

Abstract
Aims
Membrane distillation bioreactors (MDBR) have potential for industrial applications where wastewater is hot or waste heat is available, but the role of micro-organisms in MDBRs has never been determined, and thus was the purpose of this study.
Methods and Results
Microbial communities were characterized by bacterial and archaeal 16S and eukaryotic 18S rRNA gene tag-encoded pyrosequencing of DNA obtained from sludge. Taxonomy-independent analysis revealed that bacterial communities had a relatively low richness and diversity, and community composition strongly correlated with conductivity, total nitrogen and bound extracellular polymeric substances (EPS). Taxonomy-dependent analysis revealed that Rubrobacter and Caldalkalibacillus were abundant members of the bacterial community, but no archaea were detected. Eukaryotic communities had a relatively high richness and diversity, and both changes in community composition and abundance of the dominant genus, Candida, correlated with bound EPS.
Conclusions
Thermophilic MDBR communities were comprised of a low diversity bacterial community and a highly diverse eukaryotic community with no archea detected. Communities exhibited low resilience to changes in operational parameters. Specifically, retenatate nutrient composition and concentration was strongly correlated with the dominant species.
Significance and Impact of the Study
This study provides an understanding of microbial community diversity in an MDBR, which is fundamental to the optimization of reactor performance.
Keywords: community composition, community diversity, membrane biofouling, membrane distillation bioreactor, thermophiles
Introduction
Increased demand for water worldwide has led to the proposed use of industrial and domestic wastewater as a potential resource to relieve water scarcity. Various technologies have been developed to recycle and recover wastewater, including the membrane bioreactor (MBR), which has attracted much attention. MBRs combine biodegradation with membrane filtration and retains contaminants in the reactor, producing high quality product water (Stamper et al. 2003). There is an emerging interest in novel MBR systems that retain persistent and low molecular weight contaminants, thus achieving higher quality product water than conventional MBRs (Lay et al. 2011).
The membrane distillation bioreactor (MDBR) is a novel MBR system that is driven by evaporative pressure, allowing only volatile components such as water vapour to permeate the membrane (Phattaranawik et al. 2008). The membrane distillation (MD) process is operated under moderately high temperatures (45–80°C) and thus, unlike conventional MBRs, heat tolerant organisms would be required to perform biodegradation.
Efforts have been made to identify microbial community composition and functional microbial groups in different MBR systems. Miura et al.(2007) analysed the bacterial community structure in both conventional and hybrid MBRs combined with precoagulation/sedimentation steps, using denaturing gradient gel electrophoresis (DGGE) and fluorescence in situ hybridization (FISH). A marked change in community structure was detected when the MBRs did not perform stably, while a perpetual fluctuation of the bacterial community was observed when stable performance was achieved. Their results suggest that both the composition and stability of microbial communities in the MBR were important for maintenance of stable performance when treating complex municipal wastewater. The investigation of bacterial communities in a greywater MBR also demonstrated that community structure was affected by operational conditions, and in particular by the nutrient concentration of the feed solution (Stamper et al. 2003). Similarly, the investigation of the influence of operational parameters and environmental variables on the structure of microbial communities in an MBR showed that hydraulic retention time (HRT), volatile suspended solids and temperature strongly influenced the community structure (Molina-Munoz et al. 2009). As demonstrated by these studies, to optimize the biodegradation of contaminants in wastewater in MBRs, it is important to determine the effects of operational conditions on the structure and stability of microbial communities.
Culture-independent technologies such as FISH and DGGE, have provided insights into microbial communities in wastewater treatment systems, but have limitations, e.g. DGGE only detects the most abundant populations in the system (Lu et al. 2012; Zhang et al. 2012). Recently, techniques such as pyrosequencing have been used to analyse these systems, providing a more comprehensive assessment of the community (Hu et al. 2012; Lu et al. 2012; Wang et al. 2012). For example, thousands of operational taxonomic units (OTUs) were detected in activated sludge from 14 sewage treatment plants and in the bacterial community of a conventional MBR (3% cut-off level) (Zhang et al. 2012; Ma et al. 2013). Here, we characterized the overall microbial community composition and dynamics in an MDBR by 16S and 18S rRNA gene-tagged pyrosequencing. Correlation analysis was done to determine the environmental parameters that influence community composition.
Materials and methods
Submerged MDBR set up and operation
A flat sheet polyvinylidene fluoride (PVDF) membrane module with a 0·22 μm pore size, 125 μm thickness and 75% porosity, was submerged in the MDBR. Synthetic concentrated feed solution (12·4 g l−1 chemical oxygen demand (COD), 5·2 g l−1 total organic carbon (TOC), 0·33 g l−1 total nitrogen (TN) was placed at 4°C and diluted with heated distilled water before addition to the bioreactor. The reactor temperature was maintained at 55 ± 1°C by a hot water jacket as described previously (Goh et al. 2013). An air diffuser was installed beneath the membrane module to create airlift for liquid circulation and membrane scouring. Water level sensors were used to detect decreases in the feed solution, and synthetic feed was automatically added into the feed tank if necessary. The permeate was connected to a cooling tank to create thermal pressure between the two sides of membrane. Overflow of the permeate side was collected by a parallel tank located on a mass balance. The labview data logging system (National Instruments™, Singapore PCI 6229, software: labview 7) was integrated for acquisition of various parameters, including temperature, conductivity, pH and dissolved oxygen (DO).
The MDBR was operated at stable sludge retention time (SRT) and temperature for 202 days. Forty millilitres of sludge were discharged every day so the SRT was 112·5 days. The HRT was determined by permeate flux and membrane area, and varied with flux. Other operational parameters are listed in Table1. The pH of the mixed liquor automatically increased due to the stripping of CO2 (which is caused by the low solubility of CO2 in water at high temperature). At an alkaline pH, most of the ammonium generated by biological nutrient degradation would be in the form of ammonia gas which can diffuse across the membrane into the permeate. To reduce ammonia permeation, the pH was adjusted to maintain the retentate in the range 7·5–8·5. The MDBR was therefore operated in two phases, the first without pH adjustment (with an average retentate pH of 10, days 0–105) and the second phase with pH adjustment (average retentate pH of 8, days 106–202).
Table 1.
Membrane distillation bioreactor operational parameters
| Operational parameters | Values |
|---|---|
| Feed side | |
| Reactor temperature, Tr (°C) | 55·5 |
| Cross-sectional area (m2) | 0·8 × 10−3 |
| Dissolved oxygen (DO, mg l−1) | 3–4 |
| Airflow rate (L min−1) | 12 |
| Permeate side | |
| Permeate temperature, Tp,in (°C) | 19·5 |
| Cross-sectional area (m2) | 1·68 × 10−3 |
| Permeate recirculation flowrate, Qp (ml min−1) | 350 |
Analytical methods for assessment of MDBR sludge
Sludge biomass was collected by centrifugation at 18 920 g (Hitachi, Tokyo, Japan CT15RE) for 20 min, for the measurement of TOC and TN (Shimadzu TOC Analyser Model: TOC-V CSH). Biomass concentration was determined via the total suspended solids (TSS) method (American Public Health Association, American Water Works Association, Water Pollution Control Federation, and Water Environment Federation 1965). The extraction of loose and bound extracellular polymeric substances (EPS) was conducted as previously reported (Zhang et al. 2006) and the analysis of carbohydrates and proteins were conducted by the phenol-sulphuric method and Bradford test, respectively. The bound EPS refers to the EPS on the biomass surface extracted via the centrifugation-NaOH method (Liu and Fang 2002) while the loose EPS refers to the EPS in the supernatant.
Analysis of microbial community composition
Samples were collected every week for analysis by DGGE. According to the DGGE profile, eight samples from the MDBR system which were dissimilar to each other were further analysed:
Without pH adjustment: 13th (Md13), 23rd (Md23), 40th (Md40), 82th (Md82), 103rd (Md103) day
With pH adjustment: 110th (Md110), 172nd (Md172) and 201th (Md201) day
DNA was extracted from sludge samples by a modified cetyltrimethyl ammonium bromide (CTAB) method (Lay et al. 2012). The purified DNA solution was amplified by three fusion primer pairs: Gray 28F (GAGTTTGATCN TGGCTCAG)—Gray 518R (GTNTTACNGCGGCKGC TG) for amplification of bacterial 16S rDNA, Arch349F (GYGCASCAGKCGMGAAW) —Arch806R (GGACTACV SGGGTATCTAAT) for archaeal 16S rDNA and Euk SSU euk7F (AACCTGGTTGATCCTGCCAGT)-euk570R (GCT ATTGGAGCTGGAATTA) for 18S rRNA. All sequence reads were analysed in Mothur using the commands for trimming, alignment, chimeric reads check and removal, clustering and generating an OTU abundance matrix. Then both the OTU-based and phylotype-based approaches were taken to analyse the community diversity. As described in our previous study (Zhang et al. 2014), rarefaction curves obtained from Mothur were plotted in R 2.15.0. The alpha diversity was evaluated by numbers of OTUs, Chao 1 and Inversimpson estimators, and Shannon Weaver index. Resemblance analysis based on the algorithm of Bray-Curtis similarity was presented as nonmetric multidimentional scaling (NMDS) plots in Primer V6 (PRIMER-E Ltd, Lutton, UK). Furthermore, Bio-Env tool of Primer correlated the datasets of biological communities with the environmental variables. The phylotype-based approach assigned the effective sequences to taxonomic units based on classification against the SILVA database.
Results
Performance of biological processes in MDBR system
Flux fluctuated over the entire 202 days of operation (Fig.1a). The membranes were changed at a frequency of 20–30 days due to fouling and wetting (Goh et al. 2012>). During the first phase of operation, the flux increased to 7·91 Lm−2 h−1 after a new membrane was installed on day 1, and declined to as low as 2·2 Lm−2 h−1 after 27 days of continuous operation. After 70 days, a clean membrane was introduced and the flux was <6 Lm−2 h−1. During the second phase of operation, the initial flux reduced from 5·78 (105th day) to 3·08 Lm−2 h−1 (185th day) on the day a new membrane was installed, indicating that the permeate product was decreasing over time. However, water quality as measured by TOC removal efficiency showed that this MDBR was able to remove more than 98% of the hydrocarbon compounds. TN removal efficiency was 86·7% on average during the first phase and reached an average of 99·8% during the second phase. Therefore, the pH adjustment from 9 to 10 (1st phase) to 7–8 (2nd phase) allowed effective removal of ammonia from the evaporated water.
Figure 1.
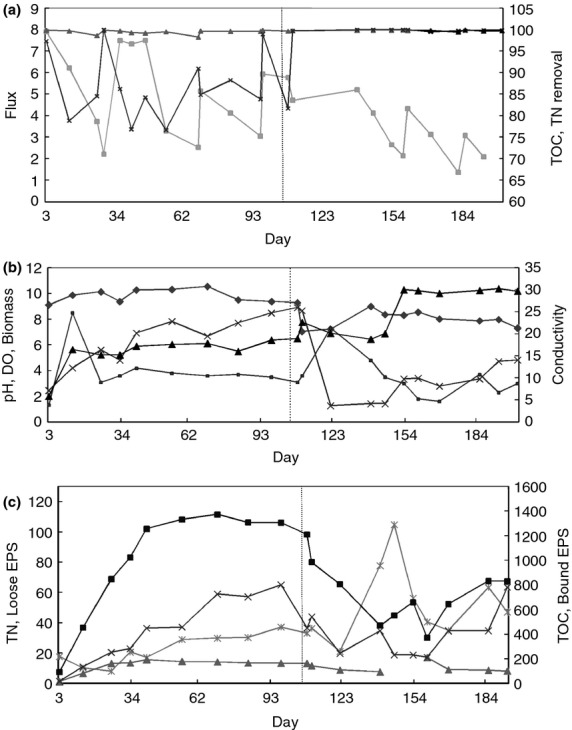
Membrane distillation bioreactor (MDBR) performance and environmental variables of an MDBR over 202 days of operation. (a) Values of flux (LMH, ▪) are plotted on the primary vertical axis; total organic carbon (TOC) removal efficiency (%, ▴) and total nitrogen (TN) removal efficiency (%, ×) are plotted on the secondary axis. (b) pH (♦), dissolved oxygen (DO) (mg l−1, ▪), biomass (g l−1, ×) are plotted on the primary vertical axis, and conductivity (mS cm−1, ▴) on the secondary axis. (c) TN (mg l−1, ▴) and soluble extracellular polymeric substances (EPS) (mg l−1, ×) are plotted on the primary axis while TOC (mg l−1, ▪) and bound EPS (mg l−1, *) are plotted on the secondary axis. The pH was adjusted from the 106th day onwards (dash line).
Throughout the entire MDBR operation period (202 days), parameters such as pH, DO, salinity, TOC, TN, biological biomass, soluble EPS (in the mixed liquor) and bound EPS (attached to microorganisms) were periodically measured (Fig.1b,c). The DO fluctuated and then stabilized after 30 days during phase 1 (days 1–105; Fig.1b). Salinity increased from 5 to 16·5 mS cm−1 in phase 1 due to the accumulation of residual undigested inorganic matters, before stabilizing at c. 17 mS cm−1. As can be seen by TOC and TN values (Fig.1c), the concentrations of carbohydrates and proteins increased during the first 40 days before stabilizing at c. 1300 and 170 mg l−1, respectively. The biomass concentration increased with time and reached a maximum of 8·96 g l−1 (Fig.1b), indicating that the feed adequately supported the growth of the microorganisms. Like the biomass concentration, both the soluble and bound EPS concentrations were observed to increase with time.
In the second phase of operation (days 106–202), HCl was added to adjust the pH to 7·5–8·5. Salinity increased gradually in the first 10 days of phase 2, then decreased to below 20 mS cm−1, before a large increase from 20 to 30 mS cm−1 between the 145th and 153rd day, after which it stabilized at c. 30 mS cm−1 (Fig.1b). The addition of HCl on day 106 resulted in the addition of chloride ions which increased conductivity. In addition, the lower pH likely increased the solubility of inorganic matters, resulting in an increase in conductivity. The sudden change of pH on days 103–110 resulted in a sharp decline in TOC, TN, biomass and related EPS production. Once the pH had stabilized, biomass, bound EPS and soluble EPS increased, however, the values were slightly lower than before pH adjustment. The slightly lower retentate TOC and TN concentrations may be attributed to greater TOC and TN removal efficiency at pH 8.
Analysis of bacterial communities using a taxonomy-independent approach
Surprisingly, no archaea were detected in the reactor throughout the entire 202 days of operation. The bacterial (Md13B etc) and eukaryotic communities (Md13E etc) were analysed by both taxonomy-independent and taxonomy-dependent approaches. The bacterial communities yielded 4195 reads per sample after processing, with an average length of 362 bp (Table2). All the datasets were normalized to 3034 reads as the minimum size. The bacterial community was comprised of 200 observed OTUs at a clustering distance of 3% at the beginning of operation, and then fluctuated between 117 and 138 in the first phase. Despite the increase in biomass and EPS concentrations with time, the diversity of the bacterial communities did not change significantly, but remained relatively stable at c. 125 OTUs. In contrast, while the biomass concentration in the mixed liquor after pH adjustment was much lower in the second phase than the first phase, the bacterial community diversity was observed to increase throughout the second phase of operation, reaching a high of 319 OTUs.
Table 2.
Number of sequences and alpha diversity of all bacterial and eukaryotic communities
| Sample | Effective reads | Operational taxonomic units | Chao 1 | Invsimpson | ACE | Shannon | Good's coverage (%)* |
|---|---|---|---|---|---|---|---|
| Md13B | 3944 | 200 | 681 | 5·84 | 1611 | 2·53 | 95·19 |
| Md23B | 3769 | 136 | 462 | 4·54 | 1005 | 2·08 | 96·84 |
| Md40B | 4687 | 123 | 328 | 2·82 | 602 | 1·75 | 97·40 |
| Md82B | 4107 | 138 | 485 | 4·76 | 1357 | 2·08 | 96·74 |
| Md103B | 4880 | 117 | 315 | 3·03 | 535 | 1·69 | 97·53 |
| Md110B | 4318 | 156 | 548 | 3·88 | 1021 | 2·02 | 96·41 |
| Md172B | 4827 | 162 | 664 | 5·30 | 1763 | 2·25 | 96·08 |
| Md201B | 3034 | 319 | 816 | 5·04 | 1552 | 2·77 | 92·78 |
| Md13E | 3108 | 370 | 728 | 17·3 | 1086 | 3·88 | 93·15 |
| Md23E | 5932 | 387 | 736 | 15·5 | 1065 | 3·82 | 92·83 |
| Md40E | 4496 | 451 | 977 | 20·8 | 1370 | 4·21 | 91·70 |
| Md82E | 6834 | 401 | 742 | 12·4 | 1189 | 3·81 | 92·76 |
| Md103E | 7427 | 331 | 567 | 10·0 | 773 | 3·58 | 94·66 |
| Md110E | 5010 | 408 | 717 | 13·1 | 1017 | 3·87 | 92·92 |
| Md172E | 9846 | 260 | 480 | 7·3 | 697 | 3·06 | 95·56 |
| Md201E | 6165 | 312 | 581 | 8·1 | 734 | 3·33 | 94·76 |
Coverage is C = 1 − n/N, where n is the number of operational taxonomic units that have been sampled once, and N is the total number of individuals in the sample.
To determine the diversity of the bacterial communities, various estimation indices were calculated and are listed in Table2. Shannon Weaver index is independent of the size of sequencing datasets, and therefore, was used for the comparison between different communities. Compared with other thermophilic studies for pollutant removal where a Shannon index of more than 3·0 were reported (Mardanov et al. 2010; Li et al. 2013), the lower Shannon index observed in the MDBR in this study revealed less richness and diversity, implying that the conditions in this MDBR were more restrictive or due to simpler substrates in the synthetic wastewater. The coverage of most samples was saturated (>96%) except the first (Md13B) and the last (Md201B) samples (Fig.2a).
Figure 2.
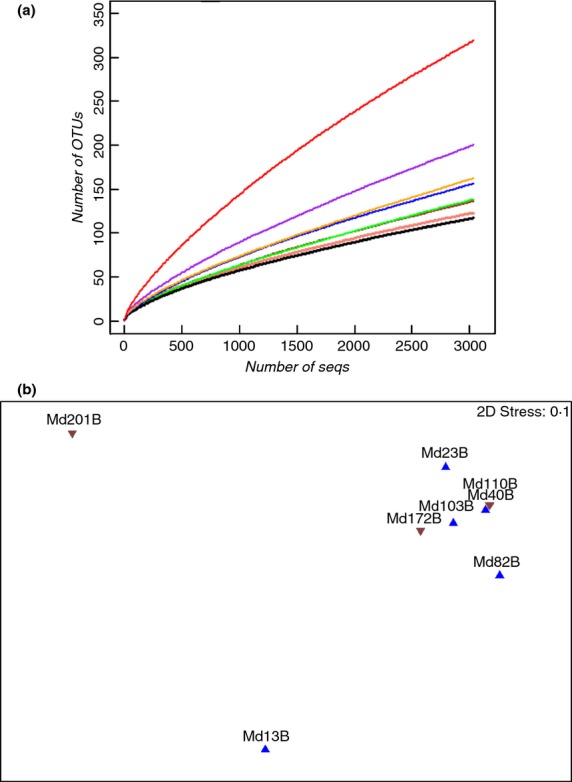
Rarefaction curves (a) and nonmetric multidimentional scaling (NMDS) (b) of eight MD bacterial communities. The samples before pH adjustment are shown as “ ” and after pH adjustment are represented by ‘
” and after pH adjustment are represented by ‘ ’. All the datasets were normalized to have the same number of reads (3034), and an operational taxonomic unit was defined to be no more than 3% distance within one unit. The stress of NMDS plot was set to be 0·1 at minimum to discern the position of all the samples. (
’. All the datasets were normalized to have the same number of reads (3034), and an operational taxonomic unit was defined to be no more than 3% distance within one unit. The stress of NMDS plot was set to be 0·1 at minimum to discern the position of all the samples. ( ) Md13B; (
) Md13B; ( ) Md23B; (
) Md23B; ( ) Md40B; (
) Md40B; ( ) Md82B; (
) Md82B; ( ) Md103B; (
) Md103B; ( ) Md110B; (
) Md110B; ( ) Md172B and (
) Md172B and ( ) Md20lB
) Md20lB
The OTU abundance matrix was generated by the clustering of all the effective reads with a cut-off of 0·03. NMDS plots demonstrated that the eight bacterial communities fell into three groups (Fig.2b). Samples from day 13 (Md13B) and day 201 (Md201B) clustered as separate groups while the remaining six samples clustered as one group. The dissimilarity among the six samples was <50%; the sample on the 13th day had a distance of 62% and the sample on the 201st day a distance of 81% from the other six samples. From the 13th to the 25th day, environmental factors such as pH and salinity were stable (Fig.1b), while the TOC and TN almost doubled (Fig.1c). Similarly, from the 172nd to the 201st day, the pH and salinity were stable, but the retentate TOC doubled while the TN decreased by half. Although a large change in pH and salinity was observed during the transition period from the 103rd to 110th day, which may have indirectly resulted in decreases in the retentate TOC and TN, the dissimilarity of the community was low. In contrast, the largest dissimilarities in the communities occurred at the beginning (62%) and the end (81%) of the operation.
Multivariate analysis was used to determine if there were correlations between the biological communities and the abiotic parameters. The subset of environmental variables that had the highest Spearman correlation coefficient were conductivity, TN and bound EPS with a coefficient (Rho) of 0·619. TN correlated the strongest (Rho = 0·515), while TOC was the least correlated with the community change (Rho = −0·002). Therefore, it is likely that TN and thus, protein concentration played a significant role in influencing the bacterial community composition.
Analysis of bacterial communities using a taxonomy-dependent approach
All the effective reads were mapped to taxa based on the SILVA (v119) database. In total there were 14 taxonomic classes present among the eight bacterial communities (Fig.3a). Most of the communities were dominated by the classes Actinobacteria and Bacilli. Most notably, Actinobacteria had a relative abundance of 7·4% in the first sample collected on the 13th day and increased to 82% on the 40th day, before decreasing in abundance when the pH was reduced to 8, reaching a low of 6·2% by day 201. Actinobacteria contributed significantly to the dissimilarity between bacterial communities from day 13 (Md13B) and other days (day 40–day 172), and also the dissimilarity between day 201(Md201B) and other samples (day 40–day 172). On average, the relative abundance of Actinobacteria after pH adjustment (32%) was much lower than before pH adjustment (56%), indicating its preference for more alkaline environments.
Figure 3.
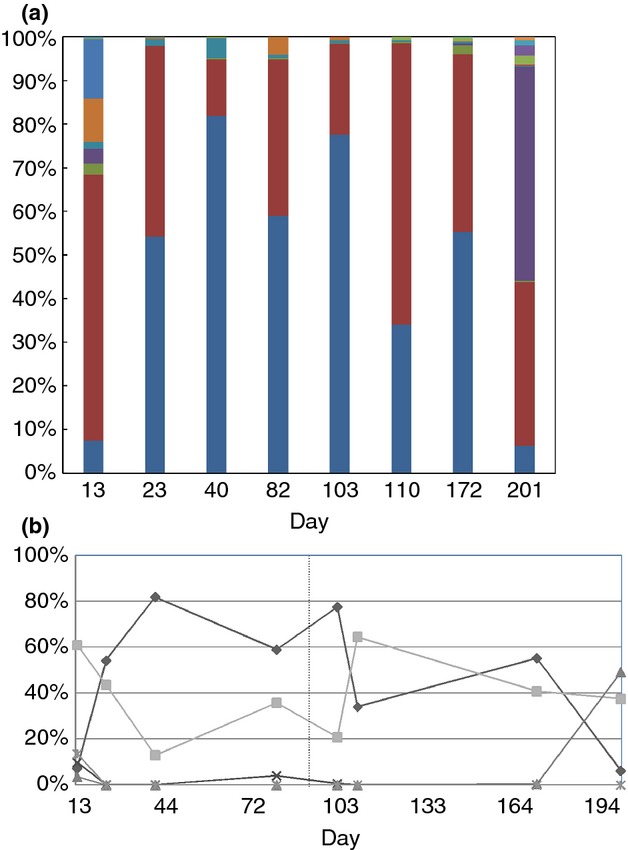
Relative abundance of 18 bacterial classes in eight membrane distillation bioreactor (MDBR) samples (a). The change of abundance of five dominant classes over the two phases represented by percentage of total community (b). The classes are: Actinobacteria (♦), Bacilli (▪), Chloroflexi (▴), Gammaproteobacteria (×) and Thermales (*). ( ) Deferribacterales; (
) Deferribacterales; ( ) Candidatus_Thiobios; (
) Candidatus_Thiobios; ( ) Arctic97B-4; (
) Arctic97B-4; ( ) Deltaproteobacteria; (
) Deltaproteobacteria; ( ) Subsectionl; (
) Subsectionl; ( ) Fusobacteria; (
) Fusobacteria; ( ) Flavobacteria; (
) Flavobacteria; ( ) SHA-26; (
) SHA-26; ( ) Themomicrobia; (
) Themomicrobia; ( ) Alphaproteobacteria; (
) Alphaproteobacteria; ( ) Bacteroidia; (
) Bacteroidia; ( ) Thermales; (
) Thermales; ( ) Gammaproteobacteria; (
) Gammaproteobacteria; ( ) Clostridia; (
) Clostridia; ( ) Chloroflexi; (
) Chloroflexi; ( ) Betaproteobacteria; (
) Betaproteobacteria; ( ) Bacilli and (
) Bacilli and ( ) Actinobacteria
) Actinobacteria
Bacilli dominated the day 13 sample, before it was outcompeted by Actinobacteria when the reactor pH was 10 (Fig.3b). After pH adjustment, it became the dominating class again, implying its resilience in varying environmental conditions. These two classes represented more than 90% of all reads in almost all the bacterial samples, with the exceptions of day 13 (Md13B) and day 201 (Md201B). The day 201 community (Md201B) was dominated by the class Chloroflexi (49%), and the day 13 community (Md13B) was comprised of Gammaproteobacteria and Thermales at c. 10%. Samples from these 2 days had a more uniform distribution of taxa, and thus had higher community diversities (Table1). With the exception of Thermales, the bacterial classes identified are not typical thermophiles and so analysis at the genera level was undertaken.
One hundred twenty-one genera were detected in the eight MDBR samples. Ten, 25 and 14 genera were detected in the classes of Actinobacteria,Bacilli and Clostridi, respectively. However, only one or two genera predominated in the community while others were present in relatively low abundance within the specific samples (Table3). Within Actinobacteria, Rubrobacter represented more than 99% of all the reads. Thermophilic species belonging to the genus Rubrobacter identified from the literature include Arthrobacter radiotolerans isolated from hot springs (Suzuki et al. 1988), Rubrobacter xylanophilus from hot runoff of a carpet factory (Carreto et al. 1996), Rubrobacter taiwanensis from hot springs in Central Taiwan (Chen et al. 2004) and Rubrobacter bracarensis from a biodeteriorated monument (Jurado et al. 2012). The MDBR sludge was pinkish-red indicating that the Rubrobacter species in the MDBR may be the red pigmented R. taiwanensis or R. xylanophilus, however, this may also be a novel Rubrobacter species.
Table 3.
Relative abundance (%) of major bacterial genera detected in membrane distillation bioreactor suspended sludge
| Taxonomy | Md13B | Md23B | Md40B | Md82B | Md103B | Md110B | Md172B | Md201B |
|---|---|---|---|---|---|---|---|---|
| Rubrobacter* | 7·38 | 54·18† | 81·76 | 58·97 | 77·56 | 33·86 | 55·25 | 5·90 |
| Caldalkalibacillus | 51·19 | 40·99 | 11·35 | 35·55 | 20·39 | 63·59 | 39·28 | 9·03 |
| Hydrogenophaga | 2·36 | –‡ | 0·30 | 0·32 | 0·08 | 0·25 | 1·84 | 0·26 |
| Roseiflexus | 3·07 | 0·03 | – | – | – | – | 0·52 | 44·79 |
| Anaerobranca | 1·17 | 1·51 | 4·14 | 0·49 | 0·49 | 0·09 | 0·17 | 0·13 |
| Lactococcus | 9·13 | 2·65 | 1·51 | 0·05 | 0·10 | 0·14 | 1·20 | – |
| Jeotgalibacillus | 0·30 | – | – | – | – | – | 0·08 | 18·98 |
| Meiothermus | 13·54 | 0·03 | – | – | – | – | 0·04 | – |
| Pseudomonas | 9·36 | – | – | – | 0·02 | – | 0·02 | – |
| Prevotella | – | – | – | 0·02 | – | – | – | 0·23 |
| Xenophilus | 0·18 | – | 0·02 | 0·05 | – | 0·05 | 0·19 | 0·07 |
| Cycloclasticus | 0·23 | – | – | 4·04 | – | – | – | – |
| Chlorothrix | 0·43 | – | – | – | – | – | – | 4·38 |
| Bacillus | 0·13 | – | – | 0·05 | 0·20 | 0·09 | 0·10 | 3·33 |
| Anoxybacillus | – | – | 0·04 | – | 0·02 | – | – | 2·90 |
| AKYG1722 | 0·13 | – | – | – | 0·02 | – | 0·06 | 2·34 |
| Kurthia | 0·10 | – | – | – | – | – | 0·02 | 1·55 |
| Clostridiaceae | 0·35 | – | 0·13 | 0·15 | 0·04 | 0·05 | 0·10 | 0·03 |
| SHA-26 | 0·20 | – | – | – | – | – | 0·02 | 1·19 |
| Glaciecola | – | 0·16 | – | – | 0·64 | – | – | – |
Names in bold are those taxa abundant in MD bacterial communities.
Values in italic are the relative abundance of dominant genera.
Absence of genera in the sample.
Of the 25 Bacilli genera detected, Caldalkalibacillus was the most abundant in the majority of the MDBR samples while Jeotgalibacillus was the dominant Bacilli on day 201 (Md201B). The abundance of Caldalkalibacillus decreased to 9·03% in the final sample after pH adjustment, indicating its preference for the alkaline environment (Xue et al. 2006). Jeotgalibacillus was present in low abundance in the first 172 days, but its relative abundance increased to 19% by day 201. The abundance of Jeotgalibacillus in the MDBR sludge increased when the retentate conductivity increased during the last few days of MDBR operation, indicating that the accumulation of salts in the reactor may have resulted in an environment favourable for the growth of the Jeotgalibacillus. However, as it has not been reported that species in this genus are thermophiles, the species detected here may be a novel species.
Thermales is a class of thermophiles, and Meiothermus was the only one genus identified in our study. Meiothermus were abundant on the 13th day. Many Meiothermus species grow well at salinities <1%. As the salinity in the reactor increased to 1·4% at 20 days of operation, there was a corresponding reduction in the abundance of Meiothermus, indicating that the Meiothermus was outcompeted at elevated salinity. The optimum pH for Roseiflexus is 7·5–8·0, and as shown by the results presented in Table3, its abundance increased significantly after pH adjustment. There were weak correlations between the abundance of every genus in the microbial community with the parameters of pH, DO, biomass, salinity and EPS. The strongest correlation occurred between Rubrobacter and TN (Fig.4b, R = 0·854), suggesting that proteins in the MDBR retentate enhanced growth of this genera. The second strongest correlation was between the abundance of Rubrobacter and the retentate TOC (Fig.4a, R = 0·562), indicating that the growth of Rubrobacter was limited by organic carbon and nutrient concentrations in the system. In contrast, the abundance of Caldalkalibacillus was negatively correlated with retentate TOC (Fig.4c, R = 0·36) and had weak correlation with retentate TN (Fig.4d, R = 0·14).
Figure 4.
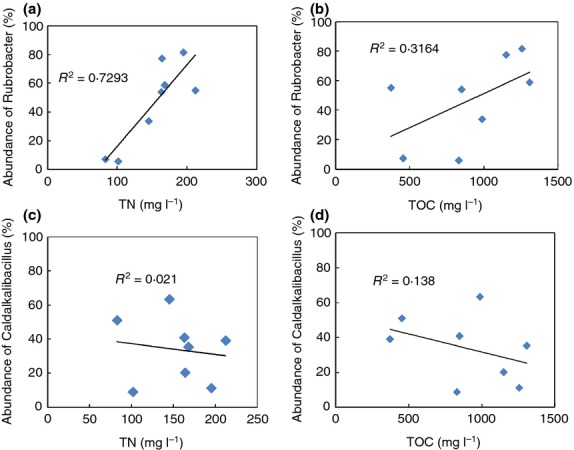
Corelation between relative abundance of the most abundant taxa, Rubrobacter, (a, b) and Caldalkalibacillus (c, d) and total nitrogen (TN) (a, c) and total organic carbon (TOC) (b, d).
Analysis of eukaryotic communities: diversity, composition and correlation
Analysis of eukaryotic communities in the MDBR over 202 days (Md13E, Md23E etc) at the distance of 3% revealed that most of the eukaryotic OTUs were detected on days 40 and 110 (Table2), which were during the middle of the first phase and the beginning of the second phase of operation. At the end of the MDBR operation (days 172, 201), the eukaryotic community diversity was at its lowest (Table2 and Fig.5a), implying that the neutral pH may have led to a reduction in diversity. The Shannon Weaver index of the eukaryotic community (3·06–4·21) was generally higher than the corresponding bacterial community H index (1·69–2·77) in the same sample.
Figure 5.
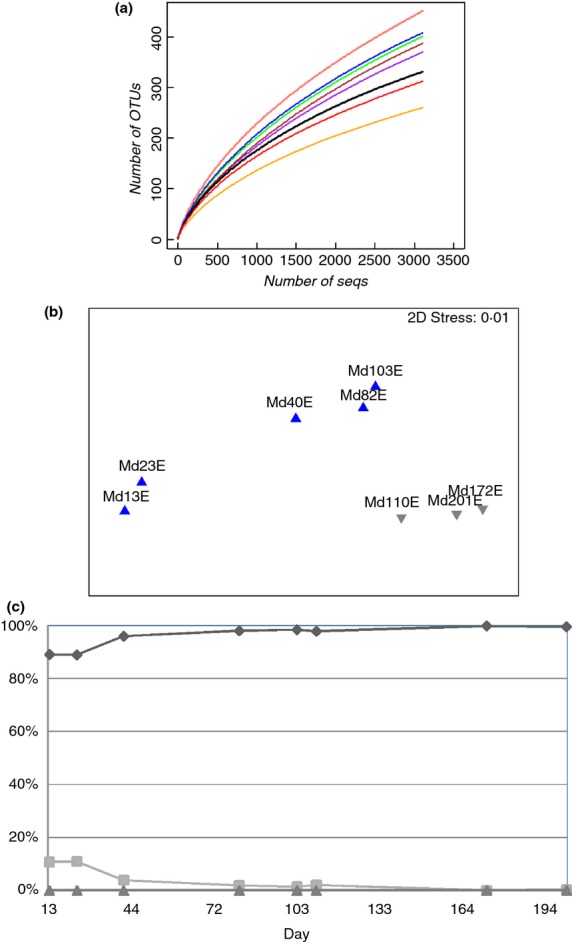
Rarefaction curves (a), nonmetric multidimentional scaling (b) of eight eukaryotic communities at a 3% cut-off and community composition at the kingdom level (c). All the datasets were normalized to have 3108 reads. The samples before pH adjustment are shown as ‘ ’ and after pH adjustment are represented by ‘
’ and after pH adjustment are represented by ‘ ’ (b). (c) All the reads which obtained using the primer set euk7F-euk570R were aligned to the SILVA database, and are shown as relative percent abundance. The eukaryotic groups are: Fungi (♦), Amoebozoa (▴) and Unclassified (▪). (
’ (b). (c) All the reads which obtained using the primer set euk7F-euk570R were aligned to the SILVA database, and are shown as relative percent abundance. The eukaryotic groups are: Fungi (♦), Amoebozoa (▴) and Unclassified (▪). ( ) Md13E; (
) Md13E; ( ) Md23E (
) Md23E ( ) Md40E (
) Md40E ( ) Md82E; (
) Md82E; ( )Md103E; (
)Md103E; ( ) Md110E; (
) Md110E; ( ) Md172E and (
) Md172E and ( ) Md201E.
) Md201E.
An NMDS plot of the Bray-Curtis similarities of the eukaryotic communities revealed that days 13 and 23 formed a group and all other communities in phase 1 (days 40–103) formed a second group (Fig.5b). The remaining three samples collected after pH adjustment formed a third group. Thus, the greatest dissimilarity between communities occurred between days 23 and 40 at the beginning and between days 103 and 110 at the end of phase 1 of operation. It was observed that a decrease in DO and an increase in TOC occurred from days 23 to 40, while all other environmental factors remained stable. During this period, bacterial communities were relatively stable (Fig.2b), thus it is possible that the reduction in DO affected the eukaryotic communities. When the pH in the bioreactor was reduced, there was a significant shift in the eukaryotic community, indicating that pH had a large effect on eukaryotic community composition. Bio-Env analysis indicated that the dissimilarities between the eukaryotic communities were strongly correlated with the concentration of bound EPS (Rho = 0·823). The eukaryotic community shift may have an effect on the EPS production if some eukaryotic species secreted EPS. Furthermore, the predation of bacteria by predatory eukaryotes could have an effect on the concentration of bound EPS, especially since some of these predators exhibit selective feeding. For example, selective feeding on EPS producing isolates would result in a decrease in the concentration of bound EPS.
While the phylotype-independent clustering analysis showed high eukaryotic community diversity, the assignment of taxa using the SILVA database showed that the eukaryotic community was comprised of two kingdoms (in addition to an unclassified group (Fig.5c)). Fungi dominated in all samples with the abundances of 89–99·9%. Amoebozoa were present at low levels in the last sample (day 201). Only eight OTUs were classifiable at the genus level and the remaining 12 OTUs listed in Table4 were assigned to the lowest common ancestor (LCA). Most of the identifiable Fungi belonged to the subkingdom Dikarya with the exception of Blakeslea and Choanephoraceae. The genus Candida dominated in many samples. A pairwise comparison between the abundance of Candida and environmental parameters was performed here. Five of the eight operational parameters were found to be correlated with the abundance of Candida (Fig.6). pH and DO were negatively correlated with abundance of Candida (R = 0·709, 0·693 respectively, Fig.6a,b), while salinity was positively correlated (R = 0·677, Fig.6c). The abundance of Candida was also positively correlated with bound EPS production (R = 0·936, Fig.6d). There was no correlation between the abundance of eukaryotic and bacterial genera.
Table 4.
Relative abundance (%) of identified eukaryotic phylotypes
| Taxonomy | Md13E | Md23E | Md40E | Md82E | Md103E | Md110E | Md172E | Md201E |
|---|---|---|---|---|---|---|---|---|
| Acanthamoeba_g* | –† | – | – | – | – | – | – | 0·02 |
| Otidea_g | 0·03 | – | – | 0·01 | – | – | 0·01 | – |
| Pezizales_o | – | – | 0·09 | 0·09 | 0·01 | – | 0·01 | 0·03 |
| Sordariomycetes_c | – | – | 0·02 | – | – | – | 0·01 | – |
| Pezizomycotina_subp | – | 0·07 | 1·31 | 1·54 | 1·37 | 1·18 | 1·60 | 1·54 |
| Metschnikowia_g | 0·68 | 0·24 | 0·20 | 0·12 | – | 0·02 | – | – |
| Candida_g‡ | 0·58 | 1·38 | 32·54§ | 46·31 | 55·81 | 45·77 | 66·39 | 60·58 |
| Saccharomycetales_o | 0·03 | 0·07 | 1·49 | 2·06 | 1·98 | 2·18 | 2·53 | 3·00 |
| Saccharomyces_g | 0·06 | 0·08 | 0·29 | 0·01 | 0·15 | 0·18 | 0·21 | 0·06 |
| Saccharomycetaceae_f | 0·03 | 0·07 | 0·11 | 0·15 | 0·28 | 0·30 | 0·31 | 0·29 |
| Saccharomycetales_o | 0·29 | 0·56 | 13·28 | 18·57 | 18·06 | 18·60 | 19·41 | 20·60 |
| Saccharomycetes_c | – | 0·02 | – | – | 0·01 | – | – | 0·03 |
| Ascomycota_p | 0·23 | 0·34 | 6·69 | 9·57 | 9·10 | 9·18 | 9·13 | 10·49 |
| Rhodotor_g | 0·10 | 0·07 | 0·04 | 0·01 | – | 0·08 | 0·02 | – |
| Dikarya_subk | 67·54 | 70·23 | 34·01 | 17·47 | 9·52 | 17·17 | 0·23 | 2·60 |
| Blakeslea_g | 5·47 | 0·35 | 1·22 | 0·01 | 0·24 | 0·86 | 0·02 | – |
| Choanephoraceae_f | – | – | 0·02 | – | – | 0·04 | – | – |
| Fungi_k | 14·22 | 15·54 | 4·76 | 2·21 | 2·05 | 2·42 | 0·07 | 0·45 |
| Eukaryota_d | 10·75 | 10·99 | 3·91 | 1·86 | 1·41 | 2·04 | 0·02 | 0·29 |
The phylogenetic levels that operational taxonomic units were assigned: domain (_d), kingdom (_k), subkingdom (_subk), phylum (_p), subphylum (_subp), class (_c), order (_o), family (_f), genus (_g).
Absence of genera in the sample.
Names in bold are those taxa abundant in MD bacterial communities.
Values in italic are the relative abundance of dominant taxa.
Figure 6.
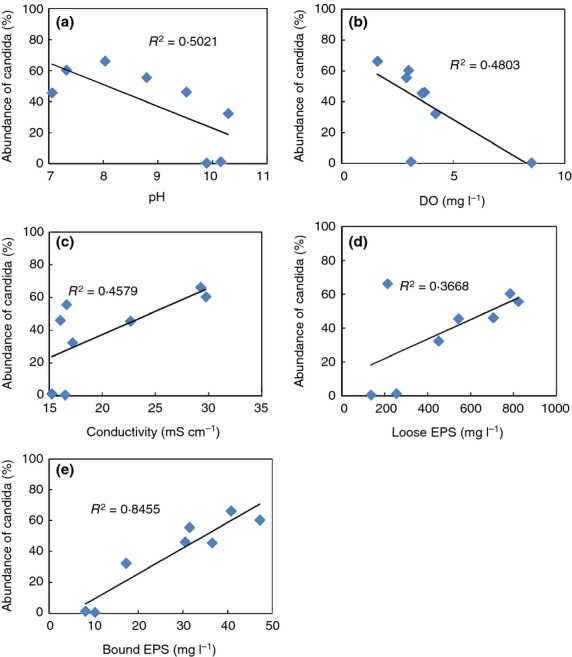
Pairwise comparisons between relative abundance of Candida and environmental parameters (a = pH, b = dissolved oxygen, c = conductivity) and microbial products (e = loose and d = bound extracellular polymeric substances). Their linear relationships were measured by the value of R.
Discussion
Here, we investigated the microbial community composition during two phases of MDBR operation under different pH conditions. The bacterial communities in the mixed liquor had relatively low richness and diversity and were dominated by Actinobacteria (genus Rubrobacter) and Bacilli (genus Caldalkalibacillus). The strongest correlation between environmental variables and individual genera occurred between TN and Rubrobacter for the bacterial community and between bound EPS and Candida for the eukaryotic community.
The dominant taxa in suspended sludge
To our knowledge, this study is the first to investigate the microbial community composition in an MDBR. The genera detected in our study for the most part, have not been identified in similar high temperature aerobic systems. Bacilli, Actinobacteria and Chloroflexi were the three dominant classes of the bacterial communities at different time points (Fig.3b). Bacillus spp. and Bacillus-like species have been isolated from thermophilic aerobic wastewater treatment reactors as early as 1962 (Tischer et al. 1962). Bacillus was present in the MDBR used in this study at abundances of 0–3·33%. However, Caldalkalibacillus, also in the Bacillaceae family, was dominant in many samples, accounting for 9·03–63·6% of bacterial genera in all the suspended bacterial communities. Only two Caldalkalibacillus species have been previously isolated, but they were not isolated from engineered systems (Xue et al. 2006; Zhao et al. 2008). Actinobacteria and Chloroflexi have been detected in aerobic thermophilic digesters in low abundance (Piterina et al. 2012; Sundberg et al. 2013).
Fungi were the dominant eukaryotes in the suspended sludge. The genus Candida of the class Saccharomycetes was the most abundant eukaryote in the MDBR communities. As seen from Table4, the reads classified to the LCAs in this lineage accounted for 55·6–98% of the eukaryotic communities. The fungal community in heated wastewater treatment systems has not been explored, however, fungi is a major group in composting municipal wastes (De Gannes et al. 2013). Interestingly, Ascomycota represented 93% of the fungal sequences from composts of lignocellulosic wastes (De Gannes et al. 2013), and two Candida species were detected (Bonito et al. 2010). Composting is also considered to be an aerobic thermal microbial process, which is similar to conditions in the MDBR in our study. However, more studies are needed to elucidate the microbial community diversity and composition in thermal aerobic microbial waste or wastewater treatment systems.
Low microbial community diversity led to low resilience to pH fluctuation
A decrease in pH in the MDBR system resulted in a decrease in sludge biomass, indicating that overall, the microbial populations were negatively affected. Rubrobacter was the dominant bacterial genus and Candida was the dominant eukaryotic genus on day 103 before pH adjustment. After adjustment of the pH on day 106, the bacterial community was dominated by Caldalkalibacillus, while Candida was still the dominant eukaryote. Thus, a sudden decrease in pH had a negative influence on the growth of Rubrobacter, which resulted in an increase in Caldalkalibacillus and had little effect on Candida. However, since the abundances of microbes were normalized to 100%, this is not a reflection of total cell numbers. In fact the absolute abundance of most microbes was largely reduced as the biomass decreased by 85% within 7 days. Thus the stability of the ecosystem was significantly affected by the shift in pH.
Some studies suggest that the stability of an ecosystem is determined by the resistance of the dominant species to environmental stresses (Tilman et al. 2006), and that the initial community evenness is a key factor in the maintenance of ecosystem function (Wittebolle et al. 2009). According to the OTU-dependent analysis, the diversity of the bacterial communities was relatively low (Shannon = 1·75–2·53) compared with mesophilic communities from other MBRs and other thermophilic communities such as biogas reactors (Li et al. 2013). The phylotype-based analysis also showed that the bacterial community was dominated by two genera (Rubrobacter and Caldalkalibacillus) and the eukaryotic community was dominated by one genus (Candida or the unknown Dikarya). The entire microbial community had a low resistance to the changing parameters in the reactor. Furthermore, the diversity of the bacterial communities increased steadily from days 103 to 201, implying that previously low abundant species were able to grow at the lower pH. Notably, the biomass also increased in the second phase. Therefore, the microbial community became more evenly distributed under the less restrictive condition of a more neutral pH.
Genera which strongly correlated with environmental variables
The correlation between the abundance of individual bacterial taxa and environmental variables showed that the strongest correlation occurred between Rubrobacter and TN in the retentate (Fig.4a, R = 0·854), and the second strongest was between the abundance of Rubrobacter and the retentate TOC (Fig.4b, R = 0·562). Thus, the carbon and nitrogen concentrations affected the growth of Rubrobacter and concomitantly led to the changes in the bacterial community composition. Four Rubrobacter species have been well-described (Jurado et al. 2012), and R. xylanophilus and R. taiwanensis have a pH optimum of 7·5–8·0, are halotolerant and are able to grow on Thermus basal salts medium containing only (NH4)2SO4 and an appropriate carbon source. It was observed that during second phase of operation of the MDBR used in this study, the pH decreased to 8·0 and the salinity increased to 2%. These conditions are similar to the optimum growth conditions for this genus, however the abundance of Rubrobacter did not increase. In fact, the abundance of Rubrobacter only increased when there was a slight decrease in TN and large decrease in TOC. Therefore, under the conditions used here, nutrient plays the most important role in determining the abundance of Rubrobacter.
The relative abundance of Candida strongly correlated with many environmental variables, i.e. pH, DO, salinity and EPS (Fig.6). Candida is a highly diverse genus containing more than 50 species, many of which are thermophiles (e.g. Candida hallenica, Candida ishiwadae (Ayachi 2013), Candida tropicalis (Dey and Maiti 2013) and Candida slooffii). Candida can grow on various carbon sources, but glucose has been found to be the best for the production of biomass (Dey and Maiti 2013). As seen from Table1, the main component of the MD feed solution was glucose, which may have favoured Candida dominance. The abundance of Candida was negatively correlated with pH and DO (Fig.6a,b). Most of the Candida spp. have an optimum growth pH that is neutral or slightly acidic, but the increased pH in the MDBR did not inhibit its growth.
The microorganisms detected in the MDBR used for this study are novel as few of them have been isolated from thermal aerobic systems. The thermophilic communities in the MDBR were comprised of a low diversity bacterial community, a highly diverse eukaryotic community and no archaea. These communities had a low resilience to changes in operational parameters. Specifically, nutrient composition and concentration in the retentate was strongly correlated with the dominant species.
Acknowledgments
This research was supported by a research grant (MEWR C651/06/177) from the Environment and Water Industry Programme Office of Singapore.
Conflict of Interest
The authors declare that there are no conflicts of interest.
References
- American Public Health Association, American Water Works Association, Water Pollution Control Federation, and Water Environment Federation. Standard Methods for the Examination of Water and Wastewater. Washington DC, USA: American Public Health Association; 1965. [Google Scholar]
- Ayachi R. Study the activity of thermophilic yeasts on wheat flour media: production of extra cellular raw starch digestive amylase enzymes. IJMEMR. 2013;1:5. [Google Scholar]
- Bonito G, Isikhuemhen OS, Vilgalys R. Identification of fungi associated with municipal compost using DNA-based techniques. Bioresour Technol. 2010;101:1021–1027. doi: 10.1016/j.biortech.2009.08.109. [DOI] [PubMed] [Google Scholar]
- Carreto L, Moore E, Nobre MF, Wait R, Riley PW, Sharp RJ, Dacosta MS. Rubrobacter xylanophilus sp nov: a new thermophilic species isolated from a thermally polluted effluent. Int J Syst Bacteriol. 1996;46:460–465. [Google Scholar]
- Chen MY, Wu SH, Lin GH, Lu CP, Lin YT, Chang WC, Tsay SS. Rubrobacter taiwanensis sp. nov., a novel thermophilic, radiation-resistant species isolated from hot springs. Int J Syst Evol Microbiol. 2004;54:1849–1855. doi: 10.1099/ijs.0.63109-0. [DOI] [PubMed] [Google Scholar]
- De Gannes V, Eudoxie G, Hickey WJ. Insights into fungal communities in composts revealed by 454-pyrosequencing: implications for human health and safety. Front Microbiol. 2013;4:164. doi: 10.3389/fmicb.2013.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey P, Maiti MK. Molecular characterization of a novel isolate of Candida tropicalis for enhanced lipid production. J Appl Microbiol. 2013;114:1357–1368. doi: 10.1111/jam.12133. [DOI] [PubMed] [Google Scholar]
- Goh S, Zhang J, Liu Y, Fane AG. Fouling and wetting in membrane distillation (MD) and MD-bioreactor (MDBR) for wastewater reclamation. Desalination. 2013;323:39–47. [Google Scholar]
- Hu M, Wang XH, Wen XH, Xia Y. Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour Technol. 2012;117:72–79. doi: 10.1016/j.biortech.2012.04.061. [DOI] [PubMed] [Google Scholar]
- Jurado V, Miller AZ, Alias-Villegas C, Laiz L, Saiz-Jimenez C. Rubrobacter bracarensis sp. nov., a novel member of the genus Rubrobacter isolated from a biodeteriorated monument. Syst Appl Microbiol. 2012;35:306–309. doi: 10.1016/j.syapm.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Lay WC, Zhang QY, Zhang JS, Mcdougald D, Tang CY, Wang R, Liu Y, Fane AG. Study of integration of forward osmosis and biological process: membrane performance under elevated salt environment. Desalination. 2011;283:123–130. [Google Scholar]
- Lay WC, Zhang QY, Zhang JS, Mcdougald D, Tang CY, Wang R, Liu Y, Fane AG. Effect of pharmaceuticals on the performance of a novel osmotic membrane bioreactor (OMBR) Sep Sci Technol. 2012;47:543–554. [Google Scholar]
- Li A, Chu Y, Wang X, Ren L, Yu J, Liu X, Yan J, Zhang L, et al. A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol Biofuels. 2013;6:3. doi: 10.1186/1754-6834-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fang HH. Extraction of extracellular polymeric substances (EPS) of sludges. J Biotechnol. 2002;95:249–256. doi: 10.1016/s0168-1656(02)00025-1. [DOI] [PubMed] [Google Scholar]
- Lu L, Xing DF, Ren NQ. Pyrosequencing reveals highly diverse microbial communities in microbial electrolysis cells involved in enhanced H-2 production from waste activated sludge. Water Res. 2012;46:2425–2434. doi: 10.1016/j.watres.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Ma JX, Wang ZW, Yang Y, Mei XJ, Wu ZC. Correlating microbial community structure and composition with aeration intensity in submerged membrane bioreactors by 454 high-throughput pyrosequencing. Water Res. 2013;47:859–869. doi: 10.1016/j.watres.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Mardanov AV, Gumerov VM, Beletsky AV, Bonch-Osmolovskaya EA, Ravin NV, Skryabin KG. Characteristic of biodiversity of thermophilic microbial community by parallel pyrosequencing method. Dokl Biochem Biophys. 2010;432:110–113. doi: 10.1134/s160767291003004x. [DOI] [PubMed] [Google Scholar]
- Miura Y, Hiraiwa MN, Ito T, Itonaga T, Watanabe Y, Okabe S. Bacterial community structures in MBRs treating municipal wastewater: relationship between community stability and reactor performance. Water Res. 2007;41:627–637. doi: 10.1016/j.watres.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Molina-Munoz M, Poyatos JM, Sanchez-Peinado M, Hontoria E, Gonzalez-Lopez J, Rodelas B. Microbial community structure and dynamics in a pilot-scale submerged membrane bioreactor aerobically treating domestic wastewater under real operation conditions. Sci Total Environ. 2009;407:3994–4003. doi: 10.1016/j.scitotenv.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Phattaranawik J, Fane AG, Pasquier ACS, Bing W. A novel membrane bioreactor based on membrane distillation. Desalination. 2008;223:386–395. [Google Scholar]
- Piterina AV, Bartlett J, Pembroke JT. Phylogenetic analysis of the bacterial community in a full scale autothermal thermophilic aerobic digester (ATAD) treating mixed domestic wastewater sludge for land spread. Water Res. 2012;46:2488–2504. doi: 10.1016/j.watres.2012.01.045. [DOI] [PubMed] [Google Scholar]
- Stamper DM, Walch M, Jacobs RN. Bacterial population changes in a membrane bioreactor for graywater treatment monitored by denaturing gradient gel electrophoretic analysis of 16S rRNA gene fragments. Appl Environ Microbiol. 2003;69:852–860. doi: 10.1128/AEM.69.2.852-860.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg C, Al-Soud WA, Larsson M, Alm E, Shakeri Yekta S, Svensson BH, Sorensen SJ, Karlsson A. 454-Pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol Ecol. 2013;85:612–626. doi: 10.1111/1574-6941.12148. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Collins MD, Iijima E, Komagata K. Chemotaxonomic characterization of a radiotolerant bacterium, Arthrobacter radiotolerans – description of Rubrobacter radiotolerans gen-nov, comb nov. FEMS Microbiol Lett. 1988;52:33–39. [Google Scholar]
- Tilman D, Reich PB, Knops JMH. Biodiversity and ecosystem stability in a decade-long grassland experiment. Nature. 2006;441:629–632. doi: 10.1038/nature04742. [DOI] [PubMed] [Google Scholar]
- Tischer RG, Brown LR, Cook DW. Decomposition of wastewater by thermophilic microorganisms. J Water Pollut Control Fed. 1962;34:1244–1255. [Google Scholar]
- Wang XH, Hu M, Xia Y, Wen XH, Ding K. Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in China. Appl Environ Microbiol. 2012;78:7042–7047. doi: 10.1128/AEM.01617-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittebolle L, Marzorati M, Clement L, Balloi A, Daffonchio D, Heylen K, De Vos P, Verstraete W, et al. Initial community evenness favours functionality under selective stress. Nature. 2009;458:623–626. doi: 10.1038/nature07840. [DOI] [PubMed] [Google Scholar]
- Xue YF, Zhang XQ, Zhou C, Zhao YJ, Cowan DA, Heaphy S, Grant WD, Jones BE, et al. Caldalkalibacillus thermarum gen. nov., sp nov., a novel alkalithermophilic bacterium from a hot spring in China. Int J Syst Evol Microbiol. 2006;56:1217–1221. doi: 10.1099/ijs.0.64105-0. [DOI] [PubMed] [Google Scholar]
- Zhang J, Chua HC, Zhou J, Fane AG. Factors affecting the membrane performance in submerged membrane bioreactors. J Memb Sci. 2006;284:54–66. [Google Scholar]
- Zhang T, Shao MF, Ye L. 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012;6:1137–1147. doi: 10.1038/ismej.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Jie YW, Loong WLC, Zhang J, Fane AG, Kjelleberg S, Rice SA, Mcdougald D. Characterization of biofouling in a lab-scale forward osmosis membrane bioreactor (FOMBR) Water Res. 2014;58:141–151. doi: 10.1016/j.watres.2014.03.052. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang CL, Romanek CS, Wiegel J. Description of Caldalkalibacillus uzonensis sp. nov. and emended description of the genus Caldalkalibacillus. Int J Syst Evol Microbiol. 2008;58:1106–1108. doi: 10.1099/ijs.0.65363-0. [DOI] [PubMed] [Google Scholar]