

Abstract
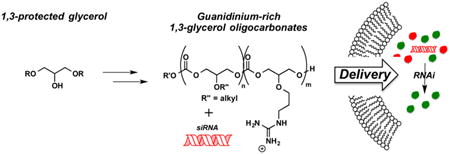
A highly versatile and step-economical route to a new class of guanidinium-rich molecular transporters and evaluation of their ability to complex, deliver, and release siRNA are described. These new drug/probe delivery systems are prepared in only two steps, irrespective of length or composition, using an organocatalytic ring-opening co-oligomerization of glycerol-derived cyclic carbonate monomers incorporating either protected guanidine or lipid side chains. The resultant amphipathic co-oligomers are highly effective vehicles for siRNA delivery, providing an excellent level of target protein suppression (>85%). These new oligocarbonates are nontoxic at levels required for cell penetration and can be tuned for particle size. Relative to the previously reported methyl(trimethylene)carbonate (MTC) scaffold, the ether linkage at C2 in the new transporters markedly enhances the stability of the siRNA/co-oligomer complexes. Both hybrid co-oligomers, containing a mixture of glycerol- and MTC-derived monomers, and co-oligomers containing only glycerol monomers are found to provide tunable control over siRNA complex stability. On the basis of a glycerol and CO2 backbone, these new co-oligomers represent a rapidly tunable and biocompatible siRNA delivery system that is highly effective in suppressing target protein synthesis.
Keywords: guanidinium-rich molecular transporters; siRNA delivery; organocatalytic ring-opening oligomerization; oligocarbonates; 1,3-glycerol carbonate
Introduction
Precise control over gene expression is invaluable in basic research and pathway elucidation and is recognized for its immense potential in the treatment of human disease.1–3 The ability of small interfering RNA (siRNA) to engage the RNA interference (RNAi) pathway and induce post-transcriptional gene silencing through the suppression of protein synthesis in a sequence-specific manner allows for potent and selective inhibition of expression for nearly any gene of interest.4 While the use of siRNA to suppress protein synthesis offers a powerful and quickly implemented alternative to the development of small molecule protein inhibitors, clinical application of this technology has been hampered principally by a single major problem: delivery of siRNA across cell membranes.2,5–8 siRNA is double-stranded and generally composed of 19–23 base pairs, with two overhanging nucleotides at the 3′ ends. Its size, susceptibility to enzymatic degradation, and hydrophilic, polyanionic character collectively prevent its diffusion across nonpolar cellular membranes, as is required to engage the cytosolic machinery of the RNAi pathway.9
As part of our work on drug delivery systems, we previously showed that the ability of the HIV-Tat 9-mer, a highly polar and water-soluble polycation, to enter cells is determined by its arginine content, more specifically, by the number and spatial array of its guanidinium groups.10 We and others have since shown that guanidinium-rich transporters enable or enhance the uptake of small molecules,11,12 probes,13,14 peptides,15,16 proteins,17–19 siRNA,20–22 and plasmids23,24 into cells.25–29 They also facilitate passage of agents across skin, buccal, lung, brain, and even cell wall (algal30) barriers. They can be targeted31,32 and also used to overcome export pump-based resistance, a major cause of chemotherapy failure.33,34
Since our initial studies, homooligomers of arginine have been synthesized on GMP scale and evaluated in the clinic for dermatological applications.11 However, because the number of steps required for the synthesis of oligoarginines is determined by their length, typically requiring 16 steps to prepare an octamer via solid-phase synthesis or nine steps using a segment doubling strategy, certain research and clinical applications have been hampered by cost and time concerns.35 To address these problems and thereby more quickly access and screen transporter compositions for optimal cellular uptake, we reported in 2009 a two-step, metal-free living oligomerization that enables rapid access to cell-penetrating, guanidinium-rich oligocarbonates based on a methyl(trimethylene)carbonate (MTC) scaffold (Figure 1).36 This new class of rapidly accessible molecular transporters was shown, using a real-time living cell uptake quantification procedure, to efficiently deliver fluorescent and bioluminescent cargos into cells.13,36,37 We and others have further advanced oligomerization strategies for synthesizing guanidinium-rich molecular transporters and have shown their use for both covalent and noncovalent cargo association and delivery.38–42 More recently, we reported the first amphipathic oligocarbonate systems derived from oligomerization of lipid and guanidinylated MTC monomers that can noncovalently complex, deliver, and release siRNA in vitro with over 85% silencing efficiencies.21
Figure 1.
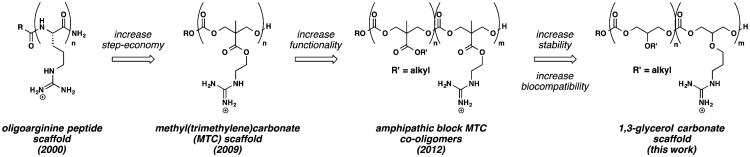
Development of step-economical routes to guanidinium-rich molecular transporters since 2000. In this work, glycerol and carbon dioxide are utilized as biocompatible building blocks for the synthesis of guanidinium-rich oligocarbonate molecular transporters for siRNA delivery.
This current study was directed at determining whether the siRNA silencing efficiency of oligocarbonate delivery systems could be retained while enhancing their biocompatibility and stability. Toward these ends, a new class of oligocarbonates derived from functionalized 1,3-glycerol carbonate monomers was designed. Glycerol-based polymers have attracted interest ranging from pharmaceutical to industrial applications.43–46 In the context of biomedical applications, the ubiquity of glycerol in living systems suggests that glycerol-based materials should have excellent biocompatibility.47 To create an siRNA delivery system, we have functionalized 1,3-glycerol carbonate monomers with guanidine- or lipid-containing side chains to produce, upon ring-opening oligomerization and deprotection, amphipathic co-oligomers that noncovalently complex and deliver siRNA into cells (Figure 1). We hypothesized that this change in transporter structure, particularly the C2-ether linkage of the guanidine and lipid side chains, would enhance the stability of siRNA-containing complexes relative to our previously reported MTC scaffold while retaining functional delivery. Indeed, here we show that these glycerol-derived oligocarbonates effectively complex, deliver, and release siRNA in cells, with over 85% suppression of target protein production in some cases. Moreover, through selective incorporation of appropriately functionalized monomers derived from either glycerol or MTC, exquisite control over physical properties, such as the half-life of the siRNA/co-oligomer complexes, is achieved while maintaining both function and cell viability. This ability to control by design the physical properties of these noncovalent complexes could be leveraged for different therapeutic applications of oligonucleotide delivery. Our studies on this new siRNA delivery system are described herein.
Experimental Section
Materials
Chemical reagents were purchased from Sigma-Aldrich and were used as received unless otherwise indicated. A lithium naphthalenide solution,48 1-(3,5-bis-trifluoromethylphenyl)-3-cyclohexyl-thiourea,49 MTC-guanidine monomer,36 and MTC-dodecyl monomer21 were all prepared according to literature procedures. siRNAs were synthesized by Thermo Fisher Scientific, Dharmacon Products. CBL350 and K6a51 siRNA sequences have been previously reported. Silencer FAM-labeled siRNA was purchased from Life Technologies. Regenerated cellulose dialysis membranes (Spectra/Por 6 Standard RC; MWCO 1000) were purchased from Spectrum Laboratories, Inc. PBS buffer was prepared from RNase-free 10× PBS solution (Fisher Scientific). Dulbecco's modified Eagle's medium (DMEM) was purchased from Invitrogen and supplemented with 10% FBS and 1% penicillin/streptomycin. Lipofectamine 2000 was purchased from Life Technologies. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Fluka.
Instrumentation
1H NMR and 13C NMR were recorded on a Varian Inova 500 (1H at 500 MHz, 13C at 125 MHz) or Varian Inova 600 (1H at 600 MHz, 13C at 150 MHz) spectrometer. Infrared spectra were measured on a PerkinElmer 1600 Series Fourier transform spectrometer (FTIR). High-resolution mass spectra (HRMS) were obtained from the Vincent Coates Foundation Mass Spectrometry Laboratory at Stanford University. Gel permeation chromatography (GPC) was performed with a Viscotek S3580 refractive index detector and Viscotek GPCmax autosampler. The system was calibrated using monodisperse polystyrene standards (Polymer Laboratories). Particle size and zeta potential were measured by dynamic light scattering on a Malvern Zetasizer Nano ZS90. Flow cytometry analysis was performed on a BD LSRII FACS Analyzer (LSRII, LSRII.UV, Stanford University Shared FACS Facility).
Synthesis of Glycerol-Derived, C2-Ether Monomers
Glycerol-derived monomers 3 and 6 were both prepared from commercially available (Aldrich) 1,3-dibenzyloxy-2-propanol (Scheme 1). Dodecyl monomer 3 was prepared by alkylation of protected glycerol 1, Pd-mediated hydrogenation of the benzyl ethers, and phosgenation. The synthesis of guanidine monomer 6 began with conjugate addition of 1 into acrylonitrile52 to provide nitrile 4 in near-quantitative yield. Cobalt(II)-assisted NaBH4 reduction53 of nitrile 4 followed by guanidinylation afforded acyclic protected guanidine 5 in 75% yield over two steps. Deprotection of the benzyl ethers with lithium naphthalenide afforded the desired diol, which was then converted to the cyclic carbonate with triphosgene, generating guanidine monomer 6 in 54% overall yield over 5 steps.
Scheme 1. Synthesis of (A) Glycerol-Derived Dodecyl Monomer 3 and (B) Guanidinylated Glycerol-Derived Monomer 6a.

aReagents and conditions: (A) (a) KOtBu, 1-bromododecane, CH2Cl2, reflux 20 h, 58%; (b) H2 (1 atm), Pd/C, EtOAc, 24 h, >99%; (c) triphosgene, pyridine, CH2Cl2, –78 °C to rt, 19 h, 70%. (B) (d) Triton B, acrylonitrile, CH2Cl2, rt, 18 h, >99%; (e) CoCl2·6H2O, NaBH4, MeOH, 0 °C, 1 h; (f) N,N′-Di-Boc-1H-pyrazole-1-carboxamidine, THF, 12 h, 75% over 2 steps; (g) lithium naphthalenide, THF, –25 °C, 3 h; (h) triphosgene, pyridine, CH2Cl2, –78 °C to rt, 3 h, 72% over 2 steps.
Synthesis of Glycerol-Derived Amphipathic Oligocar-bonates
Amphipathic co-oligomers 10–13 were synthesized using an organocatalytic oligomerization previously used to make MTC oligomers (Scheme 2).21,36,54 This method relies on selective hydrogen-bond mediated activation of the cyclic carbonate monomers with thiourea catalyst 7 in conjunction with a DBU co-catalyst to facilitate reaction with an alcohol initiator. The acyclic carbonate products do not undergo similar activation.55 The general method for the preparation of block co-oligomers 10–13 is as follows: a solution of lipidated monomer (3 or 821) in methylene chloride was added to a vial charged with alcohol initiator (R–OH, see Scheme 2A) in the presence of both the thiourea catalyst 7 and catalytic DBU under a nitrogen atmosphere at room temperature. After 75 min, guanidine-protected monomer (6 or 921) was added for an additional 75 min, and then the reaction was quenched with 10 mg of benzoic acid. The reaction mixture was dialyzed (MWCO 1000) against methanol for 20 h total; the methanol solution was changed after the first 6 h. Global deprotection of the guanidine functionality with trifluoroacetic acid (TFA) yielded the amphipathic carbonate co-oligomers 10–13 (Scheme 2). The degree of polymerization (DP) was determined by end-group analysis using 1H NMR.
Scheme 2. Preparation of Amphipathic Carbonate Co-oligomersa.
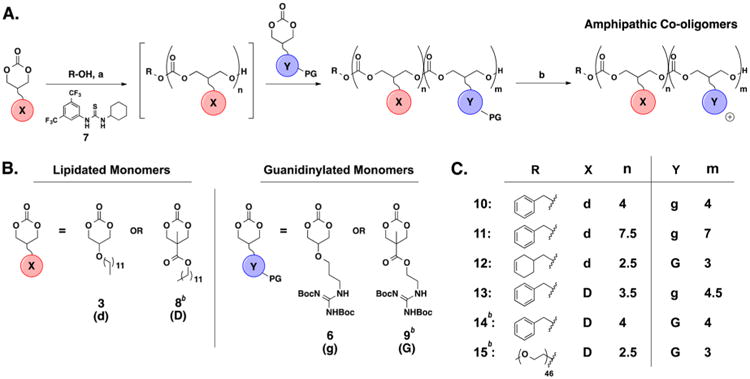
a(A) Synthesis by ring-opening oligomerization, followed by guanidine deprotection. Reagents and conditions: (a) (i) R–OH, 7 (5 mol %), DBU (5 mol %), lipidated monomer 3 or 8, CH2Cl2, rt, (ii) guanidinylated monomer 6 or 9; (b) TFA, CH2Cl2, rt; yields (over 2 steps): 10 = 45%, 11 = 48%, 12 = 75%, 13 = 34%. (B) Co-oligomers were synthesized with one of two possible lipidated monomers and guanidine monomers. (C) Structures (X, Y) and DP (n, m) of co-oligomers. bFrom ref 21.
Gel Stability Assay
Agarose gels (2% w/v) were prepared with 25 μL of ethidium bromide stock solution (10 mg/mL), cast, and allowed to set for 1 h. siRNA/co-oligomer solutions were made in 1× PBS at a constant ± charge ratio of 4.77:1 by mixing varying amounts of PBS (5.2–6.8 μL) and co-oligomer solution (1.25 mM, 1.2–2.8 μL) with K6a siRNA (2.0 μL of a 25 μM stock in RNase-free PBS pH 7.4). This charge ratio was selected based on previous optimization studies.21 The siRNA/co-oligomer solutions were incubated at room temperature for 30 min and then transferred to a heating block and incubated at 37 °C for varying amounts of time (0–96 h) before being loaded onto the gel. TAE buffer was added to the gel until covered (∼1 L), and 2.0 μL of a 50:50 glycerol/H2O solution was added to each sample before loading onto the gel. Twenty-five microliters of additional ethidium bromide stock was added to the TAE buffer, and the gel was run at 106 V for 1 h, after which band migrations were visualized with a UV illuminator. Gel band intensities were analyzed by ImageJ software.
Cell Lines
Untransfected immortalized human keratinocyte (HaCaT) cells and dual-fluorescent reporter HaCaT cells, transduced with two lentiviral vectors containing the fluorescent proteins tdTomato and EGFP, were used.21 The tdTomato vector contained a Luciferase2 cDNA fused upstream of tdTomato with the CBL3 target site cloned into the 3′ UTR under the ubiquitin C promoter. HaCaT cells were maintained in DMEM at 37 °C in a 5% CO2 atmosphere.
Flow Cytometry
tdTomato/EGFP expressing HaCaT cells were seeded at 20 000 cells per well and cultured in a 24-well plate for 24 h at 37 °C. siRNA/co-oligomer complexes were formed at a ± charge ratio of 4.77:1 and were prepared by mixing various amounts of co-oligomer from a 1.25 mM stock solution (3.0–7.0 μL in RNase-free PBS pH 7.4) with CBL3 siRNA (5.0 μL of a 25 μM stock in RNase-free PBS pH 7.4) and RNase-free PBS pH 7.4 (50.5–54.5 μL). The complexes were allowed to form at room temperature for 30 min. The Lipofectamine 2000 control was prepared in OptiMEM according to the manufacturer's instructions. The cells were washed with ∼0.5 mL of serum-free DMEM medium, and then 375 μL of serum-free DMEM medium was added to the wells with untreated cells, 337.5 μL to the wells treated with Lipofectamine 2000/siRNA, and 356.25 μL was added to the wells treated with siRNA/co-oligomer complex Then, 37.5 μL Lipofectamine/siRNA and 18.75 μL siRNA/co-oligomer complex were added to each of the appropriate wells; all conditions were performed in triplicate. The cells were incubated at 37 °C for 4 h, the medium was replaced with ∼1.0 mL of fresh serum-containing DMEM medium, and the cells incubated for 68 h at 37 °C. The medium was then removed, and the cells were washed with 1.0 mL of PBS. 0.4 mL EDTA trypsin was added, and the cells were incubated for 15 min at 37 °C. Next, 0.6 mL of serum-containing DMEM medium was added, and the contents of each well were transferred to a 15 mL centrifuge tube and centrifuged (1200 rpm for 5 min). The cells were collected and redispersed in 250 μL of PBS, transferred to FACS tubes, and read on a flow cytometry analyzer. Results were analyzed using FlowJo software. The data presented are the mean fluorescent signals from 10 000 cells analyzed. The normalized percent tdTOM expression was calculated with the following formulate: (mean fluorescence tdTOMtreated cells/mean fluorescence EGFPtreated cells)/(mean fluorescence tdTOMuntreated cells/mean fluorescence EGFPuntreated cells) × 100.21 The knockdown values are the average of a minimum of three different trial runs (n ≥ 3). Error expressed as ± standard deviation (SD).
For studies at reduced temperature, untransfected HaCat cells were seeded at 40 000 cells per well, and cells were incubated at 4 °C for 30 min before treatment with complex, Lipofectamine 2000, or siRNA alone. siRNA/co-oligomer complex was prepared by mixing co-oligomer 12 (7.0 μL of a 1.25 mM stock solution in RNase-free PBS pH 7.4), Silencer FAM siRNA (5.0 μL of a 25 μM stock in nuclease free water), and RNase-free PBS pH 7.4 (50.5 μL). siRNA alone was prepared by adding Silencer FAM siRNA (5 μL of a 25 μM stock in nuclease-free water) to RNase-free PBS pH 7.4 (57.5 μL). All washes were done with ice-cold media. The cells were treated on ice and incubated for 30 min at 4 °C before analysis by flow cytometry. The fold-increase in fluorescence was calculated from the mean fluorescence of treated cells divided by the mean fluorescence of untreated cells. The fluorescence values are the average of three different trial runs. Error is expressed as ±SD.
Cell Viability Assays
HaCaT cells were seeded at 5000 cells per well and cultured in a 96-well plate for 24 h at 37 °C. siRNA/co-oligomer complexes were formed at a charge ratio of 4.77:1 and were prepared by mixing various amounts of co-oligomer from a 1.25 mM stock solution (0.96–2.24 μL in RNase-free PBS pH 7.4) with K6a siRNA (1.6 μL of a 25 μM stock in RNase-free PBS pH 7.4) and RNase-free PBS pH 7.4 (16.16–17.44 μL). The complexes were allowed to incubate at room temperature for 30 min. Two microliters of 10 mM colchicine was added to 1 mL of serum-free medium. Cells were washed with serum-free DMEM medium, and 100 μL of serum-free DMEM medium was added to the untreated wells, 95 μL of serum-free DMEM medium was added to the wells treated with siRNA/co-oligomer complex, and 150 μL of serum-free media was added to wells treated with colchicine. Then, 5 μL of siRNA/co-oligomer or co-oligomer alone was added to each respective well and 200 μL colchicine to one well, followed by a serial dilution of 50 μL across 10 wells. The cells were incubated with the compounds for 4 h at 37 °C, and then the medium was removed and replaced with 150 μL of fresh, serum-containing DMEM medium. The cells were incubated for a further 68 h. Viability was assayed by adding 10 μL of MTT solution (5 mg/mL in DMEM medium). After 2 h of incubation at 37 °C, 100 μL of solubilizing solution (10% Triton-X-100, 90% 0.1 N HCl in isopropanol) was added to each well, and colorimetry data was obtained on a plate reader. Percent viability was determined by dividing the average colorimetric value obtained for a treated sample by the average colorimetric value obtained for untreated cells. The values reported are the average of a minimum of three different trial runs (n ≥ 3). Error is expressed as ± SD.
Dynamic Light Scattering and Zeta Potential
Dynamic light scattering (DLS) was used to determine the size of the siRNA/co-oligomer complexes. siRNA/co-oligomer solutions were made by mixing RNase-free PBS pH 7.4 (101–109 μL), various amounts of co-oligomer from a 1.25 mM stock solution (6.0–14 μL in RNase-free PBS pH 7.4), and 10 μL of K6a siRNA (25 μM stock in RNase-free PBS pH 7.4) to achieve charge ratios of 4.77:1. The solution was immediately transferred to a disposable clear plastic cuvette, and the size was measured. Size measurements were taken at the initial time (0 min), 30 min, and 60 min. The sizes reported are the z-averages based on the intensity measurements and are reported in diameter nm (dnm). The numbers reported are the average of three different trial runs. Error is expressed as ± SD. Zeta potential measurements were taken by diluting the siRNA/co-oligomer complexes formulated for DLS into 800 μL of PBS pH 7.4, transferring to zeta cell (DTS1060), and measuring zeta potential. The values reported are the average of a minimum of three trial runs. Error is expressed as ± SD.
Statistical Analysis
All data are represented as the mean ± standard deviation. Statistical differences between conditions were determined using a one-way ANOVA with a Newman–Keuls posthoc test, performed using GraphPad Prism software. To compare significance between two groups, a t-test was performed. For all statistical tests, a p-value less than 0.05 indicated significance.
Results and Discussion
We have developed a new class of cell-penetrating, guanidinium-rich molecular transporters that are capable of noncovalently complexing, delivering, and releasing siRNA in vitro. These oligomeric transporters are derived from glycerol. While aliphatic polycarbonates from simple derivatives of 1,3-glycerol carbonate have been reported,56–61 this study represents the first synthesis of guanidinium-rich, amphipathic, glycerol-derived 1,3-oligocarbonates and their first evaluation for use in siRNA delivery.
To evaluate the functional differences influenced by the C2-ether linkage of the glycerol-derived oligocarbonates, the relative stability of the resulting siRNA/co-oligomer complexes was examined under biological assay conditions by gel electrophoresis (Figures 2 and S1). siRNA complexes of the previously reported oligomer 1421 released siRNA with a half-life of 12 h, whereas those of the hybrid co-oligomer 12 had a release half-life of 16 h. In comparison, siRNA complexes formed with the C2-ether co-oligomers 10 and 11 displayed half-lives of greater than 96 h (Figures 2C and S1). Upon evaluating guanidinium homo-oligomers derived from glycerol, MTC, or a hybrid, it was found that their relative hydrolytic stabilities correlated with the stabilities of siRNA-containing complexes derived from oligomers of the same backbone composition (Table S2). This may indicate that the release of siRNA could be, at least in part, driven by a degradation mechanism. While determination of the mechanism(s) of release, i.e., oligocarbonate degradation, dissociation, or a combination of the two, is beyond the scope of this initial study, functional intracellular release of siRNA is nevertheless achieved. This finding, in turn, provides the basis for controlling cargo release rates. Despite significant differences in half-lives (hours vs days), the varying compositions of siRNA/co-oligomer complexes retain the ability to inhibit target protein production, thus providing the ability to control the lifetime of the electrostatic (noncovalent) complexes. The ability to tune the stability of the siRNA-encapsulating complex through selective incorporation of different cyclic carbonate monomers is an attractive attribute of this delivery system that could potentially be used to control the rate of release for different in vivo applications.
Figure 2.
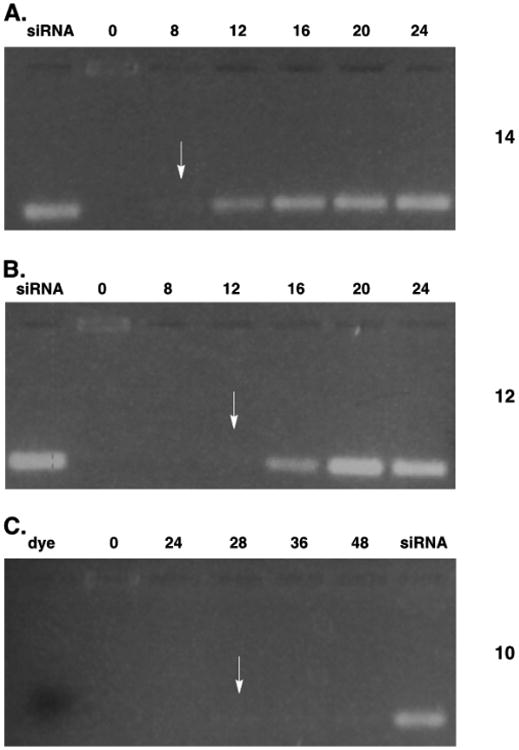
Assessment of the relative stability of siRNA/co-oligomer complexes formed with (A) MTC co-oligomer 14, (B) hybrid glycerol–MTC co-oligomer 12, or (C) glycerol co-oligomer 10. Complexes were formulated in PBS, pH 7.4, at a ± ratio of 4.77:1, incubated at room temperature for 30 min, and then placed in a 37 °C heat block for the indicated amount of time (in hours) before loading onto the gel. Reappearance of the siRNA band indicates release of siRNA from the complex; the time corresponding to first reappearance is denoted with an arrow.
A dual fluorescent protein reporter assay was used to evaluate the ability of the amphipathic co-oligomers to deliver and release siRNA that targets expression of the red fluorescent protein, tdTOM, in human keratinocytes.21 All of the co-oligomers tested were capable of delivering and releasing siRNA but exhibited varying efficacies at 100 nM siRNA concentrations (Figure 3). In a comparison study, the most effective MTC co-oligomer 1421 outperformed the corresponding, glycerol-derived 10 in protein suppression over 72 h (p < 0.01). Significantly, however, the efficacies of hybrid constructs 12 and 13 were comparable to that of 14, exhibiting, in the case of 12, over 86% silencing of the target protein in cells. Exchange of the counterion from TFA to chloride minimally impacts protein suppression. Formulation with 5% of a PEG-initiated amphipathic co-oligomer, a strategy that is known to decrease particle size and add stability, reduced the protein suppression, presumably due to the well-established shielding effect of PEG,62 yet it still achieved protein suppression levels of over 60%. As expected, neither siRNA alone nor the delivery of a control siRNA (K6a) with oligomer 12 resulted in the suppression of tdTOM expression.
Figure 3.
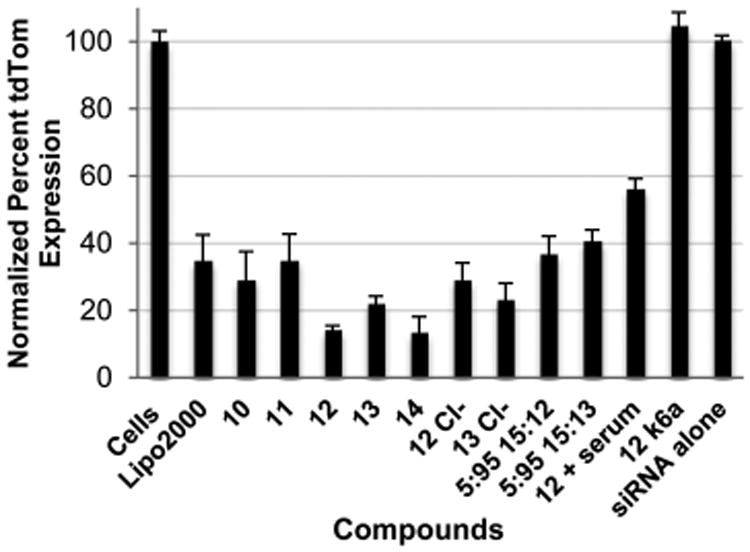
Reduction of tdTOM fluorescence normalized to EGFP fluorescence in dual fluorescence reporter HaCaT cells by glycerol-derived, amphipathic co-oligomers, as measured by flow cytometry. All treatments were 100 nM with respect to siRNA. Particles formulated at 4.77:1 ± charge ratio. 12 Cl− denotes oligomer 12 after exchange of the TFA counterion to chloride. 5:95 15:12 represents a mixture containing 5 mol % PEG-initiated oligomer 15 and 95 mol % oligomer 12.
To explore the mechanism of cellular uptake of the siRNA/co-oligomer complexes, a fluorescently labeled control siRNA was delivered to nonfluorescent HaCaT cells using co-oligomer 12 at 4 °C (a condition that inhibits endosomal pathways),26 and the relative mean fluorescence observed was compared to treatment at 37 °C (Figure 4). Endocytosis inhibition under these conditions resulted in a 57% reduction in mean fluorescence when compared to uptake at 37 °C (p < 0.01). This decrease suggests that endocytotic mechanisms are a main contributor to cellular uptake of the complexes, but other processes may also be operative.
Figure 4.
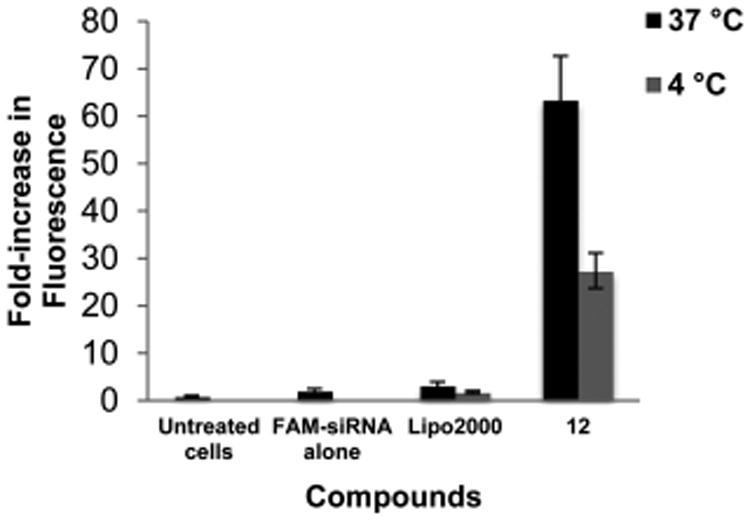
Comparison of cellular uptake of a fluorescently labeled siRNA after 30 min incubation with HaCaT cells at 37 or 4 °C. Values reflect the fold-increase in fluorescence observed in treated cells relative to untreated cells. The 57% reduction in mean fluorescence for complexes treated at 4 °C indicates that endocytotic mechanisms of uptake for the siRNA/co-oligomer complexes are in effect.
The relative cytotoxicity of the glycerol-derived transporters was evaluated by analyzing the mitochondrial staining of MTT in treated cells relative to untreated cells. The co-oligomers displayed little to no cytotoxicity to HaCaT cells, either in treatment alone or when complexed with siRNA at concentrations of 100 nM siRNA (Figure 5). This is in agreement with the relative nontoxicity of the previously reported MTC co-oligomers, indicating that the increase in stability associated with the C2-ether linkage does not result in increased cell toxicity. Additionally, exchange of the counterion from TFA to chloride results in oligomers with virtually no cytotoxicity at the tested concentrations (cell viability ≥99% in some experiments).
Figure 5.
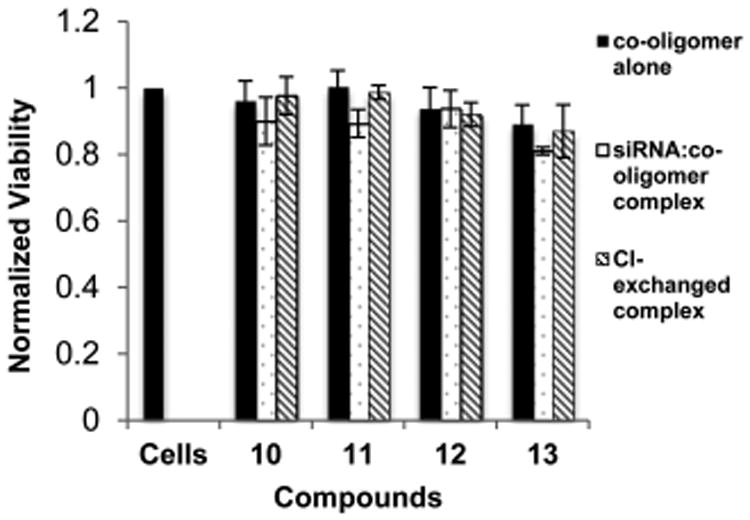
Normalized viability of HaCaT cells treated with siRNA/co-oligomer complexes relative to untreated cells, as determined by MTT assay over 72 h. Complexes tested at concentrations of 100 nM with respect to siRNA. Particles formulated at 4.77:1 ± charge ratio.
Lastly, it was shown that the size of the siRNA/co-oligomer complexes can be tuned from aggregates to discrete particles with stable size profiles through formulation with a PEG-initiated amphipathic co-oligomer or through counterion exchange from TFA to chloride (Figures 6 and S2).
Figure 6.
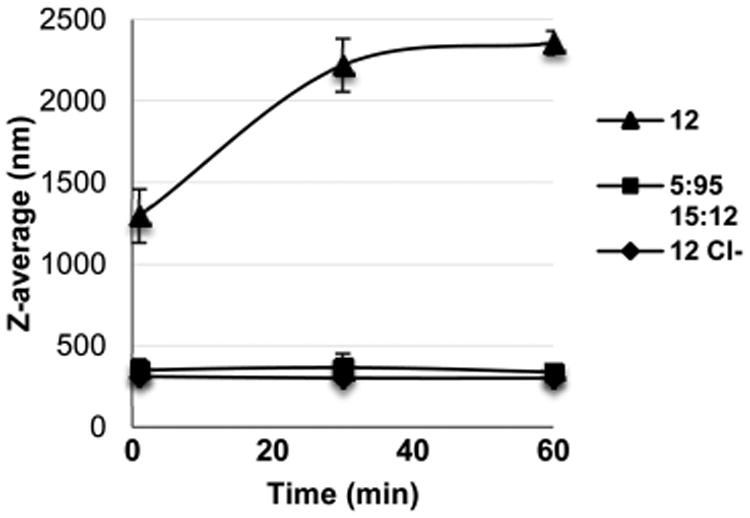
DLS measurements of siRNA/co-oligomer complexes formed with 12 over time. Particle size and stability over time of the noncovalent complexes can be controlled by counterion exchange from TFA to chloride or with 5 mol % PEG-initiated 15 incorporation. Particles were formulated at 4.77:1 ± charge ratio.
Importantly, these formulation conditions result in a significant reduction in particle size and an increase in the stability of that size profile over time, while minimally impacting protein suppression. As expected, the incorporation of 5 mol % PEG-initiated 15 also resulted in a decrease in surface charge, as measured by zeta potential (Table S1). The ability to tune the size and stability of the particles is important for certain in vivo applications in order to potentially take advantage of the enhanced permeation and retention (EPR) effect in tumor tissue and to achieve optimal clearance and circulation in an organism.63
The ability to tune function by controlling the initiator, size, stability, length, and composition of the oligomers in this study, and by extension other oligomers accessible through this two-step strategy, allows one to create a diverse library of nontoxic amphipathic co-oligomers with distinct but variable physical properties, which can be used to effectively complex, deliver, and release siRNA into cells (here, human keratinocytes).
Conclusions
The safe and effective delivery of oligonucleotides such as siRNA remains a significant challenge in research and in the development of oligonucleotide therapeutics. The objective of the current work has been to design and evaluate new delivery systems, in this case, resulting in cell-penetrating, guanidinium-rich oligocarbonates that can be rapidly synthesized (two steps) and that noncovalently complex and deliver siRNA utilizing the abundant and biocompatible molecules, glycerol and CO2, as the oligomer backbone. The synthesis of functionalized glycerol 1,3-carbonates has been achieved, and the resulting C2-etherified cyclic monomers undergo organocatalytic ring-opening oligomerization to rapidly afford guanidinium-rich, amphipathic delivery vectors that effectively complex, deliver, and release siRNA into cells.
We have shown that siRNA/co-oligomer complexes with the C2-ether scaffold display prolonged stability relative to previously reported carbonate complexes, demonstrating the importance of the side chain linkage in the stability of guanidinium-rich oligocarbonates. This extended stability allows one to tune the half-life of the particles, and thus the rate at which siRNA is released, from hours to days by selectively incorporating either glycerol- or MTC-derived monomers in the transporter synthesis. This control could potentially be exploited in different therapeutic applications. Additionally, the glycerol-derived oligocarbonates were observed to be nontoxic at concentrations needed for delivery, despite this increase in stability. Furthermore, the glycerol-derived monomers in this work establish a foundation for the further exploration of transporters with highly biocompatible, nontoxic degradation products. Finally, the ability to mix and match on demand various glycerol and MTC cyclic carbonate monomers with control of oligomer length and stability provides the basis for the broader use of this strategy in research, potentially even in kit form.
Supplementary Material
Supporting Information: Figures S1: Representative gel shift assays to determine relative hydrolytic stability of siRNA/co-oligomer complexes. Figure S2: DLS measurements of siRNA/co-oligomer complexes formed with glycerolcontaining co-oligomers over time. Figure S3: Gel shift assays to compare siRNA binding efficiency of glycerol co-oligomer 10 to glycerol guanidinium monomer 16. Table S1: Zeta potential measurements of siRNA/co-oligomer complexes with and without 5 mol % PEG-initiated 15. Table S2: Characterization and relative hydrolytic stabilities of guanidinium-rich oligocarbonates derived from glycerol, MTC, or both. General methods, synthetic procedures, and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
Support of this work through Grants NIH-CA031841 and NIH-CA031845 from the National Institutes of Health is acknowledged. Support of this work through fellowships from the National Science Foundation (M.A.H., J.R.V.), the Stanford-NIH Training Program in Biotechnology (M.A.H.), the Amgen Graduate Research Fellowship (D.S.), and the Stanford Chemistry Undergraduate Summer Research Fellowship and Chemical Engineering Undergraduate Summer Research Program (A.F.X.) is also acknowledged. The LSRII.UV was obtained by the Stanford Shared FACS Facility by using funds from NIH Shared Instrumentation Grant S10RR027431-01. We gratefully acknowledge Prof. Bob Waymouth for sharing his lab space and equipment and for insightful discussions; Prof. Chris Contag and Prof. Lynette Cegelski for materials, tissue culture equipment, and a plate reader; Prof. Richard Zare for the use of the Malvern Zetasizer; Prof. Matt Kanan for the use of the analytical HPLC; Tyler Stukenbroeker for his assistance with acquiring GPC traces; and the Stanford NMR Facility and Stanford Shared FACS Facility for instrumentation. We thank Dr. Erika Geihe Stanzl for insightful discussion. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions: All authors participated in the design of the project. M.A.H., D.S., J.R.V., and A.F.X. conducted the work. All authors participated in the writing of the manuscript and have given approval to its final version.
Notes: The authors declare no competing financial interest.
References
- 1.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral Vectors for Gene-Based Therapy. Nat Rev Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 2.Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene Therapy Clinical Trials Worldwide to 2012 — an Update. J Gene Med. 2013;15:65–77. doi: 10.1002/jgm.2698. [DOI] [PubMed] [Google Scholar]
- 3.Martínez T, Wright N, López-Fraga M, Jiménez AI, Pañeda C. Silencing Human Genetic Diseases with Oligonucleotide-Based Therapies. Hum Genet. 2013;132:481–493. doi: 10.1007/s00439-013-1288-1. [DOI] [PubMed] [Google Scholar]
- 4.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 5.Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA Interference in the Clinic: Challenges and Future Directions. Nat Rev Cancer. 2011;11:59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davidson BL, McCray PB., Jr Current Prospects for RNA Interference-Based Therapies. Nat Rev Genet. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takefman D, Bryan W. The State of Gene Therapies: The FDA Perspective. Mol Ther. 2012;20:877–878. doi: 10.1038/mt.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitehead KA, Langer R, Anderson DG. Knocking down Barriers: Advances in siRNA Delivery. Nat Rev Drug Discovery. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaynor JW, Campbell BJ, Cosstick R. RNA Interference: A Chemist's Perspective. Chem Soc Rev. 2010;39:4169–4184. doi: 10.1039/b920362c. [DOI] [PubMed] [Google Scholar]
- 10.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The Design, Synthesis, and Evaluation of Molecules That Enable or Enhance Cellular Uptake: Peptoid Molecular Transporters. Proc Natl Acad Sci USA. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothbard JB, Garlington S, Lin Q, Kirschberg T, Kreider E, McGrane PL, Wender PA, Khavari PA. Conjugation of Arginine Oligomers to Cyclosporin A Facilitates Topical Delivery and Inhibition of Inflammation. Nat Med. 2000;6:1253–1257. doi: 10.1038/81359. [DOI] [PubMed] [Google Scholar]
- 12.Luedtke NW, Carmichael P, Tor Y. Cellular Uptake of Aminoglycosides, Guanidinoglycosides, and Poly-arginine. J Am Chem Soc. 2003;125:12374–12375. doi: 10.1021/ja0360135. [DOI] [PubMed] [Google Scholar]
- 13.Jones LR, Goun EA, Shinde R, Rothbard JB, Contag CH, Wender PA. Releasable Luciferin-Transporter Conjugates: Tools for the Real-Time Analysis of Cellular Uptake and Release. J Am Chem Soc. 2006;128:6526–6527. doi: 10.1021/ja0586283. [DOI] [PubMed] [Google Scholar]
- 14.Puckett CA, Barton JK. Fluorescein Redirects a Ruthenium–Octaarginine Conjugate to the Nucleus. J Am Chem Soc. 2009;131:8738–8739. doi: 10.1021/ja9025165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Wright LR, Chen CH, Oliver SF, Wender PA, Mochly-Rosen D. Molecular Transporters for Peptides: Delivery of a Cardioprotective εPKC Agonist Peptide into Cells and Intact Ischemic Heart Using a Transport System, R7. Chem Biol. 2001;8:1123–1129. doi: 10.1016/s1074-5521(01)00076-x. [DOI] [PubMed] [Google Scholar]
- 16.Jameson KL, Mazur PK, Zehnder AM, Zhang J, Zarnegar B, Sage J, Khavari PA. IQGAP1 Scaffold-Kinase Interaction Blockade Selectively Targets RAS-MAP Kinase-Driven Tumors. Nat Med. 2013;19:626–630. doi: 10.1038/nm.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In Vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y, Siuzdak G, Scholer HR, Duan L, Ding S. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, Kim KS. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N. Transvascular Delivery of Small Interfering RNA to the Central Nervous System. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 21.Geihe EI, Cooley CB, Simon JR, Kiesewetter MK, Edward JA, Hickerson RP, Kaspar RL, Hedrick JL, Waymouth RM, Wender PA. Designed Guanidinium-Rich Amphipathic Oligocarbonate Molecular Transporters Complex, Deliver and Release siRNA in Cells. Proc Natl Acad Sci USA. 2012;109:13171–13176. doi: 10.1073/pnas.1211361109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tezgel AÖ, Gonzalez-Perez G, Telfer JC, Osborne BA, Minter LM, Tew GN. Novel Protein Transduction Domain Mimics as Nonviral Delivery Vectors for siRNA Targeting NOTCH1 in Primary Human T Cells. Mol Ther. 2013;21:201–209. doi: 10.1038/mt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siprashvili Z, Scholl FA, Oliver SF, Adams A, Contag CH, Wender PA, Khavari PA. Gene Transfer via Reversible Plasmid Condensation with Cysteine-Flanked, Internally Spaced Arginine-Rich Peptides. Hum Gene Ther. 2003;14:1225–1233. doi: 10.1089/104303403767740768. [DOI] [PubMed] [Google Scholar]
- 24.Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D'Souza GGM. Cell Transfection in Vitro and in Vivo with Nontoxic TAT Peptide–Liposome–DNA Complexes. Proc Natl Acad Sci USA. 2003;100:1972–1977. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanzl EG, Trantow BM, Vargas JR, Wender PA. Fifteen Years of Cell-Penetrating, Guanidinium-Rich Molecular Transporters: Basic Science, Research Tools, and Clinical Applications. Acc Chem Res. 2013;46:2944–2954. doi: 10.1021/ar4000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The Design of Guanidinium-Rich Transporters and Their Internalization Mechanisms. Adv Drug Delivery Rev. 2008;60:452–472. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wender PA, Cooley CB, Geihe EI. Beyond Cell Penetrating Peptides: Designed Molecular Transporters. Drug Discovery Today Technol. 2012;9:e49–e55. doi: 10.1016/j.ddtec.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torchilin VP. Tat Peptide-Mediated Intracellular Delivery of Pharmaceutical Nanocarriers. Adv Drug Delivery Rev. 2008;60:548–558. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 29.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-Rich Peptides: An Abundant Source of Membrane-Permeable Peptides Having Potential as Carriers for Intracellular Protein Delivery. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 30.Hyman JM, Geihe EI, Trantow BM, Parvin B, Wender PA. A Molecular Method for the Delivery of Small Molecules and Proteins across the Cell Wall of Algae Using Molecular Transporters. Proc Natl Acad Sci USA. 2012;109:13225–13230. doi: 10.1073/pnas.1202509109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor Imaging by Means of Proteolytic Activation of Cell-Penetrating Peptides. Proc Natl Acad Sci USA. 2004;101:17867–17872. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goun EA, Shinde R, Dehnert KW, Adams-Bond A, Wender PA, Contag CH, Franc BL. Intracellular Cargo Delivery by an Octaarginine Transporter Adapted To Target Prostate Cancer Cells through Cell Surface Protease Activation. Bioconjugate Chem. 2006;17:787–796. doi: 10.1021/bc0503216. [DOI] [PubMed] [Google Scholar]
- 33.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming Multidrug Resistance of Small-Molecule Therapeutics through Conjugation with Releasable Octaarginine Transporters. Proc Natl Acad Sci USA. 2008;105:12128–12133. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wender PA, Galliher WC, Bhat NM, Pillow TH, Bieber MM, Teng NNH. Taxol–Oligoarginine Conjugates Overcome Drug Resistance in-Vitro in Human Ovarian Carcinoma. Gynecol Oncol. 2012;126:118–123. doi: 10.1016/j.ygyno.2012.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wender PA, Jessop TC, Pattabiraman K, Pelkey ET, VanDeusen CL. An Efficient, Scalable Synthesis of the Molecular Transporter Octaarginine via a Segment Doubling Strategy. Org Lett. 2001;3:3229–3232. doi: 10.1021/ol0161108. [DOI] [PubMed] [Google Scholar]
- 36.Cooley CB, Trantow BM, Nederberg F, Kiesewetter MK, Hedrick JL, Waymouth RM, Wender PA. Oligocarbonate Molecular Transporters: Oligomerization-Based Syntheses and Cell-Penetrating Studies. J Am Chem Soc. 2009;131:16401–16403. doi: 10.1021/ja907363k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wender PA, Goun EA, Jones LR, Pillow TH, Rothbard JB, Shinde R, Contag CH. Real-Time Analysis of Uptake and Bioactivatable Cleavage of Luciferin-Transporter Conjugates in Transgenic Reporter Mice. Proc Natl Acad Sci USA. 2007;104:10340–10345. doi: 10.1073/pnas.0703919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolonko EM, Kiessling LL. A Polymeric Domain That Promotes Cellular Internalization. J Am Chem Soc. 2008;130:5626–5627. doi: 10.1021/ja8001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolonko EM, Pontrello JK, Mangold SL, Kiessling LL. General Synthetic Route to Cell-Permeable Block Copolymers via ROMP. J Am Chem Soc. 2009;131:7327–7333. doi: 10.1021/ja809284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gabriel GJ, Madkour AE, Dabkowski JM, Nelson CF, Nusslein K, Tew GN. Synthetic Mimic of Antimicrobial Peptide with Nonmembrane-Disrupting Antibacterial Properties. Biomacromolecules. 2008;9:2980–2983. doi: 10.1021/bm800855t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hennig A, Gabriel GJ, Tew GN, Matile S. Stimuli-Responsive Polyguanidino-Oxanorbornene Membrane Transporters as Multicomponent Sensors in Complex Matrices. J Am Chem Soc. 2008;130:10338–10344. doi: 10.1021/ja802587j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gasparini G, Bang EK, Molinard G, Tulumello DV, Ward S, Kelley SO, Roux A, Sakai N, Matile S. Cellular Uptake of Substrate-Initiated Cell-Penetrating Poly(disulfide)s. J Am Chem Soc. 2014;136:6069–6074. doi: 10.1021/ja501581b. [DOI] [PubMed] [Google Scholar]
- 43.Xu J, Feng E, Song J. Renaissance of Aliphatic Polycarbonates: New Techniques and Biomedical Applications. J Appl Polym Sci. 2014;131:39822. doi: 10.1002/app.39822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calderón M, Quadir MA, Sharma SK, Haag R. Dendritic Polyglycerols for Biomedical Applications. Adv Mater. 2010;22:190–218. doi: 10.1002/adma.200902144. [DOI] [PubMed] [Google Scholar]
- 45.Wilms D, Stiriba SE, Frey H. Hyperbranched Polyglycerols: From the Controlled Synthesis of Biocompatible Polyether Polyols to Multipurpose Applications. Acc Chem Res. 2010;43:129–141. doi: 10.1021/ar900158p. [DOI] [PubMed] [Google Scholar]
- 46.Feng J, Zhuo RX, Zhang XZ. Construction of Functional Aliphatic Polycarbonates for Biomedical Applications. Prog Polym Sci. 2012;37:211–236. [Google Scholar]
- 47.Tian H, Tang Z, Zhuang X, Chen X, Jing X. Biodegradable Synthetic Polymers: Preparation, Functionalization and Biomedical Application. Prog Polym Sci. 2012;37:237–280. [Google Scholar]
- 48.Wender PA, Schrier AJ. Total Synthesis of Bryostatin 9. J Am Chem Soc. 2011;133:9228–9231. doi: 10.1021/ja203034k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pratt RC, Lohmeijer BGG, Long DA, Lundberg PNP, Dove AP, Li H, Wade CG, Waymouth RM, Hedrick JL. Exploration, Optimization, and Application of Supramolecular Thiourea–Amine Catalysts for the Synthesis of Lactide (Co)polymers. Macromolecules. 2006;39:7863–7871. [Google Scholar]
- 50.Gonzalez-Gonzalez E, Ra H, Hickerson RP, Wang Q, Piyawattanametha W, Mandella MJ, Kino GS, Leake D, Avilion AA, Solgaard O, Doyle TC, Contag CH, Kaspar RL. siRNA Silencing of Keratinocyte-Specific GFP Expression in a Transgenic Mouse Skin Model. Gene Ther. 2009;16:963–972. doi: 10.1038/gt.2009.62. [DOI] [PubMed] [Google Scholar]
- 51.Smith FJD, Hickerson RP, Sayers JM, Reeves RE, Contag CH, Leake D, Kaspar RL, McLean WHI. Development of Therapeutic siRNAs for Pachyonychia Congenita. J Invest Dermatol. 2008;128:50–58. doi: 10.1038/sj.jid.5701040. [DOI] [PubMed] [Google Scholar]
- 52.Wender PA, Horan JC. Synthesis and PKC Binding of a New Class of A-Ring Diversifiable Bryostatin Analogues Utilizing a Double Asymmetric Hydrogenation and Cross-Coupling Strategy. Org Lett. 2006;8:4581–4584. doi: 10.1021/ol0618149. [DOI] [PubMed] [Google Scholar]
- 53.Satoh T, Suzuki S, Suzuki Y, Miyaji Y, Imai Z. Reduction of Organic Compounds with Sodium Borohydride–Transition Metal Salt Systems: Reduction of Organic Nitrile, Nitro and Amide Compounds to Primary Amines. Tetrahedron Lett. 1969;10:4555–4558. [Google Scholar]
- 54.Pratt RC, Nederberg F, Waymouth RM, Hedrick JL. Tagging Alcohols with Cyclic Carbonate: A Versatile Equivalent of (Meth)acrylate for Ring-Opening Polymerization. Chem Commun. 2007:114–116. doi: 10.1039/b713925j. [DOI] [PubMed] [Google Scholar]
- 55.Lohmeijer BGG, Pratt RC, Leibfarth F, Logan JW, Long DA, Dove AP, Nederberg F, Choi J, Wade C, Waymouth RM, Hedrick JL. Guanidine and Amidine Organo-catalysts for Ring-Opening Polymerization of Cyclic Esters. Macromolecules. 2006;39:8574–8583. [Google Scholar]
- 56.Simon J, Olsson JV, Kim H, Tenney IF, Waymouth RM. Semicrystalline Dihydroxyacetone Copolymers Derived from Glycerol. Macromolecules. 2012;45:9275–9281. [Google Scholar]
- 57.Parzuchowski PG, Jaroch M, Tryznowski M, Rokicki G. Synthesis of New Glycerol-Based Hyperbranched Polycarbonates. Macromolecules. 2008;41:3859–3865. [Google Scholar]
- 58.Liu J, Zeng F, Allen C. Influence of Serum Protein on Polycarbonate-Based Copolymer Micelles as a Delivery System for a Hydrophobic Anti-cancer Agent. J Controlled Release. 2005;103:481–497. doi: 10.1016/j.jconrel.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 59.Ray WC, Grinstaff MW. Polycarbonate and Poly(carbonate-ester)s Synthesized from Biocompatible Building Blocks of Glycerol and Lactic Acid. Macromolecules. 2003;36:3557–3562. [Google Scholar]
- 60.He F, Wang YP, Liu G, Jia HL, Feng J, Zhuo RX. Synthesis, Characterization and Ring-Opening Polymerization of a Novel Six-Membered Cyclic Carbonate Bearing Pendent Allyl Ether Group. Polymer. 2008;49:1185–1190. [Google Scholar]
- 61.Guerin W, Helou M, Slawinski M, Brusson JM, Carpentier JF, Guillaume SM. Macromolecular Engineering via Ring-Opening Polymerization (3): Trimethylene Carbonate Block Copolymers Derived from Glycerol. Polym Chem. 2014;5:1229–1240. [Google Scholar]
- 62.Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew Chem, Int Ed. 2010;49:6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 63.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in Medicine: Therapeutic Applications and Developments. Clin Pharmacol Ther. 2008;83:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information: Figures S1: Representative gel shift assays to determine relative hydrolytic stability of siRNA/co-oligomer complexes. Figure S2: DLS measurements of siRNA/co-oligomer complexes formed with glycerolcontaining co-oligomers over time. Figure S3: Gel shift assays to compare siRNA binding efficiency of glycerol co-oligomer 10 to glycerol guanidinium monomer 16. Table S1: Zeta potential measurements of siRNA/co-oligomer complexes with and without 5 mol % PEG-initiated 15. Table S2: Characterization and relative hydrolytic stabilities of guanidinium-rich oligocarbonates derived from glycerol, MTC, or both. General methods, synthetic procedures, and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.