

Abstract
Reactive lipid aldehydes are implicated in the pathogenesis of various oxidative stress-mediated diseases, including non-alcoholic steatohepatitis, atherosclerosis, Alzheimer’s and cataract. In the present study, we sought to define which hepatic Aldh isoform plays a major role in detoxification of lipid-derived aldehydes, such as acrolein and HNE by enzyme kinetic and gene expression studies. The catalytic efficiencies for metabolism of acrolein by Aldh1a1 was comparable to that of Aldh3a1 (Vmax/Km = 23). However, Aldh1a1 exhibits far higher affinity for acrolein (Km = 23.2 μM) compared to Aldh3a1 (Km = 464 μM). Aldh1a1 displays a 3-fold higher catalytic efficiency for HNE than Aldh3a1 (218 vs 69 ml/min/mg). The endogenous Aldh1a1 gene was highly expressed in mouse liver and a liver-derived cell line (Hepa-1c1c7) compared to Aldh2, Aldh1b1 and Aldh3a1. Aldh1a1 mRNA levels was 34-fold and 73-fold higher than Aldh2 in mouse liver and Hepa-1c1c7 cells respectively. Aldh3a1 gene was absent in mouse liver, but moderately expressed in Hepa-1c1c7 cells compared to Aldh1a1. We demonstrated that knockdown of Aldh1a1 expression by siRNA caused Hepa-1c1c7 cells to be more sensitive to acrolein-induced cell death and resulted in increased accumulation of acrolein-protein adducts and caspase 3 activation. These results indicate that Aldh1a1 plays a major role in cellular defense against oxidative damage induced by reactive lipid aldehydes in mouse liver. We also noted that hepatic Aldh1a1 mRNA levels were significantly increased (≈ 3 fold) in acrolein-fed mice compared to control. In addition, hepatic cytosolic ALDH activity was induced by acrolein when 1 mM NAD+ was used as cofactor, suggesting an Aldh1a1-protective mechanism against acrolein toxicity in mice liver. Thus, mechanisms to induce Aldh1a1 gene expression may provide a useful rationale for therapeutic protection against oxidative stress-induced pathologies.
Keywords: aldehyde dehydrogenase 1a1, aldehydes, 4-hydroxy-2-nonenal, acrolein, toxicity
1. INTRODUCTION
The liver being the major site for metabolism and biotransformation of drugs and foreign compounds is constantly exposed to reactive oxygen species (ROS), resulting in oxidative stress. During oxidative stress, these ROS which include hydrogen peroxide (H2O2), superoxide radicals (O2−) and hydroxyl radicals (OH.) can covalently modify proteins, lipids and DNA. The peroxidation of polyunsaturated fatty acids in membrane lipid by ROS produces unstable lipid hydroperoxides which decompose into aldehydes such as malondialdehyde (MDA), hexanal, trans-2-hexenal, propen-2 al (acrolein) and 4-hydroxy-2-nonenal (HNE) [1–3]. Among these aldehydes, HNE and acrolein are highly electrophilic α,β-unsaturated aldehyde and can undergo Schiff base and Michael addition reactions with nucleophilic groups on proteins and DNA [4]. There is increasing evidence that the pathophysiological effects of ROS in cells is mediated by these cytotoxic aldehydes because they are very stable and can diffuse to distant sites of formation [5;6].
In addition, humans are exposed to these aldehydes as environmental pollutants and by endogenous processes that generate reactive aldehydes in the liver. For example, chronic alcohol consumption, high fat diet or exposure to foreign compounds such as carbon tetrachloride (CCl4), allyl alcohol and the widely used anticancer drug cyclophosphamide markedly elevate the intracellular concentrations of cytotoxic aldehydes [7–10]. Reactive α,β-unsaturated aldehyde such as acrolein and crotonaldehyde are also present in cigarette smoke, vehicle exhaust emission, overheated foods and oil, drinking water and in effluents from industrial plants [10;11]. It is estimated that the maximum daily human consumption of unsaturated aldehydes is 5 mg/kg, while the total aldehyde consumption has been suggested to be ≈ 7 mg/kg [12;13].
The levels of reactive lipid aldehydes are elevated in various oxidative stress-mediated diseases, including steatohepatitis [14], atherosclerosis [15], Alzheimer’s [16], cataract [17], diabetes [18] and cancer [19]. In fact, accumulation of reactive aldehydes is associated with the pathogenesis of these diseases. The toxicity of α,β-unsaturated aldehyde lies in their ability to form Michael adducts with thiol and amino groups of proteins resulting in alteration of several cellular processes. For example, cellular enzymes such as glyceraldehyde-3-phosphate, glucose-6-phosphate dehydrogenases and cytochrome c oxidase containing lysine and cysteine residues in their active sites are readily inactivated by reactive aldehydes [20–22]. In addition, α,β-unsaturated aldehyde can induce oxidative stress in cells by depleting cellular reduced glutathione, thereby altering signal transduction pathways in cells. At low concentrations, acrolein is known to trigger apoptotic cell death by mechanisms that involve activation of mitochondrial death pathways and caspases [10;23;24]. Caspases, particularly caspase 3, which can cleave substrates such as poly(ADP-ribose) polymerase (PARP), actin, laminin are widely used as marker for apoptosis in different cell types. However at high concentrations acrolein causes necrotic cell death. This phenomenon has also been observed with HNE [25–27].
Despite their toxicity, many cytotoxic lipid-derived aldehydes can be successfully metabolized to less toxic compounds by the action of oxidative, reductive and conjugative enzymes. These enzymes include glutathione S-transferases (GST), aldehyde dehydrogenases (ALDH), aldo-keto reductases (AKR), alcohol dehydrogenases (ADH) and cytochrome P450 (CYP) [28–31]. The relative importance of these enzymes in reactive aldehydes metabolism is cell type- and species-dependent. It is now known that the conjugation of acrolein with glutathione is not a true detoxification process, since acrolein-glutathione conjugates can undergo renal processing to form reactive species [32]. In addition, the conjugation process is compromised when GSH concentrations are depleted, such as may occur during oxidative stress. ALDH oxidizes a range of toxic aldehydes to their corresponding non toxic carboxylic acids using either NAD+ or NADP+ as cofactors. They play a critical role in the cellular protection against these toxic species. There are 19 ALDH genes in the human genome [30]. To date three isozymes, the cytosolic ALDH1A1 and ALDH3A1, and the mitochondrial ALDH2 are the main lipid aldehyde-oxidizing enzymes expressed in the mouse liver.
The role of these enzymes in the cellular defense against oxidative damage induced by cytotoxic aldehydes is controversial. Previous studies by Townsend et al revealed that ALDH1A1 overexpression provides only moderate protection against trans-2-nonenal and not against other lipid aldehydes [33]. However, ALDH3A1 could protect cells against HNE-induced apoptosis. Aldh3a1 is poorly expressed in normal liver and highly expressed in cancerous cells [34]. It is also abundantly expressed in the cornea and protects the cornea against cytotoxic lipid peroxidation-derived aldehydes [35]. However, recent experiments with Aldh1a1−/− mice indicate that Aldh1a1 protects the eye from cataract formation induced by oxidative stress by detoxifying cytotoxic lipid aldehydes [36;37]. Moreover, the human lens ALDH1A1 efficiently oxidizes lipid-derived aldehydes, including HNE (Km 4.8 μM), trans-2-heptenal (Km 177.1 μM) and MDA (Km 3.5 μM) [38]. In addition, overexpression of ALDH1 in neuroblastoma cells reduces production of protein-HNE adducts and activation of caspase-3 [27]. Aldh1a1 is known to decrease the effectiveness of the anticancer drugs cyclophosphamide by detoxifying its major active metabolites acrolein [39]. These results indicate that Aldh1a1 has the potential to protect against aldehyde produced as a result of lipid peroxidation. However, it is unknown whether Aldh1a1 can protect against acrolein-induced toxicity in mouse liver. We hypothesized that Aldh1a1 is the major enzymes involved in acrolein and other lipid-derived aldehyde detoxification in mouse liver.
The relative contribution of different Aldh isozymes in detoxification of reactive aldehydes in mice liver is unknown. Moreover to date, the kinetic properties of murine Aldh orthologs for reactive lipid aldehydes oxidation especially acrolein have not been biochemically characterized. Mouse is a more common laboratory model for research in medicine because of the availability of transgenic and gene-knockout mice. In the present study, we examine the role of hepatic Aldh isozymes in detoxification of lipid-derived aldehydes such as acrolein and HNE by enzyme kinetic and gene expression studies. We showed by substrate preferences, gene expression patterns and in vitro knockdown experiments in Hepa-1c1c7 that murine Aldh1a1 is the major cytosolic Aldh in mice liver involved in cellular defense against reactive aldehydes-induced toxicity.
2. MATERIALS AND METHODS
2.2. Chemicals and Reagents
Propionaldehyde, benzaldehyde, trans-2-hexenal, acetaldehyde, dithiothreitol, 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), N-ethylmaleimide (NEM), propen-2-al (acrolein) and malondialdehyde (MDA) were purchased from Sigma Chemical Company, Inc. (St Louis, MO). 4-hydroxy-2-nonenal (HNE) was obtained from Cayman Chemical Co (Ann Arbor, MI). Oxidized β-NAD+ and β-NADP+ were purchased from Codexis (Redwood City, CA). Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody, clone 6C5 (MAB374) was purchased from Millipore (Temecula, CA). Rabbit polyclonal antibody against caspase 3 (H-277; sc-7148) was purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Cleaved caspase 3 (Asp175) antibodies (#9661), and cytochrome c treated and untreated Jurkat cell extracts (#9663) were from Cell Signaling Technology (Danvers, MA). ALDH1 antibody against purified rat ALDH1 was produced in rabbits [40]. Rabbit polyclonal antibody against acrolein-protein adducts was provided by Aruni Bhatnagar, Department of Medicine/Cardiovascular Medicine, University of Louisville. Stealth™ RNAi was synthesized by Invitrogen (Carlsbad, CA).
2.3. Cloning, Expression and Purification of Recombinant Aldh1a1 and Aldh3a1
The pCMV6-Aldh1a1 plasmid (MC202273) containing the full length of mouse Aldh1a1 cDNA (NM_013467) was purchased from OriGene (Rockville, MD). The Open Reading Frame (ORF) of Aldh1a1 was amplified by PCR using the following primers (forward: 5′-CATATGTCTTCGCCTGCACAACCTGCA-3′; reverse: 5′-GGTACCGGAGTTCTTCTGAGATATCTTCA-3′). The forward primer contains an Nde1 site for cloning into the start codon (ATG) of pET30b vector (Novagen, Inc., Madison, WI) to ensure correct initiation of translation in E coli. The reverse primer was designed to contain a Kpn1 site with deletion of the stop codon (TAA) to express a full length Aldh1a1 with a C-terminal his-tag. The 1.5 kb PCR product was then cloned into pCR2.1 vector using a TA cloning vector kit (Invitrogen, Carlsbad, CA). The sequence of the mouse Aldh1a1 ORF was confirmed by sequence analysis at the University of Louisville, Center for Genetics and Molecular Medicine Nucleic Acid Core Facility. The PCR product was then digested with Nde1 and Kpn1, and subcloned into the pET30b expression plasmid (Novagen, Inc., Madison, WI). To generate the full length of mouse Aldh3a1 cDNA (NM_007436), total RNA was isolated from mouse primary hepatocytes treated with 50 μM 1,2-benzanthracene (BA) for 24 h. Total RNA was reverse transcribed to cDNA with Oligo-dT primers using Advantage RT-for-PCR kit (Clontech, BD Bioscience, Palo Alto, CA). The ORF of Aldh3a1 was generated by PCR using as forward primer 5′-GGTACCAGCAATATCAGTAGCATCG-3′ and as reverse primer 5′-CTCGAGTGAAGTAGCCCTCTCAATGC-3′.
The sequence of the mouse Aldh3a1 ORF was confirmed by sequence analysis. The 1.5 kb PCR product was ligated into pCR2.1 vector and subsequently digested with Kpn1 and Xho1, and subcloned into pET30a (Novagen) expression vector allowing synthesis of a full-length Aldh3a1 with an N-terminal his-tag. One Shot® BL21(DE3)pLysS competent E coli cells (Invitrogen, Carlsbad, CA) were subsequently transformed with pET30b-Aldh1a1 or pET30a-Aldh3a1 plasmid and were grown until the OD600 ≈ 0.6. The recombinant protein expression was induced by addition of 0.5 mM isopropylthiogalactoside overnight. Cells were lysed with Bugbuster® Protein extraction reagent (Novagen, Inc., Madison, WI) containing 2 mM DTT and 10% glycerol, and purified using nickel nitrilotriacetic acid (Ni-NTA) column (Qiagen, Valencia, CA). Proteins were eluted with 400 mM imidazole and concentration was determined by Pierce® BCA Protein Assay (Pierce, Rockford, IL). The purity of the recombinant protein was determined by SDS-PAGE electrophoresis stained with coomassie blue. Recombinant proteins were transferred into nitrocellulose membrane and probed with anti-ALDH1A1 antibodies as described below.
2.3. Aldehyde Dehydrogenase Enzymatic Assay
The enzymatic activities of Aldh’s were measured spectrophotometrically using recombinant his-tag proteins by monitoring the reduction of NAD+ or NADP+ at 340 nm as previously described by Lindahl and Petersen [41] using the molar absorptivity value of 6220 M−1cm−1. Enzyme activity was assayed at 25°C in 50 mM sodium phosphate buffer (pH 7.4), containing 1 mM EDTA and 0.01% BME with either 1 mM NAD+ or NADP+ as cofactor. The reaction was initiated by addition of varying concentrations of aldehyde substrate into a 1 ml cuvette. The kinetic constants (Km, Vmax and Vmax/Km) of recombinant mouse Aldh1a1 and Aldh3a1 for oxidative metabolism of various aldehydes were examined. All reactions were performed in triplicate. The cofactor preference of recombinant mouse ALDH was measured using varying concentrations of either NAD+ or NADP+. The Km of Aldh1a1 for NAD+ and NADP+ was defined using 1 mM propionaldehyde while the cofactor preference of Aldh3a1 was assessed by measuring its Km for either NAD+ or NADP+ using 2.5 mM benzaldehyde as substrate.
2.3 Stealth™ siRNA knockdown of mouse Aldh1a1
Stealth™ RNA oligonucleotides are 25 bp double-stranded RNA oligonucleotides and were designed using BLOCK-iT™ RNAi Designer (Invitrogen, Carlsbad, CA). The Stealth siRNA designed to target mouse Aldh1a1 (NM_013467) was synthesized by Invitrogen (sense strand, 5′-UAAAGAUGCCAGGUGAAGAGCCGUG-3′; antisense strand, 5′-CACGGCUCUUCACCUGGCAUCUUUA-3′). The sequences of the Stealth siRNA control are: sense strand, 5′-CACUCUCCUCAUCGGACCUUGGUUA-3′; antisense strand, 5′-UAACCAAGGUCCGAUGAGGAGAGUG-3′). Before transfection, mouse hepatoma (Hepa1c1c7) cells (American Type Culture Collection, Rockville, MD) were maintained in Dulbellco modified Eagle’s media (DMEM) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic solution (Invitrogen; Carlsbad CA). Cells were transfected with Stealth™ siRNA using Lipofectamine™ RNAiMAX according to the manufacturer’s instructions. Cells were transfected with either 150 pmol (50 nM) siRNA control (siControl) or stealth siRNA specific to Aldh1a1 (siAldh1a1) using Lipofectamine™ RNAiMAX (Invitrogen). To test the effect of stealth siRNA on Aldh1a1 gene expression, the mRNA levels and protein levels were analyzed after 48 h by qRT-PCR and western blot respectively.
2.4 Measurement of cell viability by MTT assay
Hepa-1c1c7 cell viability was evaluated using an MTT assay according to manufacturer’s instruction. Hepa-1c1c7 cells (5000 cells/100 μl) were plated in a 96 well plate. Cells were treated with increasing concentrations of acrolein in serum free media for 24 h. Cells were then incubated with MTT (0.2 mg/ml) for 2 h and Cell viability was determined by measuring the absorbance at 570 nm.
2.5 RNA Isolation and Quantitative Real Time PCR (QRT-PCR)
RNA was isolated from Hepa-1c1c7 cells or mice liver using Tri reagent (Molecular Research Center, Inc., Cincinnati, OH). The mRNA levels of Aldh1a1, Aldh1a7, Aldh1b1, Aldh2 and Aldh3a1 were assessed by qRT-PCR. Total RNA isolated from cells was reverse transcribed to cDNA with random hexamer primers using the Advantage RT-for-PCR kit (Clontech, BD Bioscience, Palo Alto, CA). RNase H was then used to degrade any residual RNA in the cDNA mix. QRT-PCR was performed using the ABI 7900HT Sequence Detector System (Applied Biosystems, Foster City, CA) with gene specific FAM-labeled LUX primers synthesized by Invitrogen (Carlsbad CA). A plot of the CT versus quantity of RNA was generated to verify linearity of amplification. All qRT-PCR experiments were performed in triplicate using cDNA sample from independent RNA sets and analyzed by the absolute quantitation standard curve method. The gene expression levels were normalized to 18S rRNA as endogenous control and data were expressed as the mean ± SD. Samples were analyzed by Student’s t test and values of p < 0.05 were considered to be statistically significant.
2.6 Western Immunoblotting
Whole cell extracts were prepared from Hepa-1c1c7 cells using 1X RIPA buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin; Cell Signaling Technology, Beverly, MA) containing protease inhibitors (Protease Inhibitor Cocktail Set III, EDTA free; Calbiochem, La Jolla, CA). For determination of acrolein-protein adducts in cells a modified 1X RIPA Lysis buffer that contains in addition 50 mM NEM was used. Protein concentration was determined using Pierce® BCA Protein Assay (Pierce, Rockford, IL). Lysates were resolved on 4–12% NuPAGE® Novex® Bis-Tris mini gels and transferred onto Hybond™-ECL nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ). Membranes were probed with antibodies against rat ALDH1A1 (1:2000), Caspase 3 (1:1000), GAPDH (1:10000) at room temperature for 2 h or acrolein-protein adducts (1:2000) and cleaved Caspase 3 (1:1000) at 4°C overnight. Proteins were visualized by incubation with horseradish peroxidase-conjugated goat anti-rabbit (Sc-2004, Santa Cruz Biotechnology) or goat anti-mouse (Sc-2005, Santa Cruz Biotechnology, Santa Cruz, CA) secondary antibody and enhanced Chemiluminescence (Pierce, Rockford, IL) detection systems.
2.6 Liver Cytosolic ALDH Activity
The hepatic Aldh activity was examined in the cytosolic fractions isolated from the liver of male C57BL/6J mice (Jackson Laboratories, Bar Harbor, MA) treated daily with acrolein (5 mg/kg body weight) for 7 consecutive days and sacrificed 24 h after the last treatment. The liver cytosolic fraction was isolated following protocol [42]. Briefly, the liver were excised and cut into small pieces in a beaker of saline with scissors. The liver was then homogenized in 0.25 M sucrose buffered with 50 mM potassium phosphate buffer, pH 7.4. The homogenized liver was then sedimented at 1000 rpm for 10 min. Further sedimentation at 12000 rpm for 20 min was then carried in the same tube. The supernatant was then ultracentrifuged at 108,000 × g for 1 h. 20% glycerol was then added to the supernatant or cytosolic fractions and stored at −80°C. The Aldh activity was measured spectrophotometrically as described above using 1 mM propionaldehyde (substrates for Aldh1a1 or 2.5 mM benzaldehyde (substrate for Aldh3a1) as substrates with either 1 mM NAD+ or NADP+ at pH 7.4.
3. RESULTS
3.1. Expression, Purification and Biochemical Characterization of Recombinant Aldh1a1 and Aldh3a1
We cloned the open reading frame of Aldh1a1 and Aldh3a1 into a pET expression vector allowing us to express the recombinant mouse Aldhs in E coli with an N-terminal and C-terminal his-tag respectively. The recombinant enzymes were purified using Ni-NTA resin and the purity of the enzymes was assessed by SDS-PAGE electrophoresis. Cell extracts and eluate fractions (40 μg) were resolved on 4–12% SDS-PAGE gels and gels were stained with coomassie blue (Figure 1). We noted a single protein band ≈ 55 and 50 KDa in the eluate fractions of the pET30b-Aldh1a1 and pET30-Aldh3a1 transformed E coli cells, respectively. To further confirm the identity of Aldh1a1, extracts from BL21(DE3)pLysS cells transformed with pET30b-Aldh1a1, and purified rat ALDH1 [40] were resolved on SDS-PAGE gel, transferred onto nitrocellulose membrane and probed with antibody against purified rat ALDH1A1. We observed immunoreactivity of a protein band at ≈55 KDa, which migrated at comparable position to the purified rat ALDH1A1. Thus, we successful cloned, expressed and purified recombinant mouse Aldh1a1 and Aldh3a1 in E coli.
Figure 1. SDS-PAGE analyses of purified recombinant mouse Aldh1a1 and Aldh3a1.
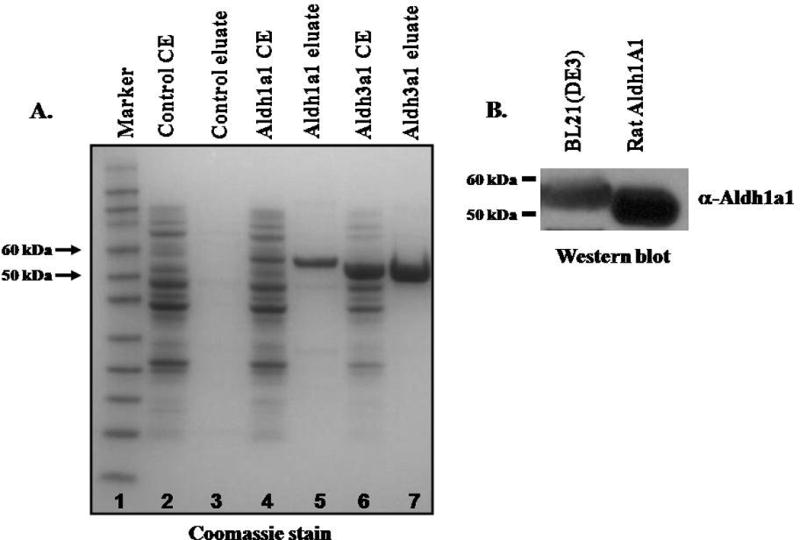
(A) Cell extracts (CE) from pET30a, pET30b-Aldh1a1 or pET30a-Aldh3a1 transformed BL21(DE3)pLysS E coli and eluate fractions purified using Ni-NTA resin were resolved on 4–12% SDS-PAGE and stained with coomassie blue. Lane1: Prestained Marker; Lane 2: CE from pET30a transformed E coli; Lane 3: eluate fractions from pET30a vector transformed E coli; Lane 4: Lysates from pET30b-Aldh1a1 transformed E coli; Lane 5: eluate fractions from pET30b-Aldh1a1 transformed E coli; Lane 6: CE from pET30a-Aldh3a1 transformed E coli; Lane 7: eluate fractions from pET30b-Aldh1a1 transformed E coli. The open reading frame of mouse Aldh3a1 and Aldh1a1 cDNA encodes a protein of 490 (Mr= 54.5 KDa) and 534 amino acids (58.0 KDa) respectively that include his-tag. (B) Western blot analysis of recombinant Aldh1a1. Extracts from BL21(DE3)pLysS transformed with pET30b-Aldh1a1, and purified rat liver ALDH1A1 were resolved by 4–12% SDS-PAGE gels. Proteins were transferred onto nitrocellulose membrane and immunoblotted with antibody against rat ALDH1. Lane 1: Eluate fractions from BL21(DE3)pLysS transformed with pET30b-Aldh1a1 plasmid; Lane 2: ALDH1A1 purified from rat liver.
We examined the kinetic parameters (Km, Vmax and Vmax/Km) of recombinant mouse Aldh1a1 and Aldh3a1 (Table 1) for oxidative metabolism of a wide range of aldehyde substrates using either NAD+ or NADP+ as pyridine nucleotide cofactor. The cofactor preference of recombinant mouse ALDH was also assessed using varying concentrations of either NAD+ or NADP+. Kinetic parameters indicate that mouse Aldh1a1 exhibited high affinity for short chain aldehydes, such as propionaldehyde (Km = 141 μM) and acetaldehyde (Km = 202 μM) and the lipid aldehydes; acrolein (Km = 23.2 μM), trans-2-hexenal (Km = 31.2 μM), HNE (Km = 2.4 μM) and MDA (Km = 7.5 μM) as shown in Table 1. The catalytic efficiency for Aldh1a1 was highest for HNE (Vmax/Km = 218) and lowest for benzaldehyde (Vmax/Km = 0.3). Mouse Aldh1a1 had an apparent Km of 752 μM for benzaldehyde, indicating that aromatic aldehydes are poor substrates for Aldh1a1. Aldh1a1 preferred NAD+ (Km = 50.2 μM) as cofactor, and displays no catalytic activity with NADP+. The order of the catalytic efficiency of mouse Aldh1a1 for aldehyde substrates, as reflected by Vmax/Km was HNE > MDA > acrolein > trans-2-hexenal > propionaldehyde > acetaldehyde > benzaldehyde. Due to the extremely high Vmax of Aldh3a1 (μmol/min/mg) for most aldehyde substrates compared to that of Aldh1a1 (nmol/min/mg), the catalytic efficiency of Aldh3a1 was higher for most aldehyde substrates except for the lipid aldehydes. In contrast to Aldh1a1, mouse Aldh3a1 exhibit high affinity for aromatic aldehydes such as benzaldehyde (Km = 37 μM) followed by the lipid aldehydes and showed poor affinity for short chain aldehydes, such as propionaldehyde (Km = 3380 μM) and acetaldehyde (Km = 1000 μM). Aldh3a1 was capable of metabolizing lipid aldehydes, such as acrolein, HNE and MDA with low affinity for substrate binding (Km > 300 μM) compared to Aldh1a1. In addition, Aldh3a1 used both NAD+ and NADP+ as cofactor for oxidative metabolism of aldehydes, but prefers NAD+ (Vmax/Km = 484) over NADP+ (Vmax/Km = 87). The catalytic efficiencies for metabolism of acrolein is comparable between Aldh1a1 and Aldh3a1 (Vmax/Km = 23). However, Aldh1a1 exhibits far higher affinity for acrolein (Km = 23.2 μM) compared to Aldh3a1 (Km = 464 μM). Furthermore, the Aldh1a1 displays high catalytic efficiency for HNE compared Aldh3a1, suggesting that reactive LPO-derived aldehydes are preferred substrates for Aldh1a1.
Table 1.
Kinetic properties of recombinant mouse Aldhs
| Aldh1a1 | Aldh3a1 | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Substrate | Vmaxa | Kmb | Vmax/Kmc | Vmaxd | Kmb | Vmax/Kmc |
| Propionaldehyde | 799 ± 22 | 141 ± 30 | 5.6 | 24.7 ± 1.2 | 3380 ± 38 | 7 |
| Acetaldehyde | 910 ± 36 | 202 ± 17 | 4.5 | 32.4 ± 1.0 | 1000 ± 20 | 32 |
| Benzaldehyde | 256 ± 14 | 752 ± 90 | 0.3 | 28.6 ± 1.6 | 37 ± 6 | 773 |
| Trans-2-hexenal | 390 ± 37 | 31.2 ± 4.1 | 12.5 | 28.1 ± 1.1 | 236 ± 40 | 119 |
| HNE | 522 ± 26 | 2.4 ± 0.9 | 218 | 29.5 ± 3.7 | 425 ± 94 | 69 |
| Acrolein | 527 ± 22 | 23.2 ± 4.3 | 23 | 10.8 ± 1.1 | 464 ± 85 | 23 |
| Malondialdehyde | 408 ± 15 | 7.5 ± 1.2 | 54 | 8.3 ± 0.7 | 302 ± 52 | 27 |
| NAD+ | 751 ± 7 | 50.2 ± 4 | 15 | 26.9 ± 5.9 | 55.5 ± 2.3 | 484 |
| NADP+ | 0 | 0 | 0 | 28 ± 7.5 | 320 ± 18 | 87 |
Aldh enzyme activity was determined by monitoring the formation of NADH at 340 nm. Enzyme activities were measured spectrophotometrically at 25°C in a reaction mixture containing 1 mM NAD+ at pH 7.4. The kinetic parameters were determined using non-linear regression software Kineti77. The values represent the means ± SD from 3–4 assays. The kinetic parameters for NAD+ and NADP+ were determined using 1 mM propionaldehyde (Aldh1a1) or 2.5 mM benzaldehyde (Aldh3a1) as the substrate.
Vmax values are expressed as nmol of NADH/min/mg of protein
Km are expressed in μM
Vmax/Km values are expressed as ml/min/mg (after normalization of Vmax to μmol product/min/mg)
Vmax is expressed in μmol/min/mg
3.2. Expression of Aldehyde dehydrogenases in mouse liver and mouse hepatoma cell lines
To assess which Aldh isoform plays a prominent role in oxidative metabolism and detoxification reactive lipid aldehydes in mice liver, we assessed the mRNA levels of the major Aldhs expressed in mice liver and Hepa-1c1c7 cells by qRT-PCR. Aldh1a1 gene was highly expressed in mouse liver and Hepa-1c1c7 cells compared to Aldh2, Aldh1b1 and Aldh3a1 (Figure 2). The mRNA levels of Aldh2 were expressed at comparably levels in both mice liver and Hepa-1c1c7 cells. However, Aldh1b1 mRNA expression was higher in Hepa-1c1c7 compared to mouse liver (≈2 fold). As previously demonstrated, Aldh3a1 mRNA expression was absent in mouse liver, but was moderately expressed in Hepa-1c1c7 cells compared to Aldh1a1. Aldh1a1 mRNA levels was 34 fold and 73-fold higher than the Aldh2 mRNA levels in mouse liver and Hepa-1c1c7 cells respectively. The order of mRNA expression of the major Aldhs involved in lipid aldehyde metabolism in mice liver and Hepa-1c1c7 was Aldh1a1 > Aldh2 > Aldh1b1 > Aldh3a1. We can conclude from enzyme kinetics and gene expression that Aldh1a1 plays a prominent role in the cellular defense against oxidative damage induced by cytotoxic aldehydes in mice liver.
Figure 2. Endogenous levels of Aldehyde dehydrogenase gene expression in mouse liver and mouse hepatoma cells.
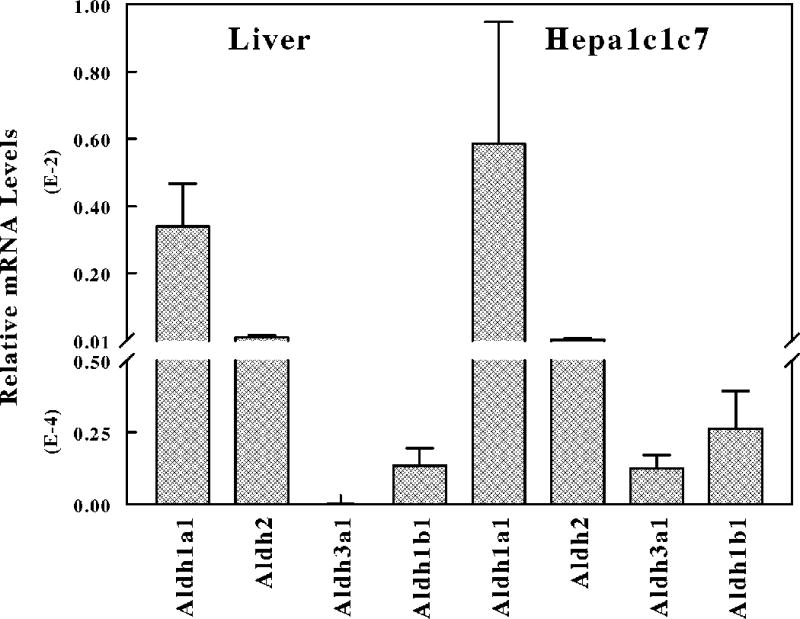
Total RNA extracted from the liver of C57BL6 mice or Hepa-1c1c7 cells was reverse transcribed to cDNA with random hexamer primers using Advantage RT-for-PCR kit. cDNA was used for analysis of gene expression of Aldh1a1, Aldh2, Aldh1b1 and Aldh3a1 by qRT-PCR.
3.3. Knockdown of Aldh1a1 gene expression sensitizes liver-derived cell lines to acrolein-induced cell death
To specifically examine the role of Aldh1a1 in cellular protection against reactive lipid-derived aldehydes such as acrolein, we assess whether knockdown of Aldh1a1 gene expression sensitizes Hepa-1c1c7 cells to acrolein-induced cell death. Hepa-1c1c7 cells were transfected with either Stealth control or Stealth Aldh1a1-specific siRNA. The effect of knockdown of Aldh1a1 gene expression was assessed by qRT-PCR and western blot. RNA was isolated from cells 48 h after transfection and was reverse transcribed to cDNA. The cDNA was then used to assess Aldh1a1 mRNA expression normalized to 18S endogenous control by qRT-PCR. In Figure 3A, we observe > 80% reduction in Aldh1a1 mRNA levels in stealth Aldh1a1-specific siRNA transfected cells compared to untransfected control. Cell extracts (40 μg) prepared from Hepa-1c1c7 cells transfected with Stealth control siRNA or Stealth Aldh1a1-specific siRNA were separated on SDS-PAGE gels and transferred onto nitrocellulose membrane. The membranes were probed with antibodies against rat ALDH1A1 and GAPDH as endogenous control (Figure 3B). Densitometry analysis of Aldh1a1 protein levels corrected for GADPH showed ≈ 85% decrease Aldh1a1 protein level in cells transfected with Stealth Aldh1a1-specific siRNA compared to untransfected control (Figure 3C). Subsequently, we assessed whether Hepa-1c1c7 cells with knockdown of Aldh1a1 gene exhibit higher sensitivity to acrolein-induced cell death. Twenty four hours after transfection of Hepa-1c1c7 cells with either stealth control or Stealth Aldh1a1 siRNA, cells were exposed to a range of acrolein concentrations (0–100 μM) for 24 h and cell viability was examined by MTT assay. Figure 3D showed that Hepa-1c1c7 cells exhibit higher sensitivity to acrolein, with 25 μM and higher concentrations producing concentration-dependent decrease in cell viability. We also observed a comparable dose-dependent decrease in viability between untransfected and control siRNA transfected cells. However, knockdown of Aldh1a1 gene expression resulted in marked decreased in cell viability compared to WT cells. The half maximal effective concentrations (EC50) of acrolein for cells with low expression of Aldh1a1 (EC50≈ 22 μM) was significantly reduced compared to that of the untransfected and control siRNA transfected cells (EC50≈ 35 μM). These results indicate that cells in which Aldh1a1 expression was decreased are highly vulnerable to acrolein-induced cell death, suggesting that Aldh1a1 protects cells from reactive aldehydes-induced cell death by enhanced metabolism of these reactive species.
Figure 3. Hepa-1c1c7 cells with knockdown of Aldh1a1 gene expression by stealth specific siRNA are highly sensitive to acrolein-induced cell death compared to control.
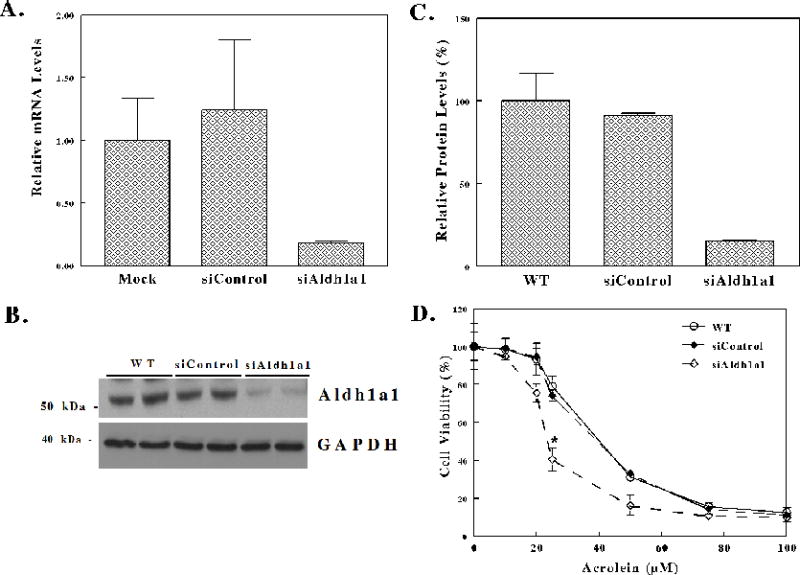
(A–C) Stealth siRNA knockdown of Aldh1a1 gene in Hepa-1c1c7 cells. Hepa-1c1c7 cells were transfected with either 150 pmol (50 nM) Stealth control (siControl) or Stealth Aldh1a1-specific siRNA (siAldh1a1) using Lipofectamine RNAiMAX. (A) qRT-PCR analysis of Aldh1a1 gene expression in Hepa1c1c7 cells. Total RNA isolated from WT (untransfected cells), Stealth control (siControl) or Aldh1a1-specific Stealth siRNA (siAldh1a1) transfected cells was converted to cDNA using random hexamer primers. The mRNA levels of Aldh1a1 were examined by qRT-PCR. (B) Western blot analysis of Aldh1a1 expression in Hepa-1c1c7 cells. Cell extracts prepared from WT, siControl and siAldh1a1 cells were separated on 4–12% SDS-PAGE gel and transferred onto nitrocellulose membrane. Membranes were probed with antibodies against ALDH1A1. Membranes were stripped and then probed with antibodies against GAPDH. (C) Densitometry analysis of Aldh1a1 protein levels normalized to GAPDH in WT, siControl and siAldh1a1 Hepa1c1c7 cells. (D) Knockdown of Aldh1a1 sensitizes Hepa-1c1c7 cells to acrolein treatment. Twenty four h after transfection with Stealth siRNA control or Stealth Aldh1a1 specific siRNA, cells were treated with increasing concentrations of acrolein for 24 h and cell viability assessed by MTT (0.2 mg/ml for 2 h) at 570 nm as described in materials and methods. The half maximal effective concentrations (EC50) were as follows; WT, untransfected cells (35.61 ± 1.64), Stealth control siRNA transfected cells (35.72 ± 1.62), and Stealth Aldh1a1 siRNA transfected cells (22.92 ± 0.32). The values are expressed as the mean values ± S.E.
3.4. Increased acrolein-protein adducts in Hepa1c1c7 cells with knockdown of Aldh1a1 gene expression
Acrolein and other highly reactive α,β-unsaturated aldehydes readily alkylate proteins forming Michael addition adducts with a free carbonyl group. It is now known that protein carbonylation by reactive aldehydes precedes the toxic process leading to cell death [23]. We examine whether acrolein-protein adducts are increased in cells with knockdown of Aldh1a1 after acrolein treatment. Hepa-1c1c7 cells were transfected with either stealth control siRNA (siControl) or Aldh1a1 specific stealth siRNA (siAldh1a1). Twenty four hours after transfection, cells were exposed to either 10 μM or 25 μM acrolein for 24 h. Lysates prepared from cells were separated on SDS-PAGE and transferred onto nitrocellulose membrane. The membrane was probed with antibodies against acrolein-protein adducts and GAPDH (Figure 4A). The level of acrolein-protein adducts of individual proteins was analyzed by densitometry corrected for GADPH using UN-SCAN-IT gel 6.1 software. Three protein bands labeled P1, P2 and P3 were chosen for analysis of acrolein-protein adducts accumulation in the siControl and siAldh1a1 cells following exposure to acrolein (10 μM and 25 μM) (Figure 4B). Densitometry analysis showed that acrolein-protein adducts content of P1 and P3 continued to increase in both siControl and siAldha1-treated cells up to 25 μM acrolein. We also noticed accumulation of endogenous acrolein-protein adducts for protein P1, P2 and P3 in siAldh1a1-treated cells compared to siControl cells. In protein 1 and 3 (P1 and P3), the level of acrolein-protein adducts were increased in Aldh1a1-specific Stealth siRNA transfected cells compared to control siRNA transfected cells after treatment with 10 μM acrolein. However, for P1, P2 and P3, we observed elevated levels of acrolein-protein adducts in siAldh1a1 cells compared to siControl cells at 25 μM acrolein treatment. These results indicate that when Aldh1a1 protein levels are low, the deficiency in acrolein detoxification led to accumulation of acrolein-protein adducts.
Figure 4. The levels of acrolein-protein adducts in response to acrolein treatment are increased in Hepa-1c1c7 cells with knockdown of Aldh1a1 gene compared to control siRNA transfected cells.
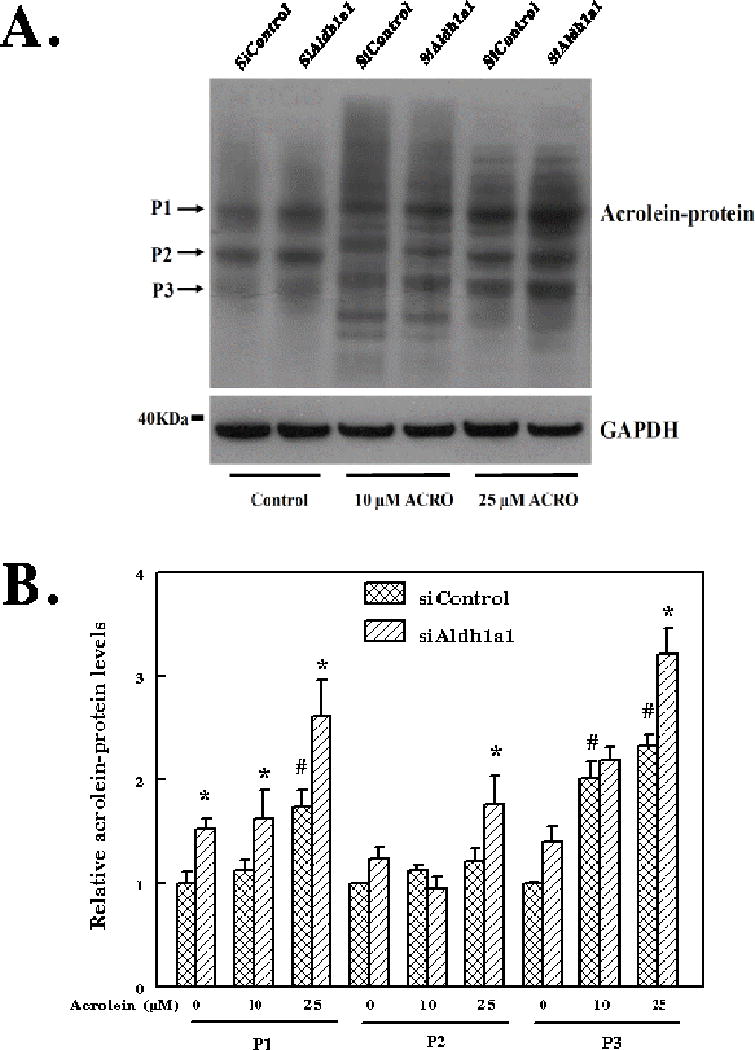
(A) Western blot analysis of acrolein-protein adducts in Hepa1c1c7 cells exposed to acrolein. Lanes 1–2: untreated cells. Lanes 3–4: cells exposed to 10 μM acrolein for 24 h and Lanes 5–6: cells exposed to 25 μM acrolein for 24 h. (B) Densitometry analysis of the levels of individual protein-acrolein adducts in siControl (Stealth control siRNA transfected cells) and siAldh1a1 (Stealth Aldh1a1 specific siRNA transfected cells) in response to 10 μM and 25 μM acrolein.
3.5. Increased acrolein-induced Caspase 3 activation and apoptosis in Hepa1c1c7 cells after knockdown of Aldh1a1 gene expression
In addition to protein-acrolein adducts accumulation and cell viability, we investigated whether knockdown of Aldh1a1 sensitizes cells to acrolein-induced caspase 3 activation, a marker of apoptosis. Reactive lipid aldehydes are known to induce oxidative stress by depletion of GSH. Oxidative stress and acrolein or HNE-protein adducts have been implicated in induction of apoptosis [24;43]. Previous studies demonstrated that HNE induces activation of caspase 3 in human lens epithelia cells (HLECs), human leukemia cells (HL-60) and neuroblastoma cell line (SH-SY5Y) [25;27;44]. Inactive procaspase 3 (32 KDa) zymogen is activated by proteolytic cleavage adjacent to Asp175 into activated 17- and 19-KDa fragments. Thus, the activation of caspase 3 is monitored by western blot analysis of the cleaved fragments. To examine whether Aldh1a1 can protect cells against acrolein-induced apoptosis, we transfected Hepa-1c1c7 cells with Stealth Aldh1a1-specific siRNA (siAldh1a1) or Stealth control siRNA (siControl) for 24 h and cells were subsequently exposed to 25 μM acrolein for 24 h. Cell extracts separated by SDS-PAGE gels were transferred onto nitrocellulose membrane and probed for cleaved caspase 3 fragments, caspase 3 and GADPH as endogenous control (Figure 5A). The levels of procaspase (Figure 5B) and cleaved caspase 3 (17 and 19 KDa fragments) (Figure 5C) in untransfected cell (WT), siControl- and siAldh1a1-treated Hepa1c1c7 cells exposed to 25 μM acrolein were assessed by densitometry corrected for GADPH using UN-SCAN-IT gel 6.1 software. The levels of procaspase 3 were moderately increased in WT and siAldh1a1 cells after acrolein treatment. However, procaspase 3 protein levels were slightly decreased in siAldh1a1 cells in the absence of acrolein treatment. As expected, the levels of the 17 and 19 KDa fragments of caspase 3 (active) were robustly increased (≈ 40-fold) when cells were exposed to acrolein compared to untreated WT cells or control siRNA transfected cells. The 19 KDa fragment was moderately increased in siAldh1a1 cells compared to WT and siControl cells after acrolein (25 μM) exposure (Figure 5C). However, we observe elevated levels (≈ 3-fold) of the 17 KDa fragment in siAldh1a1 cells compared WT and siControl cells after acrolein (25 μM) exposure. These results indicate that Aldh1a1 knockdown cells exhibit high sensitivity to acrolein-induced caspase 3 initiated apoptosis. We can conclude from these data that Aldh1a1 by enhanced detoxification and metabolism of acrolein protects cells against apoptosis induced by acrolein.
Figure 5. Transient knockdown of Aldh1a1 gene expression sensitizes Hepa-1c1c7 cells to acrolein-induced caspase 3 activation and apoptosis.
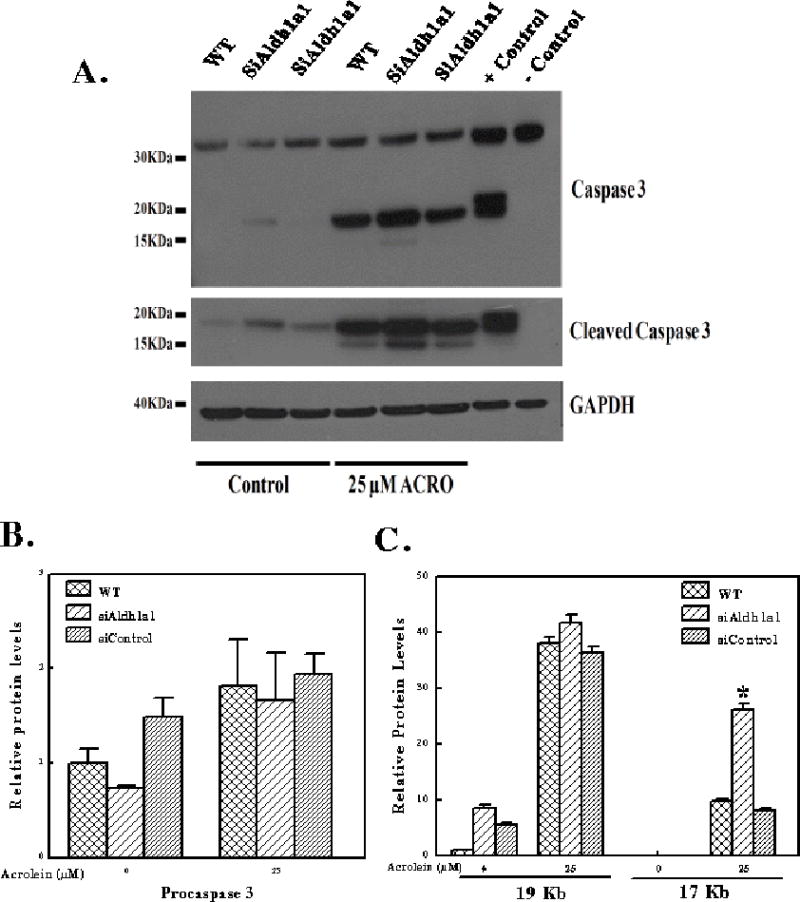
(A) Western blot analysis of acrolein-induced caspase 3 activation in Hepa-1c1c7 cells. Hepa-1c1c7 cells were transfected with Stealth specific Aldh1a1 siRNA (siAldh1a1) or Stealth control siRNA (siControl) for 24 h and cells were subsequently exposed to 25 μM acrolein for 24 h. Cell extracts prepared from cells with 1X RIPA buffer were separated on SDS-PAGE gel and transferred onto membranes. The membranes were probed for cleaved caspase 3 with anti-cleaved caspase 3 antibodies, caspase 3 with anti-Caspase 3 antibodies. Anti-cleaved caspase 3 (Asp175) antibody detects levels of large fragment (17/19 KDa) of activated caspase 3 resulting from cleavage adjacent to Asp175. Densitometry normalized to GAPDH of procaspase 3 (B) and Cleaved Caspase 3 (17 and 19 KDa fragments) (C) in WT, siControl and siAldh1a1-treated Hepa1c1c7 cells exposed to 25 μM acrolein. Positive (+) control for caspase 3 cleavage is extracts from cytochrome c treated Jurkat cell (#9663) and negative (−) control is extract from untreated Jurkat cells.
3.6. Acrolein induces the expression of Aldh1a1 gene in mice liver and hepatoma cell
The expression of Aldh1a1 gene is regulated by a wide variety of endogenous and exogenous stimuli. Microsomal and other chemical inducers were shown to modulate the expression of cytoprotective and antioxidant genes including Aldh1a1 [34;45]. Recent studies indicate that oxidative stress induces the expression of antioxidant and phase II detoxifying genes, including Aldh1a1 in an in vivo model of renal ischemia/reperfusion injury [45]. There is increasing evidence that acrolein and related aldehydes, such as HNE and crotonaldehyde at sublethal concentrations, are important signaling molecules and can induce the expression of cytoprotective genes encoding antioxidant and electrophile detoxification enzymes [46–48]. Interestingly, many of these cytoprotective genes are involved in acrolein and reactive aldehydes metabolism and detoxification.
We examined whether acrolein at sublethal concentration can induce the expression of Aldhs in mice liver. C57BL/6 mice were treated with saline (control) or 5 mg/kg acrolein by gavage daily for 7 days. The daily human consumption of unsaturated aldehydes is ≈ 5 mg/kg and no hepatocellular damage was observed in mice exposed to this dose of acrolein [13]. Total RNA was extracted from the liver, reverse transcribed to cDNA and used for qRT-PCR analysis of gene expression of Aldh1a1, Aldh2, Aldh3a1 and Aldh1b1. Aldh1a1 mRNA levels were significantly increased (≈ 3 fold) in acrolein exposed mice compared to control mice. Acrolein treatment did not caused induction of Aldh2, Aldh3a1 or Aldh1b1 gene in mice liver. Hepatic cytosolic Aldh activity was induced in mice treated with acrolein with benzaldehyde or propionaldehyde as substrate and NAD+ as cofactor. However, acrolein-induced hepatic cytosolic Aldh activity was absent with both substrates when NADP+ was used as pyridine nucleotide cofactor. Since Aldh3a1 gene expression in mice liver is negligible, the major isoform that contributes to acrolein-induced Aldh activity is Aldh1a1. This is the first report to demonstrate induction of Aldh gene expression by acrolein in mouse liver.
4.0 Discussion
4.1 Comparison of Substrate Specificity for murine Aldh’s
The relative contribution of Aldh isozymes in protection against aldehyde toxicity is controversial. While earlier studies indicated that ALDH3A1 but not ALDH1A1 protects against cytotoxic lipid derived-aldehydes, most recent studies in eye lens and neuroblastoma cell lines showed that ALDH1A1 can protect against these reactive aldehydes [33;37]. The contribution of the different isozymes of Aldh to oxidation of lipid derived-aldehydes has not been examined in the liver with the liver being the major site for metabolism and biotransformation of foreign compounds. In this study, we sought to clearly define which Aldh isoform play a major role in oxidation and detoxification of cytototic lipid-derived aldehydes in mouse liver by enzyme kinetics and gene expression pattern. Having obtained purified recombinant murine Aldh’s expressed in E. coli cells, we performed enzyme kinetic analysis, in order, to compare their relative capacity for clearance of various aldehydes. As seen in Table 1, the catalytic efficiencies can best be compared using the Vmax/Km parameters. Aldh3a1 displays highest preference for aromatic aldehyde substrates and utilizes both NAD+ and NADP+ as oxidizing pyridine nucleotice cofactors with preference for NAD+. Although Aldh3A1 displays 20–112 fold higher Vmax values for most aldehyde substrates, the Vmax/Km parameters demonstrate that Aldh1a1 is a better catalyst than Aldh3a1 for oxidative clearance of α,β-unsaturated aldehydes. The human and murine Aldh1a1 proteins exhibit similar Vmax/Km parameters, with some differences [38]. The human ALDH2 [49] and ALDH1B1 [50] proteins display significantly lower Vmax/Km parameters for the lipid-derived aldehydes than Aldh1a1. The conclusion taken from the data in this report and the literature is that the catalytic efficiency for lipid aldehyde oxidation by Aldh1a1 is the highest reported, suggesting its importance for clearance of these toxic compounds.
4.2 Aldh1a1 is highly expressed Aldh in mouse liver and Hepa1c1c cells
In Figure 2, we show that Aldh1a1 is highly expressed in the livers of male C57BL/6 mice and in mouse hepatoma Hepa-1c1c cells, relative to other Aldh enzymes. The levels of Aldh1a1 mRNA were about 34-fold higher than that of the next more abundant message, Aldh2. There was much less Aldh1b1 and Aldh3A1 message expressed in either liver or Hepa-1c1c cells. This observation is supported by the data in Figure 6, which further demonstrates that acrolein caused a 3-fold induction of Aldh1a1 mRNA in male mouse livers, while other forms of Aldh were not inducible by treatment with acrolein, providing a mechanism for the liver cell to respond to toxic aldehydes. In addition, it is known that Aldh1a1 is also induced by animal treatment with butylated hydroxyanisole [34] and phenobarbital [51], activators of transcription factors such as activator protein 1 (AP1), nuclear factor-erythroid 2 related factor 2 (Nrf2), and constitutive androstane receptor. These observations clearly demonstrate that mouse Aldh1a1 and probably in the human ortholog are the major protective systems against the products of lipid peroxidation and other toxic aldehydes in the environment.
Figure 6. Acrolein induces the expression of Aldh1a1 gene and cytosolic Aldh activity in mice liver.
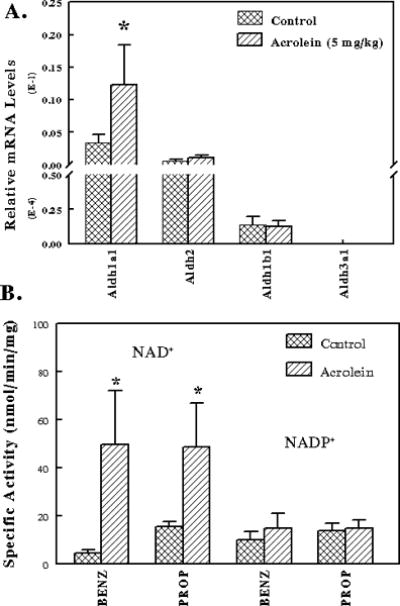
(A) C57BL/6 mice were treated with saline (control, n=5) or 5 mg/kg acrolein (treated, n=5) by gavage daily for 7 days. Total RNA was extracted from the liver, reverse transcribed to cDNA and used for qRT-PCR analysis for gene expression of Aldh1a1, Aldh2, Aldh3a1 and Aldh1b1. (B) The effect of acrolein on hepatic cytosolic ALDHs. C57BL/6 mice were treated with saline (control, n=5) or 5 mg/kg acrolein (treated, n=5) by gavage daily for 7 days. The liver was extracted and the cytosol obtained by centrifugation at 108,000 × g for 1 h. Aldh activity assay was determined by monitoring the formation of NAD(P)H at 340 nm. The reactions rates were assayed spectrophotometrically at 37°C in a reaction mixture containing 1 mM propionaldehyde, 2.5 mM benzaldehyde with either 1 mM NAD+ or NADP+ at pH 7.4. The reaction was initiated by addition of aldehyde substrates into a 1 ml cuvette. * Significantly different from controls (p< 0.05; 2 tailed t test).
4.3 Ablation of Aldh1a1 gene expression increases acrolein cell death, acrolein protein adducts, and caspase cleavage
As a method of assessing the role of specific gene products in toxicological processes, an in vitro method using gene ablation allows one to test the role of specific proteins in response to toxic agents. Since Hepa-1c1c cells appeared to recapitulate the spectrum of Aldh’s expressed in mouse liver, we chose this cell system to test whether ablation of Aldh1a1 protein and enzyme activity increases the sensitivity of these cells to acrolein (Figure 3). Using Stealth™ siRNA knockdown technology, we documented that transfection of Aldh1a1-specific siRNA suppresses the levels of Aldh1a1 mRNA and protein in Hepa1c1c cells (Figure 3A–C). We then challenged these cells with increasing concentrations of acrolein to assess the relative sensitivity to acrolein toxicity (Figure 3D). While the Control siRNA had no affect on acrolein toxicity curve that measured MTT-dependent cell viability, transfection of Hepa-1c17 cells with Aldh1a1-specific siRNA caused a shift to the left in the acrolein toxicity curve, clearly demonstrating that cells with Aldh1a1 ablated were statistically more sensitive to acrolein toxicity than control siRNA transfected or untransfected cells. These results provide evidence of an important protective role for Aldh1a1.
Previous experiments demonstrate that treatment of cells with acrolein led to increased acrolein-protein adducts and enhanced caspase 3 activation resulting in proteolytic cleavage of PARP [23;24]. We measured the levels of acrolein-protein adduct in cells transfected with Aldh1a1-specific siRNA and noted that the levels of acrolein-protein adducts were significantly increased relative to control siRNA transfected cells (Figures 4A, B). In addition, caspase 3 cleavage to a 17 KDa fragment was strikingly increased in siAldh1a1 transfected Hepa-1c1c7 cells exposed to 25 μM acrolein, relative to untransfected or siControl transfected cells (Figure 5). These results clearly demonstrate that the pro-apoptotic changes caused by acrolein and levels of acrolein-protein adducts are increase under conditions of Aldh1a1 depletion.
5.0 Conclusion
Our studies confirm that Aldh1a1 quantitatively is a major Aldh in mouse liver at mRNA and protein level. Its kinetic parameters also indicates that catalytically it is a major factor in cytosolic oxidation of α,β-unsaturated aldehydes in liver. Ablation of Aldh1a1 increases sensitivity of Hepa1c1c cells to acrolein toxicity, acrolein-protein adducts and caspase 3 cleavage leading to apoptosis. Induction of Aldh1a1, on the other hand, significantly decreased acrolein toxicity. Since Aldh1a1 is an inducible enzyme in the liver by compounds such as butylated hydroxyanisol and phenobarbital, ALDH1A1 induction by these compounds may provide a useful rationale for therapeutic protection against oxidative stress-induced pathologies.
Acknowledgments
Supported in part from NIH Grants ES11860(DJC/RAP), HL89380 (DJC), and 5P30ES014443. This publication is in partial completion of the Ph.D. degree for N.L. Makia and he was also supported by USPHS NIH grant R13-AA019612 to present this work at the 15th International Meeting on Enzymology and Molecular Biology of Carbonyl Metabolism in Lexington, KY, USA. The authors are indebted to Sanjay Srivastava, Ph.D., Department of Medicine/Cardiovascular Medicine for providing substrates and serving as a consultant.
Abbreviations
- Aldh
aldehyde dehydrogenases
- 4-HNE
4-Hydroxy-2-nonenal
- LPO
Lipid peroxidation
- MDA
malondialdehyde
- WT
wild type
- ORF
open reading frame
- CYP
Cytochrome P450
- ROS
reactive oxygen species
- MTT
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Reference List
- 1.Esterbauer H, Cheeseman KH, Dianzani MU, Poli G, Slater TF. Separation and characterization of of the aldehydic products of lipid peroxidation stimulated by ADP-Fe2+ in rat liver hepatocytes. Biochem J. 1982;208:129–140. doi: 10.1042/bj2080129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 3.Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- 4.Esterbauer H. Cytotoxicity and genotoxicity of lipid-oxidation products. Am J Clin Nutr. 1993;57:779S–785S. doi: 10.1093/ajcn/57.5.779S. [DOI] [PubMed] [Google Scholar]
- 5.Ellis EM. Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther. 2007;115:13–24. doi: 10.1016/j.pharmthera.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 6.Uchida K. Role of reactive aldehyde in cardiovascular diseases. Free Radic Biol Med. 2000;28:1685–1696. doi: 10.1016/s0891-5849(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 7.Dwivedi S, Sharma R, Sharma A, Zimniak P, Ceci JD, Awasthi YC, Boor PJ. The course of CCl4 induced hepatotoxicity is altered in mGSTA4-4 null (−/−) mice. Toxicology. 2006;218:58–66. doi: 10.1016/j.tox.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Nanji AA, Zhao S, Sadrzadeh SM, Dannenberg AJ, Tahan SR, Waxman DJ. Markedly enhanced cytochrome P450 2E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol Clin Exp Res. 1994;18:1280–1285. doi: 10.1111/j.1530-0277.1994.tb00119.x. [DOI] [PubMed] [Google Scholar]
- 9.Sampey BP, Stewart BJ, Petersen DR. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J Biol Chem. 2007;282:1925–1937. doi: 10.1074/jbc.M610602200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stevens JF, Maier CS. Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol Nutr Food Res. 2008;52:7–25. doi: 10.1002/mnfr.200700412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feron VJ, Til HP, de VF Woutersen RA, Cassee FR, van Bladeren PJ. Aldehydes: occurrence, carcinogenic potential, mechanism of action and risk assessment. Mutat Res. 1991;259:363–385. doi: 10.1016/0165-1218(91)90128-9. [DOI] [PubMed] [Google Scholar]
- 12.Wang GW, Guo Y, Vondriska TM, Zhang J, Zhang S, Tsai LL, Zong NC, Bolli R, Bhatnagar A, Prabhu SD. Acrolein consumption exacerbates myocardial ischemic injury and blocks nitric oxide-induced PKCepsilon signaling and cardioprotection. J Mol Cell Cardiol. 2008;44:1016–1022. doi: 10.1016/j.yjmcc.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 13.Conklin DJ, Barski OA, Lesgards JF, Juvan P, Rezen T, Rozman D, Prough RA, Vladykovskaya E, Liu S, Srivastava S, Bhatnagar A. Acrolein consumption induces systemic dyslipidemia and lipoprotein modification. Toxicol Appl Pharmacol. 2010;243:1–12. doi: 10.1016/j.taap.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen JJ, Schenker S, Henderson GI. 4-hydroxynonenal levels are enhanced in fetal liver mitochondria by in utero ethanol exposure. Hepatology. 1997;25:142–147. doi: 10.1002/hep.510250126. [DOI] [PubMed] [Google Scholar]
- 15.Uchida K, Toyokuni S, Nishikawa K, Kawakishi S, Oda H, Hiai H, Stadtman ER. Michael addition-type 4-hydroxy-2-nonenal adducts in modified low-density lipoproteins: markers for atherosclerosis. Biochemistry. 1994;33:12487–12494. doi: 10.1021/bi00207a016. [DOI] [PubMed] [Google Scholar]
- 16.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 17.Srivastata SK, Awasthi S, Wang L, Bhatnagar A, Awasthi YC, Ansari NH. Attenuation of 4-hydroxynonenal-induced cataractogenesis in rat lens by butylated hydroxytoluene. Curr Eye Res. 1996;15:749–754. doi: 10.3109/02713689609003458. [DOI] [PubMed] [Google Scholar]
- 18.Toyokuni S, Yamada S, Kashima M, Ihara Y, Yamada Y, Tanaka T, Hiai H, Seino Y, Uchida K. Serum 4-hydroxy-2-nonenal-modified albumin is elevated in patients with type 2 diabetes mellitus. Antioxid Redox Signal. 2000;2:681–685. doi: 10.1089/ars.2000.2.4-681. [DOI] [PubMed] [Google Scholar]
- 19.Hammer A, Ferro M, Tillian HM, Tatzber F, Zollner H, Schauenstein E, Schaur RJ. Effect of oxidative stress by iron on 4-hydroxynonenal formation and proliferative activity in hepatomas of different degrees of differentiation. Free Radic Biol Med. 1997;23:26–33. doi: 10.1016/s0891-5849(96)00630-2. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Henderson GI, Freeman GL. Role of 4 hydroxynonenal in modification of cytochrome c oxidase in the ischemic/reperfused rat heart. J Mol Cell Cardiol. 2001;33:1919–1927. doi: 10.1006/jmcc.2001.1454. [DOI] [PubMed] [Google Scholar]
- 21.Szweda LI, Uchida K, Tsai L, Stadtman ER. Inactivation of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Selective modification of an active-site lysine. J Biol Chem. 1993;268:3342–3347. [PubMed] [Google Scholar]
- 22.Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction. J Biol Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 23.Burcham PC, Fontaine F. Extensive protein carbonylation precedes acrolein-mediated cell death in mouse hepatocytes. J Biochem Mol Toxicol. 2001;15:309–316. doi: 10.1002/jbt.10007. [DOI] [PubMed] [Google Scholar]
- 24.Tanel A, Averill-Bates DA. Inhibition of acrolein-induced apoptosis by the antioxidant N-acetylcysteine. J Pharmacol Exp Ther. 2007;321:73–83. doi: 10.1124/jpet.106.114678. [DOI] [PubMed] [Google Scholar]
- 25.Cheng JZ, Singhal SS, Sharma A, Saini M, Yang Y, Awasthi S, Zimniak P, Awasthi YC. Transfection of mGSTA4 in HL-60 cells protects against 4-hydroxynonenal-induced apoptosis by inhibiting JNK-mediated signaling. Arch Biochem Biophys. 2001;392:197–207. doi: 10.1006/abbi.2001.2452. [DOI] [PubMed] [Google Scholar]
- 26.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 27.Zhang M, Shoeb M, Goswamy J, Liu P, Xiao TL, Hogan D, Campbell GA, Ansari NH. Overexpression of aldehyde dehydrogenase 1A1 reduces oxidation-induced toxicity in SH-SY5Y neuroblastoma cells. J Neurosci Res. 2010;88:686–694. doi: 10.1002/jnr.22230. [DOI] [PubMed] [Google Scholar]
- 28.Amunom I, Stephens LJ, Tamasi V, Cai J, Pierce WM, Jr, Conklin DJ, Bhatnagar A, Srivastava S, Martin MV, Guengerich FP, Prough RA. Cytochromes P450 catalyze oxidation of alpha, beta-unsaturated aldehydes. Arch Biochem Biophys. 2007;464:187–196. doi: 10.1016/j.abb.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srivastava S, Liu SQ, Conklin DJ, Zacarias A, Srivastava SK, Bhatnagar A. Involvement of aldose reductase in the metabolism of atherogenic aldehydes. Chem Biol Interact. 2001;130–132:563–571. doi: 10.1016/s0009-2797(00)00299-4. [DOI] [PubMed] [Google Scholar]
- 30.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4:697–720. doi: 10.1517/17425250802102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava S, Chandra A, Wang LF, Seifert WE, Jr, DaGue BB, Ansari NH, Srivastava SK, Bhatnagar A. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J Biol Chem. 1998;273:10893–10900. doi: 10.1074/jbc.273.18.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramu K, Fraiser LH, Mamiya B, Ahmed T, Kehrer JP. Acrolein mercapturates: synthesis, characterization, and assessment of their role in the bladder toxicity of cyclophosphamide. Chem Res Toxicol. 1995;8:515–524. doi: 10.1021/tx00046a005. [DOI] [PubMed] [Google Scholar]
- 33.Townsend AJ, Leone-Kabler S, Haynes RL, Wu Y, Szweda L, Bunting KD. Selective protection by stably transcfected human ALDH3A1 (but not human ALDH1A1) against toxicity of aldehydes in V79 cells. Chem Biol Interact. 2001;130:261–273. doi: 10.1016/s0009-2797(00)00270-2. [DOI] [PubMed] [Google Scholar]
- 34.Alnouti Y, Klaassen CD. Tissue distribution, ontogeny, and regulation of aldehyde dehydrogenase (Aldh) enzymes mRNA by prototypical microsomal enzyme inducers in mice. Toxicol Sci. 2008;101:51–64. doi: 10.1093/toxsci/kfm280. [DOI] [PubMed] [Google Scholar]
- 35.Lassen N, Bateman JB, Estey T, Kuszak JR, Nees DW, Piatigorsky J, Duester G, Day BJ, Huang J, Hines LM, Vasiliou V. Multiple and additive functions of ALDH3A1 and ALDH1A1: cataract phenotype and ocular oxidative damage in Aldh3a1(−/−)/Aldh1a1(−/−) knock-out mice. J Biol Chem. 2007;282:25668–25676. doi: 10.1074/jbc.M702076200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choudhary S, Srivastava S, Xiao T, Andley UP, Srivastava SK, Ansari NH. Metabolism of lipid derived aldehyde, 4-hydroxynonenal in human lens epithelial cells and rat lens. Invest Ophthalmol Vis Sci. 2003;44:2675–2682. doi: 10.1167/iovs.02-0965. [DOI] [PubMed] [Google Scholar]
- 37.Choudhary S, Xiao T, Vergara LA, Srivastava S, Nees D, Piatigorsky J, Ansari NH. Role of aldehyde dehydrogenase isozymes in the defense of rat lens and human lens epithelial cells against oxidative stress. Invest Ophthalmol Vis Sci. 2005;46:259–267. doi: 10.1167/iovs.04-0120. [DOI] [PubMed] [Google Scholar]
- 38.Xiao T, Shoeb M, Siddiqui MS, Zhang M, Ramana KV, Srivastava SK, Vasiliou V, Ansari NH. Molecular cloning and oxidative modification of human lens ALDH1A1: implication in impaired detoxification of lipid aldehydes. J Toxicol Environ Health A. 2009;72:577–584. doi: 10.1080/15287390802706371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreb JS, Mohuczy D, Ostmark B, Zucali JR. RNAi-mediated knockdown of aldehyde dehydrogenase class-1A1 and class-3A1 is specific and reveals that each contributes equally to the resistance against 4-hydroperoxycyclophosphamide. Cancer Chemother Pharmacol. 2007;59:127–136. doi: 10.1007/s00280-006-0233-6. [DOI] [PubMed] [Google Scholar]
- 40.Tweedie DJ, Fernandez D, Spearman ME, Feldhoff RC, Prough RA. Metabolism of azoxy derivatives of procarbazine by aldehyde dehydrogenase and xanthine oxidase. Drug Metab Dispos. 1991;19:793–803. [PubMed] [Google Scholar]
- 41.Lindahl R, Petersen DR. Lipid aldehyde oxidation as a physiological role for class 3 aldehyde dehydrogenases. Biochem Pharmacol. 1991;41:1583–1587. doi: 10.1016/0006-2952(91)90157-z. [DOI] [PubMed] [Google Scholar]
- 42.Remmer H, Schenkman J, Estabrook RW, Sasame H, Gillette J, Narasimhulu S, Cooper DY, Rosenthal O. Drug interaction with hepatic microsomal cytochrome. Mol Pharmacol. 1966;2:187–190. [PubMed] [Google Scholar]
- 43.Xiao T, Choudhary S, Zhang W, Ansari NH, Salahudeen A. Possible involvement of oxidative stress in cisplatin-induced apoptosis in LLC-PK1 cells. J Toxicol Environ Health A. 2003;66:469–479. doi: 10.1080/15287390306449. [DOI] [PubMed] [Google Scholar]
- 44.Choudhary S, Zhang W, Zhou F, Campbell GA, Chan LL, Thompson EB, Ansari NH. Cellular lipid peroxidation end-products induce apoptosis in human lens epithelial cells. Free Radic Biol Med. 2002;32:360–369. doi: 10.1016/s0891-5849(01)00810-3. [DOI] [PubMed] [Google Scholar]
- 45.Leonard MO, Kieran NE, Howell K, Burne MJ, Varadarajan R, Dhakshinamoorly S, Porter AG, O’Farrelly C, Rabb H, Taylor CT. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. Faseb J. 2006;20:E2166–E2176. doi: 10.1096/fj.06-5097fje. [DOI] [PubMed] [Google Scholar]
- 46.Liu XY, Yang ZH, Pan XJ, Zhu MX, Xie JP. Gene expression profile and cytotoxicity of human bronchial epithelial cells exposed to crotonaldehyde. Toxicol Lett. 2010;197:113–122. doi: 10.1016/j.toxlet.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 47.Ranganna K, Yousefipour Z, Nasif R, Yatsu FM, Milton SG, Hayes BE. Acrolein activates mitogen-activated protein kinase signal transduction pathways in rat vascular smooth muscle cells. Mol Cell Biochem. 2002;240:83–98. doi: 10.1023/a:1020659808981. [DOI] [PubMed] [Google Scholar]
- 48.Tirumalai R, Rajesh KT, Mai KH, Biswal S. Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicol Lett. 2002;132:27–36. doi: 10.1016/s0378-4274(02)00055-3. [DOI] [PubMed] [Google Scholar]
- 49.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4:697–720. doi: 10.1517/17425250802102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stagos D, Chen Y, Brocker C, Donald E, Jackson BC, Orlicky DJ, Thompson DC, Vasiliou V. Aldehyde dehydrogenase 1B1 (ALDH1B1): Molecular cloning and characterization of a novel mitochondrial acetaldehyde metabolizing enzyme. Drug Metab Dispos. 2010 doi: 10.1124/dmd.110.034678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deitrich RA, Bludeau P, Stock T, Roper M. Induction of different rat liver supernatant aldehyde dehydrogenases by phenobarbital and tetrachlorodibenzo-p-dioxin. J Biol Chem. 1977;252:6169–6176. [PubMed] [Google Scholar]