

Abstract
Pseudomonas aeruginosa is an opportunistic bacterium that can cause serious infection in immunocompromised individuals. Although autophagy may augment immune responses against P. aeruginosa (Pa) infection in macrophages, the critical components and their role of autophagy in host defense are largely unknown. Here, we show that Pa infection-induced autophagy activates JAK2/STAT1α and increases nitric oxide (NO) production. Knocking down Atg7 resulted in increased IFN-γ release, excessive reactive oxygen species (ROS), and increased SHP2 (Src homology-2 domain-containing phosphatase 2) activity, which led to lowered phosphorylation of JAK2/STAT1α and subdued expression of NOS2 (NO Synthase 2). In addition, we demonstrated the physiological relevance of dysregulated NO under Atg7 deficiency as atg7−/− mice were more susceptible to Pa infection with increased mortality and severe lung injury than wild-type (WT) mice. Furthermore, Pa infected-atg7−/− mice exhibited increased oxidation but decreased bacterial clearance in the lung and other organs compared to WT mice. Mechanistically, atg7 deficiency suppressed NOS2 activity by down-modulating JAK2/STAT1α, leading to decreased NO both in vitro and in vivo. Taken together, these findings revealed that the JAK2/STAT1α/NOS2 dysfunction leads to dysregulated immune responses, and worsened disease phenotypes.
Keywords: Inflammatory responses, autophagy, Gram-negative bacterial infection, bioluminescence in vivo imaging, reactive oxygen species, oxidation
Introduction
Pseudomonas aeruginosa, thereafter Pa, is a ubiquitous Gram-negative opportunistic bacterium and is the most common cause of community-acquired pneumonias (1–3). An improved understanding of the molecular pathogenesis of Pa is urgently needed for developing novel strategies to combat its infection. Autophagy involves mammalian lysosomal degradation and is recently implicated in the degradation of intracellular bacteria, which protects host organisms against diverse pathogens (4). Atg7, an E1 like ubiquitin, is a critical factor for the formation of an Atg12-Atg5-Atg16L1 complex (5), and has been recently shown critical for immune response against bacterial infections (6–8).
Alveolar macrophages (AM), the resident mononuclear phagocyte in the respiratory tract, are part of the first line of host defenses against inhaled organisms by secreting chemokines and phagocytizing pathogens. Two antimicrobial mechanisms of tissue macrophages are production of reactive oxygen intermediates (ROI) by the phagocyte oxidase (phox) and reactive nitrogen intermediates (RNI) by NOS2. Once phagocytized, macrophages will produce a spectrum of reactive oxygen species (ROS, mainly hydrogen peroxide, hydroxyl radical and superoxide anion) and reactive nitrogen species (RNS, derived from nitric oxide or superoxide) to eradicate bacteria in lysosomes with the help of lysosome enzymes (9). The indigestible debris and excess ROS and RNS are subsequently evacuated from macrophages (9). However, the production of oxidative molecules needs to be tightly regulated as excessive ROS may impede the immune defense and hampering bacterial clearance, ultimately leading to tissue injury. Despite being involved in infection, the molecular mechanism of Atg7 during bacterial infection and clearance by AM is largely unknown.
IFN-γ interacts with interferon receptor (IFNR) complex, which activates the JAK/STAT pathway leading to synthesis of the transcription factor interferon response factor-1 (IRF1) and stimulation of NOS2 mRNA transcription. STAT1 is the most important IFN-γ-activated transcription factor for regulation of this response. JAK2 is activated to elicit phosphorylation of STAT1 (Tyr701). Phosphorylated STAT1 forms homodimers and translocates into the nucleus to bind the promoter of NOS2.
To investigate the molecular mechanism in oxidation regulation, we examined autophagy relevant proteins and found the involvement of Atg7 with NO levels. Using specific up- or down- regulation approaches, we set out to elucidate the regulatory role of Atg7 in NO production and its relevance to bacterial killing in vitro and in vivo. Our investigation suggests that Atg7 may be critical for controlling Pa infection progression through the JAK2/STAT1/NOS2 pathway to differentially impact NO production and H2O2 release.
Materials and methods
Mouse and Cells
atg7f/f-deficient (atg7−/−) mice (C57BL/6J) were provided by Drs. Youwen He at Duke University and these mice were originally generated by Masaaki Komatsu at Tokyo Metropolitan Institute of Medical Science. Exon 14 encoded the active site cysteine residue, which was disrupted to generate atg7−/− mice. To conditionally delete the target gene, atg7−/− mice were bred with estrogen receptor (ER) cre mice and were injected with 0.1 mg/kg of tamoxifen (Sigma, St Louis, MO) daily for 5 days before experiments (10). The KO mice were based on C57BL/6J genetic background, so normal C57BL/6J mice were used as wild-type controls. Mice were kept and bred in the animal facility at the University of North Dakota, and the animal experiments were performed in accordance with the NIH guidelines and approved by the institutional animal care and use committee (IACUC) (10). MLE-12 and MH-S cells were obtained from ATCC and cultured in HITES medium (MLE-12) and RPMI 1640 medium (MH-S) supplemented with 5% fetal bovine serum (HyClone Laboratories, Logan, UT) and 100 U/ml of penicillin/streptomycin (Life Technologies, Rockville, MD) antibiotics in a 37°C incubator with 5% CO2. Mouse alveolar macrophage (AM) cells were isolated by bronchoalveolar lavage (BAL). After centrifugation at 2000 rpm, AM cells were resuspended and cultured in RPMI 1640 medium supplemented with 5% fetal bovine serum for evaluating phagocytosis and superoxide production ability. MH-S and MLE-12 cells were transfected with corresponding siRNA (Santa Cruz, Biotechnology, Santa Cruz, CA) or LC3-RFP and achieved high efficiency in transfection using LipofectAmine 2000 reagent (Invitrogen, Carlsbad, CA) in serum-free HITES medium according to the manufacturer’s instructions for transient expression.
Bacterial Infection
Pseudomonas aeruginosa strain PAO1 WT was provided by Dr. S. Lory (Harvard Medical School, Boston, MA). PAO1-GFP was obtained from Dr. G. Pier (Channing Laboratory, Harvard Medical School). Pa Xen-41 expressing luciferase bioluminescence was bought from Caliper Company (PerkinElmer, Waltham, MA). After culturing in Luria-Bertani (LB) broth at 37°C with vigorous shaking overnight, the bacteria were centrifuged at 6000×g for 5 min, and then resuspended in 5 ml fresh LB broth to allow growing till mid-logarithmic phase. The concentration of the bacteria was counted by reading at OD600 (0.1 OD=1×108 cells/ml).
After anesthesia with 40 mg/kg ketamine, mice were given with 1×107 (6 mice/group) colony-forming units (CFU, suspended in 50 μl PBS) of Pa by intranasal instillation, and sacrificed when they were moribund. If indicated, 1 h before infection the mice were given intraperitoneal injections of the NOS2 inhibitors Aminoguanidine (AG, 100 mg/kg body weight), or the NO donor NOC-18 (10 mg/kg body weight). Survival was determined using Kaplan-Meier curve. After BAL procedures, lung and other tissues were fixed in 10% formalin using a routine histological procedure. The formalin-fixed tissues were used for H&E staining to examine tissue damage post infection (11). The lung, spleen, liver, and kidney were homogenized with PBS. The homogenates were used for counting the colony forming units (CFUs).
Before infection, cells were washed once with PBS, and replaced with serum and antibiotic-free medium immediately. Cells were infected by Pa at multiplicity of infection (MOI) of 10: 1 (bacteria-cell ratio) for 1 h and then washed 3 times with PBS to remove the floating bacteria. For required groups, 100 μM AG or NOC-18 was added 30 min before infection. Bacteria on the surface of the cells were killed by adding 100 μg/ml of polymyxin B and left in incubation for another 1 h. Cells were lysed with 1% Triton X-100 dissolved in PBS. Cell homogenates were used for CFU counts.
In Vivo Imaging
Mice were infected with 1×107 of CFU Pa Xen-41 following anesthesia using ketamine. At various time points post infection, whole body of the infected mice was imaged under an IVIS XRII system following the user guides provided by the company (PerkinElmer-Caliper) (12).
Cell Death and Oxidation Assays
AM isolated from lavage fluid were cultured in 96-well plates overnight. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (MTT) assay, dihydro-dichlorofluorescein diacetate (H2DCF-DA, to detect reactive oxygen species, primarily hydrogen peroxide) assay, EuTc (europium tetracycline hydrogen peroxide quantification) assay, mitochondrial membrane potential (JC-1) assay were applied following the manufacturer’s instructions (13–15). NO production was determined using Griess reagent (Sigma) analysis. Lung and other organ tissues were homogenized and equal protein amounts were used for MPO and lipid peroxidation assay. Cytokine concentrations in the first 0.6 ml BAL fluid collected at the indicated times after infection were measured by standard ELISA kits following the manufacturer’s instructions (eBioscience, San Diego, CA) (16–19).
Immunoblotting
Samples taken from cells or lung tissues from same batch of mice infected as above were lysed with RIPA buffer (30 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol, and complete cocktail (Roche) and phosphatases (Sigma). Lysates were centrifuged at 14000×g for 15 min, the supernatants were collected and the concentration was quantitated. The Samples were boiled for 10 min, and equal amount was applied to 12% SDS-polyacrylamide minigels and electrophoresed. The proteins in the gel were then transferred to nitrocellulose filter membranes (Thermo, Rockford, IL). Horseradish peroxidase (HRP)-linked secondary antibody (Rockland, Gilbertsville, PA) and X-ray film (Kodak) were used for exposure (20, 21). Mouse polyclonal antibody anti-LC3, Beclin1, Bcl-2, Bax, Cytochrome C, rabbit polyclonal antibody anti-IFN-γ, pJAK2, JAK2, STAT1α, IRF1, SHP2, β-actin and goat polyclonal antibody anti-Atg5-Atg12 and pSTAT1α were bought from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit monoclonal antibody anti-cleaved-PARP and cleaved-caspase3 were bought from Cell Signaling Technology (Danvers, MA).
Cytokine Profiling
Cytokine concentrations in cell culture supernatant or BAL fluid were measured by ELISA kits following the manufacturer’s instructions (eBioscience Inc., San Diego, CA).
Measurement of mRNA Expression
Total RNA was extracted using the TRIzol (Invitrogen, Grand Island, NY) according to the manufacturer’s instructions. RNA was eluted in RNase-free water and stored at −70°C. The expression of NOS2 mRNA was detected by QuantiTect SYBR Green RT-PCR Kit (Qiagen, Valencia, CA) (Forward TCCTGGACATTACGACCCCT; Reverse AGGCCTCCAATCTCTGCCTA). The separate well 2−ΔΔCt cycle threshold method was used to determine relative quantitative levels of mRNA, and these were expressed as the fold difference to GAPDH (Forward CAGGTTGTCTCCTGCGACTT; Reverse TATGGGGGTCTGGGATGGAA).
Confocal Laser Scanning Microscopy
AM, MH-S and MLE-12 cells were cultured in glass bottom dishes (MatTek, Ashland, MA). The fluorescence images were obtained by LSM 510 Meta confocal microscope (Carl Zeiss Micro Imaging, Thornwood, NY) (22).
Statistical Analyses
Each experiment was conducted in triplicate or repeated independently at least 3 times. The differences in outcomes of atg7−/− mice are presented as percent or amount changes compared with WT controls after Pa infection. Data were analyzed by Mann-Whitney U Test using Prism 5.0 statistical software (GraphPad Software, La Jolla, CA) (23). The survival percentage was generated using Kaplan-Meier curve, with p<0.05 from a log-rank test.
Results
Atg7 knockdown aggravated apoptosis in lung cells after Pa infection
To determine whether Atg7 is involved in Pa pathogenesis, we employed siRNA silencing to repress gene expression in murine alveolar macrophage cell line (MH-S). We first measured cell viability by an MTT assay and found that Pa infection resulted in more cell death which is associated with Atg7 loss and infection extent (Fig. 1A). Similar results were also observed in primary alveolar macrophages (AM). In both cell types, Atg7 deficiency aggravated infection-induced cell death (Fig. 1A). Next, we analyzed cell death patterns and noticed that Pa infection induced apoptosis in both MH-S and primary AM by measuring mitochondrial potential (Fig. 1B). We also evaluated apoptosis-associated signals and found that Pa infection increased cleavage of PARP and caspase3 in MH-S cells vs. non-infection controls, which was further intensified by Atg7 siRNA transfection (Fig. 1C, S1A). In addition, Atg7 siRNA interference significantly facilitated the release of cytochrome C from mitochondria (Fig. 1C). Consistent with this observation, TUNEL assay revealed that Pa infection resulted in markedly increased apoptosis in Atg7-deficient cells (Fig. 1D). To demonstrate the induction of autophagy by infection, we examined the punctate foci following infection to indicate LC3-phospholipid conjugation using fluorescence microscopy. As shown in Fig. S1B, Atg7 knockdown resulted in less LC3 puncta than control siRNA in MH-S cells upon PAO1-GFP infection (MOI=10: 1), suggesting that an impaired autophagy due to Atg7 loss may be associated with increased host cell death (19).
FIGURE 1. Pa induces apoptosis of macrophages in a dose dependent manner.

MH-S cells were transfected with control (Ctrl) siRNA or Atg7 siRNA for 24 h. Murine primary macrophages were isolated using bronchoalveolar lavage (BAL) procedures. Cells were infected with different amounts of PAO1 for 2 h. (A) Cell viability index was measured by MTT assay. (B) Mitochondrial potential was assessed by the JC-1 fluorescence assay (0.1 μg/ml). The fluorescence was quantified at 532 nm with a fluorimeter. (C) Immunoblotting analysis of PARP, caspase3, Bcl-2, Bax and cytochrome C using lysates of MH-S cells after infected with various amounts of Pa. β-actin was used as the loading control throughout the manuscript. (D) TUNEL assay was performed to measure the ratio of apoptotic cells. TUNEL positive cells were counted from at least 100 random fields. Scale bars=100 μm. Data are representative as means±SD of three independent experiments (*, p<0.05; **, p<0.01). RFU, relative fluorescence units.
Atg7 deficiency promoted reactive oxygen species (ROS) release while limited nitric oxide (NO) release
ROS and RNS serve as important intracellular signaling molecules that influence cell metabolism and survival (24). We sought to determine levels of ROS and RNS in MH-S cells by H2DCF (to detect primarily hydrogen peroxide) and Griess test, respectively, and observed that ROS generation was increased in Atg7 knockdown groups upon Pa infection (Fig. 2A), while NO release was suppressed by Atg7 siRNA interference (Fig. 2B). After treatment with ROS inhibitor diphenyleneiodonium (DPI) and NOC-18, a diazeniumdiolate (NONOate) small molecule NO donor, the survival of Pa-infected Atg7 deficient cells was restored (Fig. 2C). While the addition of NOC-18 did not affect total ROS production in Atg7-deficient cells upon Pa infection (Fig. 2D), addition of DPI increased NO release in these cells (Fig. 2E). Although the overall levels of ROS were not affected by NOC-18, insufficient NO release may be related to the infection-induced cell death. Next, we used a bactericidal killing assay to measure the ex vivo clearance of intracellular Pa by macrophages. We found that Atg7 siRNA silencing led to decreased Pa clearance in MH-S cells after 2 h infection by colony forming units (CFU) (Fig. 2F). Importantly, DPI or NOC-18 was found to be able to augment bactericidal activities (Fig. 2F). There is no difference in uptake (phagocytosis) of Pa between WT and Atg7-deficient cells (Fig. S1C). Note the uptake assay was performed at 30 min upon Pa infection while the clearance assay was performed at 2 h after Pa infection, in which the cells were treated with polymyxin B to remove the surface bacteria and then detect the survived intracellular bacteria. These findings indicate that Atg7-deficiency dampens the innate immunity of macrophages by affecting ROS and NO production and bacterial eradication.
FIGURE 2. ROS and NO production are reversely correlated with atg7 deficiency in MH-S cells upon Pa infection.

MH-S cells were transfected with Ctrl siRNA or Atg7 siRNA for 24 h, and then infected with PAO1 at different MOI for 2 h. ROS generation (A) and NO release (B) in supernatant were determined by H2DCF assay or Griess test, respectively. (C–E) Cells were pretreated with DPI (5 μM) or NOC-18 (100 μM) for 30 min, then infected with PAO1 for 2 h. (C) Cell viability was measured by MTT assay. ROS generation (D) and NO release (E) were determined by an H2DCF assay or Griess test, respectively. (F) Bacterial killing of Pa by MH-S cells treated with DPI or NOC-18. Data are representative as means±SD of three independent experiments (*, p<0.05; **, p<0.01). RFU, relative fluorescence units.
Induction of excessive NO synthase (iNOS, also known as NOS2) often occurs in an oxidative environment, and thus high levels of NO have the opportunity to react with superoxide, leading to peroxynitrite formation and cell toxicity (25). NOS2 was found to be inhibited by Atg7 knock-out with Pa infection (Fig. 3A, S2A). Besides, knockdown of NOS2 significantly diminished the production of NO (Fig. 3B, S2B). When treated with DPI or NOC-18, NOS2 mRNA expression showed a strong correlation with NO production (Fig. 3C).
FIGURE 3. NOS2 contributes to NO release upon Pa infection.
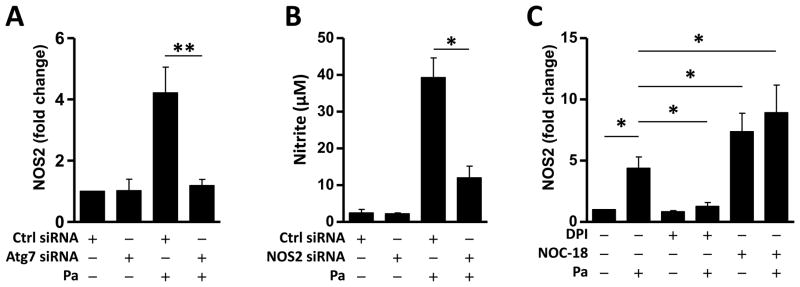
24 hours after the transfection of the MH-S cells with Ctrl siRNA or Atg7 siRNA, the cells were infected with Pa as above. (A) NOS2 mRNA abundance was detected by qPCR. (B) NO release was determined by Griess test. (C) Cells were pretreated with DPI or NOC-18 as above, then infected with Pa. NOS2 mRNA abundance was detected by qPCR. Data are representative as means+SD of three independent experiments (*, p<0.05; **, p<0.01).
Autophagy is required to activate JAK2/STAT1/NOS2 signaling upon Pa infection
Microorganisms use an array of mechanisms to survive in host environments, including ROS, NO and cytokines (IFN-γ) (9, 26). NOS2 produces large quantities of NO upon stimulation by pro-inflammatory cytokines or other mediators. To define how Atg7 influences host defense, we assessed the production of IFN-γ, a critical cytokine for resistance against acute bacterial infection (27). As shown in Fig. 4A, Pa infection induced IFN-γ release in a dose-dependent manner, and after Atg7 knockdown, the production of IFN-γ was further increased (Fig. 4B). However, knocking down NOS2, and adding DPI or NOC-18 did not affect the IFN-γ production (Fig. S2C–E).
FIGURE 4. Effects of autophagy on the expression of IFN-γ and the activation of JAK2/STAT1 pathway upon Pa infection.

(A) MH-S cells were infected with different amount of PAO1 for 2 h. ELISA was used to measure IFN-γ in supernatants. (B) MH-S cells were transfected with Ctrl siRNA or Atg7 siRNA for 24 h, then infected with PAO1 (MOI=10, 2 h). IFN-γ in supernatant was detected by ELISA. (C) Cells lysates were collected for immunoblotting. (D) Before Pa infection, Cells were pretreated with Rapamycin (500 nM, 12 h) or 3-MA (5 mM, 3 h). Cells lysates were collected for immunoblotting. (E) MH-S cells were transfected with Ctrl siRNA, Atg5 siRNA or Beclin1 siRNA, respectively. Levels of IFN-γ, (p)JAK2(Tyr1007/Tyr1008), (p)STAT1α(Tyr701), IRF1 and NOS2 were detected in whole cell lysates by immunoblotting. (F) MH-S cells were pre-neutralized with 10 μg/ml rabbit-anti-IFN-γ or control IgG antibody for 30 min, and then infected with IFN-γ (100 ng/ml, 2 h) or PAO1 (MOI=10, 2 h). The expression of IRF1 and NOS2, as well as phosphorylated JAK2 and STAT1α were detected by immunoblotting. (G) Griess reagent was used to detect the generation of nitrite in supernatants after IFN-γ or PAO1 treatment as above. Data are representative as means±SD of three independent experiments (*, p<0.05; **, p<0.01).
Binding of IFN-γ to its receptor IFNR results in rapid autophosphorylation and activation of IFNR-mediated JAK2, which in turn regulates the phosphorylation and activation of STAT1. STAT1 modulates inflammatory responses through the regulation of NOS2 production (28). Here, we found that JAK2/STAT1 signaling was significantly activated in MH-S cells upon Pa infection. Despite a higher IFN-γ production, the JAK2/STAT1/NOS2 pathway was inhibited by Atg7 knockdown (Fig. 4C, S2F), indicating a link of Atg7 to STAT1 activation. To determine whether JAK2/STAT1 activation was dependent on integrity of autophagy, we used rapamycin and 3-methyl adenine (3-MA) and found that 3-MA could effectively inhibit Pa induced JAK2/STAT1 activation (Fig. 4D). Additionally, similar results were found in Atg5- and Beclin1-siRNA transfected cells (Fig. 4E, S3A, B). To further study the specific role of JAK2/STAT1/NOS2 activation by IFN-γ, we treated the cells with IFN-γ as a positive control and IFN-γ neutralizing antibody as a blocker. As shown in Fig. 4F, Pa-induced JAK2/STAT1/NOS2 activation was inhibited by an anti-IFN-γ antibody. Finally, NO production detected by Griess test recapitulated the above results (Fig. 4G). These data indicate that autophagy blockade may hamper the JAK2/STAT1/NOS2 pathway, thereby impairing IFN-γ activation.
Autophagy facilitated IFN-γ signaling by negatively regulating ROS
Since autophagy deficiency was shown to cause the accumulation of ROS, we surmised that ROS may play a role in Pa-activated STAT1 signaling. To determine whether infection specifically induces high levels of hydrogen peroxide (H2O2, the most important ROS in regards to pathogen stimulation), we used EuTc assay to quantify the level of H2O2, and found that Pa infection did cause H2O2 production 2 h post infection (Fig. 5A) and Atg7 deficiency enhanced this process (Fig. 5B). In addition, 4-amino benzoyl hydrazide (ABH, mechanism-based inhibitor of peroxidase), apocynin (a potent inhibitor of NADPH-dependent ROS production, APO), N-acetyl-cysteine (NAC, a scavengers of hydrogen peroxide [H2O2]) could all abolish Pa-induced H2O2, while DPI showed the best inhibitory efficiency in all ROS including H2O2 (Fig. S3C). Further, IFN-γ induced H2O2 similarly as Pa did, and these processes were inhibited by DPI (Fig. 5C). Next, we used exogenous H2O2 to mechanistically define the role of ROS against Pa infection. Fig. 5D showed that NO production induced by IFN-γ or Pa was inhibited by H2O2. We first showed that DPI further boosted activation of STAT1 and NOS2 (Fig. 5E), while exogenous H2O2 inhibited it. These results indicate that Atg7-deficiency put a brake on NO production (while increasing H2O2), thereby suppressing Pa-induced STAT1 signaling.
FIGURE 5. ROS-regulated SHP2 inhibited Pa-activated STAT1 in the absence of Atg7.

(A) MH-S cells were infected with PAO1 (MOI=10) for different times. H2O2 production was determined using EuTc assay (absorbance at 617 nm). (B) MH-S cells were transfected with Ctrl siRNA or Atg7 siRNA for 24 h, then infected with PAO1 (MOI=10, 2 h). H2O2 was measured as above. (C) MH-S cells were pretreated with DPI (5 μM) for 30 min. EuTc assay was used to measure H2O2 after INF-γ or Pa infection. (D) MH-S cells were pretreated with H2O2 (10 mM) or DPI (5 μM) for 30 min. Griess reagent was used to detect the generation of nitrite after IFN-γ or Pa infection. (E) Immunoblotting was used to determine NOS2 and phosphorylation of STAT1α after Pa infection. (F, G) 24 h after transfected with Ctrl siRNA or SHP2 siRNA, or double knocked down using Atg7 siRNA and SHP2 siRNA, MH-S cells were infected with Pa as above. Immunoblotting shows the expression of NOS2 and phosphorylation of STAT1α. (H) Bacterial killing of Pa by MH-S cells treated with H2O2 either with Ctrl siRNA, SHP2 siRNA or Atg7 siRNA transfection. (I) Cell viability was measured by MTT assay. Data are representative as means±SD of three independent experiments (*, p<0.05; **, p<0.01).
SHP2 is an important regulator in the IFN-γ feedback loop, which inactivates the JAK2-STAT1 pathway (29). We hypothesized that ROS-mediated SHP2 activation is critical for inhibiting Pa-induced STAT1 in Atg7-deficient macrophages. Immunoblotting was used to analyze the feedback regulation of SHP2 and showed that Atg7 siRNA inhibited SHP2 expression (Fig. 5F, G). Exogenous H2O2 effectively inhibited Pa-induced STAT1 and NOS2 (Fig. 5F, S3D), showing that inhibiting Pa-induced STAT1 in autophagy-deficient cells is dependent upon ROS-mediated SHP2 activation. Thus, Atg7/SHP2 knockdown reactivated STAT1 under Pa infection (Fig. 5G).
To determine the role of SHP2 in Atg7 associated immune response and bacterial clearance, we used bacterial killing assay to measure intracellular Pa in MH-S cells. Consistent with the aforementioned data, bacterial burdens decreased in Atg7/SHP2 siRNA-cotransfected cells compared with Atg7 siRNA-transfected groups, while bacterial burdens further decreased with only SHP2 knock down in a phagocytosis assay (Fig. 5H). Besides, SHP2 siRNA interference restored viability under Atg7 knockdown upon Pa infection (Fig. 5I). These findings indicate that autophagy negatively regulates ROS-activated SHP2, which, in turn, facilitates Pa-induced STAT1/NOS2 activation.
atg7 deficiency led to increased lung injury and bacterial dissemination
AMs are the first line of innate host defense to eradicate bacteria in early infection in the lung (30). To confirm the essential role of AM in bacterial clearance, we infected atg7−/− mice (31). atg7−/− mice and WT mice were infected with 1×107 CFU of PAO1. Primary AMs were isolated by bronchoalveolar lavage (BAL) to evaluate viability using an MTT assay. As shown in Fig. 6A, survival of AMs decreased by approximately 60% in atg7−/− mice compared to that of WT mice 24 h post infection, suggesting that AM phagocytic function may be impaired in atg7−/− mice. Mitochondrial membrane potential was also found to be decreased in atg7−/− AM using a JC-1 fluorescence assay (Fig. 6B). To investigate whether atg7 deficiency is associated with ROS production in vivo during infection, we examined levels of superoxide in AMs. AMs of atg7−/− mice showed about a 2.3-fold increase in oxidative stress at 24 h post infection compared to those of WT, as determined by an H2DCF-DA assay (Fig. 6C). Besides, H2O2 production in KO mice was higher than that in WT mice (Fig. 6D). NO release in BAL was similarly increased as determined using the Griess reagent (Fig. 6E). By immunoblotting of lung homogenates, Atg7 deficiency disrupted the formation of Atg5-Atg12 complex both with and without infection, and reduced conversion of LC3-I to LC3-II upon Pa infection (Fig. 6F). Due to inflammatory responses, we assessed signaling proteins in lung tissues and found that INF-γ markedly increased in the lungs of atg7−/− mice as compared to those of WT mice upon Pa infection; on the contrary, the phosphorylation of JAK2 and STAT1α increased in WT mice compared to atg7−/− mice upon Pa infection (Fig. 6F). Besides, the levels of IRF1 and NOS2 were found to be increased in WT mice but not in KO mice which is consistent with the in vitro data (Fig. 6F).
FIGURE 6. atg7−/− mice exhibited exacerbated lung infection and bacterial burdens against Pa.

WT mice and atg7−/− mice were infected with 1×107 CFU of PAO1. After 24 h, AMs were isolated from BAL fluids. (A) Cell viability was tested by MTT assay. (B) Mitochondrial potential was assessed by the JC-1 fluorescence assay. (C) Superoxide production was determined using H2DCF assay. (D) H2O2 production was determined using EuTc assay. (E) NO production was detected using Griess reagent. (F) The lung homogenates were used for immunoblotting to determine the relevant signaling pathways and phosphorylation of signaling proteins. (G) Lungs were removed for histological analysis (inset showing the typical tissue injury and inflammatory influx). (H) The leukocyte infiltration score in lungs was determined by blindly (for the samples) enumerating the lymphocytes. (I) Lungs were homogenized in PBS and were used for assessing bacterial colonies. Data are shown as means±SEM from three independent experiments. (One-way ANOVA (Tukey’s post hoc); *, p<0.05; **, p<0.01).
We also performed histological studies and showed that although Pa infection caused tissue damage and inflammatory response in WT mice, the pathophysiological changes in atg7−/− mice were more severe than those in WT mice, indicating that WT mice are more resistant to Pa than atg7−/− mice. Furthermore, we have not observed histological differences between uninfected atg7−/− and WT mice (Fig. 6G), excluding the potential pathophysiological alterations due to the loss of atg7 gene per se. Together, both atg7−/− and WT mice exhibited signs of pneumonia, while more severe histological alterations and higher leukocytes recruitment (Fig. 6H) combined with higher pro-inflammatory cytokines release (Fig. S3E) occurred in the lungs of atg7−/− mice. Besides, atg7−/− mice showed elevated bacterial dissemination after Pa infection, and CFU numbers in the lung increased significantly as compared to WT mice in a time-dependent manner (Fig. 6I). Similar results were found in the liver, kidney and spleen (Fig. S4A). Taken together, these findings suggest that Atg7 is crucial for resistance to Pa in the respiratory tract.
Impaired NO generation contributed to immune impotency in atg7−/− mice against Pa infection
To assess the physiological significance of Atg7 in Pa infection, we intranasally infected atg7−/− and WT mice with 1×107 CFUs of Pa Xen-41 strain (derived from parental strain PAO1) through the intratracheal (i.t.) route (22). Xen-41 emits bioluminescence for in vitro and in vivo imaging using Caliper Xenogen IVIS XRII. The dissemination of infection illuminated by bioluminescence was monitored for 7 days post infection (Fig. 7A, B). atg7−/− mice were highly susceptible to Pa infection and died as early as 15 h after infection, with 66.7% mortality within 27 h, and finally only 16.7% of mice survived to day 7. However, 83.3% of WT mice survived up to 7 days after Pa infection (Fig. 7C). The increased mortality was associated with uncontrolled bacterial growth as atg7−/− mice exhibited wider dissemination of bioluminescence in the area of thoracic cavity 12 h post infection. In contrast, WT mice showed significant initial clearance of the instilled bacteria, whereas the dissemination areas were more constrained than those in atg7−/− mice as reflected by decreased bioluminescence starting 24 h post infection (Fig. 7B). These data indicate that resistance to Pa profoundly decreased in atg7−/− mice.
FIGURE 7. NO generation contributes to resistance to Pa infection in atg7−/− mice.

WT mice and atg7−/− mice were infected with 1×107 CFU of Pa-Xen41 (6 mice/group). (A) Images of the lung in different time points were obtained using digital camera in an IVIS XRII system. (B) Statistic analysis of lung infection level by measure luminescence signal using IVIS XRII software. Data are presented as means±SD from 6 mice. (C) Survival test was represented by Kaplan-Meier survival curves (p<0.05, 95% confidence interval, log rank test). (D, E) Survival of atg7−/− or WT mice treated with NOC-18, AG or PBS after pulmonary Pa infection. *, p<0.05; **, p<0.01; ***, p<0.001. (F) A schematic diagram showing how Atg7 activates NOS2 and regulates the JAK2/STAT1 pathway to modulate the inflammatory response to Pa infection.
We next detected myeloperoxidase (MPO) activity of the lung and other organs. As expected, increased MPO in the lung, liver, spleen, and kidney suggests that oxidative stress has resulted from systemic spread of the invading bacteria (Fig. S4B) or higher leukocyte recruitment and cytokine production. Lipid peroxidation indicates oxidative degradation of lipids. We detected lipid peroxidation in the lungs, liver, kidneys, and spleen and noticed that lipid peroxidation increased significantly in all Pa-infected organs of atg7−/− mice compared to those of WT mice (Fig. S4C), suggesting that superoxide release may be related to the systemic spread of the invading Pa bacteria.
In macrophages, NOS2 is produced following exposure to endotoxins or cytokines to increase NO levels in order to control invading microorganisms or inhibit neoplasm growth (25, 32). To confirm that the impairment of NO release during infection contributes to the impaired host defense against Pa, we elucidated whether exogenous supplementation of NO enhances the bactericidal capability to increase host defense. Indeed, we found that addition of NOC-18 significantly reduced the mortality of atg7−/− mice infected with Pa (Fig. 7D, S4D, E). Treatment with aminoguanidine (AG, an irreversible and selective inhibitor of NOS2) impeded bacterial clearance in WT mice, whereas combined treatment with AG and NOC-18 prior to infection substantially increased mouse survival rates after infection (Fig. 7E, S4D, E). These findings indicate that NOS2 expression and NO release induced by NOC-18 at least partially restored the bacterial killing capability of AMs in atg7−/− mice and protect mice from Pa-inflicted death. To summarize the discoveries of this study, Fig. 7F illustrates a model delineating the role of Atg7 in Pa-infection and the underlying cell signaling process.
Discussion
In this study, we demonstrate that Pa infection of atg7−/− mice exhibits a severe disease phenotype, implying a crucial role of this gene in host defense against infection. We find that atg7 deficiency significantly increases ROS release but decreases NO production and bacterial clearance. The majority of previous work has focused on the role of Atg7 in cellular process induced by starvation or various other stress factors (31, 33); however, Atg7-dependent autophagy may also serve as an innate immunologic effector in antimicrobial responses. Different bacteria may have divergent pathogenesis because bacterial pathogens have developed a variety of virulence factors to subvert host defense to establish persistent infection (34). Previous studies have revealed that Atg7 contributes to plant basal immunity towards fungal infection (35), accompanied by production of reactive oxygen intermediates. The literature demonstrated that atg7 deletion in the hematopoietic system resulted in loss of hematopoietic stem cell functions, severe myeloproliferation, and mortality within weeks (36). Another report, however, showed that knockdown of autophagy enhances the innate immunity in hepatitis C virus-infected hepatocytes (37). Our recent report revealed a role of Atg7 in inflammatory response against K. pneumoniae infection (38). Autophagy has also been reported to enhance bacterial clearance against Pa infection in in vitro models (39). These observations laid out the foundation for us to dissect the molecular mechanism for Atg7, which may be also pathogen specific.
Autophagy protein Rubicon has been reported to mediate phagocytic NADPH oxidase activation upon microbial infection (40), but the role of autophagy in regulating oxidation in Pa remains undemonstrated. Our previous data have demonstrated that autophagy plays an essential role in Pa clearance by alveolar macrophages (19). However, detailed pathogenic molecular mechanisms of Pa remain to be learned. In NOS2 KO mice, it has been demonstrated that reactive nitrogen intermediate contributes to host defense against a restricted set of pathogens (41, 42). IFN-γ could induce MD-2 protein expression in corneal epithelial cells, which is mediated by JAK/STAT1 signaling during Pa infection. However, whether this process is similar in macrophages is still unknown. In this report, we linked these processes and reveal that Atg7, perhaps to a bigger perspective, autophagy is indeed essential in immune response against bacterial infection. Atg7 loss leads to a spontaneous inhibition of critical innate immunity and severe lung injury, which may be due to the impairment of both epithelial and macrophage functions (43). However, tissues of uninfected-atg7−/− mice display no significant pathological signs. We speculate that Pa infection may trigger complex pathogenesis using multiple virulence factors (44), which is even more complex when intertwined with host multilayer signaling pathways, and thereby leading to the inflammatory cascade. Our recent studies indicate that critical cysteines in Pa are responsible for the oxidative sensing (44, 45). Elevated levels of IFN-γ and MPO in atg7−/− mice may be due to higher leukocyte recruitment and stronger pro-inflammatory cytokines (46). One of the striking findings of this study is that atg7 deficiency could contribute to uncontrolled inflammatory responses, such as TNF-α, IL-1β, IL-6 and IFN-γ secretion. A sudden rise of IFN-γ may activate JAKs/STATs and their related host response (NO release) to help clear bacteria. NO may be an immunity player in host defense by blocking the synthesis of microorganism DNA or other mechanisms (47). Cell wall components of bacteria and fungi, pathogen associated molecular patterns (PAMPs), trigger innate immunity, leading to increased expression of NOS2. LPS-mediated TLR4 activation functions on releasing transcription factor NF-κB, which interacts with IκB elements in the NOS2 5′, triggering NOS2 transcription. Cytokines, including TNF-α and IL-1β, also activate NO production (32, 48, 49). Here we demonstrated that under Pa infection, IFN-γ could interact with the IFNR1 and IFNR2 complex that activates JAK/STAT pathways, resulting in the synthesis of transcription factor IRF1 and stimulation of NOS2 transcription. Hydrogen peroxide is a factor that going too far is as bad as not going far enough. Our current study analyzed the involvement of H2O2 in detail using several critical specific inhibitors together with exogenous ROS specifies, suggesting that Atg7 loss augmented H2O2 release while reduced NO. A previous report demonstrated that atg7−/− MEFs had higher levels of ROS under basal and starved conditions (50), but the authors did not determine the role and changes in each subtype of ROS. Despite its importance in killing bacteria, uncontrolled accumulation of ROS may cause lung injury, and bacteria also have oxidation-sensing mechanisms modulate the oxidation-mediated host response (51). For example, excessive ROS inhibits IFN-γ-induced JAK2/STAT1 activation in neurons (52). In astrocytes and BCR/ABL-chronic myelogenous leukemia, ROS-mediated signaling positively regulates SHP2. ROS-generating mitochondria accumulates in MEFs with an autophagy deficiency, and ROS-regulated SHP2 inhibits JAK2/STAT1 signaling (53).
The in vivo imaging technology allowed us to monitor the pathophysiology and dynamics of Pa infection in real-time. This animal imaging revealed infection dynamic, and showed a wider and quicker infection spread with Pa Xen-41 bioluminescence strain in atg7−/− mice. The animal imaging may be useful for further studying infection in a physiological relevant manner (12). We revealed a novel role of autophagy against bacterial infection by showing a severe phenotype in Pa-challenged atg7 deficient mice, including a worsened disease, increased mortality, and intensified inflammatory response. These results indicate that Atg7 may serve as a negative regulator of inflammatory response. Our in vivo and in vitro results suggest that Atg7 is critical for controlling Pa infection progression by regulating the JAK2/STAT1 pathway. This is consistent with recent reports about the anti-inflammatory role of Atg7 signaling during Herpes simplex virus infection (54, 55). In particular, we identify that NOS2 is responsible for the failure of Pa killing in atg7−/− mice as well as siRNA silenced cells.
Atg7 deficiency could severely compound the function of autophagy, resulting in general autophagy deficiency. Thus, our overall results suggest that autophagy deficiency may severely dampen immunity against Pa and, perhaps broadly, Gram-negative bacterial infection. In addition, other autophagy factors may also be involved in regulating oxidative products and impacting bacterial infection. Although further work needs to be done to completely establish the molecular role for Atg7 in this model, our studies provide critical new insight into the understanding of molecular mechanism for Atg7 and autophagic process during Pa infection. Thus further understanding the molecular mechanism for inflammatory regulation and oxidation in autophagy context may identify novel therapeutic strategies for this infection.
Supplementary Material
Acknowledgments
We thank S. Rolling of UND imaging core for help with confocal imaging.
Financial support. This work was supported by Flight Attendant Medical Research Institute (FAMRI, Grant #103007), National Institute of Health AI109317-01A1, AI101973-01, and AI097532-01A1 to M. W. This work was also supported by grants from the National 973 Basic Research Program of China (2013CB911300), the National Science and Technology Major Project (2012ZX09501001-003) and Chinese NSFC (81072022, 81172173) to C.H.
Abbreviations
- Pa
P. aeruginosa
- Atg7
autophagy-related protein 7
- AM
alveolar macrophages
- atg7−/−
atg7-dificient
- NOS2
nitric oxide synthase 2
- iNOS
inducible NO synthase
- BAL
bronchoalveolar lavage
- MOI
multiplicity of infection
- MTT
3-(4, 5-dimethylthiazol-2-yl)-2, 5-dimethyltetrazolium bromide
- H2DCF-DA
dihydro dichlorofluorescein diacetate
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- HSC
hematopoietic stem cell
- NO
nitric oxide
- MPO
myeloperoxidase
Footnotes
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. The Journal of infectious diseases. 2001;183:1767–1774. doi: 10.1086/320737. [DOI] [PubMed] [Google Scholar]
- 2.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D’Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 4.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. The Journal of biological chemistry. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 6.Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, Hutt J, Lyons CR, Dobos KM, Deretic V. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E3168–3176. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell host & microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. The Journal of biological chemistry. 2006;281:11374–11383. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Noel JM, Velmurugan J, Nogala W, Mirkin MV, Lu C, Guille Collignon M, Lemaitre F, Amatore C. Nanoelectrodes for determination of reactive oxygen and nitrogen species inside murine macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11534–11539. doi: 10.1073/pnas.1201552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou X, Wang L, Hasegawa H, Amin P, Han BX, Kaneko S, He Y, Wang F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9424–9429. doi: 10.1073/pnas.0914725107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu M, Hussain S, He HY, Pasula R, Smith PA, Martin WJ., II Genetically engineered macrophages expressing IFN-γ restore alveolar immune function in scid mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14589–14594. doi: 10.1073/pnas.251451498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Zhou X, Ye Y, Li Y, Li J, Privratsky B, Wu E, Gao H, Huang C, Wu M. Lyn regulates inflammatory responses in Klebsiella pneumoniae infection via the p38/NF-kappaB pathway. European journal of immunology. 2014;44:763–773. doi: 10.1002/eji.201343972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K, Liu R, Li J, Mao J, Lei Y, Wu J, Zeng J, Zhang T, Wu H, Chen L, Huang C, Wei Y. Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-mTOR- and hypoxia-induced factor 1alpha-mediated signaling. Autophagy. 2011;7:966–978. doi: 10.4161/auto.7.9.15863. [DOI] [PubMed] [Google Scholar]
- 14.Yuan K, Huang C, Fox J, Gaid M, Weaver A, Li G, Singh BB, Gao H, Wu M. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB) The Journal of biological chemistry. 2011;286:21814–21825. doi: 10.1074/jbc.M111.237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribeiro JP, Magalhaes LM, Segundo MA, Reis S, Lima JL. Hydrogen peroxide, antioxidant compounds and biological targets: an in vitro approach for determination of scavenging capacity using fluorimetric multisyringe flow injection analysis. Talanta. 2010;81:1840–1846. doi: 10.1016/j.talanta.2010.03.049. [DOI] [PubMed] [Google Scholar]
- 16.Guo Q, Shen N, Yuan K, Li J, Wu H, Zeng Y, Fox J, 3rd, Bansal AK, Singh BB, Gao H, Wu M. Caveolin-1 plays a critical role in host immunity against Klebsiella pneumoniae by regulating STAT5 and Akt activity. European journal of immunology. 2012;42:1500–1511. doi: 10.1002/eji.201142051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu M, Huang H, Zhang W, Kannan S, Weaver A, McKibben M, Herington D, Zeng H, Gao H. Host DNA repair proteins in response to Pseudomonas aeruginosa in lung epithelial cells and in mice. Infection and immunity. 2011;79:75–87. doi: 10.1128/IAI.00815-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kannan S, Huang H, Seeger D, Audet A, Chen Y, Huang C, Gao H, Li S, Wu M. Alveolar epithelial type II cells activate alveolar macrophages and mitigate P. Aeruginosa infection. PloS one. 2009;4:e4891. doi: 10.1371/journal.pone.0004891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, Yin Q, Gao H, Wu M. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. Journal of cell science. 2012;125:507–515. doi: 10.1242/jcs.094573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang D, Wu M, Nelson DE, Pasula R, Martin WJ., 2nd Alpha-1-antitrypsin expression in the lung is increased by airway delivery of gene-transfected macrophages. Gene therapy. 2003;10:2148–2152. doi: 10.1038/sj.gt.3302121. [DOI] [PubMed] [Google Scholar]
- 21.Wisniowski PE, Spech RW, Wu M, Doyle NA, Pasula R, Martin WJ., 2nd Vitronectin protects alveolar macrophages from silica toxicity. American journal of respiratory and critical care medicine. 2000;162:733–739. doi: 10.1164/ajrccm.162.2.9808015. [DOI] [PubMed] [Google Scholar]
- 22.Wu M, Pasula R, Smith PA, Martin WJ., 2nd Mapping alveolar binding sites in vivo using phage peptide libraries. Gene therapy. 2003;10:1429–1436. doi: 10.1038/sj.gt.3302009. [DOI] [PubMed] [Google Scholar]
- 23.He YH, Wu M, Kobune M, Xu Y, Kelley MR, Martin WJ., 2nd Expression of yeast apurinic/apyrimidinic endonuclease (APN1) protects lung epithelial cells from bleomycin toxicity. American journal of respiratory cell and molecular biology. 2001;25:692–698. doi: 10.1165/ajrcmb.25.6.4550. [DOI] [PubMed] [Google Scholar]
- 24.Wang T, Gu J, Wu PF, Wang F, Xiong Z, Yang YJ, Wu WN, Dong LD, Chen JG. Protection by tetrahydroxystilbene glucoside against cerebral ischemia: involvement of JNK, SIRT1, and NF-kappaB pathways and inhibition of intracellular ROS/RNS generation. Free radical biology & medicine. 2009;47:229–240. doi: 10.1016/j.freeradbiomed.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 25.Colton CA, Vitek MP, Wink DA, Xu Q, Cantillana V, Previti ML, Van Nostrand WE, Weinberg JB, Dawson H. NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12867–12872. doi: 10.1073/pnas.0601075103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nature reviews. Microbiology. 2004;2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 27.Diel R, Loddenkemper R, Meywald-Walter K, Niemann S, Nienhaus A. Predictive value of a whole blood IFN-gamma assay for the development of active tuberculosis disease after recent infection with Mycobacterium tuberculosis. American journal of respiratory and critical care medicine. 2008;177:1164–1170. doi: 10.1164/rccm.200711-1613OC. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Li X, Carpinteiro A, Goettel JA, Soddemann M, Gulbins E. Kinase suppressor of Ras-1 protects against pulmonary Pseudomonas aeruginosa infections. Nature medicine. 2011;17:341–346. doi: 10.1038/nm.2296. [DOI] [PubMed] [Google Scholar]
- 29.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nature reviews. Immunology. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 30.Nau GJ, Richmond JF, Schlesinger A, Jennings EG, Lander ES, Young RA. Human macrophage activation programs induced by bacterial pathogens. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1503–1508. doi: 10.1073/pnas.022649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. The Journal of cell biology. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis RL, Sanchez AC, Lindley DJ, Williams SC, Syapin PJ. Effects of mechanistically distinct NF-kappaB inhibitors on glial inducible nitric-oxide synthase expression. Nitric oxide : biology and chemistry/official journal of the Nitric Oxide Society. 2005;12:200–209. doi: 10.1016/j.niox.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 34.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nature immunology. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 35.Lenz HD, Vierstra RD, Nurnberger T, Gust AA. ATG7 contributes to plant basal immunity towards fungal infection. Plant signaling & behavior. 2011;6:1040–1042. doi: 10.4161/psb.6.7.15605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J, Knight S, Jacobsen SE, Kranc KR, Simon AK. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. The Journal of experimental medicine. 2011;208:455–467. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology. 2011;53:406–414. doi: 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye Y, Li X, Wang W, Ouedraogo KC, Li Y, Gan C, Tan S, Zhou X, Wu M. Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. American journal of physiology. Lung cellular and molecular physiology. 2014;307:L355–363. doi: 10.1152/ajplung.00046.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Junkins RD, Shen A, Rosen K, McCormick C, Lin TJ. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PloS one. 2013;8:e72263. doi: 10.1371/journal.pone.0072263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang CS, Lee JS, Rodgers M, Min CK, Lee JY, Kim HJ, Lee KH, Kim CJ, Oh B, Zandi E, Yue Z, Kramnik I, Liang C, Jung JU. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell host & microbe. 2012;11:264–276. doi: 10.1016/j.chom.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murray HW, Xiang Z, Ma X. Responses to Leishmania donovani in mice deficient in both phagocyte oxidase and inducible nitric oxide synthase. The American journal of tropical medicine and hygiene. 2006;74:1013–1015. [PubMed] [Google Scholar]
- 42.Shiloh MU, MacMicking JD, Nicholson S, Brause JE, Potter S, Marino M, Fang F, Dinauer M, Nathan C. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 43.Kannan S, Wu M. Respiratory stem cells and progenitors: overview, derivation, differentiation, carcinogenesis, regeneration and therapeutic application. Current stem cell research & therapy. 2006;1:37–46. doi: 10.2174/157488806775269115. [DOI] [PubMed] [Google Scholar]
- 44.Zhao K, Deng X, He C, Yue B, Wu M. Pseudomonas aeruginosa outer membrane vesicles modulate host immune responses by targeting the Toll-like receptor 4 signaling pathway. Infection and immunity. 2013;81:4509–4518. doi: 10.1128/IAI.01008-13. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Deng X, Liang H, Ulanovskaya OA, Ji Q, Zhou T, Sun F, Lu Z, Hutchison AL, Lan L, Wu M, Cravatt BF, He C. Steady-state hydrogen peroxide induces glycolysis in Staphylococcus aureus and Pseudomonas aeruginosa. Journal of bacteriology. 2014;196:2499–2513. doi: 10.1128/JB.01538-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Speyer CL, Neff TA, Warner RL, Guo RF, Sarma JV, Riedemann NC, Murphy ME, Murphy HS, Ward PA. Regulatory effects of iNOS on acute lung inflammatory responses in mice. The American journal of pathology. 2003;163:2319–2328. doi: 10.1016/S0002-9440(10)63588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bogdan C. Nitric oxide and the immune response. Nature immunology. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 48.Kadowaki S, Chikumi H, Yamamoto H, Yoneda K, Yamasaki A, Sato K, Shimizu E. Down-regulation of inducible nitric oxide synthase by lysophosphatidic acid in human respiratory epithelial cells. Molecular and cellular biochemistry. 2004;262:51–59. doi: 10.1023/b:mcbi.0000038215.89821.7f. [DOI] [PubMed] [Google Scholar]
- 49.Mizel SB, Honko AN, Moors MA, Smith PS, West AP. Induction of macrophage nitric oxide production by Gram-negative flagellin involves signaling via heteromeric Toll-like receptor 5/Toll-like receptor 4 complexes. Journal of immunology. 2003;170:6217–6223. doi: 10.4049/jimmunol.170.12.6217. [DOI] [PubMed] [Google Scholar]
- 50.Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira, Gutkind S, Daniels MP, Komatsu M, Finkel T. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging. 2009;1:425–437. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yi C, Jia G, Hou G, Dai Q, Zhang W, Zheng G, Jian X, Yang CG, Cui Q, He C. Iron-catalysed oxidation intermediates captured in a DNA repair dioxygenase. Nature. 2010;468:330–333. doi: 10.1038/nature09497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaur N, Lu B, Monroe RK, Ward SM, Halvorsen SW. Inducers of oxidative stress block ciliary neurotrophic factor activation of Jak/STAT signaling in neurons. Journal of neurochemistry. 2005;92:1521–1530. doi: 10.1111/j.1471-4159.2004.02990.x. [DOI] [PubMed] [Google Scholar]
- 53.Chang YP, Tsai CC, Huang WC, Wang CY, Chen CL, Lin YS, Kai JI, Hsieh CY, Cheng YL, Choi PC, Chen SH, Chang SP, Liu HS, Lin CF. Autophagy facilitates IFN-gamma-induced Jak2-STAT1 activation and cellular inflammation. The Journal of biological chemistry. 2010;285:28715–28722. doi: 10.1074/jbc.M110.133355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nature reviews. Immunology. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.