses statistical analyses to correlate the quantitative variance of adaptive traits with molecular markers distributed across the genome and identify chromosomal regions contributing to phenotype differentiation (Ellegren & Sheldon 2008; Mackay, Stone & Ayroles 2009; Nadeau & Jiggins 2010; Stapley et al. 2010; Slate 2013) (Table 1). In sticklebacks, for example, pelvic structures and pigmentation play adaptive roles as defensive structures against predation and crypsis, respectively, and the genetic regions controlling natural adaptive variation at these two characters were both found by QTL mapping with microsatellites (Shapiro et al. 2004; Miller et al. 2007). In a similar way, QTL mapping using AFLPs revealed that only three genomic intervals modulate most of the observed adaptive wing colour variation in the butterfly Heliconius erato (Papa et al. 2013). A growing number of studies have applied next-generation sequencing techniques to QTL mapping. In the perennial ryegrass Lolium perenne, the identification of three QTL that explained nearly 40% of the resistance to stem rust was achieved using RAD-seq (Pfender et al. 2011). Similarly, the identification of a QTL involved in spinosad resistance in the diamondback moth Plutella xylostella applied a RAD-seq approach (Baxter et al. 2011).
Although QTL mapping has been historically useful in narrowing broad regions associated with traits of interest, their power relies on obtaining large families. Unfortunately, obtaining enough recombinant offspring is not always possible due to the nature of many organisms, and thus, the power and applicability of QTL may be limited. Also, this method faces some limitations related to estimating effect size, number of loci [or Quantitative Trait Nucleotides (QTNs)] and their interactions contributing to adaptation. First, the null hypothesis for QTL mapping is the absence of a QTL and not the presence of infinitesimal QTL, so it is not so applicable for the study of adaptive traits shaped by many minimal effect loci (Rockman 2011). In line with this argument, empirical data show that QTL mapping generally finds a skewed L-distribution of effect sizes (with few large-effect loci accounting for most of the variation). It is also widely known that if QTL mapping is performed with small sample sizes, the magnitude of the identified QTL is likely to be overestimated (‘Beavis effect’), the power of detecting medium to small effect QTL is limited, and the signature of several linked QTL may be blurred into a single large QTL (Mackay, Stone & Ayroles 2009; Nadeau & Jiggins 2010; Rockman 2011; Slate 2013). A recent review of QTL mapping studies showed that there is clear evidence for an upward bias in the magnitude of QTL estimates, particularly when sample sizes are small, but possibly even when they are as large as 1000 individuals (Slate 2013). Nonetheless, QTL mapping is often an important initial step towards gene discovery, which can lead to further genomic scans (such as GWAS) to establish whether QTL can be replicated (Slate 2013). This is exemplified by a study of horn morphology in Soay sheep, where a QTL was first finely mapped (Johnston et al. 2010) and then confirmed using an association study. This identified the gene RXFP2 as the gene control variation in horn phenotype (Johnston et al. 2011).
Reverse genetics approaches
Reverse genetics (or genome scan) approaches refer to the genome-wide sampling of loci in order to detect regions with footprints of selection and thus detect selective (adaptive) loci even without a prior knowledge of their associated phenotypic trait(s) (Luikart et al. 2003; Storz 2005; Martin & Jiggins 2013). This approach benefits from the fact that no experimental crosses are needed but instead, variation in natural populations can be used (Schlötterer 2003; Stinchcombe & Hoekstra 2007) and also, it should be less biased towards large-effect loci (as it identifies actual targets of selection) (Martin & Jiggins 2013).
The reverse genetics strategy involves a comprehensive sampling of independent loci across the entire genome and the application of statistical analyses to identify loci with genetic variation indicative of selection that are possibly involved in adaptation (Balding & Nichols 1995; Nicholson et al. 2002; Luikart et al. 2003; Vitalis et al. 2003; Joost et al. 2007; Foll & Gaggiotti 2008; Coop et al. 2010; Nunes et al. 2012; De Mita et al. 2013; Frichot et al. 2013; Martin & Jiggins 2013; de Villemereuil et al. 2014). These statistics can be applied both at intra- and inter-population level.
Within a single population, measures of genetic diversity and distribution of genetic polymorphism (or allele frequency spectrum, AFS) with statistics such as π and Tajima's D are expected to be influenced by selection fixing an advantageous allele. This fixation also leads to a reduction of genetic diversity in the surrounding sequences creating a selective sweep, due to genetic hitchhiking. The location of such selective sweeps can be detected by evaluating patterns of linkage disequilibrium (LD) (Kim & Nielsen 2004). The detection of hard sweeps (i.e. new mutations that arise and quickly go to fixation) is facilitated by the pattern of strong reduced nucleotide polymorphism at the selected locus and its neighbour regions (Pritchard, Pickrell & Coop 2010; Olson-Manning, Wagner & Mitchell-Olds 2012). In contrast, detecting the signal of soft sweeps (when the selected allele has been already segregating in the population before being swept) is more difficult because the selected haplotype may be impossible to differentiate from the genetic background (Pritchard, Pickrell & Coop 2010; Olson-Manning, Wagner & Mitchell-Olds 2012). Furthermore, the signal of a selective sweep may be lost fairly rapidly over time especially in large populations. According to simulation studies, this limitation can be overcome with the combination of LD scans with AFS scans (Pavlidis, Jensen & Stephan 2010). Nonetheless, false positives remain a problem. For example, Pavlidis et al. (2012) used simulated data under neutrality in Drosophila and still detected false positive sweeps with misleading biological functions (Pavlidis et al. 2012). Due to these constraints, genomic scans of selective sweeps are less commonly used as a first step in identifying adaptive loci, yet they are useful to confirm sweep signals at loci involved in adaptations (Hohenlohe et al. 2010; Nadeau et al. 2013).
Demographic factors can also affect nucleotide polymorphism and produce patterns of summary statistics (like π and Tajima's D) easily confused with those expected by the action of selection (Stinchcombe & Hoekstra 2007). However, it is expected that demography affects the genome as a whole. The problem can therefore be ameliorated to some degree by using genome-wide data to evaluate the demographic history of the species and provide a neutral expectation against which particular regions can be tested for the influence of selection (McVicker et al. 2009; Pool et al. 2010; Li & Durbin 2011). Selective sweeps also influence haplotype structure, and inferences based on more complex measures, not based on SNPs alone, are likely to be more powerful (Gusev et al. 2009, 2011; Pavlidis et al. 2013).
Inter-population measures can also be used to detect footprints of selection across the genome. The most common approach is to apply tests based on FST to reveal loci where the fixation of alternative alleles in each population results in greater differences in allele frequency than expected under neutral evolution (Beaumont & Nichols 1996; Bonhomme et al. 2010; Narum & Hess 2011; Martin & Jiggins 2013; de Villemereuil et al. 2014; Lotterhos & Whitlock 2014). Alternatively, there are methods to detect selection based on correlations of genetic data with environmental changes (Joost et al. 2007; Coop et al. 2010; De Mita et al. 2013). Simulation studies under a monogenic scenario have shown that FST-based methods are able to consider complex population history and structure (when known) and are less prone to detect false positives; however, they fail to detect true selection outliers when selection is not strong and their results are strongly dependent on the demographic model implemented (De Mita et al. 2013; Lotterhos & Whitlock 2014). Additionally, it has been recently pointed out that the use of relative measures in detection of outliers (e.g. FST) may be misleading when searching regions in the genome involved in adaptation and speciation (Noor & Bennett 2010; Martin et al. 2013; Cruickshank & Hahn 2014). In some genomic regions, for example, genetic divergence between species of recent origin may lead to a decrease in genetic variability and recombination rate as consequence of the speciation process, which can be misinterpreted as signatures of positive or negative selection acting within species. Similarly, regions of the genome with restricted gene flow compared to the genomic background (measured with FST) are usually interpreted as islands of divergence (Turner, Hahn & Nuzhdin 2005; Ellegren et al. 2012). However, this signal also may be the result of positive or background selection that acted, in the past, on the ancestral population (Noor & Bennett 2010; Cruickshank & Hahn 2014). Thus, the misinterpretation of FST patterns can confuse processes of adaptation with those of speciation. It has been suggested that absolute measures of genetic divergence (that do not rely on allelic frequencies), such as DXY distance, may be better, but these also fail to detect some regions known to be under selection (Cruickshank & Hahn 2014). A related factor to consider is the relationship between recombination and selection. Divergence and low diversity is commonly higher in regions with low recombination (e.g. centromeres), which may result from natural selection or just be a consequence of limited recombination (Turner & Hahn 2010). There may also be a positive relationship between recombination and mutation, which would increase the variation available for selection to act upon (Cutter & Payseur 2013). However, to date, studies are still needed that investigate how recombination rate varies across the genome and in candidate regions for adaptation and speciation.
Environment–genetic correlation methods, on the other hand, are more powerful and work well under different strengths of selection. On the other hand, they may have a high false discovery rate (FDR) if genetic correlations between populations are not accounted for (De Mita et al. 2013). In some cases, correct estimation of the environmental variables can be challenging, potentially limiting application of this latter method. In polygenic scenarios, where multiple loci of small effect underlie a single trait, both environment–genetic correlation methods and FST-based methods show lower power of detection compared to major locus genetic architecture (de Villemereuil et al. 2014). Demographic effects (such as a high level of hierarchical population structure) and its correlation with an environmental variable underlying the selective pressure also affect the power of detection in these polygenic cases (de Villemereuil et al. 2014).
Despite the limitation of environment–genetic correlation methods and FST-based methods, both have helped in identifying outlier loci associated with adaptations in natural populations. Examples include the identification of whole-genome outlier SNPs associated with climatic adaptations in Arabidopsis thaliana (Hancock et al. 2011a), transcriptome-derived SNPs correlated with stress tolerance in coral reefs (Lundgren et al. 2013), outlier AFLP loci correlated with insecticide resistance in mosquitoes (Paris & Despres 2012), SNPs associated with adaptation to coastal environments in Senecio lautus (Roda et al. 2013) and SNPs associated with climatic adaptations in humans (Hancock et al. 2011b). A combination of population genetics and environmental correlations can help to reduce the number of false positives (de Villemereuil et al. 2014). This has been rarely done, although one exception is the identification of loci involved in host plant use in the large pine weevil Hylobius abietis (Manel, Conord & Despres 2009). Yet, ‘new more general and robust likelihood test are needed that are flexible enough to accommodate departures from classical demographic models’ (de Villemereuil et al. 2014).
Regardless of the method used, it is fundamental to validate that outliers are genuinely implicated in adaptation (Luikart et al. 2003; Barrett & Hoekstra 2011). This can be achieved by combining population genetics both within and between populations, and/or complementing forward genetics with reverse genetics (Stinchcombe & Hoekstra 2007; Butlin 2010; Hohenlohe, Phillips & Cresko 2010). The latter combination has been possible in wild populations of threespine sticklebacks where the confident identification of the Eda gene, responsible for the adaptive reduction of armour plates, was achieved by combining QTL mapping with SNP typing in wild populations (Colosimo et al. 2005) and also in Heliconius butterflies where the identification of the transcription factor optix controlling red adaptive wing pattern variation was possible thanks to the application of AFLP mapping (Baxter et al. 2008) followed by population genetic analyses on the focal region (Baxter et al. 2010b; Counterman et al. 2010; Reed et al. 2011).
Candidate genes
The knowledge of candidate genes derived from other organisms can be combined with these approaches to identify genes that are associated with a particular adaptive phenotype in a different species (Table 1). Thus, either in crosses or in naturally varying phenotypes, candidate genes can be examined for evidence of their association with the trait of interest (Shimizu & Purugganan 2005; Stinchcombe & Hoekstra 2007). In this way, the variation in plumage colour in natural populations of the flycatcher Monarcha castaneiventris was shown to be associated with a single mutation of the previously identified pigmentation gene melanocortin-1 receptor (MC1R) (Mundy 2005; Uy et al. 2009). It is important to be selective at the time of applying the candidate gene approach. If there are many candidate genes associated with a phenotype, this might not be as fruitful as when there is a handful of strong candidates (Luikart et al. 2003). Furthermore, a major drawback of a candidate gene approach is that the literature becomes biased towards a few well-known candidate genes, which may not therefore be representative of their actual importance in evolutionary change (Mundy 2005). In addition, this approach makes the assumption that the genes that matter for evolution are necessarily few and of large effect.
As genomic technologies become more widely available and more information on gene interactions and pathways exists, researchers are moving away from a simplistic candidate gene approach and are applying larger studies that evaluate, at once, the role of multiple genes in a pathway suspected to affect the formation of the adaptive trait (candidate pathway approach) (Suh & Vijg 2005). For example, the flavonoid pathway has been a model system in plants that has helped understanding the genetics underlying flower coloration and other evolutionary processes, including the role of gene duplication in the evolution of novel phenotypes (Des Marais & Rausher 2008), causes of evolutionary rate variation among genes (Lu & Rausher 2003) and the relative importance of coding vs. regulatory mutations in the evolution of ecologically relevant traits (Wessinger & Rausher 2012). Flower coloration is an adaptive trait (Kopp 2009) caused by anthocyanin pigments, whose production requires at least six sequential reactions catalysed by six different enzymes in the anthocyanin pathway (Rausher 2006). The pathway candidate approach in flowers has not only led to identification of the particular enzymes involved in synthesizing red/orange, blue/magenta and blue/purple pigments (Zufall & Rausher 2003; Rausher 2006) but also helped in the identification of the transcriptional complex, composed by bHLH and MYB domain transcription factors, responsible for natural variation in flower coloration among many plant species (Rausher 2006; Kopp 2009). Despite the potential of the candidate pathway analysis in the study of natural adaptation, to date, it is poorly applied to evolutionary studies and largely remains restricted to studies on the genetics of human diseases (Suh & Vijg 2005).
Characterizing regions narrowed by forward or reverse genetics
Outlier loci have been identified in a wide range of species (Bonin et al. 2006; Minder & Widmer 2008; Apple et al. 2010), but fewer studies have moved from their detection to the characterization of underlying QTNS, genes or networks controlling adaptation (Minder & Widmer 2008; Wood et al. 2008; Paris et al. 2010; Midamegbe et al. 2011; Rockman 2011; Kunte et al. 2014). Following up on a particular outlier locus can be time-consuming and technically demanding, but will be a necessary step in order to find the genes or regulatory elements involved in adaptation. One of the most popular approaches is a library-based search of genomic regions flanked by outliers, followed by positional cloning using BACs and sequencing of the genetic interval (Butlin 2010; Nunes et al. 2012). This has been successfully applied in the identification of several wing colour pattern loci in Heliconius butterflies (Baxter et al. 2010b; Counterman et al. 2010), in Papilio polytes to find mimicry ‘supergenes’ (Kunte et al. 2014) and, in the marine gastropod Littorina saxatilis, it has been used to pinpoint candidate loci for local adaptation (Wood et al. 2008).
Pathway analysis (where multiple ‘outlier’ SNPs are analysed jointly) also offers an interesting, yet underutilized, tool to discover gene sets likely involved in the formation of a trait of interest (Pan et al. 2014). This is because although the detection of multiple outlier SNPs associated with a trait (with GWAS, for example) offers an insight into its underlying genetics, this alone may not be very informative in the case of quantitative polygenic traits, because individual SNPs only account for a small part of the trait variance (Mokry et al. 2013). However, pathway analysis is challenging, because a large number of SNPs per individual need to be considered in predictive models. Machine learning methods such as multi-dimensional reduction (MDR), support vector machines (SVM), neural networks (NN) and random forest (RF) are capable of dealing with this dimensionality problem in a flexible manner and can effectively select important variables from irrelevant ones (Goldstein et al. 2010; Gonzalez-Recio & Forni 2011; González-Recio, Rosa & Gianola 2014; Yang & Charles Gu 2014). In particular, RF analysis has been particularly useful in pathway analysis because interactions are implicitly modelled (De Lobel et al. 2010; Chung & Chen 2012) and it is straightforward to understand and interpret (Goldstein et al. 2010; González-Recio, Rosa & Gianola 2014). Nonetheless, this methodology has been primarily applied to the study of complex diseases in humans, considering a small number of SNPs (Chang et al. 2008; Ballard et al. 2010). Its application to large SNP data sets (such as those produced by next-generation techniques) is more complicated and requires the modification of certain standard assumptions (Goldstein et al. 2010; Chen & Ishwaran 2012). Still, this methodology offers an interesting option to the study of genetic pathways shaping natural adaptations. Pioneering work on this includes the search for loci involved in environmental adaptation in Senecio lautus. By comparing genomes of phenotypically contrasting parapatric populations, researchers assessed genetic association at different levels, from SNPs to physiological pathways (Roda et al. 2013).
Alternatively, when ‘outlier’ markers associated with a trait have been identified, they can be mapped back to a reference genome or to available linkage maps (in the same species or a closely related one), to detect whether they fall in or near protein-coding genes possibly affecting the trait. For example, in the rainbow and steelhead trout, species where no reference genome exists but linkage maps do, GWAS coupled with SNP mapping discovered that migration in these species has a complex quantitative genetic basis, resulting from many loci of small effect (Hecht et al. 2013). A similar approach was used to study the genetics of the adaptive natural variation in female abdominal pigmentation in Drosophila melanogaster and determined that variation in this trait is under the control of cis-regulatory regions of the genes tan and bric-à-brac (Bastide et al. 2013). With the increasing availability of whole-genome sequences in a wide variety of taxa and the possibility to develop genomic resources at a reasonable cost in species that lack them, it is becoming more feasible to apply this strategy in non-model species.
Gene expression profiling
When DNA variation associated with phenotypic change does not occur in protein-coding regions, examining the expression pattern of genes provides an important complementary method to test for regulatory change (Rockman & Kruglyak 2006; Hoekstra & Coyne 2007; Hofmann et al. 2009). Although the detection of one or more differentially expressed genes provides information about potential candidates involved in the production of the trait of interest, this does not necessarily imply that all (or any) of them bear the causal variants. Differential expression may result from gene regulation due to upstream mutation(s), which may lie elsewhere in the genome (Stern & Orgogozo 2008; Kopp 2009; Stern & Orgogozo 2009). Thus, a combination of DNA data with expression data is often needed to determine whether the trait has a cis-regulatory basis (i.e. when differential expression and DNA polymorphism associated with phenotype both reside in the same locus) or it is trans-regulated. To this end, allele-specific expression (ASE) assays testing single or multiple genes offer a useful alternative to uncover the respective contributions of cis- and trans-regulatory variation (Knight 2004; Gilad, Rifkin & Pritchard 2008; Main et al. 2009; Wittkopp 2011).
Studies of gene expression can be conducted at either individual candidate loci (e.g. in situ hybridization, reverse-transcriptase quantitative PCR, immunochemistry) or many loci at once (e.g. microarrays, RNA-seq) depending on the information and resources available (Pavey et al. 2010). Studying thousands of transcripts allows a detailed and unbiased description of the genes involved in shaping natural evolution, and has the potential to identify entire genetic and developmental pathways driving adaptive variation. Thus, transcriptomic approaches can catalyse the discovery of multiple components of these gene networks, from the genes regulating phenotypic ‘switches’ to the repertoire of molecules responding to such genes of major effect using, for example, the recently developed Weighted Gene Co-Expression Network Analysis (WGCNA). This method offers the possibility to identify networks of co-expressed genes participating in the formation of a trait while at the same time points to candidate nodal ‘key’ genes likely controlling phenotypic variation only using gene expression data (Oldham, Horvath & Geschwind 2006; Filteau et al. 2013). This approach, however, does not identify the actual mutations controlling trait variance, and therefore, complementary DNA characterization on these candidates should also be carried out.
Microarrays have pioneered the genome-wide characterization of transcripts associated with adaptive natural variation. Almost a decade ago, microarrays were used in one of the first attempts to identify genes controlling beak morphology variation in Darwin's finches and showed that the calmodulin (CaM)-dependent pathway is a key component of the evolution of beak variation in these birds (Abzhanov et al. 2006). Similarly, microarrays showed that in limnetic Coregonine fishes, the parallel phenotypic evolution towards using the same ecological niche involves similar changes in expression at the same genes (Derome & Bernatchez 2006). Nonetheless, microarrays suffer from several limitations. Background levels of hybridization, differences in hybridization properties among probes and the restriction of interrogating only the transcripts included in the array are among the most common problems of this technique (Marioni et al. 2008).
Nowadays, high-throughput mRNA sequencing technology (RNA-seq) has the potential to overcome some of these limitations (Marioni et al. 2008; Wang, Gerstein & Snyder 2009). By using next-generation sequencing technologies, RNA-seq allows for a direct estimation of relative transcript abundance across the entire genome (Cheviron & Brumfield 2012) keeping the background noise low where sequences can be unambiguously mapped to unique regions of the genome (Marioni et al. 2008; Wang, Gerstein & Snyder 2009). This technique is not limited to detecting transcripts in organisms with a reference genome as the same RNA-seq data can be used to create a transcriptome assembly which is then used as a reference for read mapping (Grabherr et al. 2011), thus being particularly attractive for non-model organisms. This approach has recently been applied to naturally varying organisms such as the intertidal copepod Tigriopus californicus, where differences in thermal tolerance were associated with differential expression of heat-shock proteins (Hsp) and genes involved in ubiquitination and proteolysis (Schoville et al. 2012) and, in eucalyptus, RNA-seq has provided insights into the molecular mechanisms underlying the adaptation to water shortage (Villar et al. 2011). RNA-seq, as any other next-generation sequencing technique, presents limitations in terms of data storage, analysis and cost. Nonetheless, with RNA-seq, it is especially important to consider sequence coverage (directly related to the sequencing cost) because in organisms with large genomes and complex transcriptomes, more sequencing depth will be required for an adequate coverage (Wang, Gerstein & Snyder 2009) and/or where multiple replicates and comparisons will be needed to correctly tackle a particular trait.
Alternatively, when strong single candidate genes have been isolated at the DNA level or have been derived from transcriptomics studies, their further and complementary characterization can be done using reverse-transcriptase quantitative PCR (RT-qPCR). Several studies have applied RT-qPCR to profile transcription levels of particular candidate loci. For example, measuring the expression levels of the gene Agouti with RT-qPCR (implicated in producing pheomelanin in Mus musculus) demonstrated that cis-regulatory evolution at this gene was involved in adaptive variation in cryptic colouration of deer mice (Linnen et al. 2009). Similarly, adaptive differential retinal sensitivity in African cichlids inhabiting clear vs. turbid water relates to differences in opsin gene expression measured with RT-qPCR (Hofmann et al. 2009). However, RT-qPCR depends on the performance and specificity of primers so only the gene of interest is quantified (Busk 2014), making optimization a time-consuming process. Also, its accuracy is strongly reliant on the use of multiple control genes (e.g. reference or housekeeping genes) for normalization and correction of the multiple variation sources. A correct choice of control genes is not a trivial task. It is desirable to use more than one, as a single control gene can lead to normalization biases. Also, it is necessary to be sure that they are equally expressed across all the tissue types and species interrogated, as it can affect the accuracy of the calculation of relative expression differences between samples (Fedrigo et al. 2010).
The spatial distribution of expression patterns of candidate genes can be visualized using in situ hybridization (ISH), and this approach has provided the foundation for much of the field of ‘evo-devo’. The technique involves hybridizing an antisense RNA probe to an mRNA transcript, and it is a powerful method to characterize gene expression in tissues. The ISH procedure follows five major steps: (i) sample preparation, including fixation, mounting and ISH pre-treatment, (ii) probe preparation, (iii) hybridization, (iv) probe removal and (v) detection (Apostolopoulos 2001). Several studies have exemplified the usefulness of this technique to characterize the loci of adaptation. Shapiro et al. (2004) used ISH to compare profiles of expression of the gene Pitx1 between benthic and marine sticklebacks and thus showed that a cis-regulatory element of Pitx1 is responsible for pelvic size variation in fishes of the two environments (Shapiro et al. 2004). In the Darwin's finches, ISH patterns of expression of the genes TGFβIIr, β-catenin and Dickkopf-3 are differentially expressed in the developing pre-maxillary bone of embryos of species with different beak shapes, a trait associated with the exploitation of multiple ecological niches (Mallarino et al. 2011). In a similar way, ISH showed that in Heliconius butterflies, cis-regulatory evolution of the transcription factor optix drives the convergent evolution of red wing patterns in distantly related species (Reed et al. 2011). Nonetheless, ISH protocols are not easy to establish because the technique can be challenging to optimize (Abzhanov et al. 2008). Furthermore, it requires enough supply of organismal tissue at different developmental stages. This therefore requires raising a large number of individuals in a controlled environment or sampling enough individuals in the wild, specifically at the developmental points required (Abzhanov et al. 2008), which already imposes a limitation for many natural systems.
The use of immunohistochemistry provides an alternative to spatially localize the products of gene expression, and it is a much more forgiving technique than ISH (Abzhanov et al. 2008). It has been successfully applied to document the genetics of adaptive wing radiation in Heliconius (Martin et al. 2014) and the genes controlling male wing pigmentation in Drosophila (Gompel et al. 2005; Prud'homme et al. 2006). However, immunohistochemistry depends on the development of a species-specific antibody targeting the protein of interest, which can be time-consuming and expensive, or the availability of a cross-reactive antibody in a different species (Abzhanov et al. 2008). Just as ISH, immunohistochemistry also requires enough supply of organismal tissue at specific developmental points, limiting its application in many natural organisms.
Nonetheless, care should be taken in design and interpretation of gene expression assays. Variation in ecologically relevant traits is sometimes due to phenotypic plasticity (Hoffman & Goodisman 2007; Bossdorf, Richards & Pigliucci 2008; Whiteman & Agrawal 2009) which can be confounded with adaptive heritable variation. In this way, if the adaptive relevance of a trait has not been experimentally tested and it turns out to be a plastic phenotype, comparing gene expression in different conditions will yield a set of genes that do not contain the causal adaptive variants. Of course this is not to deny the importance of phenotypic plasticity in adaptation, but this is a subject beyond the scope of this paper (Ghalambor et al. 2007; Bossdorf, Richards & Pigliucci 2008; Hughes 2012).
Assays of molecular function
The implementation of assays of molecular function such as transgenics, knockouts, knockdowns (with RNA interference (RNAi), for example) and gene replacement constitutes an important test to prove that a gene actually underlies natural variation (Shimizu & Purugganan 2005; Hoekstra & Coyne 2007; Pavey et al. 2012) and establish whether it is required and/or sufficient for the development of the adaptive trait (Abzhanov et al. 2008). Functional tests of candidate genes are well implemented in model organisms like Drosophila, yeast, nematodes and mice (Feder & Mitchell-Olds 2003; Heffer & Pick 2013) and provide the standard evidence for the genetic basis of trait variation. In these organisms, for example, the application of functional tests has confirmed the identification of the genes, and even mutations, controlling adaptive natural variation. In Drosophila, the application of a set of transgenic reporter assays found the actual mutations in the regulatory elements of the ebony gene controlling adaptive abdominal pigmentation in African natural populations (Rebeiz et al. 2009). Similarly, in the deer mouse Peromyscus maniculatus, the generation of Agouti knockouts confirmed the involvement of this gene in adaptive melanism (Kingsley et al. 2009).
Over the past several years, new functional protocols have been developed for a wide range of emerging organisms including Daphnia, wasps, crickets, ladybirds, cavefish and sticklebacks (Osanai-Futahashi et al. 2012; Pavey et al. 2012) and, although this is a field under active development, these experiments are still not feasible in all organisms and therefore impose a limitation in several non-model systems. Nonetheless, when functional assays are impossible in the target organism, it is still possible to use closely related species as ‘model’ organisms. For example, use of the retroviral vector RCAS in the chicken embryo implicated the genes CaM, TGFβIIr, β-catenin and Dkk3 in controlling beak development which may imply a role in the evolution of Darwin's finches (Abzhanov et al. 2006; Mallarino et al. 2011). However, it is important to bear in mind that such experiments in ‘model’ species do not directly identify the role of natural variants. Similarly, in the sticklebacks, a functional test of the Ectodysplasin-A (EDA) gene using transgenics showed how this gene controls adaptive plate variation in natural populations (Colosimo et al. 2005), but the transgenic construct carried a mouse EDA-A1 cDNA rather than the native stickleback ‘complete’ EDA allele. Although changes in plate phenotype were indeed observed, the results were variable. Three out of fourteen transgenic ‘low-plated’ fishes developed extra plates on their sides, but not in a consistent manner; the number and type of extra plates developed varied between and within individuals (different in each side) (Colosimo et al. 2005). In the future, it is hoped that experiments can be developed that more directly replicate the role of naturally occurring variants in their native species.
With the constant development of functional tools that were previously only available in more traditional ‘model’ organisms, now it is possible not only to pinpoint genes and mutations shaping natural adaptations but also to establish new organisms in which to study the genetics underlying evolution. Nonetheless, the task of developing more functional assays applicable to a wider range of organism is still needed. RNA interference (RNAi), for example, is a method for knocking down expression of a target gene and appeared to be easily accessible. However, in some taxa such as the Lepidoptera, it has proved to be highly problematic (Terenius et al. 2011). Responding to this need, recently developed approaches commonly referred as ‘genome editing tools’ and based on the use of engineered nucleases coupled to DNA recognition domains have been developed; these include zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and the clustered regulatory interspaced short palindromic repeats (CRISPR)/Cas9 endonuclease system (Gaj, Gersbach & Barbas Iii 2013; Wei et al. 2013). In all these ‘genome editing tools’, the DNA-binding module recognizes and binds the target gene while the nuclease module induces DNA double-strand breaks (Bassett et al. 2013; Gaj, Gersbach & Barbas Iii 2013; Wei et al. 2013). This activates either error-prone non-homologous end joining, which commonly introduces indels or frameshifts leading to the knockout of gene function, or homology directed repair, which allows the introduction of changes from single nucleotide changes to entire transgenes (Gaj, Gersbach & Barbas Iii 2013). These emerging technologies have already started to expand the ability to manipulate genes in less traditional organisms. For example, a related technique using zinc-finger nucleases (ZFNs) has allowed the mutation of genes related to the circadian clockwork in the monarch butterfly (Danaus plexippus) (Merlin et al. 2012). Interestingly, the (CRISPR)/Cas9 system is now viewed as a more attractive technical choice as their DNA recognition potential is bigger than that of ZFNs or TALENs, although given its recent development, at the moment it only has been applied to ‘model’ organisms (Bassett et al. 2013; Chang et al. 2013; Gaj, Gersbach & Barbas Iii 2013; Wei et al. 2013). Yet, the potential of (CRISPR)/Cas9 in the study of the genetics of adaptation in natural systems is very promising since it could be combined with classical genetic approaches (such as genetic complementation) to simultaneously map natural variation and functionally test the genes harbouring causal alleles (Turner 2014). This great advance in genome editing technologies opens new opportunities to decipher and test the molecular underpinnings of adaptations in a wider range of organisms and exemplifies how active research and development of tools broadens our methodological possibilities to answer longstanding questions.
In the future, transgenic tests should ideally involve replacement of alternate natural alleles at a locus, in order to demonstrate the functional value of particular substitutions. To date, this has only rarely, if ever, been achieved. Methods that involve knockouts or experiments involving ‘model’ species should be seen as a complement to other DNA or RNA approaches narrowing candidate genes likely shaping adaptive variation, but not a definitive test of adaptive function.
Assays of ecological function
Although all the methods described above help to identify genes (or QTNs) contributing to adaptive phenotypic variation, it is crucial to perform field experiments either in nature or in conditions that closely mimic naturally occurring events, to evaluate that the trait of interest is indeed adaptive and, also, to test the fitness consequences of allelic substitutions at the causal genes (Barrett & Hoekstra 2011). Experimental evolution studies that link genes, phenotype and fitness have been possible under laboratory conditions and using organisms such as virus, bacteria and yeast, with very short generation times and where the replicated sequencing of whole genomes is feasible (Rokyta et al. 2005; Barrick et al. 2009; Araya et al. 2010; Brockhurst, Colegrave & Rozen 2011; Barrick & Lenski 2013). However, potential confounding effects or artefacts in those artificial systems make it hard to extrapolate the patterns and conclusions derived from them to a context of natural adaptation, highlighting the need to perform this experiments in natural systems (Barrett & Hoekstra 2011).
The most common approach to evaluate whether a trait has a direct impact on fitness in nature is by testing cause-and-effect relationships in a planned field experiment comparing different conditions (i.e. varying the suspected natural selection agent). There, phenotypic variation in the trait driven by differences in environment should be observed. Field experiments to test the adaptive value of traits in the wild come in a great variety and complexity of forms including QST–FST comparison (Leinonen et al. 2013), reciprocal transplants of hybrid individuals (Lowry et al. 2009), controlled introduction of live organisms to new environments (Reznick et al. 1997; Kapan 2001; Barrett & Schluter 2008; Irschick & Reznick 2009; Gompert et al. 2014) and replacing of the real organisms with synthetic replicas in nature primarily to quantify the impact of predation (Irschick & Reznick 2009; Merrill et al. 2012; Linnen et al. 2013). For example, Linnen et al. (2013) used plasticine mice models of two different colours in the field and found that light-coloured models matching light-coloured soil were less attacked by visually hunting predators (Linnen et al. 2013). Similarly, when Anolis lizards were introduced from the mainland into a series of islands, the introduced population evolved a different hind limb phenotype potentially as an adaption to the use of narrow surfaces (Losos, Warheitt & Schoener 1997). While these examples show that a trait affects organismal fitness in the wild, they do not tell us about how the genetic variation in the genes shaping those adaptive traits evolves in response to the experimental treatments, and thus, no connection between genotype, phenotype and fitness can be established.
In addition, QTL mapping has been useful to identify regions associated with habitat adaptation using reciprocal transplants of hybrid individuals [F2, backcross, recombinant inbred lines (RILs), near isogenic lines (NILs)] in contrasting environments (Bradshaw & Schemske 2003; Lowry et al. 2009). This approach allows evaluating both genotype–environment interactions and the effects of epistasis in fitness (Barrett & Hoekstra 2011).
To overcome this, when the identity of the gene underlying the formation of the adaptive trait is known (using the methods described above), one should study whether genetic variation in the causal gene evolves in the expected direction in response to differential treatments and also presents signatures of selection (Weinig et al. 2003). For instance, hybrid sticklebacks between a lake and a river population were transplanted into river and lake environments that differ in their parasitic diversity. Under the hypothesis that in order to survive and reproduce a host should resist to local parasites and pathogens, researchers measured allele diversity at the major histocompatibility complex (MHC), involved in the recognition of parasite-specific antigens. After one generation, diversity of MHC alleles was higher at the lake environment (which bears a broader range of parasites than the river environment), thus providing lake sticklebacks the advantage of fighting a more diverse set of parasites (Eizaguirre et al. 2012). In a similar experiment, researchers measured selection on natural allelic variants of the (Eda) locus, known to control adaptive differences in armour plates in sticklebacks (Colosimo et al. 2005). By transplanting marine sticklebacks harbouring both the low-plate and high-plate alleles of Eda into freshwater ponds and studying genotype frequency variations in one generation, researchers found that the low-plate allele was positively selected once lateral plates developed, likely because it provides a growth advantage in freshwater environments. However, the same allele was negatively selected before the plates were formed, indicating that either the Eda gene affects additional unmeasured traits under selection, or that tightly linked loci also have effects on fitness (Barrett & Schluter 2008). This last observation suggests that, when possible, tests of selection should be performed genome-wide using next-generation sequencing techniques in order to generate a high density of markers. This resolution can permit not only to confirm/detect selection signatures on the adaptive gene itself but also to detect the loci controlling unmeasured traits with fitness effects, and determine whether they are shaped by the same pleiotropic gene or by multiple linked loci (Barrett & Hoekstra 2011). To date, only a handful of studies have explored the genomic consequences of contemporary selection in the field using whole-genome data. In one study, researchers transplanted stick insects to native and novel host plants and measured allele frequency changes within a generation at genome-wide level (Gompert et al. 2014). In another study, patterns of genome-wide selection in purple sea urchins were evaluated under different ocean acidification levels (Pespeni et al. 2013). Both studies detect changes in allele frequencies driven by selection at multiple loci across the genome. However, as there was no previous knowledge of gene(s) underlying the formation of the adaptive traits directly affecting fitness, these results do not directly lead to conclusions about how selection affects allelic variance in a causal gene and the possible explanations for observing selection in non-causal regions of the genome (i.e. pleiotropy, indirect selection, linkage).
In addition, the combination of genomic data with selection experiments also gives the opportunity to evaluate the role of epistasis in adaptation. This has been applied to the study of laboratory adaptation using yeast, bacteria and viruses. These laboratory-based studies show a global pattern of diminishing returns epistasis (i.e. where the more mutations that accumulate, the weaker their fitness effect), which impedes the rate of ongoing adaptation relative to a null model of independent mutational effects (Chou et al. 2011; Khan et al. 2011; Kryazhimskiy et al. 2014). As the evolutionary patterns observed in small laboratory populations may not be the same as those contributing to natural evolution, the confirmation of these results still needs to be carried out in natural populations in order to determine the genomic effect of epistasis and its overall contribution to natural adaptation.
Conclusions
Recent research has led to a remarkable growth in our understanding of the molecular basis of adaptive evolution (Table 1). Altogether, these studies have provided important insights into the genetic basis of adaptations and also the methodological approaches needed to answer this evolutionary problem. Nonetheless, it has become clear that the characterization of the genes underlying adaptive traits is not an easy task because factors such as demography, epistasis and pleiotropy can introduce confounding effects that will complicate any clear genetic signal. Also, methodological bias can mislead findings by pointing to large-effect loci and missing the detection of genes with small effect, thus complicating the description of ‘all’ important variants contributing to natural adaptation. Still, the search for the loci of evolution can benefit from following an organized and complementary methodology. First, it is necessary to corroborate that a trait affects fitness in the field and is in fact adaptive. Then, the region(s) of the genome in which genotypes are correlated with adaptive phenotypes should be defined either with classical genetic tools or applying new genomic approaches. Next, when DNA polymorphism associated with phenotype in the candidate genes does not occur in protein-coding regions, the expression pattern of such genes must be analysed for in order to test whether the trait has a cis- or trans-regulatory basis. Ultimately, functional experiments are required to prove that a gene or mutation is actually responsible for the phenotype observed. Once individual genes or SNPs have been identified, it is important to quantify their effect in the ‘trait value’ (i.e. how much variation in the phenotype is explained by the candidate SNPs/genes). Finally, the genetic variation in the genes shaping those adaptive traits should be evaluated in field selection experiments in order to establish a definite connection between genotype, phenotype and fitness.
A comprehensive review of the conclusions of such studies is beyond the scope of this article. However, some of the major findings include the following. First, the evolution of similar adaptive traits in different lineages commonly involves the action of the same genes (Colosimo et al. 2005; Nadeau & Jiggins 2010; Reed et al. 2011). Secondly, both cis-regulatory changes and coding changes contribute to adaptive variation (Mundy et al. 2004; Colosimo et al. 2005; Chan et al. 2010; Kunte et al. 2014). However, cis-regulatory changes may be more frequently involved in the evolution of morphological traits compared to physiological traits and in the evolution of morphological interspecific differences compared to the evolution of morphological intraspecific variation (Stern & Orgogozo 2008). Thirdly, the position of an adaptive gene in a regulatory network matters, as mutation in upstream patterning (input) genes is likely to affect the development of several body structures, while mutations in downstream (responsive) genes will influence the form of all incidences of the particular structure (Stern & Orgogozo 2008). However, cis-regulatory mutations in input/output genes provide great precision in evolutionary change with minimal pleiotropic effects (if any) (Stern & Orgogozo 2008; Gompel & Prud'homme 2009). Fourthly, adaptations can evolve from standing genetic variation, de novo mutations and adaptive introgression (Hermisson & Pennings 2005; Feldman, Brodie & Pfrender 2009; Hedrick 2013). Finally, most of the adaptations reported to date seem to arise through few initial mutations of major effect followed by many small effect mutations on minor genes (Orr 2005; Rockman 2011; Olson-Manning, Wagner & Mitchell-Olds 2012). Nonetheless, this last observation may be tainted by our experimental bias towards detecting large-effect alleles, so there is a likely ascertainment bias in the literature (Rockman 2011).
The search for the loci of evolution will be surely fuelled by the continuous increase in genomic and transcriptomics resources in natural populations, along with the development of novel methodologies applicable to such organisms. This offers exciting opportunities for testing new predictions and understanding how evolution proceeds.
Acknowledgments
This work was supported by BBSRC grant number BB/K019945/1 to CJ.
Data accessibility
This manuscript does not use data.
References
- Abzhanov A, Kuo WP, Hartmann C, Grant BR, Grant PR. Tabin CJ. The calmodulin pathway and evolution of elongated beak morphology in Darwin's finches. Nature. 2006;442:563–567. doi: 10.1038/nature04843. [DOI] [PubMed] [Google Scholar]
- Abzhanov A, Extavour CG, Groover A, Hodges SA, Hoekstra HE, Kramer EM. Monteiro A. Are we there yet? Tracking the development of new model systems. Trends in Genetics. 2008;24:353–360. doi: 10.1016/j.tig.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Albertson R, Yan Y, Titus T, Pisano E, Vacchi M, Yelick P, Detrich Hr. Postlethwait J. Molecular pedomorphism underlies craniofacial skeletal evolution in Antarctic notothenioid fishes. BMC Evolutionary Biology. 2010;10:4. doi: 10.1186/1471-2148-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhaddad H, Khan R, Grahn RA, Gandolfi B, Mullikin JC, Cole SA, et al. Extent of linkage disequilibrium in the domestic cat, Felis silvestris catus, and its breeds. PLoS ONE. 2013;8:e53537. doi: 10.1371/journal.pone.0053537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L. Lander ES. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- Amemiya CT, Alfoldi J, Lee AP, Fan S, Philippe H, MacCallum I, et al. The African coelacanth genome provides insights into tetrapod evolution. Nature. 2013;496:311–316. doi: 10.1038/nature12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P, Davison D, Erezyilmaz D, Hu TT, Mast J, Sunayama-Morita T. Stern DL. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Research. 2011;21:610–617. doi: 10.1101/gr.115402.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolopoulos J. Nonradioactive in situ hybridization in atherosclerotic tissue. In: Drew A, editor. Atherosclerosis. New York: Humana Press; 2001. pp. 195–206. [DOI] [PubMed] [Google Scholar]
- Apple JL, Grace T, Joern A, Amand PS. Wisely SM. Comparative genome scan detects host-related divergent selection in the grasshopper Hesperotettix viridis. Molecular Ecology. 2010;19:4012–4028. doi: 10.1111/j.1365-294X.2010.04792.x. [DOI] [PubMed] [Google Scholar]
- Aranzana M, Kim S, Zhao K, Bakker E, Horton M, Jakob K, et al. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genetics. 2005;1:e60. doi: 10.1371/journal.pgen.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya C, Payen C, Dunham M. Fields S. Whole-genome sequencing of a laboratory-evolved yeast strain. BMC Genomics. 2010;11:88. doi: 10.1186/1471-2164-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult L, Su KFY, Manoel D, Minervino C, Magriña J, Gompel N. Prud'homme B. Emergence and diversification of fly pigmentation through evolution of a gene regulatory module. Science. 2013;339:1423–1426. doi: 10.1126/science.1233749. [DOI] [PubMed] [Google Scholar]
- Assunção A, Pieper B, Vromans J, Lindhout P, Aarts M. Schat H. Construction of a genetic linkage map of Thlaspi caerulescens and quantitative trait loci analysis of zinc accumulation. New Phytologist. 2006;170:21–32. doi: 10.1111/j.1469-8137.2005.01631.x. [DOI] [PubMed] [Google Scholar]
- Balding D. Nichols R. A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica. 1995;96:3–12. doi: 10.1007/BF01441146. [DOI] [PubMed] [Google Scholar]
- Ballard D, Abraham C, Cho J. Zhao H. Pathway analysis comparison using Crohn's disease genome wide association studies. BMC Medical Genomics. 2010;3:25. doi: 10.1186/1755-8794-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RDH. Hoekstra HE. Molecular spandrels: tests of adaptation at the genetic level. Nature Reviews Genetics. 2011;12:767–780. doi: 10.1038/nrg3015. [DOI] [PubMed] [Google Scholar]
- Barrett RDH. Schluter D. Adaptation from standing genetic variation. Trends in Ecology & Evolution. 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Barrick JE. Lenski RE. Genome dynamics during experimental evolution. Nature Reviews Genetics. 2013;14:827–839. doi: 10.1038/nrg3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick J, Yu D, Yoon S, Jeong H, Oh T, Schneider D, Lenski R. Kim J. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP. Liu J-L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Reports. 2013;4:220–228. doi: 10.1016/j.celrep.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastide H, Betancourt A, Nolte V, Tobler R, Stöbe P, Futschik A. Schl√∂tterer C. A genome-wide, fine-scale map of natural pigmentation variation in Drosophila melanogaster. PLoS Genetics. 2013;9:e1003534. doi: 10.1371/journal.pgen.1003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter SW, Papa R, Chamberlain N, Humphray SJ, Joron M, Morrison C, ffrench-Constant RH, McMillan WO. Jiggins CD. Convergent evolution in the genetic basis of Müllerian mimicry in Heliconius butterflies. Genetics. 2008;180:1567–1577. doi: 10.1534/genetics.107.082982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter I, Brazelton JN, Yu D, Huang YS, Lahner B, Yakubova E, et al. A coastal cline in sodium accumulation in Arabidopsis thaliana is driven by natural variation of the sodium transporter AtHKT1;1. PLoS Genetics. 2010a;6:e1001193. doi: 10.1371/journal.pgen.1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter SW, Nadeau NJ, Maroja LS, Wilkinson P, Counterman BA, Dawson A, et al. Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in the Heliconius melpomene clade. PLoS Genetics. 2010b;6:e1000794. doi: 10.1371/journal.pgen.1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter SW, Davey JW, Johnston JS, Shelton AM, Heckel DG, Jiggins CD. Blaxter ML. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS ONE. 2011;6:e19315. doi: 10.1371/journal.pone.0019315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu J, Doerksen T, Boyle B, Clément S, Deslauriers M, Beauseigle S, et al. Association genetics of wood physical traits in the conifer white spruce and relationships with gene expression. Genetics. 2011;188:197–214. doi: 10.1534/genetics.110.125781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont MA. Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London. Series B: Biological Sciences. 1996;263:1619–1626. [Google Scholar]
- Beldade P, Brakefield PM. Long AD. Contribution of distal-less to quantitative variation in butterfly eyespots. Nature. 2002;415:315–318. doi: 10.1038/415315a. [DOI] [PubMed] [Google Scholar]
- Beraldi D, McRae AF, Gratten J, Slate J, Visscher PM. Pemberton JM. Development of a linkage map and mapping of phenotypic polymorphisms in a free-living population of Soay Sheep (Ovis aries. Genetics. 2006;173:1521–1537. doi: 10.1534/genetics.106.057141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson J. Roux F. Towards identifying genes underlying ecologically relevant traits in Arabidopsis thaliana. Nature Reviews Genetics. 2010;11:867–870. doi: 10.1038/nrg2896. [DOI] [PubMed] [Google Scholar]
- Bodmer W. Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nature Genetics. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhomme M, Chevalet C, Servin B, Boitard S, Abdallah J, Blott S. SanCristobal M. Detecting selection in population trees: the Lewontin and Krakauer test extended. Genetics. 2010;186:241–262. doi: 10.1534/genetics.110.117275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin A. Population genomics: a new generation of genome scans to bridge the gap with functional genomics. Molecular Ecology. 2008;17:3583–3584. doi: 10.1111/j.1365-294X.2008.03854.x. [DOI] [PubMed] [Google Scholar]
- Bonin A, Taberlet P, Miaud C. Pompanon F. Explorative genome scan to detect candidate loci for adaptation along a gradient of altitude in the common frog (Rana temporaria. Molecular Biology and Evolution. 2006;23:773–783. doi: 10.1093/molbev/msj087. [DOI] [PubMed] [Google Scholar]
- Bossdorf O, Richards CL. Pigliucci M. Epigenetics for ecologists. Ecology Letters. 2008;11:106–115. doi: 10.1111/j.1461-0248.2007.01130.x. [DOI] [PubMed] [Google Scholar]
- Bradley Shaffer H, Minx P, Warren D, Shedlock A, Thomson R, Valenzuela N, et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biology. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw HD. Schemske DW. Allele substitution at a flower colour locus produces a pollinator shift in monkeyflowers. Nature. 2003;426:176–178. doi: 10.1038/nature02106. [DOI] [PubMed] [Google Scholar]
- Bradshaw HD, Otto KG, Frewen BE, McKay JK. Schemske DW. Quantitative trait loci affecting differences in floral morphology between two species of monkeyflower (Mimulus. Genetics. 1998;149:367–382. doi: 10.1093/genetics/149.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhurst MA, Colegrave N. Rozen DE. Next-generation sequencing as a tool to study microbial evolution. Molecular Ecology. 2011;20:972–980. doi: 10.1111/j.1365-294X.2010.04835.x. [DOI] [PubMed] [Google Scholar]
- Busk P. A tool for design of primers for microRNA-specific quantitative RT-qPCR. BMC Bioinformatics. 2014;15:29. doi: 10.1186/1471-2105-15-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butlin R. Population genomics and speciation. Genetica. 2010;138:409–418. doi: 10.1007/s10709-008-9321-3. [DOI] [PubMed] [Google Scholar]
- Cai Q, Qian X, Lang Y, Luo Y, Pan S, Hui Y, et al. The genome sequence of the ground tit Pseudopodoces humilis provides insights into its adaptation to high altitude. Genome Biology. 2013;14:R29. doi: 10.1186/gb-2013-14-3-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YF, Marks ME, Jones FC, Villarreal G, Jr, Shapiro MD, Brady SD, et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science. 2010;327:302–305. doi: 10.1126/science.1182213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JS, Yeh R-F, Wiencke JK, Wiemels JL, Smirnov I, Pico AR, et al. Pathway analysis of single-nucleotide polymorphisms potentially associated with glioblastoma multiforme susceptibility using Random Forests. Cancer Epidemiology Biomarkers & Prevention. 2008;17:1368–1373. doi: 10.1158/1055-9965.EPI-07-2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong J-W. Xi JJ. Genome editing with RNA-guided Cas9 nuclease in Zebrafish embryos. Cell Research. 2013;23:465–472. doi: 10.1038/cr.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. Ishwaran H. Random forests for genomic data analysis. Genomics. 2012;99:323–329. doi: 10.1016/j.ygeno.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheviron ZA. Brumfield RT. Genomic insights into adaptation to high-altitude environments. Heredity. 2012;108:354–361. doi: 10.1038/hdy.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou H-H, Chiu H-C, Delaney NF, Segré D. Marx CJ. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science. 2011;332:1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung R-H. Chen Y-E. A two-stage random forest-based pathway analysis method. PLoS ONE. 2012;7:e36662. doi: 10.1371/journal.pone.0036662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin H. Fumagalli L. Evidence for morphological and adaptive genetic divergence between lake and stream habitats in European minnows (Phoxinus phoxinus, Cyprinidae) Molecular Ecology. 2011;20:4490–4502. doi: 10.1111/j.1365-294X.2011.05284.x. [DOI] [PubMed] [Google Scholar]
- Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J, et al. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science. 2005;307:1928–1933. doi: 10.1126/science.1107239. [DOI] [PubMed] [Google Scholar]
- Coop G, Witonsky D, Di Rienzo A. Pritchard JK. Using environmental correlations to identify loci underlying local adaptation. Genetics. 2010;185:1411–1423. doi: 10.1534/genetics.110.114819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counterman BA, Araujo-Perez F, Hines HM, Baxter SW, Morrison CM, Lindstrom DP, et al. Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato. PLoS Genetics. 2010;6:e1000796. doi: 10.1371/journal.pgen.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank TE. Hahn MW. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Molecular Ecology. 2014;23:3133–3157. doi: 10.1111/mec.12796. [DOI] [PubMed] [Google Scholar]
- Cutter AD. Payseur BA. Genomic signatures of selection at linked sites: unifying the disparity among species. Nature Reviews Genetics. 2013;14:262–274. doi: 10.1038/nrg3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhoff EP. Rank NE. Functional and physiological consequences of genetic variation at phosphoglucose isomerase: heat shock protein expression is related to enzyme genotype in a montane beetle. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10056–10061. doi: 10.1073/pnas.160277697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM. Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- De Lobel L, Geurts P, Baele G, Castro-Giner F, Kogevinas M. Van Steen K. A screening methodology based on Random Forests to improve the detection of gene-gene interactions. European Journal of Human Genetics. 2010;18:1127–1132. doi: 10.1038/ejhg.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mita S, Thuillet A-C, Gay L, Ahmadi N, Manel S, Ronfort J. Vigouroux Y. Detecting selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations. Molecular Ecology. 2013;22:1383–1399. doi: 10.1111/mec.12182. [DOI] [PubMed] [Google Scholar]
- Derome N. Bernatchez L. The transcriptomics of ecological convergence between 2 limnetic coregonine fishes (Salmonidae) Molecular Biology and Evolution. 2006;23:2370–2378. doi: 10.1093/molbev/msl110. [DOI] [PubMed] [Google Scholar]
- Derome N, Bougas B, Rogers SM, Whiteley AR, Labbe A, Laroche J. Bernatchez L. Pervasive sex-linked effects on transcription regulation as revealed by expression quantitative trait loci mapping in lake whitefish species pairs (Coregonus sp., Salmonidae) Genetics. 2008;179:1903–1917. doi: 10.1534/genetics.107.086306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Des Marais DL. Rausher MD. Escape from adaptive conflict after duplication in an anthocyanin pathway gene. Nature. 2008;454:762–765. doi: 10.1038/nature07092. [DOI] [PubMed] [Google Scholar]
- Dittmar EL, Oakley CG, Ågren J. Schemske DW. Flowering time QTL in natural populations of Arabidopsis thaliana and implications for their adaptive value. Molecular Ecology. 2014;23:4291–4303. doi: 10.1111/mec.12857. [DOI] [PubMed] [Google Scholar]
- Dopman E, Robbins P. Seaman A. Components of reproductive isolation between North American pheromone strains of the European corn borer. Evolution. 2010;64:881–902. doi: 10.1111/j.1558-5646.2009.00883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin ML, Lundy KE, Morrell PL, Torres-Martinez CL. Clegg MT. Genes that determine flower color: the role of regulatory changes in the evolution of phenotypic adaptations. Molecular Phylogenetics and Evolution. 2003;29:507–518. doi: 10.1016/s1055-7903(03)00196-9. [DOI] [PubMed] [Google Scholar]
- Eizaguirre C, Lenz TL, Kalbe M. Milinski M. Divergent selection on locally adapted major histocompatibility complex immune genes experimentally proven in the field. Ecology Letters. 2012;15:723–731. doi: 10.1111/j.1461-0248.2012.01791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. Sheldon BC. Genetic basis of fitness differences in natural populations. Nature. 2008;452:169–175. doi: 10.1038/nature06737. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Smeds L, Burri R, Olason PI, Backstrom N, Kawakami T, et al. The genomic landscape of species divergence in Ficedula flycatchers. Nature. 2012;491:756–760. doi: 10.1038/nature11584. [DOI] [PubMed] [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES. Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder ME. Mitchell-Olds T. Evolutionary and ecological functional genomics. Nature Reviews Genetics. 2003;4:649–655. doi: 10.1038/nrg1128. [DOI] [PubMed] [Google Scholar]
- Fedrigo O, Warner LR, Pfefferle AD, Babbitt CC, Cruz-Gordillo P. Wray GA. A pipeline to determine RT-QPCR control genes for evolutionary studies: application to primate gene expression across multiple tissues. PLoS ONE. 2010;5:e12545. doi: 10.1371/journal.pone.0012545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman CR, Brodie ED. Pfrender ME. The evolutionary origins of beneficial alleles during the repeated adaptation of garter snakes to deadly prey. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13415–13420. doi: 10.1073/pnas.0901224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filteau M, Pavey SA, St-Cyr J. Bernatchez L. Gene co-expression networks reveal key drivers of phenotypic divergence in lake whitefish. Molecular Biology and Evolution. 2013;30:1384–1396. doi: 10.1093/molbev/mst053. [DOI] [PubMed] [Google Scholar]
- Foll M. Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics. 2008;180:977–993. doi: 10.1534/genetics.108.092221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frichot E, Schoville SD, Bouchard G. François O. Testing for associations between loci and environmental gradients using latent factor mixed models. Molecular Biology and Evolution. 2013;30:1687–1699. doi: 10.1093/molbev/mst063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA. Barbas Iii CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in Biotechnology. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo J, Grahame JW. Butlin RK. An EST-based genome scan using 454 sequencing in the marine snail Littorina saxatilis. Journal of Evolutionary Biology. 2010;23:2004–2016. doi: 10.1111/j.1420-9101.2010.02071.x. [DOI] [PubMed] [Google Scholar]
- Ghalambor CK, McKay JK, Carroll SP. Reznick DN. Adaptive versus non-adaptive phenotypic plasticity and the potential for contemporary adaptation in new environments. Functional Ecology. 2007;21:394–407. [Google Scholar]
- Gilad Y, Rifkin SA. Pritchard JK. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends in Genetics. 2008;24:408–415. doi: 10.1016/j.tig.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BA, Hubbard AE, Cutler A. Barcellos LF. An application of Random Forests to a genome-wide association dataset: methodological considerations & new findings. BMC Genetics. 2010;11:49. doi: 10.1186/1471-2156-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompel N. Prud'homme B. The causes of repeated genetic evolution. Developmental Biology. 2009;332:36–47. doi: 10.1016/j.ydbio.2009.04.040. [DOI] [PubMed] [Google Scholar]
- Gompel N, Prud'homme B, Wittkopp PJ, Kassner VA. Carroll SB. Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature. 2005;433:481–487. doi: 10.1038/nature03235. [DOI] [PubMed] [Google Scholar]
- Gompert Z, Comeault AA, Farkas TE, Feder JL, Parchman TL, Buerkle CA. Nosil P. Experimental evidence for ecological selection on genome variation in the wild. Ecology Letters. 2014;17:369–379. doi: 10.1111/ele.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Recio O. Forni S. Genome-wide prediction of discrete traits using bayesian regressions and machine learning. Genetics Selection Evolution. 2011;43:7. doi: 10.1186/1297-9686-43-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Recio O, Rosa GJM. Gianola D. Machine learning methods and predictive ability metrics for genome-wide prediction of complex traits. Livestock Science. 2014;166:217–231. [Google Scholar]
- Goodwillie C, Ritland C. Ritland K. The genetic basis of floral traits associated with mating system evolution in Leptosiphon (Polemoniaceae): an analysis of quantitative trait loci. Evolution. 2006;60:491–502. [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MR, Godiard L, Straube E, Ashfield T, Lewald J, Sattler A, Innes RW. Dangl JL. Structure of the Arabidopsis RPM1 gene enabling dual specificity disease resistance. Science. 1995;269:843–846. doi: 10.1126/science.7638602. [DOI] [PubMed] [Google Scholar]
- Gratten J, Beraldi D, Lowder B, McRae AF, Visscher P, Pemberton J. Slate J. Compelling evidence that a single nucleotide substitution in TYRP1 is responsible for coat-colour polymorphism in a free-living population of Soay sheep. Proceedings of the Royal Society B: Biological Sciences. 2007;274:619–626. doi: 10.1098/rspb.2006.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnarsson U, Hellström AR, Tixier-Boichard M, Minvielle F, Bed'hom B, Ito Si, et al. Mutations in SLC45A2 cause plumage color variation in chicken and japanese quail. Genetics. 2007;175:867–877. doi: 10.1534/genetics.106.063107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Lowe JK, Stoffel M, Daly MJ, Altshuler D, Breslow JL, Friedman JM. Pe'er I. Whole population, genome-wide mapping of hidden relatedness. Genome Research. 2009;19:318–326. doi: 10.1101/gr.081398.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Kenny EimearE, Lowe JenniferK, Salit J, Saxena R, Kathiresan S, et al. DASH: a method for identical-by-descent haplotype mapping uncovers association with recent variation. The American Journal of Human Genetics. 2011;88:706–717. doi: 10.1016/j.ajhg.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag CR, Saastamoinen M, Marden JH. Hanski I. A candidate locus for variation in dispersal rate in a butterfly metapopulation. Proceedings of the Royal Society B: Biological Sciences. 2005;272:2449–2456. doi: 10.1098/rspb.2005.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F. Bergelson J. Adaptation to climate across the Arabidopsis thaliana genome. Science. 2011a;334:83–86. doi: 10.1126/science.1209244. [DOI] [PubMed] [Google Scholar]
- Hancock AM, Witonsky DB, Alkorta-Aranburu G, Beall CM, Gebremedhin A, Sukernik R, et al. Adaptations to climate-mediated selective pressures in humans. PLoS Genetics. 2011b;7:e1001375. doi: 10.1371/journal.pgen.1001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassinen V, Tervahauta A, Halimaa P, Plessl M, Peräniemi S, Schat H, Aarts M, Servomaa K. Kärenlampi S. Isolation of Zn-responsive genes from two accessions of the hyperaccumulator plant Thlaspi caerulescens. Planta. 2007;225:977–989. doi: 10.1007/s00425-006-0403-0. [DOI] [PubMed] [Google Scholar]
- Hecht BC, Campbell NR, Holecek DE. Narum SR. Genome-wide association reveals genetic basis for the propensity to migrate in wild populations of rainbow and steelhead trout. Molecular Ecology. 2013;22:3061–3076. doi: 10.1111/mec.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology. 2013;22:4606–4618. doi: 10.1111/mec.12415. [DOI] [PubMed] [Google Scholar]
- Heffer A. Pick L. Conservation and variation in Hox genes: how insect models pioneered the evo-devo field. Annual Review of Entomology. 2013;58:161–179. doi: 10.1146/annurev-ento-120811-153601. [DOI] [PubMed] [Google Scholar]
- Hermisson J. Pennings PS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics. 2005;169:2335–2352. doi: 10.1534/genetics.104.036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines HM, Papa R, Ruiz M, Papanicolaou A, Wang C, Nijhout HF, McMillan OW. Reed RD. Transcriptome analysis reveals novel patterning and pigmentation genes underlying Heliconius butterfly wing pattern variation. BMC Genomics. 2012;13:288. doi: 10.1186/1471-2164-13-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra HE. Coyne JA. The locus of evolution: evo-Devo and the genetics of adaptation. Evolution. 2007;61:995–1016. doi: 10.1111/j.1558-5646.2007.00105.x. [DOI] [PubMed] [Google Scholar]
- Hoekstra HE. Nachman MW. Different genes underlie adaptive melanism in different populations of rock pocket mice. Molecular Ecology. 2003;12:1185–1194. doi: 10.1046/j.1365-294x.2003.01788.x. [DOI] [PubMed] [Google Scholar]
- Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA. Crossland JP. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science. 2006;313:101–104. doi: 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- Hoffman E. Goodisman M. Gene expression and the evolution of phenotypic diversity in social wasps. BMC Biology. 2007;5:23. doi: 10.1186/1741-7007-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann CM, O'Quin KE, Marshall NJ, Cronin TW, Seehausen O. Carleton KL. The eyes have it: regulatory and structural changes both underlie cichlid visual pigment diversity. PLoS Biology. 2009;7:e1000266. doi: 10.1371/journal.pbio.1000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenlohe PA, Phillips PC. Cresko WA. Using population genomics to detect selection in natural populations: key concepts and methodological considerations. International Journal of Plant Sciences. 2010;171:1059–1071. doi: 10.1086/656306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA. Cresko WA. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genetics. 2010;6:e1000862. doi: 10.1371/journal.pgen.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday J, Ritland K. Aitken S. Widespread, ecologically relevant genetic markers developed from association mapping of climate-related traits in Sitka spruce (Picea sitchensis. New Phytology. 2010;188:501–514. doi: 10.1111/j.1469-8137.2010.03380.x. [DOI] [PubMed] [Google Scholar]
- Holliday J, Wang T. Aitken S. Predicting adaptive phenotypes from multilocus genotypes in Sitka Spruce (Picea sitchensis) using random forest. G3 (Bethesda) 2012;2:1085–1093. doi: 10.1534/g3.112.002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Xu L, Liu J, Jones WD, Su Z, Ning B, et al. Technical reproducibility of genotyping SNP arrays used in genome-wide association studies. PLoS ONE. 2012;7:e44483. doi: 10.1371/journal.pone.0044483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JK, Uy JAC, Hauber ME, Hoekstra HE. Safran RJ. Vertebrate pigmentation: from underlying genes to adaptive function. Trends in Genetics. 2010;26:231–239. doi: 10.1016/j.tig.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Huggins P, Johnson CK, Schoergendorfer A, Putta S, Bathke AC, Stromberg AJ. Voss SR. Identification of differentially expressed thyroid hormone responsive genes from the brain of the Mexican Axolotl (Ambystoma mexicanum. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology. 2012;155:128–135. doi: 10.1016/j.cbpc.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Evolution of adaptive phenotypic traits without positive Darwinian selection. Heredity. 2012;108:347–353. doi: 10.1038/hdy.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter B, Wright KM. Bomblies K. Short read sequencing in studies of natural variation and adaptation. Current Opinion in Plant Biology. 2013;16:85–91. doi: 10.1016/j.pbi.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Hwang E-Y, Song Q, Jia G, Specht J, Hyten D, Costa J. Cregan P. A genome-wide association study of seed protein and oil content in soybean. BMC Genomics. 2014;15:1. doi: 10.1186/1471-2164-15-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irschick D, Reznick DN. Field experiments, introductions, and experimental evolution. In: Garland T, Rose MR, editors. Experimental Evolution: Concepts, Methods, and Applications of Selection Experiments. Oxford: Oxford University Press; 2009. pp. 173–193. [Google Scholar]
- Jarvis JP, Scheinfeldt LB, Soi S, Lambert C, Omberg L, Ferwerda B, et al. Patterns of ancestry, signatures of natural selection, and genetic association with stature in western African pygmies. PLoS Genetics. 2012;8:e1002641. doi: 10.1371/journal.pgen.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S, Rebeiz M, Andolfatto P, Werner T, True J. Carroll SB. The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell. 2008;132:783–793. doi: 10.1016/j.cell.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Jessen TH, Weber RE, Fermi G, Tame J. Braunitzer G. Adaptation of bird hemoglobins to high altitudes: demonstration of molecular mechanism by protein engineering. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:6519–6522. doi: 10.1073/pnas.88.15.6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiggins CD, Mavarez J, Beltrán M, McMillan WO, Johnston JS. Bermingham E. A genetic linkage map of the mimetic butterfly Heliconius melpomene. Genetics. 2005;171:557–570. doi: 10.1534/genetics.104.034686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SE, Beraldi D, McRae AF, Pemberton JM. Slate J. Horn type and horn length genes map to the same chromosomal region in Soay sheep. Heredity. 2010;104:196–205. doi: 10.1038/hdy.2009.109. [DOI] [PubMed] [Google Scholar]
- Johnston SE, McEwan JC, Pickering NK, Kijas JW, Beraldi D, Pilkington JG, Pemberton JM. Slate JON. Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Molecular Ecology. 2011;20:2555–2566. doi: 10.1111/j.1365-294X.2011.05076.x. [DOI] [PubMed] [Google Scholar]
- Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012;484:55–61. doi: 10.1038/nature10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost S, Bonin A, Bruford MW, DesprÉS L, Conord C, Erhardt G. Taberlet P. A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Molecular Ecology. 2007;16:3955–3969. doi: 10.1111/j.1365-294X.2007.03442.x. [DOI] [PubMed] [Google Scholar]
- Kapan DD. Three-butterfly system provides a field test of mullerian mimicry. Nature. 2001;409:338–340. doi: 10.1038/35053066. [DOI] [PubMed] [Google Scholar]
- Khan AI, Dinh DM, Schneider D, Lenski RE. Cooper TF. Negative epistasis between beneficial mutations in an evolving bacterial population. Science. 2011;332:1193–1196. doi: 10.1126/science.1203801. [DOI] [PubMed] [Google Scholar]
- Kim Y. Nielsen R. Linkage disequilibrium as a signature of selective sweeps. Genetics. 2004;167:1513–1524. doi: 10.1534/genetics.103.025387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley EP, Manceau M, Wiley CD. Hoekstra HE. Melanism in Peromyscus is caused by independent mutations in Agouti. PLoS ONE. 2009;4:e6435. doi: 10.1371/journal.pone.0006435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JC. Allele-specific gene expression uncovered. Trends in Genetics. 2004;20:113–116. doi: 10.1016/j.tig.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Kohn MH, Pelz H-J. Wayne RK. Locus-specific genetic differentiation at Rw among warfarin-resistant rat (Rattus norvegicus) populations. Genetics. 2003;164:1055–1070. doi: 10.1093/genetics/164.3.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp A. Metamodels and phylogenetic replication: a systematic approach to the evolution of developmental pathways. Evolution. 2009;63:2771–2789. doi: 10.1111/j.1558-5646.2009.00761.x. [DOI] [PubMed] [Google Scholar]
- Korte A. Farlow A. The advantages and limitations of trait analysis with GWAS: a review. Plant Methods. 2013;9:29. doi: 10.1186/1746-4811-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryazhimskiy S, Rice DP, Jerison ER. Desai MM. Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science. 2014;344:1519–1522. doi: 10.1126/science.1250939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP. Kronforst MR. doublesex is a mimicry supergene. Nature. 2014;507:229–232. doi: 10.1038/nature13112. [DOI] [PubMed] [Google Scholar]
- Lachance J, Vernot B, Elbers CC, Ferwerda B, Froment A, Bodo J-M, et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse african hunter-gatherers. Cell. 2012;150:457–469. doi: 10.1016/j.cell.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasseur R, Longin-Sauvageon C, Videmann B, Billeret M, Berny P. Benoit E. Warfarin resistance in a French strain of rats. Journal of Biochemical and Molecular Toxicology. 2006;19:379–385. doi: 10.1002/jbt.20104. [DOI] [PubMed] [Google Scholar]
- Leinonen T, McCairns RJS, O'Hara RB. Merila J. QSTFST comparisons: evolutionary and ecological insights from genomic heterogeneity. Nature Reviews Genetics. 2013;14:179–190. doi: 10.1038/nrg3395. [DOI] [PubMed] [Google Scholar]
- Lempe J, Balasubramanian S, Sureshkumar S, Singh A, Schmid M. Weigel D. Diversity of flowering responses in wild Arabidopsis thaliana strains. PLoS Genetics. 2005;1:e6. doi: 10.1371/journal.pgen.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin C. The Genetic Basis of Evolutionary Change. New York: Columbia University Press; 1974. [Google Scholar]
- Li H. Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnen CR, Kingsley EP, Jensen JD. Hoekstra HE. On the origin and spread of an adaptive allele in deer mice. Science. 2009;325:1095–1098. doi: 10.1126/science.1175826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnen CR, Poh Y-P, Peterson BK, Barrett RDH, Larson JG, Jensen JD. Hoekstra HE. Adaptive evolution of multiple traits through multiple mutations at a single gene. Science. 2013;339:1312–1316. doi: 10.1126/science.1233213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AD. Langley CH. The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Research. 1999;9:720–731. [PMC free article] [PubMed] [Google Scholar]
- Losos JB, Warheitt KI. Schoener TW. Adaptive differentiation following experimental island colonization in Anolis lizards. Nature. 1997;387:70–73. [Google Scholar]
- Lotterhos KE. Whitlock MC. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Molecular Ecology. 2014;23:2178–2192. doi: 10.1111/mec.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Hall MC, Salt DE. Willis JH. Genetic and physiological basis of adaptive salt tolerance divergence between coastal and inland Mimulus guttatus. New Phytologist. 2009;183:776–788. doi: 10.1111/j.1469-8137.2009.02901.x. [DOI] [PubMed] [Google Scholar]
- Lu Y. Rausher MD. Evolutionary rate variation in anthocyanin pathway genes. Molecular Biology and Evolution. 2003;20:1844–1853. doi: 10.1093/molbev/msg197. [DOI] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S. Taberlet P. The power and promise of population genomics: from genotyping to genome typing. Nature Reviews Genetics. 2003;4:981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- Lundgren P, Vera J, Peplow L, Manel S. van Oppen M. Genotype – environment correlations in corals from the Great Barrier Reef. BMC Genetics. 2013;14:9. doi: 10.1186/1471-2156-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri M, Sanguineti MC, Demontis A, El-Ahmed A, Garcia del Moral L, Maalouf F, et al. Association mapping in durum wheat grown across a broad range of water regimes. Journal of Experimental Botany. 2011;62:409–438. doi: 10.1093/jxb/erq287. [DOI] [PubMed] [Google Scholar]
- Mackay TFC, Stone EA. Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nature Reviews Genetics. 2009;10:565–577. doi: 10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- Main B, Bickel R, McIntyre L, Graze R, Calabrese P. Nuzhdin S. Allele-specific expression assays using Solexa. BMC Genomics. 2009;10:1–9. doi: 10.1186/1471-2164-10-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallarino R, Grant PR, Grant BR, Herrel A, Kuo WP. Abzhanov A. Two developmental modules establish 3D beak-shape variation in Darwin's finches. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4057–4062. doi: 10.1073/pnas.1011480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau M, Domingues VS, Mallarino R. Hoekstra HE. The developmental role of Agouti in color pattern evolution. Science. 2011;331:1062–1065. doi: 10.1126/science.1200684. [DOI] [PubMed] [Google Scholar]
- Manel S, Conord C. Despres L. Genome scan to assess the respective role of host-plant and environmental constraints on the adaptation of a widespread insect. BMC Evolutionary Biology. 2009;9:288. doi: 10.1186/1471-2148-9-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni J, Mason C, Mane S, Stephens M. Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Research. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH. Jiggins CD. Genomic studies of adaptation in natural populations. eLS. 2013 doi: 10.1016/j.tig.2010.08.004. John Wiley & Sons, Ltd, Chichester. doi: 10.1002/9780470015902.a9780470024613. [DOI] [PubMed] [Google Scholar]
- Martin A, Papa R, Nadeau NJ, Hill RI, Counterman BA, Halder G, et al. Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:12632–12637. doi: 10.1073/pnas.1204800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F, et al. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Research. 2013;23:1817–1828. doi: 10.1101/gr.159426.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, McCulloch K, Patel N, Briscoe A, Gilbert L. Reed R. Multiple recent co-options of optix associated with novel traits in adaptive butterfly wing radiations. EvoDevo. 2014;5:7. doi: 10.1186/2041-9139-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr E. The Growth of Biological Thought: Diversity, Evolution, and Inheritance. Cambridge, Massachusetts: Belknap Press; 1982. [Google Scholar]
- McVicker G, Gordon D, Davis C. Green P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genetics. 2009;5:e1000471. doi: 10.1371/journal.pgen.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin C, Beaver L, Taylor O, Wolfe S. Reppert S. Efficient targeted mutagenesis in the monarch butterfly using zinc-finger nucleases. Genome Research. 2012;23:159–168. doi: 10.1101/gr.145599.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill RM, Wallbank RWR, Bull V, Salazar PCA, Mallet J, Stevens M. Jiggins CD. Disruptive ecological selection on a mating cue. Proceedings of the Royal Society B: Biological Sciences. 2012;22:4907–4913. doi: 10.1098/rspb.2012.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midamegbe A, Vitalis R, Malausa T, Delava E, Cros-Arteil S. Streiff R. Scanning the European corn borer (Ostrinia spp.) genome for adaptive divergence between host-affiliated sibling species. Molecular Ecology. 2011;20:1414–1430. doi: 10.1111/j.1365-294X.2011.05035.x. [DOI] [PubMed] [Google Scholar]
- Miller CT, Beleza S, Pollen AA, Schluter D, Kittles RA, Shriver MD. Kingsley DM. cis-regulatory changes in kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 2007;131:1179–1189. doi: 10.1016/j.cell.2007.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minder AM. Widmer A. A population genomic analysis of species boundaries: neutral processes, adaptive divergence and introgression between two hybridizing plant species. Molecular Ecology. 2008;17:1552–1563. doi: 10.1111/j.1365-294X.2008.03709.x. [DOI] [PubMed] [Google Scholar]
- Mokry F, Higa R, de Alvarenga Mudadu M, Oliveira de Lima A, Meirelles SL, Barbosa da Silva MV, et al. Genome-wide association study for backfat thickness in Canchim beef cattle using Random Forest approach. BMC Genetics. 2013;14:47. doi: 10.1186/1471-2156-14-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy NI. A window on the genetics of evolution: MC1R and plumage colouration in birds. Proc Biol Sci. 2005;272:1633–1640. doi: 10.1098/rspb.2005.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy NI. Conservation and convergence of colour genetics: MC1R mutations in brown cavefish. PLoS Genetics. 2009;5:e1000388. doi: 10.1371/journal.pgen.1000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy NI, Badcock NS, Hart T, Scribner K, Janssen K. Nadeau NJ. Conserved genetic basis of a quantitative plumage trait involved in mate choice. Science. 2004;303:1870–1873. doi: 10.1126/science.1093834. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Hoekstra HE. D’ Agostino SL. The genetic basis of adaptive melanism in pocket mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5268–5273. doi: 10.1073/pnas.0431157100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau NJ. Jiggins CD. A golden age for evolutionary genetics? Genomic studies of adaptation in natural populations. Trends in Genetics. 2010;26:484–492. doi: 10.1016/j.tig.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Nadeau NJ, Martin SH, Kozak KM, Salazar C, Dasmahapatra KK, Davey JW, et al. Genome-wide patterns of divergence and gene flow across a butterfly radiation. Molecular Ecology. 2013;22:814–826. doi: 10.1111/j.1365-294X.2012.05730.x. [DOI] [PubMed] [Google Scholar]
- Narum SR. Hess JE. Comparison of FST outlier tests for SNP loci under selection. Molecular Ecology Resources. 2011;11:184–194. doi: 10.1111/j.1755-0998.2011.02987.x. [DOI] [PubMed] [Google Scholar]
- Nicholson G, Smith AV, Jónsson F, Gústafsson Ó, Stefánsson K. Donnelly P. Assessing population differentiation and isolation from single-nucleotide polymorphism data. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2002;64:695–715. [Google Scholar]
- Nielsen R, Paul JS, Albrechtsen A. Song YS. Genotype and SNP calling from next-generation sequencing data. Nature Reviews Genetics. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Korneliussen T, Albrechtsen A, Li Y. Wang J. SNP calling, genotype calling, and sample allele frequency estimation from new-generation sequencing data. PLoS ONE. 2012;7:e37558. doi: 10.1371/journal.pone.0037558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor MAF. Bennett SM. Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity. 2010;103:439–444. doi: 10.1038/hdy.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes VL, Beaumont MA, Butlin RK. Paulo OS. Challenges and pitfalls in the characterization of anonymous outlier AFLP markers in non-model species: lessons from an ocellated lizard genome scan. Heredity. 2012;109:340–348. doi: 10.1038/hdy.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Horvath S. Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson-Manning CF, Wagner MR. Mitchell-Olds T. Adaptive evolution: evaluating empirical support for theoretical predictions. Nature Reviews Genetics. 2012;13:867–877. doi: 10.1038/nrg3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. The genetic theory of adaptation: a brief history. Nature Reviews Genetics. 2005;6:119–127. doi: 10.1038/nrg1523. [DOI] [PubMed] [Google Scholar]
- Osanai-Futahashi M, Ohde T, Hirata J, Uchino K, Futahashi R, Tamura T, Niimi T. Sezutsu H. A visible dominant marker for insect transgenesis. Nature Communications. 2012;3:1295. doi: 10.1038/ncomms2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R, Voss S, Samuels A, Smith J, Putta S. Beachy C. Effect of thyroid hormone concentration on the transcriptional response underlying induced metamorphosis in the Mexican axolotl (Ambystoma. BMC Genomics. 2008;9:1–17. doi: 10.1186/1471-2164-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RB, Monaghan JR, Walker JA. Voss SR. A model of transcriptional and morphological changes during thyroid hormone-induced metamorphosis of the axolotl. General and Comparative Endocrinology. 2009;162:219–232. doi: 10.1016/j.ygcen.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R, Boley M, Smith J, Putta S. Voss S. Microarray analysis of a salamander hopeful monster reveals transcriptional signatures of paedomorphic brain development. BMC Evolutionary Biology. 2010;10:199. doi: 10.1186/1471-2148-10-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Hu T, Malley JD, Andrew AS, Karagas MR. Moore JH. A system-level pathway-phenotype association analysis using synthetic feature Random Forest. Genetic Epidemiology. 2014;38:209–219. doi: 10.1002/gepi.21794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa R, Kapan DD, Counterman BA, Maldonado K, Lindstrom DP, Reed RD, Nijhout HF, Hrbek T. McMillan OW. Multi-allelic major effect genes interact with minor effect QTLs to control adaptive color pattern variation in Heliconius erato. PLoS ONE. 2013;8:e57033. doi: 10.1371/journal.pone.0057033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchman TL, Gompert Z, Mudge J, Schilkey FD, Benkman CW. Buerkle CA. Genome-wide association genetics of an adaptive trait in lodgepole pine. Molecular Ecology. 2012;21:2991–3005. doi: 10.1111/j.1365-294X.2012.05513.x. [DOI] [PubMed] [Google Scholar]
- Paris M. Despres L. Identifying insecticide resistance genes in mosquito by combining AFLP genome scans and 454 pyrosequencing. Molecular Ecology. 2012;21:1672–1686. doi: 10.1111/j.1365-294X.2012.05499.x. [DOI] [PubMed] [Google Scholar]
- Paris M, Boyer S, Bonin A, Collado A, David J-P. Despres L. Genome scan in the mosquito Aedes rusticus: population structure and detection of positive selection after insecticide treatment. Molecular Ecology. 2010;19:325–337. doi: 10.1111/j.1365-294X.2009.04437.x. [DOI] [PubMed] [Google Scholar]
- Pavey SA, Collin H, Nosil P. Rogers SM. The role of gene expression in ecological speciation. Annals of the New York Academy of Sciences. 2010;1206:110–129. doi: 10.1111/j.1749-6632.2010.05765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavey SA, Bernatchez L, Aubin-Horth N. Landry CR. What is needed for next-generation ecological and evolutionary genomics? Trends in Ecology & Evolution. 2012;27:673–678. doi: 10.1016/j.tree.2012.07.014. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Jensen JD. Stephan W. Searching for footprints of positive selection in whole-genome SNP data from nonequilibrium populations. Genetics. 2010;185:907–922. doi: 10.1534/genetics.110.116459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Jensen JD, Stephan W. Stamatakis A. A critical assessment of story-telling: Gene Ontology categories and the importance of validating genomic scans. Molecular Biology and Evolution. 2012;29:3237–3248. doi: 10.1093/molbev/mss136. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, ZivkoviC D, Stamatakis A. Alachiotis N. SweeD: likelihood-based detection of selective sweeps in thousands of genomes. Molecular Biology and Evolution. 2013;30:2224–2234. doi: 10.1093/molbev/mst112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pespeni MH, Sanford E, Gaylord B, Hill TM, Hosfelt JD, Jaris HK, et al. Evolutionary change during experimental ocean acidification. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6937–6942. doi: 10.1073/pnas.1220673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfender WF, Saha MC, Johnson EA. Slabaugh MB. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theoretical and Applied Genetics. 2011;122:1467–1480. doi: 10.1007/s00122-011-1546-3. [DOI] [PubMed] [Google Scholar]
- Pool JE, Hellmann I, Jensen JD. Nielsen R. Population genetic inference from genomic sequence variation. Genome Research. 2010;20:291–300. doi: 10.1101/gr.079509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto-Neto L, Kijas J. Reverter A. The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genetics Selection Evolution. 2014;46:22. doi: 10.1186/1297-9686-46-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Pickrell JK. Coop G. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Current Biology. 2010;20:R208–R215. doi: 10.1016/j.cub.2009.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protas ME, Hersey C, Kochanek D, Zhou Y, Wilkens H, Jeffery WR, Zon LI, Borowsky R. Tabin CJ. Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nature Genetics. 2006;38:107–111. doi: 10.1038/ng1700. [DOI] [PubMed] [Google Scholar]
- Prud'homme B, Gompel N, Rokas A, Kassner VA, Williams TM, Yeh S-D, True JR. Carroll SB. Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature. 2006;440:1050–1053. doi: 10.1038/nature04597. [DOI] [PubMed] [Google Scholar]
- Rank N. A hierarchical analysis of genetic differentiation in a montane leaf beetle Chrysomela aeneicollis (coleoptera: chrysomelidae) Evolution. 1992;46:1097–1111. doi: 10.1111/j.1558-5646.1992.tb00622.x. [DOI] [PubMed] [Google Scholar]
- Rausher MD. The evolution of flavonoids and their genes. In: Grotewold E, editor. The Science of Flavonoids. New York: Springer; 2006. pp. 175–211. [Google Scholar]
- Rebeiz M, Pool JE, Kassner VA, Aquadro CF. Carroll SB. Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population. Science. 2009;326:1663–1667. doi: 10.1126/science.1178357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed RD, Papa R, Martin A, Hines HM, Counterman BA, Pardo-Diaz C, et al. Optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science. 2011;333:1137–1141. doi: 10.1126/science.1208227. [DOI] [PubMed] [Google Scholar]
- Renaut S, Nolte AW, Rogers SM, Derome N. Bernatchez L. SNP signatures of selection on standing genetic variation and their association with adaptive phenotypes along gradients of ecological speciation in lake whitefish species pairs (Coregonus spp.) Molecular Ecology. 2011;20:545–559. doi: 10.1111/j.1365-294X.2010.04952.x. [DOI] [PubMed] [Google Scholar]
- Reznick DN, Shaw FH, Rodd FH. Shaw RG. Evaluation of the rate of evolution in natural populations of guppies (Poecilia reticulata. Science. 1997;275:1934–1937. doi: 10.1126/science.275.5308.1934. [DOI] [PubMed] [Google Scholar]
- Roberts RB, Ser JR. Kocher TD. Sexual conflict resolved by invasion of a novel sex determiner in lake malawi cichlid fishes. Science. 2009;326:998–1001. doi: 10.1126/science.1174705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV. The QTN program and the alleles that matter for evolution: all that's gold does not glitter. Evolution. 2011;66:1–17. doi: 10.1111/j.1558-5646.2011.01486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV. Kruglyak L. Genetics of global gene expression. Nature Reviews Genetics. 2006;7:862–872. doi: 10.1038/nrg1964. [DOI] [PubMed] [Google Scholar]
- Roda F, Liu H, Wilkinson MJ, Walter GM, James ME, Bernal DM, et al. Convergence and divergence during the adaptation to similar environments by an Australian groundsel. Evolution. 2013;67:2515–2529. doi: 10.1111/evo.12136. [DOI] [PubMed] [Google Scholar]
- Rogers SM. Bernatchez L. Integrating QTL mapping and genome scans towards the characterization of candidate loci under parallel selection in the lake whitefish (Coregonus clupeaformis. Molecular Ecology. 2005;14:351–361. doi: 10.1111/j.1365-294X.2004.02396.x. [DOI] [PubMed] [Google Scholar]
- Rokyta DR, Joyce P, Caudle SB. Wichman HA. An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nature Genetics. 2005;37:441–444. doi: 10.1038/ng1535. [DOI] [PubMed] [Google Scholar]
- Rosenblum E, Hoekstra H. Nachman M. Adaptive reptile color variation and the evolution of the Mc1r gene. Evolution. 2004;58:1794–1808. doi: 10.1111/j.0014-3820.2004.tb00462.x. [DOI] [PubMed] [Google Scholar]
- Salomé PA, Bomblies K, Laitinen RAE, Yant L, Mott R. Weigel D. Genetic architecture of flowering-time variation in Arabidopsis thaliana. Genetics. 2011;188:421–433. doi: 10.1534/genetics.111.126607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen O, Lascoux M. Merila J. Ecological genomics of local adaptation. Nature Reviews Genetics. 2013;14:807–820. doi: 10.1038/nrg3522. [DOI] [PubMed] [Google Scholar]
- Scheinfeldt LB. Tishkoff SA. Recent human adaptation: genomic approaches, interpretation and insights. Nature Reviews Genetics. 2013;14:692–702. doi: 10.1038/nrg3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C. Hitchhiking mapping - functional genomics from the population genetics perspective. Trends in Genetics. 2003;19:32–38. doi: 10.1016/s0168-9525(02)00012-4. [DOI] [PubMed] [Google Scholar]
- Schoville S, Barreto F, Moy G, Wolff A. Burton R. Investigating the molecular basis of local adaptation to thermal stress: population differences in gene expression across the transcriptome of the copepod Tigriopus californicus. BMC Evolutionary Biology. 2012;12:170. doi: 10.1186/1471-2148-12-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte PM, Glémet HC, Fiebig AA. Powers DA. Adaptive variation in lactate dehydrogenase-B gene expression: role of a stress-responsive regulatory element. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6597–6602. doi: 10.1073/pnas.97.12.6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MD, Bell MA. Kingsley DM. Parallel genetic origins of pelvic reduction in vertebrates. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13753–13758. doi: 10.1073/pnas.0604706103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jónsson B, Schluter D. Kingsley DM. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature. 2004;428:717–723. doi: 10.1038/nature02415. [DOI] [PubMed] [Google Scholar]
- Shimizu K. Purugganan M. Evolutionary and ecological genomics of Arabidopsis. Plant Physiology. 2005;138:578–584. doi: 10.1104/pp.105.061655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slate J. From beavis to beak color: a simulation study to examine how much QTL mapping can reveal about the genetic architecture of quantitative traits. Evolution. 2013;67:1251–1262. doi: 10.1111/evo.12060. [DOI] [PubMed] [Google Scholar]
- Slate J, Visscher PM, MacGregor S, Stevens D, Tate ML. Pemberton JM. A genome scan for quantitative trait loci in a wild population of red deer (Cervus elaphus. Genetics. 2002;162:1863–1873. doi: 10.1093/genetics/162.4.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slate J, Santure AW, Feulner PGD, Brown EA, Ball AD, Johnston SE. Gratten J. Genome mapping in intensively studied wild vertebrate populations. Trends in Genetics. 2010;26:275–284. doi: 10.1016/j.tig.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Stapley J, Reger J, Feulner PGD, Smadja C, Galindo J, Ekblom R, et al. Adaptation genomics: the next generation. Trends in Ecology & Evolution. 2010;25:705–712. doi: 10.1016/j.tree.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Stern DL. Orgogozo V. The loci of evolution: how predictable is genetic evolution? Evolution. 2008;62:2155–2177. doi: 10.1111/j.1558-5646.2008.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL. Orgogozo V. Is genetic evolution predictable? Science. 2009;323:746–751. doi: 10.1126/science.1158997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe JR. Hoekstra HE. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity. 2007;100:158–170. doi: 10.1038/sj.hdy.6800937. [DOI] [PubMed] [Google Scholar]
- Storz JF. Using genome scans of DNA polymorphism to infer adaptive population divergence. Molecular Ecology. 2005;14:671–688. doi: 10.1111/j.1365-294X.2005.02437.x. [DOI] [PubMed] [Google Scholar]
- Stranger BE, Stahl EA. Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187:367–383. doi: 10.1534/genetics.110.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streelman J, Albertson R. Kocher T. Genome mapping of the orange blotch colour pattern in cichlid fishes. Molecular Ecology. 2003;12:2465–2471. doi: 10.1046/j.1365-294x.2003.01920.x. [DOI] [PubMed] [Google Scholar]
- Streisfeld MA. Rausher MD. Altered trans-regulatory control of gene expression in multiple anthocyanin genes contributes to adaptive flower color evolution in Mimulus aurantiacus. Molecular Biology and Evolution. 2009;26:433–444. doi: 10.1093/molbev/msn268. [DOI] [PubMed] [Google Scholar]
- Streisfeld MA, Young WN. Sobel JM. Divergent selection drives genetic differentiation in an R2R3-MYB transcription factor that contributes to incipient speciation in Mimulus aurantiacus. PLoS Genetics. 2013;9:e1003385. doi: 10.1371/journal.pgen.1003385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Terai Y, Imai H, Turner GF, Koblmüller S, Sturmbauer C, Shichida Y. Okada N. Parallelism of amino acid changes at the RH1 affecting spectral sensitivity among deep-water cichlids from Lakes Tanganyika and Malawi. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5448–5453. doi: 10.1073/pnas.0405302102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh Y. Vijg J. SNP discovery in associating genetic variation with human disease phenotypes. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;573:41–53. doi: 10.1016/j.mrfmmm.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Terai Y, Seehausen O, Sasaki T, Takahashi K, Mizoiri S, Sugawara T, et al. Divergent selection on opsins drives incipient speciation in lake Victoria cichlids. PLoS Biology. 2006;4:e433. doi: 10.1371/journal.pbio.0040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenius O, Papanicolaou A, Garbutt JS, Eleftherianos I, Huvenne H, Kanginakudru S, et al. RNA interference in Lepidoptera: an overview of successful and unsuccessful studies and implications for experimental design. Journal of Insect Physiology. 2011;57:231–245. doi: 10.1016/j.jinsphys.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Tornero P, Chao R, Luthin W, Goff S. Dangl J. Large-scale structure-function analysis of the Arabidopsis RPM1 disease resistance protein. Plant Cell. 2002;14:435–450. doi: 10.1105/tpc.010393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Torres M, Sanchez P, Fernandez-Delmond I. Grant M. Expression profiling of the host response to bacterial infection: the transition from basal to induced defence responses in RPM1-mediated resistance. Plant J. 2003;33:665–676. doi: 10.1046/j.1365-313x.2003.01653.x. [DOI] [PubMed] [Google Scholar]
- Tsai IJ, Zarowiecki M, Holroyd N, Garciarrubio A, Sanchez-Flores A, Brooks KL, et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature. 2013;496:57–63. doi: 10.1038/nature12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchin MC, Chiang CWK, Palmer CD, Sankararaman S, Reich D. Hirschhorn JN. Evidence of widespread selection on standing variation in Europe at height-associated SNPs. Nature Genetics. 2012;44:1015–1019. doi: 10.1038/ng.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TL. Fine-mapping natural alleles: quantitative complementation to the rescue. Molecular Ecology. 2014;23:2377–2382. doi: 10.1111/mec.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TL. Hahn MW. Genomic islands of speciation or genomic islands and speciation? Molecular Ecology. 2010;19:848–850. doi: 10.1111/j.1365-294X.2010.04532.x. [DOI] [PubMed] [Google Scholar]
- Turner TL, Hahn MW. Nuzhdin SV. Genomic islands of speciation in Anopheles gambiae. PLoS Biology. 2005;3:e285. doi: 10.1371/journal.pbio.0030285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uy J, Moyle R, Filardi C. Cheviron Z. Difference in plumage color used in species recognition between incipient species is linked to a single amino acid substitution in the melanocortin-1 receptor. The American Naturalist. 2009;174:244–254. doi: 10.1086/600084. [DOI] [PubMed] [Google Scholar]
- Villar E, Klopp C, Noirot C, Novaes E, Kirst M, Plomion C. Gion J. RNA-Seq reveals genotype-specific molecular responses to water deficit in eucalyptus. BMC Genomics. 2011;12:538. doi: 10.1186/1471-2164-12-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villemereuil P, Frichot É, Bazin É, François O. Gaggiotti OE. Genome scan methods against more complex models: when and how much should we trust them? Molecular Ecology. 2014;23:2006–2019. doi: 10.1111/mec.12705. [DOI] [PubMed] [Google Scholar]
- Vitalis R, Dawson K, Boursot P. Belkhir K. DetSel 1.0: a computer program to detect markers responding to selection. Journal of Heredity. 2003;94:429–431. doi: 10.1093/jhered/esg083. [DOI] [PubMed] [Google Scholar]
- Voss SR. Shaffer HB. Adaptive evolution via a major gene effect: paedomorphosis in the Mexican axolotl. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:14185–14189. doi: 10.1073/pnas.94.25.14185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M. Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JN, Peterson BK. Hoekstra HE. Discrete genetic modules are responsible for complex burrow evolution in Peromyscus mice. Nature. 2013;493:402–405. doi: 10.1038/nature11816. [DOI] [PubMed] [Google Scholar]
- Wei C, Liu J, Yu Z, Zhang B, Gao G. Jiao R. TALEN or Cas9 - Rapid, efficient and specific choices for genome modifications. Journal of Genetics and Genomics. 2013;40:281–289. doi: 10.1016/j.jgg.2013.03.013. [DOI] [PubMed] [Google Scholar]
- Weinig C, Dorn LA, Kane NC, German ZM, Halldorsdottir SS, Ungerer MC, et al. Heterogeneous selection at specific loci in natural environments in Arabidopsis thaliana. Genetics. 2003;165:321–329. doi: 10.1093/genetics/165.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessinger CA. Rausher MD. Lessons from flower colour evolution on targets of selection. Journal of Experimental Botany. 2012;63:5741–5749. doi: 10.1093/jxb/ers267. [DOI] [PubMed] [Google Scholar]
- Westram AM, Galindo J, Alm Rosenblad M, Grahame JW, Panova M. Butlin RK. Do the same genes underlie parallel phenotypic divergence in different Littorina saxatilis populations? Molecular Ecology. 2014;23:4603–4616. doi: 10.1111/mec.12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman D, Agrawal A. What is phenotypic plasticity and why is it important? In: Ananthakrishnan DWWTN, editor. Phenotypic Plasticity of Insects: Mechanisms and Consequences. Boca Raton: CRC Press; 2009. pp. 1–63. [Google Scholar]
- Wittkopp PJ. Using pyrosequencing to measure allele-specific mRNA abundance and infer the effects of cis- and trans-regulatory differences. In: Orgogozo V, Rockman MV, editors. Molecular Methods for Evolutionary Genetics. New York: Humana Press; 2011. pp. 297–317. [DOI] [PubMed] [Google Scholar]
- Wood HM, Grahame JW, Humphray S, Rogers J. Butlin RK. Sequence differentiation in regions identified by a genome scan for local adaptation. Molecular Ecology. 2008;17:3123–3135. doi: 10.1111/j.1365-294X.2008.03755.x. [DOI] [PubMed] [Google Scholar]
- Wray GA. Genomics and the evolution of phenotypic traits. Annual Review of Ecology, Evolution, and Systematics. 2013;44:51–72. [Google Scholar]
- Yang W. Charles Gu C. Random forest fishing: a novel approach to identifying organic group of risk factors in genome-wide association studies. European Journal of Human Genetics. 2014;22:254–259. doi: 10.1038/ejhg.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Qi Y, Bi K. Fu J. Toward understanding the genetic basis of adaptation to high-elevation life in poikilothermic species: a comparative transcriptomic analysis of two ranid frogs, Rana chensinensis and R. kukunoris. BMC Genomics. 2012;13:588. doi: 10.1186/1471-2164-13-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Pan S, Wang J, Dixon A, He J, Muller MG, et al. Peregrine and saker falcon genome sequences provide insights into evolution of a predatory lifestyle. Nature Genetics. 2013;45:563–566. doi: 10.1038/ng.2588. [DOI] [PubMed] [Google Scholar]
- Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, Fang X, et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2013;339:456–460. doi: 10.1126/science.1230835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuellig MP, Kenney AM. Sweigart AL. Evolutionary genetics of plant adaptation: insights from new model systems. Current Opinion in Plant Biology. 2014;18:44–50. doi: 10.1016/j.pbi.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufall RA. Rausher MD. The genetic basis of a flower color polymorphism in the common morning glory (Ipomoea purpurea. Journal of Heredity. 2003;94:442–448. doi: 10.1093/jhered/esg098. [DOI] [PubMed] [Google Scholar]
- Zufall RA. Rausher MD. Genetic changes associated with floral adaptation restrict future evolutionary potential. Nature. 2004;428:847–850. doi: 10.1038/nature02489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This manuscript does not use data.