

Background: AMP kinase is a regulator of lipid metabolism.
Results: PP2APpp2r2d regulates AMP kinase by dephosphorylating Thr-172, which is required for AMP kinase activation.
Conclusion: PP2APpp2r2d may regulate lipogenesis by negatively regulating AMP kinase.
Significance: An AMP kinase-PP2APPp2r2d axis exists that may regulate critical regulators of lipid metabolism.
Keywords: Cholesterol, Diet, Lipid, Phosphatase, Phosphorylation, Kinase
Abstract
AMP kinase is a heterotrimeric serine/threonine protein kinase that regulates a number of metabolic processes, including lipid biosynthesis and metabolism. AMP kinase activity is regulated by phosphorylation, and the kinases involved have been uncovered. The particular phosphatases counteracting these kinases remain elusive. Here we discovered that the protein phosphatase 2A heterotrimer, PP2APpp2r2d, regulates the phosphorylation state of AMP kinase by dephosphorylating Thr-172, a residue that activates kinase activity when phosphorylated. Co-immunoprecipitation and co-localization studies indicated that PP2APpp2r2d directly interacted with AMP kinase. PP2APpp2r2d dephosphorylated Thr-172 in rat aortic and human vascular smooth muscle cells. A positive correlation existed between decreased phosphorylation, decreased acetyl-CoA carboxylase Acc1 phosphorylation, and sterol response element-binding protein 1c-dependent gene expression. PP2APpp2r2d protein expression was up-regulated in the aortas of mice fed a high fat diet, and the increased expression correlated with increased blood lipid levels. Finally, we found that the aortas of mice fed a high fat diet had decreased AMP kinase Thr-172 phosphorylation, and contained an Ampk-PP2APpp2r2d complex. Thus, PP2APpp2r2d may antagonize the aortic AMP kinase activity necessary for maintaining normal aortic lipid metabolism. Inhibiting PP2APpp2r2d or activating AMP kinase represents a potential pharmacological treatment for many lipid-related diseases.
Introduction
AMP kinase is a heterotrimeric serine/threonine protein kinase that consists of α, β, and γ subunits (1–3). There are two α, two β, and three γ subunits. The α subunit harbors kinase activity, whereas β and γ subunits are regulatory. The various subunits combine to make multiple heterotrimeric isoforms. There is tissue specificity in expression; for example, α1β1γ1 is predominant in liver, α1β2γ2 is predominant in the heart, and α2β2γ1 is the major isoform seen in skeletal muscle (4). Interestingly, a dominant activated allele of the γ2 subunit of the α1β2γ2 heart isoform contributes to Wolff-Parkinson-White syndrome (5). Symptoms include cardiac ventricular pre-excitation, myocardial glycogen accumulation, and cardiac hypertrophy. Isoforms have been crystalized, and these studies have been informative in understanding AMP kinase structure/function (6–8). Biochemical studies have shown that AMP kinase regulates multiple cell pathways, including lipid synthesis and glucose metabolism (9, 10).
AMP kinase is phosphorylated and activated when the cellular AMP:ATP ratio increases (11). An increased AMP level stimulates phosphorylation of Thr-172 on the α subunit, which induces kinase activity (11) and stimulates the phosphorylation of factors involved in lipid synthesis, such as acetyl-CoA carboxylase 1 (ACC1)2 (12) and HMG-CoA reductase (HMGCR) (13). Phosphorylation of these substrates results in reduced enzymatic activity. Kinases phosphorylating Thr-172 include LKB1, CAMKKβ, and TAK1 (14). AMP kinase is negatively regulated by cAMP-stimulated PKA through phosphorylation of Ser-173 on the α subunit, which inhibits the phosphorylation of Thr-172 (11, 15). Insulin-stimulated AKT1-dependent phosphorylation of Ser-485/491 also inhibits AMP kinase activity (16). It may do so by promoting Thr-172 dephosphorylation.
PP2A is a heterotrimeric serine/threonine phosphatase that contains two regulatory subunits, A and B, and one C catalytic subunit (17) (see Table 1). Two A and two C subunits exist. The A subunit acts as a scaffold. The association of the A and C subunits constitutes the A/C holoenzyme dimer. There are four distinct B subunit families (PPP2R2, PPP2R3, PPP2R4, and PPP2R5) that contain multiple isoforms that bind the A/C dimer (18) (see Table 1). Binding of these B subunits directs PP2A substrate specificity. Ppp2r2d belongs to the PPP2R2 family, which contains four isoforms (18) (see Table 1). Various heterotrimeric PP2A species regulate signaling pathways that include mitosis, apoptosis, tumor suppression, and global metabolism (19–22).
TABLE 1.
Protein levels of PP2A subunits in rodent tissue
| PP2A subunits | Brain | Liver | Aorta |
|---|---|---|---|
| C subunits | |||
| Ppp2caa | +++ | ++ | ++b |
| Ppp2cb | +++ | + | +b |
| A subunits (2R1) | |||
| Ppp2r1a | ++ | + | +b |
| Ppp2r1b | +++ | ++ | +b |
| B subunits (2R2) | |||
| Ppp2r2a | − | − | − |
| Ppp2r2b | +c | + | − |
| Ppp2r2c | − | +++ | − |
| Ppp2r2d | ++d | − | ++b |
| B subunits (2R3) | |||
| Ppp2r3a | ++e | − | − |
| Ppp2r3b | ++f | +g | − |
| B subunits (2R4) | |||
| C14ORF10 | +++h | − | − |
| Ppp2r4 | +++ | + | − |
| B subunits (2R5) | |||
| Ppp2r5a | + | + | − |
| Ppp2r5b | − | − | − |
| Ppp2r5d | +++ | − | − |
| Ppp2r5e | ++ | + | ++b |
a Bold subunits were analyzed.
b PP2A subunits found to be highly expressed in aorta.
c Present as a higher molecular weight cross-reactive band.
d Present as two cross-reactive bands, one that migrates at a higher molecular weight and an appropriate molecular weight cross-reactive band.
e Both isoforms are present (130 and 70 kDa).
f Only the 70-kDa isoform present.
g Both isoforms are present (70 and 48 kDa).
h 75-, 70-, and 50-kDa cross-reactive bands present.
Several protein phosphatases can dephosphorylate AMP kinase at least in cell culture and in in vitro kinase dephosphorylation assays. These include PP2A, PP1, and PP2C (23–27). Very few PP2A B subunits have been elucidated that direct AMP kinase dephosphorylation. Those that are associated with the A/C dimer and act on AMP kinase include Ppp2r2d (28) and Ppp2r3a (29) (see Table 1). Heterotrimers containing these subunits are activated under conditions of metal excess, calcium release, change in glucose, and heat stress (28–31).
Mouse models have shown a correlation between loss of AMP kinase activity and the onset of diabetes (32–34), making AMP kinase an attractive target for pharmacological intervention. Metformin, one of the most used drugs to treat diabetes, targets and activates AMP kinase (35). Diabetes is a major risk factor for the occurrence of cardiovascular disease and atherosclerosis. As the aorta is a major site for lipid deposition and plaque formation, we wanted to determine how AMP kinase activity was regulated in this organ. We reasoned that treating AMP kinase at the specific plaque-forming site represents a novel approach to reducing the severity of cardiovascular disease in diabetic patients.
To date, the particular PP2A B subunit(s) dephosphorylating Thr-172 in aorta in response to diet remains to be elucidated. Here we show that PPP2APpp2r2d directly regulates lipid metabolism through its dephosphorylation of Thr-172, thus negatively regulating AMP kinase activity in the aorta. The results suggest that early activation of PPP2APpp2r2d in response to a western style diet may help to initiate aortic plaque formation and atherosclerosis.
EXPERIMENTAL PROCEDURES
Cell Lines
A7r5 (rat aortic smooth muscle) and human vascular smooth muscle (HVSM) cells were obtained from ATCC. A7r5 cells were cultured in Dulbecco's modified Eagle's medium (ATCC® 30-2002TM) modified to contain 4 mm glutamine, 4500 mg/liter glucose, 1 mm sodium pyruvate, and 1500 mg/liter sodium bicarbonate supplemented with 10% fetal bovine serum. HVSM cells were cultured in F-12K medium supplemented with 0.05 mg/ml ascorbic acid, 0.01 mg/ml insulin-transferrin-sodium selenite, 0.03 mg/ml endothelial cell growth supplement, 10% FBS, 10% HEPES, and 10 mm TES. Cells were incubated at 37 °C with 5% CO2. All cells were serum-starved overnight before initiating any experiments. For methyl-β-cyclodextrin (MCD; Sigma) and MCD-cholesterol (Sigma) treatments, cells were incubated in serum-free medium containing 50 μm MCD and 1 μg/ml MCD-cholesterol, respectively, at 37 °C for 2 h. Okadaic acid (OA) was purchased from Calbiochem (80055-324). STO-609 was purchased from Sigma.
Preparation of Mouse Aortic Lysate
Soon after the mice were euthanized, aortas were dissected and cleaned of adhering fat and soft tissues. Aortas were washed in ice-cold PBS to remove blood tissues, snap frozen in liquid nitrogen, and stored at −80 °C until further processing. Mouse aortic lysates were prepared by homogenization in radioimmuneprecipitation assay buffer containing phosphatase and protease inhibitors. Tissue and cell debris were removed by centrifugation, and protein concentration was determined using a Bradford assay (Bio-Rad).
Okadaic Acid Treatment
A7r5 cells were serum-starved overnight. The next day cells were treated with 500 pm, 1 nm, 5 nm, and 10 nm OA. Control cells were treated with DMSO. Protein concentration was determined using the Bradford assay system. Lysates were stored at −80 °C.
Protein Phosphatase 2A Assay
Phosphatase activity was determined using the DuoIC set PP2A phosphatase activity kit (R&D Systems) according to the manufacturer's instructions. Cells were rinsed two times with TBS. Cells were solubilized in 1 ml of lysis buffer (50 mm HEPES, 0.1 mm EGTA, 0.1 mm EDTA, 120 mm NaCl, 0.5% Nonidet P-40, pH 7.5, 25 μg/ml leupeptin, 25 μg/ml pepstatin, 2 μg/ml aprotinin, 1 mm PMSF)/1 × 107 cells. Cell extract was centrifuged at 2000 × g for 5 min, and sample protein concentration was quantified using a Bradford assay. 300–400 μg of the cell lysate was added to 96-well plates coated with immobilized capture antibody specific for the catalytic subunit of PP2A. After removing unbound material, a serine/threonine synthetic phosphopeptide substrate, which is dephosphorylated by active PP2A to generate free phosphate and unphosphorylated peptide, was added. The free phosphate released during the 30-min incubation was then detected by a dye binding assay using malachite green and molybdic acid. The activity of PP2A was determined by calculating the rate of phosphate release.
Western Blot Analysis
For Western analysis, cell cultures were collected by centrifugation at 2000 rpm for 5 min. Cells were washed with ice-cold PBS and centrifuged at 4000 rpm for 10 min. The cell pellet was resuspended in 150–200 μl of radioimmuneprecipitation assay buffer containing phosphatase and protease inhibitors. The cell suspension was subjected to bioruption two times for 5 min each and pelleted at high speed. Protein concentration was determined using the Bradford assay. Aliquots of cell lysate (50 μg) were stored at −80 °C or used in co-immunoprecipitation assays. Total protein from lysates or co-immunoprecipitates that were to be analyzed was resuspended in protein sample buffer and incubated at 95 °C for 10 min. All samples were subjected to 10% SDS-PAGE. Resolved proteins were transferred onto a nitrocellulose membrane. The immunoblot membranes were then blocked for 1 h with 10% milk and washed once with TBS-Tween 20. The membranes were incubated with primary antibody overnight at the appropriate dilutions. After five washes with TBS-Tween 20 for 10 min each, membranes were incubated with appropriate secondary antibody for 1 h. After five washes with TBS-Tween 20 for 10 min each, the membranes were immersed in a chemiluminescent agent and exposed for 2–5 min. Antibody dilutions were as follows: PPP2CA, 1:2000 (Abcam, ab33537); PPP2CB, 1:2000 (Abcam, ab72343); PPP2R1A, 1:1000 (Abcam, ab24728–100); PPP2R2D, 1:500 (GeneTex, GTX116609); PPP2R5E, 1:500 (GeneTex); pLKB1, 1:250 (Cell Signaling Technology); Camkkβ, 1:500 (Cell Signaling Technology); CamkII, 1:500 (Cell Signaling Technology); phosphorylated AMP kinase α (pAMPKα), 1:250 (Cell Signaling Technology); β-actin, 1:1000 (Abcam).
siRNA Transfection
A7r5 or HVSM cells were seeded in 10-cm plates and grown to 80% confluence. Cells were serum-starved the day before siRNA treatment. 100 μm siRNA stock solution was prepared in 1× reaction buffer. siRNAs that were used targeted the following genes: rat PPP2CA, PPP2CB, PPP2R1A, PPP2R1B, LKB1, and CAMKKβ; human PPP2CA, PPP2CB, PPP2R1A, PPP2R2D, LKB1, and CAMKKβ. Non-targeting siRNA was used in all cell lines as a control. For the experiment, the siRNA was diluted in serum-free medium (Opti-MEM, Invitrogen) in tube 1. In tube 2, DharmaFECT transfection reagent was diluted with Opti-MEM. Each tube was incubated for 5 min at room temperature. The content of tube 1 was added to tube 2, mixed, and incubated for 20 min at room temperature. The mixture was then added to the serum-starved cells to obtain a final concentration of 100 nm siRNA. After 24 and 48 h of siRNA treatment, cells were harvested for protein extraction. Proteins were stored at −80 °C after assaying the protein level using the Bradford assay system or were resolved by SDS-PAGE immediately for Western analysis.
Co-immunoprecipitation Experiments
The interactions between various proteins were assayed by using co-immunoprecipitation using the ProFound mammalian co-immunoprecipitation kit (Pierce) according to the manufacturer's instructions. Briefly, antibodies specific for Ppp2ca, Ppp2r1a, Ppp2r2d, Ppp2r5e, or Ampkα were treated with AminoLink® plus coupling gel slurry containing the beads overnight at 4 °C by end-over-end mixing. Subsequently, antibodies immobilized on beads were incubated with 300 μg of cell lysate at room temperature for 90 min. Beads were then extensively washed to remove all unbound proteins. Proteins bound to antibodies were separated using an elution buffer supplied by the commercial kit. The eluted proteins were further analyzed by SDS-PAGE and Western blotting. Beads that were not immobilized with target antibodies served as the control to account for any nonspecific interaction between the proteins and the beads.
Immunofluorescence Microscopy
Rat A7r5 cells (50,000) were seeded on poly-l-lysine treated coverslips in 24-well plates and cultured overnight in DMEM with 10% fetal bovine serum at 37 °C in an incubator with an atmosphere of 5% CO2. The cells then were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and washed three times followed by a permeabilization step with 0.25% Triton X-100, PBS for 8 min at room temperature. The cells were blocked in 10% donkey serum in 0.05% Triton X-100 in PBS at room temperature for 1 h and then treated with a 1:50 dilution of primary antibodies (PPP2R2D (Abcam)) and AMPKα1 antibodies (Santa Cruz Biotechnology) in 2% donkey serum, 0.05% Triton X-100, PBS for 2 h at 30 °C. After extensive washing with 0.05% Triton X-100 in PBS, the coverslips were incubated with a 1:100 dilution of secondary antibodies (Cy3-conjugated rabbit antibodies and DyLight 488-conjugated goat antibodies (Jackson ImmunoResearch Laboratories) in 2% donkey serum, 0.05% Triton X-100 in PBS at room temperature for 1 h. After extensive washing with 0.05% Triton X-100 in PBS and a final rinse with water, the coverslips were mounted in Fluoromount-G, imaged by a Leica DMI6000B microscope, and analyzed by ImageJ software.
Cholesterol Measurement in Cell Lysate
Cholesterol content in cell lysates were determined using the cholesterol assay kit supplied by Cell Biolabs Inc. according to the manufacturer's instructions. Cells treated with MCD or MCD-cholesterol were washed three times with cold PBS prior to lysis. Cells were extracted with 200 μl of chloroform:isopropanol:Nonidet P-40 (7:11:0.1) in a microhomogenizer. The extracts were centrifuged for 10 min at 15,000 × g. Without disturbing the pellet, the aqueous layer was transferred to a new tube and air-dried. The dried lipids were dissolved in 200 μl of 1× assay diluent supplied with the kit. 50 μl of cholesterol reaction reagent was added to each well of the 96-well plate containing the samples and cholesterol standards, and the plates were incubated for 45 min at 37 °C. Plates were read immediately after incubation with a fluorescence microplate reader equipped for excitation in the 530–570-nm range and for emission in the 590–600-nm range. Cholesterol standards and samples were assayed in triplicate, and a freshly prepared standard curve was used each time the assay was performed.
Mouse Feeding Studies
Wild type male C57BL/6J (B6) mice were purchased from The Jackson Laboratory and housed at Temple University, Philadelphia, PA. The Temple University Institutional Animal Care and Use Committee approved all experimental procedures. 6–8-week-old male C57BL/6J mice were fed either a normal diet (7% fat; BioServ) or a high fat diet (21% fat; BioServ) for 12 weeks. Fasted blood samples were taken every 4 weeks. Blood serum was used to measure total cholesterol, triglycerides, LDL, and HDL using the cholesterol and triglyceride assay kits (Stanbio, Boerne, TX) following the manufacturer's protocol. Mice from each group were sacrificed at weeks 4, 8, and 12, and aortas were collected for Western blot analysis.
RNA Isolation and Quantitative Real Time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen). cDNA was synthesized from total RNA using the RT Easy First Strand kit (Qiagen). Quantitative RT-PCR was carried out using a Stratagene MX3005P system. The relative mRNA levels were normalized to levels of GAPDH.
Statistical Analyses
The data shown are the average of five independent experiments. The data are the mean ± S.E. Statistical analysis was performed using Student's t test.
RESULTS
Inhibiting PP2A Activity by OA Treatment Causes an Increase in AMP Kinase Activity
The PP2A inhibitor OA was used to see whether reducing PP2A activity caused a difference in α subunit Thr-172 phosphorylation and/or AMP kinase α activity. A7r5 rat smooth muscle cells were treated with various concentrations of OA, and the phosphorylation state of Thr-172 was detected using an anti-Thr172 antibody. AMP kinase activity was assayed by determining the phosphorylation status of Acc1 (Fig. 1). Increasing concentrations of OA caused a dose-dependent decrease in PP2A activity (Fig. 1A). The addition of 10 nm OA acid, which resulted in an 80% reduction in PP2A activity, caused a significant increase in AMP kinase Thr-172 phosphorylation (Fig. 1B). This correlated with an increase in Acc1 phosphorylation status. The total protein levels of AMP kinase remained the same under either condition.
FIGURE 1.
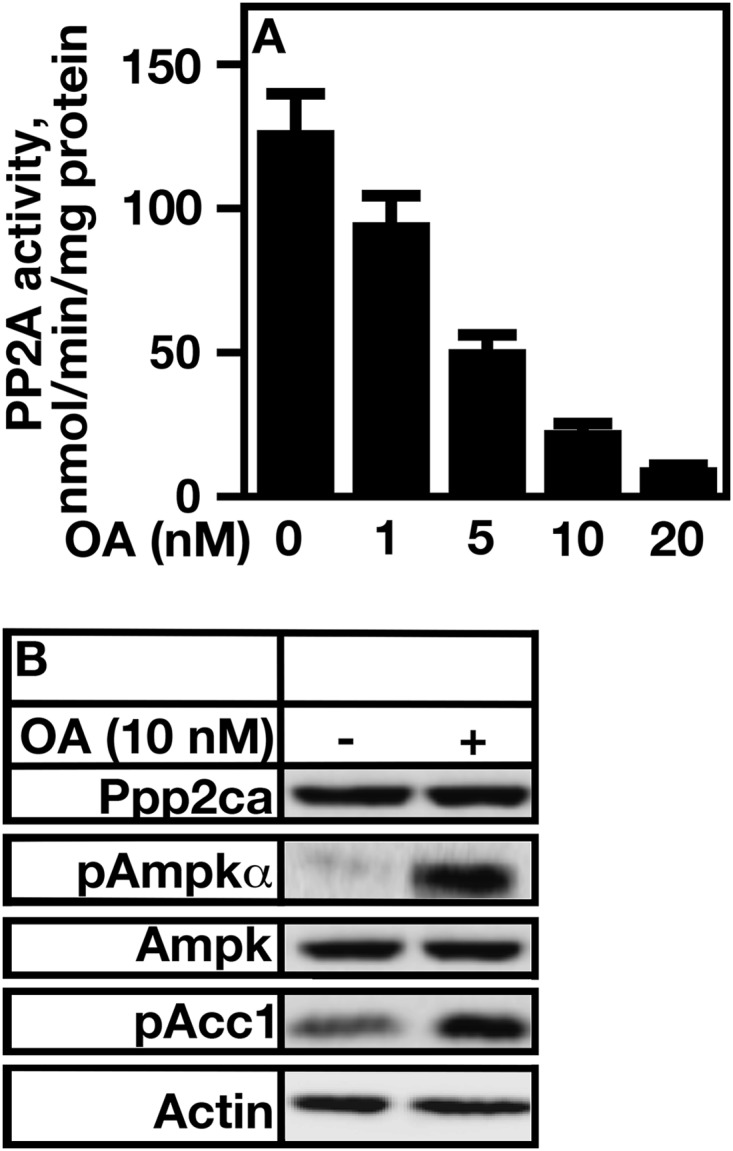
Inhibition of PP2A by okadaic acid treatment increases AMP kinase phosphorylation and activity. A, total cell extracts from A7r5 cells were obtained and treated with the concentrations of OA indicated. PPP2A activity was determined using an ELISA Ppp2ca phosphatase assay kit from R&D Systems. B, A7r5 cells were treated with 10 nm okadaic acid, and cell extracts were obtained. The levels of pAMPKα, AMP kinase, and Acc1 (pAcc1) phosphorylation were determined using specific antibodies generated to recognize the phosphorylation status of AMP kinase α Thr-172, total AMP kinase, and Acc1 Ser-79. Error bars represent S.E.
Specific PP2A Subunits Are Expressed in Various Rodent Organs
Table 1 shows the expression of various PP2A subunits in rodent brain, liver, and aorta. The major isoforms expressed in aorta were Ppp2ca/Ppp2cb, Ppp2r1a/Ppp2r1b, Ppp2r2d, and Ppp2r5E (Table 1). Ppp2r2d was expressed to a much higher extent than was Ppp2r5e. Interestingly, the major B subunit isoform expressed in liver was Ppp2r2c, whereas the brain expressed multiple B subunits to the same extent. A representative immunoblot is shown in Fig. 2. Based on these data, only the Ppp2r2d and Ppp2r5e B subunits were examined further.
FIGURE 2.

Rodent aorta expresses PP2APpp2r2d subunits. Ppp2ca, Ppp1r1a, Ppp2r2d, Ppp2r5e, and Ppp2r2b levels were determined in brain, liver, and aorta by Western analysis. Polyclonal antibodies to each specific subunit were used at concentrations described under “Experimental Procedures.”
The Protein Expression of Specific PP2A Subunits Is Responsive to Changes in Cholesterol Level
The protein levels of the specific PP2A subunits expressed in aorta were determined to see whether any were responsive to changes in cholesterol level. The level of cholesterol was reduced with MCD or saturated with MCD loaded with cholesterol. A7r5 cells were first treated with MCD (Fig. 3A) and then treated with MCD-cholesterol (Fig. 3B).
FIGURE 3.

PP2APpp2r2d protein expression is responsive to cholesterol level. A, A7r5 cells were incubated with MCD for the indicated times, and the cholesterol level was determined using a cholesterol assay kit. B, A7r5 cells that were treated with MCD for 2 h were incubated with cholesterol-loaded MCD (MCD-CHOL) for the indicated times, and the cholesterol level was determined using a cholesterol assay kit. C, the protein expression of various PP2A subunits, pAMPKα, AMP kinase, and Acc1 (pAcc1) phosphorylation was determined in A7r5 cells at the indicated times using Western analysis. The antibodies used were specific for each subunit, and they were tested for any cross-reactivity using siRNA knockdown experiments. Actin was used as a loading control. Error bars represent S.E.
Treatment of cells with the cholesterol-sequestering agent MCD caused a 3-fold decrease in cell cholesterol (Fig. 3A). Concomitant with this decrease was an increase in the expression of the PP2A catalytic subunits Ppp2ca and Ppp2cb (Fig. 3C). In addition there was 1) an increase in Ppp2r2d expression, 2) a decrease in AMP kinase Thr-172 phosphorylation, and 3) a decrease in Acc1 phosphorylation (Fig. 3C).
In contrast, MCD-cholesterol-treated cells had a 6-fold increase in cholesterol level (Fig. 3B). Ppp2ca, Ppp2cb, and Ppp2r2d expression decreased, whereas there were increases in phosphorylation of AMP kinase and ACC1. The level of total AMP kinase remained constant under either condition (Fig. 3C) as did the B subunit Ppp2r5e (Fig. 3C).
A Decrease in PP2APPP2R2D Expression Results in Increased AMP Kinase Activity
siRNA methods were used in A7r5 cells to decrease the mRNA expression of PP2APpp2r2d and PP2APpp2r5e subunits to determine whether PP2APpp2r2d and/or PP2APpp2r5e regulated AMP kinase activity. Total cell PP2A activity was first determined when PPP2R2D or PPP2R5E expression was reduced. The loss of expression of PPP2R2D reduced PP2A activity by ∼90% (Fig. 4A, Csi versus PPP2R2Dsi). A reduction in PPP2R5E expression reduced activity by ∼25% (Fig. 4A, Csi versus PPP2R5Esi). Thus, the loss of Ppp2r2d or Ppp2r5e affected PP2A activity to varying extents.
FIGURE 4.

Loss of PP2APpp2r2d, but not PP2APpp2r5e, results in increased AMP kinase Thr-172 phosphorylation and activity. A, A7r5 cells were treated with control siRNA (Csi) or siRNA directed against PPP2R2D or PPP2R5E, and PP2A activity was determined using the PP2A activity assay kit from R&D Systems. B, A7r5 cells were treated with control siRNA (Csi) or siRNA directed against PPP2CA or PPP2CB, and protein level and phosphorylation status were determined by Western analysis (WB). C, A7r5 cells were treated with control siRNA (Csi) or siRNA (si) directed against PPP2R1A or PPP2R1B, and protein level and phosphorylation status were determined by Western analysis. D, A7r5 cells were treated with control siRNA (Csi) or siRNA directed against PPP2R2D or PPP2R5E, and protein level and phosphorylation status were determined by Western analysis. Actin was used as a loading control. Error bars represent S.E.
The expression of the catalytic subunit isoforms was first reduced. Reducing the expression of Ppp2ca or Ppp2cb resulted in increases in AMP kinase Thr-172 and Acc1 phosphorylation (Fig. 4B). Similar results were obtained when the expression of either A subunit was reduced (Fig. 4C). The loss of expression of Ppp2r2d, but not Ppp2r5e, caused increases in phosphorylation of AMP kinase Thr-172 and Acc1 (Fig. 4D).
In all cases where MCD-cholesterol was used, parallel experiments using oxidized LDL were performed. Similar results were obtained (not shown).
Ppp2aPpp2r2d Directly Interacts with AMP Kinase α
Co-immunoprecipitation was used to determine which, if any, PP2A subunits directly interacted with AMP kinase. 10% of each protein input is represented in Fig. 5A. A heterotrimeric Ppp2aPpp2r2d was co-immunoprecipitated using antibodies directed against Ppp2ca, Ppp2r1a, or Ppp2r2d (Fig. 5B, IP, Ppp2ca, Ppp2r1a, and Ppp2r2d). Each individual subunit could be co-immunoprecipitated with AMP kinase (Fig. 5B, WB, Ampkα). Moreover, antibodies directed against AMP kinase brought down the Ppp2aPpp2r2d heterotrimer (Fig. 5C, IP, AMPKα). Ppp2r5e was unable to be co-immunoprecipitated with Ppp2ca, Ppp2r1a, or AMP kinase (Fig. 5C, IP, Ppp2r5e). Neither could AMP kinase pull down Ppp2r5e (Fig. 5C, IP, Ampkα).
FIGURE 5.
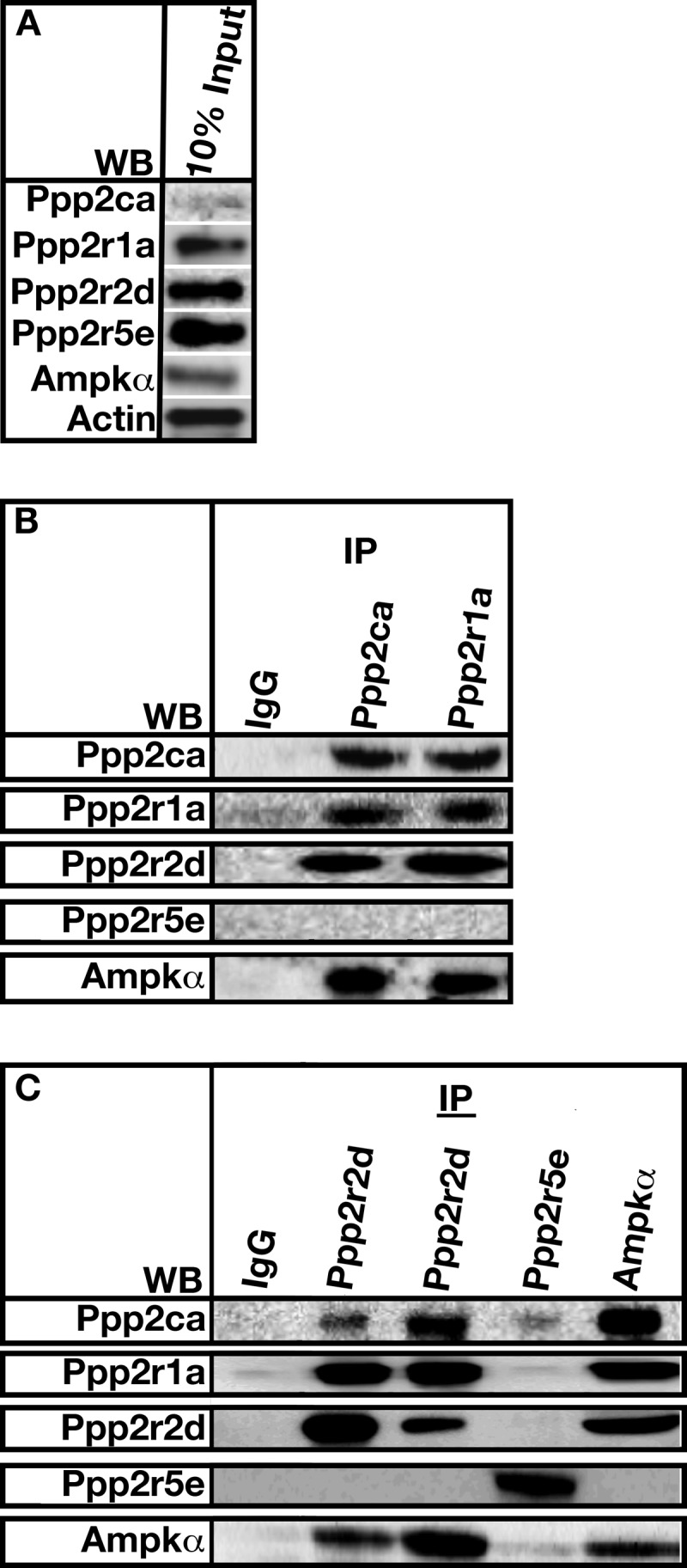
PP2APpp2r2d directly binds to AMP kinase α. A, 10% of the protein input used for co-immunoprecipitation assays. Actin was used as a loading control. B and C, cells extracts from A7r5 cells were obtained as described under “Experimental Procedures.” Extracts were incubated with the indicated antibodies (IP). Bound proteins were pulled down using Protein A-Sepharose and resolved by SDS-PAGE. Co-immunoprecipitated proteins were determined using Western analysis (WB).
We next used immunofluorescence microscopy to localize Ppp2r2d and AMP kinase α within cells. Results revealed that AMP kinase α and Ppp2r2d co-localized. The co-localization was seen within punctate structures at the cell periphery (Fig. 6).
FIGURE 6.

Ppp2r2d and Ampkα co-localize in A7r5 cells. A7r5 cells were seeded on poly-l-lysine treated coverslips and cultured in DMEM with 10% fetal bovine serum overnight. Cells were fixed with 4% paraformaldehyde in PBS followed by a permeabilization step with 0.25% Triton X-100. The cells were blocked in 10% donkey serum in 0.05% Triton X-100, PBS and then treated with a 1:50 dilution of primary antibodies (PPP2R2D (Abcam)) and AMPKα1 antibodies (Santa Cruz Biotechnology). The coverslips were incubated with secondary antibodies (Cy3-conjugated rabbit antibodies and DyLight 488-conjugated goat antibodies). Coverslips were mounted in Fluoromount-G, imaged by a Leica DMI6000B microscope, and analyzed by ImageJ software.
The Activities of Lkb1 and Camkkβ Are Not Needed for OA Regulation of Thr-172
Thr-172 on the α subunit of AMP kinase is predominately phosphorylated by LKB1 or CAMKKβ (14). We showed that addition of OA increases the phosphorylation status of Thr-172. Thus, we tested whether Lkb1 and/or Camkkβ was required for the increased phosphorylation of Thr-172 upon OA addition.
STO-609 is an inhibitor of CAMKKβ (36). CAMKII is a substrate of CAMKKβ. CAMKKβ activity is routinely assayed by determining the phosphorylation status of CAMKII. STO-609 was used to decrease CAMKKβ activity in the absence or presence of OA, and CAMKII and Ampkα phosphorylation was determined by Western analysis (Fig. 7).
FIGURE 7.

PP2APpp2r2d functions downstream of Lkb1 and Camkkβ in regulating AMP kinase activity. A, A7r5 cells were treated or not treated with 50 μm Camkkβ inhibitor STO-609, and the level of Camkkβ activity was determined by assaying for the level of CamkII phosphorylation. B, A7r5 cells were incubated in the absence or presence of STO-609 and OA acid, and the level of AMP kinase α phosphorylation was determined using Western analysis. Actin was used as a loading control. C and D, A7r5 cells were treated with control (Csi), LKB1 siRNA (LKBsi), CAMKKβ siRNA (CAMKKsi), or LKB1 and CAMKKβ siRNAs. OA was added under the indicated conditions. Protein levels were determined using Western analysis (WB). The level of the phosphorylation status of AMP kinase α was determined using antibodies directed against Thr-172. Actin was used as a loading control.
The addition of STO-609 efficiently inhibited CAMKKβ activity as evidenced by a decrease in CAMKII phosphorylation (Fig. 7A, pCamkII versus STO-609). The level of CAMKII did not change. AMP kinase α Thr-172 phosphorylation was low in the presence of STO-609 alone (Fig. 7B, STO-609 versus pAmpkα). The reduction in phosphorylation was reversed when OA was added (Fig. 7B, STO-609 OA versus pAmpkα).
There are no inhibitors for LKB1. Thus, siRNA was used to knock down LKB1 expression, and the phosphorylation status of AMP kinase Thr-172 was determined in the absence and presence of OA. siRNA against LKB1 efficiently knocked down the protein expression of Lkb1 (Fig. 7C, Csi versus LKBsi). The phosphorylation status of AMP kinase Thr-172 remained at the basal level (Fig. 7C, LKBsi, pAmpkα). The addition of OA to LKB1 siRNA-treated cells increased the level of phosphorylation of Thr-172 (Fig. 7C, LKBsi versus LKBsi + OA versus pAmpkα).
Finally, both LKB1 and CAMKKβ expression was knocked down by siRNA in the absence and presence of OA. AMP kinase α Thr-172 phosphorylation remained at the basal level in the presence of both siRNAs (Fig. 7D, LKBsi + CAMKKsi versus pAmpkα). The addition of OA increased Thr-172 phosphorylation (Fig. 7D, LKBsi + CAMKKsi + OA versus pAmpkα). Thus, the activities of both kinases are not needed for OA to increase Thr-172 phosphorylation.
Loss of PP2APpp2r2d Decreases SREBP1c Processing and Transcriptional Activity Forms a Complex with AMP Kinase and Srebp1
AMP kinase is known to inhibit Srebp1c activity by phosphorylating Ser-372 on the mature nuclear form of the protein (37). Phosphorylation causes a decrease in Srebp1c transcription factor activity and an increase in proteolytic degradation. To further define whether and how PP2APpp2r2d regulates the activity of AMP kinase, the level of nuclear Srebp1c was determined, and Srebp1c transcription factor activity was assayed by examining the level of PPARγ. Increased proteolytic degradation of mature SREBP1c was seen in the absence of PPP2R2D expression, indicating that AMP kinase was activated and caused SREBP1 degradation (Fig. 8A). Concomitant with activation was a decrease in PPARγ expression (Fig. 8B). SREBP1 regulates PPARγ expression; thus, it is a direct marker for Srebp1 function. Interestingly, PP2APpp2r2d associated with both AMP kinase and SREBP1 (Fig. 8, C and D). Ppp2r5e did not interact with either protein.
FIGURE 8.

PP2APpp2r2d forms a complex with AMP kinase α and Srebp1 and regulates Srebp1-dependent gene expression. A, A7r5 cells were treated with control (Csi) or PPP2R2D siRNA (PPP2R2Dsi). The levels of Ppp2r2d and mature Srebp1 were determined using Western analysis. Actin was used as a loading control. B, A7r5 cells were grown in the absence (Csi) or presence of PPP2R2D siRNA (PPP2R2Dsi). The level of Srebp1-dependent PPARγ mRNA expression was determined by quantitative RT-PCR. C, 10% of the total protein used for co-immunoprecipitation experiments. Actin was used as a loading control. D, cell extracts were incubated with AMP kinase or Srebp1 antibodies (IP), and associated proteins were isolated using Protein A-Sepharose beads. Co-immunoprecipitated proteins were resolved by SDS-PAGE and detected using Western analysis (WB) using the indicated antibodies.
Ppp2aPpp2r2d Regulates AMP Kinase Activity through a Direct Interaction in HVSM Cells
The interaction between AMP kinase and PP2APpp2r2d was tested in HVSM cells. HVSM cells represent an excellent human model for the study of smooth muscle cell physiology. The loss of either PPP2CA or PPP2R2D resulted in increases in AMP kinase Thr-172 and Acc1 phosphorylation (Fig. 9A). The loss of LKB1 caused a decrease in AMP kinase phosphorylation that was restored when OA was added (Fig. 9B). Moreover, an increase in AMP kinase Thr-172 phosphorylation was seen when LKB1 and CAMKKβ expression was knocked down by siRNA in the presence of OA (Fig. 9C, LKB1si + CAMKKsi + OA). A direct physical interaction was observed between PPP2APpp2r2d and AMP kinase in these cells (Fig. 9, D and E).
FIGURE 9.

PP2APpp2r2d regulates AMP kinase activity in human vascular smooth muscle cells. A, HVSMCs were grown as described under “Experimental Procedures.” Cells were treated with control siRNA (Csi) or siRNA directed against PPP2CA or PPP2R2D, and protein level and phosphorylation status were determined by Western analysis. Actin was used as a loading control. B and C, cells were treated with control (Csi) or LKB1 siRNA (LKB1si), CAMKKβ siRNA (CAMKKsi), or LKB1 and CAMKKβ siRNAs. OA was added under the indicated conditions. Protein levels were determined using Western analysis. The level of Camkkβ activity was determined by assaying for the level of CamkII phosphorylation. The phosphorylation status of AMP kinase α was determined using antibodies directed against Thr-172. Actin was used as a loading control. D, 10% of the protein input used for co-immunoprecipitation assays. Actin was used as a loading control. E, extracts were incubated with the indicated antibodies (IP). Bound proteins were pulled down using Protein A-Sepharose and resolved by SDS-PAGE. Co-immunoprecipitated proteins were determined using Western analysis (WB).
The Level of Ppp2aPpp2r2d Is Elevated in the Aortas of Mice Fed a High Fat Diet
To determine whether the protein level and phosphatase activity of Ppp2aPpp2r2d were regulated by diet, C57BL/6 mice were fed a high fat diet for 12 weeks, and PP2APpp2r2d subunit levels and AMP kinase Thr-172 phosphorylation were determined. AMP kinase activity was indirectly assayed by determining Acc1 phosphorylation.
The weights of mice fed a high fat diet doubled over the time of the study when compared with those fed normal chow (Fig. 10A). The blood levels of cholesterol (Fig. 10B), triglycerides (Fig. 10C), and LDL (Fig. 10D) increased in a time-dependent manner. There was a direct correlation between weight and lipid increases with increases in the levels of Ppp2ca and Ppp2r2d (Fig. 10, E, F, and G). The level of Ppp2r1a remained constant (Fig. 10, E, F, and G). AMP kinase Thr-172 phosphorylation was decreased as was Acc1 phosphorylation (Fig. 10, E, F, and G). Three individual aortas were analyzed (Fig. 10, E, F, and G).
FIGURE 10.

PPPPpp2r2d level increases in the aorta of mice fed a high fat diet. C57BL/6 mice were fed a high fat diet as described under “Experimental Procedures.” A, the weight of all mice was determined at the indicated times (n = 8). B, blood cholesterol levels of all mice were determined at the indicated times using a cholesterol level assay kit from Stanbio. C, blood triglyceride levels of individual mice were determined at the indicated times using a triglyceride level assay kit from Stanbio. D, blood total LDL levels of individual mice were determined at the indicated times using an LDL level assay kit from Stanbio. E, F, and G, protein expression levels of various proteins were determined in aortas at the indicated times using Western analysis in high fat-fed mice. The antibodies used were specific for each PP2A subunit, AMP kinase, phosphorylated AMP kinase, and phosphorylated Acc1 (pAcc1), and they were tested for any cross-reactivity using siRNA knockdown experiments. Actin was used as a loading control. The data from three individual mice are shown. Closed circles, WT, normal chow; open circles, WT, high fat diet. 7%, 7% fat diet; 21%, 21% fat diet. Error bars represent S.E.
PPP2APpp2r2d Directly Interacts with AMP Kinase in High Fat-fed Mouse Aorta
Co-immunoprecipitation was used to determine whether PP2APpp2r2d directly interacted with AMP kinase in aortas from high fat-fed mice. Fig. 11A shows 10% of the protein input used for the co-immunoprecipitation experiments. Polyclonal antibodies directed against AMP kinase α were able to pull down the PP2APpp2r2d heterotrimer but were unable to immunoprecipitate Ppp2r5e (Fig. 11B). We point out that cross-sectioning of aorta and immunofluorescence microscopy looking at Ppp2r2d and Ampk localization would definitively demonstrate an interaction. However, there was no interaction of AMPK with Ppp2r5e, so we believe the Ppp2r2d-AMPK interaction is real.
FIGURE 11.
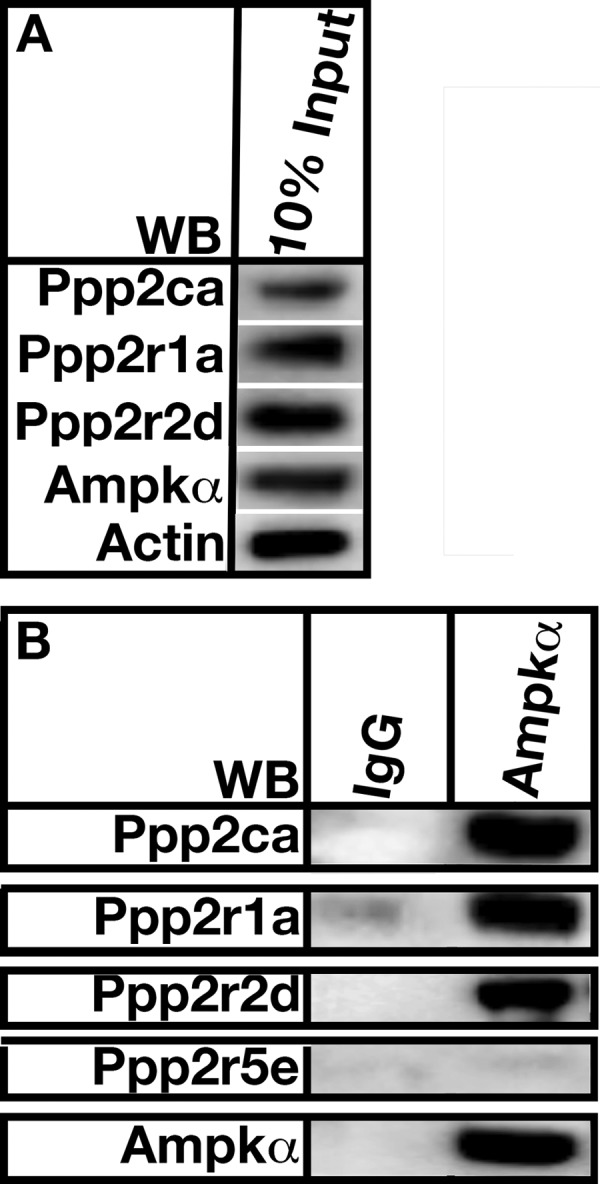
PP2APpp2r2d forms a complex with AMP kinase α in mouse aorta. A, 10% of the total protein used for co-immunoprecipitation experiments. Actin was used as a loading control. B, aortic lysate was incubated with AMP kinase antibodies (IP), and associated proteins were isolated using Protein A-Sepharose beads. Co-immunoprecipitated proteins were resolved by SDS-PAGE and detected using Western analysis (WB) using the indicated antibodies. The figure is a representation of 10 separate experiments using aortas from wild type C57BL/6 mice.
DISCUSSION
AMP kinase is an “energy-sensing” kinase that is at the center of an axis necessary for maintaining cell homeostasis and energy consumption (38). AMP kinase substrates include Acc1, Hmgcr, hormone-sensitive lipase, and Srebp1c, all of which are involved in lipogenesis (37, 39–41), indicating that proper regulation of AMP kinase activity is necessary to maintain normal lipid metabolism in response to cell energy status. AMP kinase activation is stimulated by an increase in the AMP:ATP ratio, which causes 1) an increase in AMP binding and stimulation of activity and 2) further activation through Thr-172 phosphorylation by the LKB1 and CAMKKβ kinases (14). Here, we report on the regulation of AMP kinase activity by the specific PP2A phosphatase heterotrimer, PP2APpp2r2d.
We showed that PP2APpp2r2d directly interacted with AMP kinase and dephosphorylated Thr-172, resulting in a reduction in kinase activity. Moreover, PP2APpp2r2d formed a complex with AMP kinase and Srebp1c, indicating that PP2APpp2r2d inhibited AMP kinase by dephosphorylating Thr-172 but also suggesting that SREBP1 is activated through PP2APpp2r2d-dependent dephosphorylation of an inhibitory AMP kinase site (Ser-372). We also found that the level of PP2APpp2r2d was elevated in aortas from high fat-fed mice. The elevation correlated with increased total blood cholesterol, triglyceride, and LDL. We showed that the expression of specific PP2A subunits was up-regulated in response to a high fat diet. Finally, we demonstrated that PP2APpp2r2d forms a complex with AMP kinase in the aortas of high fat-fed mice. The results presented here are significant as aberrant increased PP2APpp2r2d activity would inhibit AMP kinase at a time when kinase activity would be essential for turning down anabolic pathways (11). The data together suggest that regulation of the PP2APpp2r2d heterotrimer, either directly or indirectly, represents an avenue for drug discovery for treating diseases associated with metabolic syndrome, such as hyperlipidemia, type II diabetes, and obesity (42).
The crystal structures of α2β1γ1 and α1β1γ1 have recently been determined and have been insightful in understanding the mechanism for and regulation of Thr-172 phosphorylation (6–8). The α subunits contain traditional kinase and autoinhibitory domains, whereas the β subunits have a conserved carbohydrate-binding domain that is required for AMP kinase glycogen binding and inhibition. The β subunits act as scaffolds for tethering together the α-γ subunits. The γ subunit itself contains four nucleotide-binding sites (Bateman domains) that have varying specificities for AMP and ATP (7). AMP binding to specific AMP-binding sites activates kinase activity, whereas ATP binding is inhibitory. Phosphorylation of Thr-172 by AMP kinase kinases requires AMP binding (7). Thr-172 is found in a cleft between the α catalytic and γ nucleotide-binding domains. The movement of these domains regulates the accessibility of Thr-172 to phosphorylation/dephosphorylation. AMP binding changes the conformation of the α/γ cleft to one that allows for phosphorylation and protects Thr-172 from being dephosphorylated by phosphatases (7).
AMP kinase can be dephosphorylated by a number of phosphatases (PP2A, PP2C, and PP1) (23–27). In some cases, the specific heterotrimeric PP2A-dephosphorylating AMP kinase is known (28, 29, 31), whereas some remain to be identified. Studies during the 1980s that were aimed at identifying phosphatase activities in various tissues dephosphorylating AMP kinase revealed the existence of several PP2A isoforms in rat liver: PP2AD (A/C dimer) and several heterotrimeric species. Based on our studies, the major PP2A isoform found in liver was PP2APpp2r2c, whereas the major forms in aorta were PP2APP2r2d and PP2APpp2r5e (Table 1). We showed that only the PP2APP2r2d heterotrimer was capable of dephosphorylating AMP kinase in our cell lines. Dephosphorylation caused a reduction in Acc1 phosphorylation and an increase in Acc1 activity. These results were recapitulated in aortas from high fat-fed mice. Thus, it seems that PP2APP2r2d is the sole regulator of AMP kinase activity at a critical site for plaque formation.
There is evidence that PP2A activity is regulated by changes in lipid metabolism (43–48). For example, it can be activated by changes in free fatty acid or cholesterol levels (44, 46), whereas it can be inhibited by increased gluconeogenesis and lipogenesis (43). PP2A activity is also regulated during insulin signaling (43, 48). It has been shown that PP2A targets phosphatidylinositol 3-kinase signaling through dephosphorylating Akt (49). Finally, several studies have shown that PP2A subunit mRNA expression is regulated by diet (50, 51).
Recently, we showed that loss of Ppp2ca catalytic activity resulted in a loss of SREBP2-dependent gene expression in HepG2 cells (52). Mechanistically, Ppp2ca directly bound SREBP2 and decreased its ability to bind to promoter sterol response elements. The B subunit responsible for guiding Ppp2ca to SREBP2 was not determined. Preliminary data suggest that liver Ppp2r2c may be that B subunit; this subunit is the only one expressed in mouse and human liver (Table 1).3
Small molecule compounds that activate or inhibit AMP kinase have been generated (53–57). Preclinical trials suggest that they may be efficacious in treating diet-induced diabetes (56, 58). Moreover, activation of AMP kinase in cell culture suggests that it can be an excellent pharmacological target for treating several cancers (56). There are several inhibitors of PP2A activity, including a methyl esterase (protein phosphatase 2A demethylase 1) that demethylates the Ppp2ca catalytic subunit, inhibiting its activity (59). The stimulation of this inhibitor results in the inactivation of PP2A, which may activate AMP kinase. The caveat to this type of treatment is the lack of specificity for a particular PP2A heterotrimer. Based on our results, it may be advantageous to inhibit only PP2APPpp2r2d as a means to activate AMP kinase, giving rise to an avenue to reduce metabolic syndrome and atherosclerosis (42).
Acknowledgments
We are grateful for the many discussions with Drs. Jonathan Yavelow, Martin Adelson, and Eli Mordechai. We thank Drs. Jenny Yip and Kei Liu for helpful ideas. We appreciate our colleagues at the Institute of Metabolic Disorders for their willingness to help with all aspects of the work.
Note Added in Proof
Fig. 8 was inadvertently published as a duplicate of Fig. 9 in the version of this article that was published as a Paper in Press on February 18, 2015. The correct version of Fig. 8 is now shown.
This work was supported by the Genesis Biotechnology Group.
B. Joseph and J. T. Nickels, Jr., unpublished data.
- ACC1
- acetyl-CoA carboxylase 1
- PP2A
- protein phosphatase 2A
- SREBP
- sterol response element-binding protein
- HMGCR
- HMG-CoA reductase
- OA
- okadaic acid
- PPP2CA
- protein phosphatase 2A α catalytic subunit gene
- PPP2R2D
- protein phosphatase 2A Ppp2r2d B subunit gene
- PPP2R5E
- protein phosphatase 2A Ppp2r5e B subunit gene
- PPP2CA
- protein phosphatase 2A α catalytic subunit
- Ppp2r2d
- Ppp2r2d B subunit
- Ppp2r5e
- PPP2R5E B subunit
- LKB1
- liver kinase B1
- CAMKKβ
- Ca2+/calmodulin-dependent protein kinase kinase β
- TAK1
- transforming growth factor-β-activated kinase 1
- MCD
- methyl-β-cyclodextrin
- HVSM
- human vascular smooth muscle
- PP1
- protein phosphatase 1
- PP2C
- protein phosphatase 2C
- TES
- 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid
- AMPKα
- AMP kinase α
- pAMPKα
- phosphorylated AMP kinase α
- PPARγ
- peroxisome proliferator-activated receptor γ
- CamkII
- Ca2+/calmodulin-dependent protein kinase II.
REFERENCES
- 1. Carling D., Mayer F. V., Sanders M. J., Gamblin S. J. (2011) AMP-activated protein kinase: nature's energy sensor. Nat. Chem. Biol. 7, 512–518 [DOI] [PubMed] [Google Scholar]
- 2. Xiao B., Sanders M. J., Underwood E., Heath R., Mayer F. V., Carmena D., Jing C., Walker P. A., Eccleston J. F., Haire L. F., Saiu P., Howell S. A., Aasland R., Martin S. R., Carling D., Gamblin S. J. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardie D. G., Carling D., Gamblin S. J. (2011) AMP-activated protein kinase: also regulated by ADP? Trends Biochem. Sci. 36, 470–477 [DOI] [PubMed] [Google Scholar]
- 4. Wu J., Puppala D., Feng X., Monetti M., Lapworth A. L., Geoghegan K. F. (2013) Chemoproteomic analysis of intertissue and interspecies isoform diversity of AMP-activated protein kinase (AMPK) J. Biol. Chem. 288, 35904–35912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nagata D., Hirata Y. (2010) The role of AMP-activated protein kinase in the cardiovascular system. Hypertens. Res. 33, 22–28 [DOI] [PubMed] [Google Scholar]
- 6. Xiao B., Sanders M. J., Carmena D., Bright N. J., Haire L. F., Underwood E., Patel B. R., Heath R. B., Walker P. A., Hallen S., Giordanetto F., Martin S. R., Carling D., Gamblin S. J. (2013) Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 4, 3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen L., Xin F. J., Wang J., Hu J., Zhang Y. Y., Wan S., Cao L. S., Lu C., Li P., Yan S. F., Neumann D., Schlattner U., Xia B., Wang Z. X., Wu J. W. (2013) Conserved regulatory elements in AMPK. Nature 498, E8–10 [DOI] [PubMed] [Google Scholar]
- 8. Chen L., Jiao Z. H., Zheng L. S., Zhang Y. Y., Xie S. T., Wang Z. X., Wu J. W. (2009) Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature 459, 1146–1149 [DOI] [PubMed] [Google Scholar]
- 9. Hardie D. G., Ross F. A., Hawley S. A. (2012) AMP-activated protein kinase: a target for drugs both ancient and modern. Chem. Biol. 19, 1222–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kinney B. P., Qiao L., Levaugh J. M., Shao J. (2010) B56α/protein phosphatase 2A inhibits adipose lipolysis in high-fat diet-induced obese mice. Endocrinology 151, 3624–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hardie D. G. (2014) AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 33C, 1–7 [DOI] [PubMed] [Google Scholar]
- 12. Munday M. R., Hemingway C. J. (1999) The regulation of acetyl-CoA carboxylase—a potential target for the action of hypolipidemic agents. Adv. Enzyme Regul. 39, 205–234 [DOI] [PubMed] [Google Scholar]
- 13. Clarke P. R., Hardie D. G. (1990) Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 9, 2439–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Witczak C. A., Sharoff C. G., Goodyear L. J. (2008) AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell. Mol. Life Sci. 65, 3737–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suter M., Riek U., Tuerk R., Schlattner U., Wallimann T., Neumann D. (2006) Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 281, 32207–32216 [DOI] [PubMed] [Google Scholar]
- 16. Valentine R. J., Coughlan K. A., Ruderman N. B., Saha A. K. (2014) Insulin inhibits AMPK activity and phosphorylates AMPK Ser(485/491) through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch. Biochem. Biophys. 562, 62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cohen P. T., Brewis N. D., Hughes V., Mann D. J. (1990) Protein serine/threonine phosphatases; an expanding family. FEBS Lett. 268, 355–359 [DOI] [PubMed] [Google Scholar]
- 18. Eichhorn P. J., Creyghton M. P., Bernards R. (2009) Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 1795, 1–15 [DOI] [PubMed] [Google Scholar]
- 19. Depaoli-Roach A. A., Park I. K., Cerovsky V., Csortos C., Durbin S. D., Kuntz M. J., Sitikov A., Tang P. M., Verin A., Zolnierowicz S. (1994) Serine/threonine protein phosphatases in the control of cell function. Adv. Enzyme Regul. 34, 199–224 [DOI] [PubMed] [Google Scholar]
- 20. Mumby M. C., Walter G. (1993) Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 73, 673–699 [DOI] [PubMed] [Google Scholar]
- 21. Hardie D. G. (1990) Roles of protein kinases and phosphatases in signal transduction. Symp. Soc. Exp. Biol. 44, 241–255 [PubMed] [Google Scholar]
- 22. Sontag E. (2001) Protein phosphatase 2A: the Trojan Horse of cellular signaling. Cell. Signal. 13, 7–16 [DOI] [PubMed] [Google Scholar]
- 23. Tamura S., Tsuiki S. (1980) Purification and subunit structure of rat-liver phosphoprotein phosphatase, whose molecular weight is 260000 by gel filtration (phosphatase IB). Eur. J. Biochem. 111, 217–224 [DOI] [PubMed] [Google Scholar]
- 24. Tamura S., Kikuchi H., Kikuchi K., Hiraga A., Tsuiki S. (1980) Purification and subunit structure of a high-molecular-weight phosphoprotein phosphatase (phosphatase II) from rat liver. Eur. J. Biochem. 104, 347–355 [DOI] [PubMed] [Google Scholar]
- 25. Moore F., Weekes J., Hardie D. G. (1991) Evidence that AMP triggers phosphorylation as well as direct allosteric activation of rat liver AMP-activated protein kinase. A sensitive mechanism to protect the cell against ATP depletion. Eur. J. Biochem. 199, 691–697 [DOI] [PubMed] [Google Scholar]
- 26. Garcia-Haro L., Garcia-Gimeno M. A., Neumann D., Beullens M., Bollen M., Sanz P. (2010) The PP1-R6 protein phosphatase holoenzyme is involved in the glucose-induced dephosphorylation and inactivation of AMP-activated protein kinase, a key regulator of insulin secretion, in MIN6 β cells. FASEB J. 24, 5080–5091 [DOI] [PubMed] [Google Scholar]
- 27. Sanders M. J., Grondin P. O., Hegarty B. D., Snowden M. A., Carling D. (2007) Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 403, 139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu X. N., Chen L. P., Bai Q., Ma L., Li D. C., Zhang J. M., Gao C., Lei Z. N., Zhang Z. B., Xing X. M., Liu C. X., He Z. N., Li J., Xiao Y. M., Zhang A. H., Zeng X. W., Chen W. (2014) PP2A-AMPKα-HSF1 axis regulates the metal-inducible expression of HSPs and ROS clearance. Cell. Signal. 26, 825–832 [DOI] [PubMed] [Google Scholar]
- 29. Park S., Scheffler T. L., Rossie S. S., Gerrard D. E. (2013) AMPK activity is regulated by calcium-mediated protein phosphatase 2A activity. Cell Calcium 53, 217–223 [DOI] [PubMed] [Google Scholar]
- 30. Wang T., Yu Q., Chen J., Deng B., Qian L., Le Y. (2010) PP2A mediated AMPK inhibition promotes HSP70 expression in heat shock response. PLoS One 5, e13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gimeno-Alcañiz J. V., Sanz P. (2003) Glucose and type 2A protein phosphatase regulate the interaction between catalytic and regulatory subunits of AMP-activated protein kinase. J. Mol. Biol. 333, 201–209 [DOI] [PubMed] [Google Scholar]
- 32. O'Neill H. M., Maarbjerg S. J., Crane J. D., Jeppesen J., Jørgensen S. B., Schertzer J. D., Shyroka O., Kiens B., van Denderen B. J., Tarnopolsky M. A., Kemp B. E., Richter E. A., Steinberg G. R. (2011) AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. U.S.A. 108, 16092–16097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Viollet B., Andreelli F., Jørgensen S. B., Perrin C., Geloen A., Flamez D., Mu J., Lenzner C., Baud O., Bennoun M., Gomas E., Nicolas G., Wojtaszewski J. F., Kahn A., Carling D., Schuit F. C., Birnbaum M. J., Richter E. A., Burcelin R., Vaulont S. (2003) The AMP-activated protein kinase α2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Investig. 111, 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Viollet B., Athea Y., Mounier R., Guigas B., Zarrinpashneh E., Horman S., Lantier L., Hebrard S., Devin-Leclerc J., Beauloye C., Foretz M., Andreelli F., Ventura-Clapier R., Bertrand L. (2009) AMPK: lessons from transgenic and knockout animals. Front. Biosci. 14, 19–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pawlyk A. C., Giacomini K. M., McKeon C., Shuldiner A. R., Florez J. C. (2014) Metformin pharmacogenomics: current status and future directions. Diabetes 63, 2590–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tokumitsu H., Inuzuka H., Ishikawa Y., Ikeda M., Saji I., Kobayashi R. (2002) STO-609, a specific inhibitor of the Ca2+/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 277, 15813–15818 [DOI] [PubMed] [Google Scholar]
- 37. Li Y., Xu S., Mihaylova M. M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J. Y., Gao B., Wierzbicki M., Verbeuren T. J., Shaw R. J., Cohen R. A., Zang M. (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steinberg G. R., Macaulay S. L., Febbraio M. A., Kemp B. E. (2006) AMP-activated protein kinase—the fat controller of the energy railroad. Can. J. Physiol. Pharmacol. 84, 655–665 [DOI] [PubMed] [Google Scholar]
- 39. Carling D., Zammit V. A., Hardie D. G. (1987) A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 223, 217–222 [DOI] [PubMed] [Google Scholar]
- 40. Carling D., Clarke P. R., Zammit V. A., Hardie D. G. (1989) Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur. J. Biochem. 186, 129–136 [DOI] [PubMed] [Google Scholar]
- 41. Garton A. J., Campbell D. G., Carling D., Hardie D. G., Colbran R. J., Yeaman S. J. (1989) Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism. Eur. J. Biochem. 179, 249–254 [DOI] [PubMed] [Google Scholar]
- 42. Khoo M. C., Oliveira F. M., Cheng L. (2013) Understanding the metabolic syndrome: a modeling perspective. IEEE Rev. Biomed. Eng. 6, 143–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galbo T., Perry R. J., Nishimura E., Samuel V. T., Quistorff B., Shulman G. I. (2013) PP2A inhibition results in hepatic insulin resistance despite Akt2 activation. Aging 5, 770–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Galbo T., Olsen G. S., Quistorff B., Nishimura E. (2011) Free fatty acid-induced PP2A hyperactivity selectively impairs hepatic insulin action on glucose metabolism. PLoS One 6, e27424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dobrowsky R. T., Kamibayashi C., Mumby M. C., Hannun Y. A. (1993) Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 268, 15523–15530 [PubMed] [Google Scholar]
- 46. Wang P. Y., Liu P., Weng J., Sontag E., Anderson R. G. (2003) A cholesterol-regulated PP2A/HePTP complex with dual specificity ERK1/2 phosphatase activity. EMBO J. 22, 2658–2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kowluru A., Metz S. A. (1997) Ceramide-activated protein phosphatase-2A activity in insulin-secreting cells. FEBS Lett. 418, 179–182 [DOI] [PubMed] [Google Scholar]
- 48. Højlund K., Poulsen M., Staehr P., Brusgaard K., Beck-Nielsen H. (2002) Effect of insulin on protein phosphatase 2A expression in muscle in type 2 diabetes. Eur. J. Clin. Invest. 32, 918–923 [DOI] [PubMed] [Google Scholar]
- 49. Ugi S., Imamura T., Maegawa H., Egawa K., Yoshizaki T., Shi K., Obata T., Ebina Y., Kashiwagi A., Olefsky J. M. (2004) Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 24, 8778–8789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jun H. S., Hwang K., Kim Y., Park T. (2008) High-fat diet alters PP2A, TC10, and CIP4 expression in visceral adipose tissue of rats. Obesity 16, 1226–1231 [DOI] [PubMed] [Google Scholar]
- 51. Jump D. B. (2011) Fatty acid regulation of hepatic lipid metabolism. Curr. Opin. Clin. Nutr. Metab. Care 14, 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rice L. M., Donigan M., Yang M., Liu W., Pandya D., Joseph B. K., Sodi V., Gearhart T. L., Yip J., Bouchard M., Nickels J. T., Jr. (2014) Protein phosphatase 2A (PP2A) regulates low density lipoprotein uptake through regulating sterol response element-binding protein-2 (SREBP-2) DNA binding. J. Biol. Chem. 289, 17268–17279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sullivan J. E., Brocklehurst K. J., Marley A. E., Carey F., Carling D., Beri R. K. (1994) Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 353, 33–36 [DOI] [PubMed] [Google Scholar]
- 54. Sanders M. J., Ali Z. S., Hegarty B. D., Heath R., Snowden M. A., Carling D. (2007) Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J. Biol. Chem. 282, 32539–32548 [DOI] [PubMed] [Google Scholar]
- 55. King T. D., Song L., Jope R. S. (2006) AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem. Pharmacol. 71, 1637–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rana S., Blowers E. C., Natarajan A. (2015) Small molecule adenosine 5′-monophosphate activated protein kinase (AMPK) modulators and human diseases. J. Med. Chem. 58, 2–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lian Z., Li Y., Gao J., Qu K., Li J., Hao L., Wu S., Zhu H. (2011) A novel AMPK activator, WS070117, improves lipid metabolism discords in hamsters and HepG2 cells. Lipids Health Dis. 10, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boon H., Bosselaar M., Praet S. F., Blaak E. E., Saris W. H., Wagenmakers A. J., McGee S. L., Tack C. J., Smits P., Hargreaves M., van Loon L. J. (2008) Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 51, 1893–1900 [DOI] [PubMed] [Google Scholar]
- 59. Ogris E., Du X., Nelson K. C., Mak E. K., Yu X. X., Lane W. S., Pallas D. C. (1999) A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J. Biol. Chem. 274, 14382–14391 [DOI] [PMC free article] [PubMed] [Google Scholar]