

Background: SALL4 plays important roles in hematopoiesis and leukemogenesis, including at the stem cell level.
Results: SALL4 blocks ATRA-mediated differentiation through directly suppressing RA receptor α signaling.
Conclusion: SALL4 plays an unfavorable role in ATRA-based regimes.
Significance: SALL4 contributes to AML pathogenesis by inhibiting differentiation stimuli, reinforcing its role as a therapeutic target in leukemia.
Keywords: Hematopoietic Stem Cells, Histone Demethylase, Stem Cells, Transcription Corepressor, Tumor Therapy
Abstract
All-trans retinoic acid (ATRA) is a differentiation agent that revolutionized the treatment of acute promyelocytic leukemia. However, it has not been useful for other types of acute myeloid leukemia (AML). Here we explored the effect of SALL4, a stem cell factor, on ATRA-induced AML differentiation in both ATRA-sensitive and ATRA-resistant AML cells. Aberrant SALL4 expression has been found in nearly all human AML cases, whereas, in normal bone marrow and peripheral blood cells, its expression is only restricted to hematopoietic stem/progenitor cells. We reason that, in AMLs, SALL4 activation may prevent cell differentiation and/or protect self-renewal that is seen in normal hematopoietic stem/progenitor cells. Indeed, our studies show that ATRA-mediated myeloid differentiation can be largely blocked by exogenous expression of SALL4, whereas ATRA plus SALL4 knockdown causes significantly increased AML differentiation and cell death. Mechanistic studies indicate that SALL4 directly associates with retinoic acid receptor α and modulates ATRA target gene expression. SALL4 is shown to recruit lysine-specific histone demethylase 1 (LSD1) to target genes and alter the histone methylation status. Furthermore, coinhibition of LSD1 and SALL4 plus ATRA treatment exhibited the strongest anti-AML effect. These findings suggest that SALL4 plays an unfavorable role in ATRA-based regimes, highlighting an important aspect of leukemia therapy.
Introduction
SALL4 is a zinc finger transcription factor and is essential for embryonic development (1, 2). We and others have reported previously that SALL4 maintains the properties of ES cells by interacting with the core factors Oct4 and Nanog (3, 4). ES cells deprived of SALL4 expression differentiate spontaneously and start expressing trophectoderm markers (5). In extraembryonic endoderm (XEN)-derived ES cell-like cells, depletion of Sall4 also disrupts self-renewal and induces differentiation (5). SALL4 is also a potent tissue stem cell factor. In normal bone marrow, it is expressed in hematopoietic stem/progenitor cells but decreased in mature blood elements. Moreover, although down-regulation of SALL4 in hematopoietic stem/progenitor cells leads to loss of bone marrow stem cell markers, forced SALL4 overexpression causes drastic ex vivo cell expansion (6–8). These studies suggest a critical role for SALL4 in hematopoiesis, mediating both differentiation and self-renewal.
SALL4 could be one of a few genes that bridge the unique properties of stem cells and malignancies. Although down-regulated or absent in most adult tissues, it is highly expressed in various human tumors, including liver cancer, breast cancer, lung cancer, ovarian cancer, gastric cancer, colorectal cancer, Wilms tumor, and germ cell tumors as well as leukemia such as AML,2 B-acute lymphoblastic leukemia, and chronic myeloid leukemia (for a review, see Ref. 9). Moreover, SALL4 expression was enriched in the side population of tumor cells, implicating SALL4 in cancer initiation and drug resistance (10). In animal studies, SALL4 transgenic mice exhibit myelodysplasic syndrome-like features and AML transformation. These mice display an increased hematopoietic stem/progenitor cell population and increased serial replating potential (11), indicating the presence of leukemia-initiating cells.
More recently, several studies support a role for SALL4 in non-hematopoietic tissue development, tumorigenesis, and tumor growth. For instance, SALL4 in the adult liver is expressed in hepatic stem cells and hepatoblasts but absent in later-stage parenchymal cells (12). SALL4 is also aberrantly expressed in hepatic cancers, and although overexpression of SALL4 enhances cell proliferation, SALL4 knockdown decreases cell growth. Moreover, its expression levels are inversely correlated with hepatocytic differentiation markers (13). These findings further highlight SALL4 roles in not only protecting tumor proliferation but also inhibiting their differentiation. On the basis of all these studies, we deduce that, in human AMLs, constant SALL4 activation may prevent blast cells from differentiating and/or protect their self-renewal. Indeed, in our earlier study, we have demonstrated that loss of SALL4 in leukemic cells leads to extensive apoptosis (14).
By targeting maturation arrest instead of rapidly dividing cells, selectively inducing tumor cell differentiation may serve as an attractive alternative approach to cytotoxic regimens, which should also improve the immune status of patients (15). In this regard, ATRA has been used successfully in the treatment of APL, a distinct subtype of AML. ATRA alone can induce complete hematologic remission in nearly all APL patients and does not cause bone marrow hypoplasia or exacerbation of the frequently occurring fatal hemorrhagic syndromes (16). Unfortunately, despite the high clinical significance achieved in APL, the effects of ATRA in other types of AML and other leukemia and cancers are still largely unsatisfactory. The underlying reasons why ATRA fails to induce effective differentiation in other AMLs and cancers remain elusive.
APL cells have been characterized by the expression of a unique PML/RARα fusion protein that is absent in other AMLs. It is generally thought that ATRA-induced differentiation in APL cells is mediated by RARα and the PML-RARα fusion protein, whereas, in other AMLs, differentiation is mediated by RARα (17). Because leukemia cells share many common properties and regulatory machineries with normal stem cells and because the stem cell protein SALL4 is highly expressed in both populations, we hypothesize that SALL4 may play a counterregulatory role against the ATRA pathway and maintain cells in their undifferentiated status. To test this, in this study, we utilized both doxycycline-inducible SALL4 knockdown and overexpression strategies and tested APL and non-APL AML cells with or without ATRA treatment. Additionally, the mechanisms underlying the SALL4 and ATRA regulatory network in modulating AML phenotypes were also explored.
MATERIALS AND METHODS
Plasmids, Cell Culture, Transfections, and Luciferase Assays
Plasmids expressing HA-tagged SALL4 isoforms and shortened mutants have been described elsewhere (18). FLAG-RARα was purchased from Addgene. The TripZ-7410-, TripZ-7410–7412-, and TripZ-7410-SALL4-inducible lentiviral vectors were constructed by inserting each shRNA oligos or SALL4B cDNA into the TripZ vector (Thermo Scientific) between the EcoRI and Xhol sites. The RARβ-Luc reporter was provided by Dr. Luciano Croce (19). For cell culture, HEK293, 293T, HL60, and NB4 cells were purchased from the ATCC and cultured as described previously (14). TEX cells were grown as described elsewhere (20). Primary AML blast samples were obtained from the Marrow Stem Cell Laboratory and denoted AML1 to AML4. The study was approved by the Institutional Review Board of Stony Brook University Medical Center.
Lentivirus Production and Quantitative real-time PCR (qRT-PCR)
Lentiviruses expressing SALL4B or different shRNA oligos were produced as described previously (6, 14). Shed virus was harvested 48 h after transfection and used for transduction. Stable knockdown or overexpression clones were obtained under puromycin (0.5∼5 μg/ml) selection for 7 days. RNA preparation and qRT-PCR were performed as described previously (14). FAST qPCR Mastermix and SYBR Green reagents were purchased from Invitrogen. Tranylcypromine (TCP) and doxycycline (Dox) were purchased from Sigma.
Western Blotting and Coimmunoprecipitation (Co-IP)
These assays were conducted as described previously (18), and each assay was repeated individually at least three times. LSD1 antibody was purchased from Cell Signaling Technology. PML, RARα, and β-actin antibodies were purchased from Abcam, and SALL4 antibody was purchased from Abnova (Taipei, Taiwan). When used for Western blotting, the antibodies were diluted according to the instructions of the manufacturer.
Flow Cytometry
Flow cytometry was conducted with phycoerythrin- or allophycocyanin-conjugated antibody to CD11b and CD14 and FITC-conjugated antibody to annexin-V (all from BioLegend) and analyzed on a FACSCalibur flow cytometer (Stony Brook University Flow Core Facility).
Immunofluorescence Staining
Cells were fixed with 4% paraformaldehyde at room temperature for 10 min, permeabilized with 0.1% saponin for 5 min, blocked with 20% donkey serum for 1 h, and then incubated with primary antibodies for 30 min. Cells were washed with phosphate-buffered saline and then stained with mouse Alexa Fluor 488 and rabbit Alexa Fluor 594. For immunofluorescent analysis, we used a Nikon Eclipse 90i microscope equipped with ×63 oil immersion objectives. Images were captured with a DS-Fil camera.
Analysis of Myeloid Differentiation
Superoxide anion levels in tested cells were evaluated using the Lumimax superoxide anion detection kit (Sigma) following the instructions of the manufacturer. For morphological analysis of myeloid cell differentiation, we prepared cytospins using a Cytospin 3 centrifuge and stained the cytospin slides with May-Grünwald-Giemsa (Sigma). Cellular morphology was examined using a Nikon Eclipse 90i microscope.
ChIP
Chromatin was isolated by using a chromatin extraction kit (Epigentek). ChIP assays were conducted with a one-step ChIP kit (Epigentek). ChIP-qualified antibodies were purchased from Abcam. ChIP enrichment results were evaluated by PCR assays. The PCR products were separated by agarose gel electrophoresis, DNA band intensities were quantified, and the data were analyzed by normalization to the corresponding input values. Primers have been described elsewhere (19, 21).
Xenograft Model
Eight-week-old female nonobese diabetic/SCID mice were sublethally irradiated with 225 centigray from a 137 Cs γ irradiator and injected subcutaneously with tested cells. All mice in the same experiment received an equal numbers of cells (3–4 × 106 AML cells). Animal experiments were preapproved by the Stony Brook University Institutional Animal Care and Use Committee.
RESULTS
ATRA-induced AML Differentiation Is Largely Blocked by Forced Expression of SALL4
We have demonstrated previously that SALL4 maintains leukemic cell growth by repressing important apoptosis-inducing genes. Here we examined the potential of SALL4 in protecting AML cells against differentiation stimuli. In an initial study, we tested the effect of SALL4 on cellular differentiation and growth arrest in the presence of ATRA treatment. We generated a Dox-inducible SALL4 (B isoform)-expressing lentivirus using the TripZ vector, and the virus-transduced cell lines were established through puromycin selection. To better evaluate the effect of SALL4, we started with a low cell number culture (20,000 cells/ml) using two AML cell lines, NB4 and HL60. Dox-mediated SALL4 overexpression was validated by qRT-PCR and Western blotting (Fig. 1A). As shown in Fig. 1B, treatment of ATRA at 0.1 or 1 μm induced marked cell differentiation and cell death in a time-dependent manner. However, when the cells were cotreated with Dox to induce SALL4 overexpression, these ATRA effects were largely blocked, as demonstrated by substantially increased cell survival and reduced irregular cell appearance under the microscope. We then performed flow cytometry assays using CD11b and CD14 myeloid differentiation markers to quantify the observed blocking effect (22). SALL4 overexpression reversed the ATRA-induced differentiation by up to ∼60–70%, and this effect was also corelated with the SALL4 overexpression levels (Fig. 1C). Therefore, SALL4 overexpression protects cells from ATRA-induced cell differentiation and apoptosis.
FIGURE 1.

ATRA-induced differentiation is blocked by overexpression of SALL4. A, the indicated cell lines were transduced with lentivirus expressing TripZ-SALL4B. SALL4 expression levels following Dox treatment at different dosages were evaluated. Top panel, qRT-PCR assays showing SALL4 mRNA expression levels normalized by GAPDH housekeeping gene expression. Results represent the mean ± S.D. of three experiments. Bottom panel, Western blot results were normalized by β-actin as a loading control (Ctrl). B, 2 × 104 HL60 or NB4 cells were treated with ATRA (1 and 0.1 μm, respectively). Dox was added to cells to induce SALL4 overexpression (OE), and cell morphology was evaluated under the bright field of an AMG EVOS fl microscope (×20) on different days. C, flow cytometry assays using the indicated antibodies were performed after ATRA-induced cell differentiation with or without Dox-mediated SALL4 overexpression. Representative dot plots (measured at day 3 of treatment) and percentages of positive cells for the indicated antigens are shown. Data were analyzed by Student's t test (ATRA versus ATRA+Dox; *, p < 0.01). Values represent the mean ± S.D. of at least four independent experiments.
Reduction of SALL4 Enhances ATRA Activity in Vitro and in Vivo
To further elucidate SALL4 roles in modulating AML differentiation in the presence or absence of ATRA, we next performed an opposite loss of function study using a Dox-inducible SALL4 shRNA-expressing lentivirus. We adopted this inducible system because it permits reversible, controlled gene silencing and enables transductions of primary and non-dividing cells. The shRNA sequences, including 7410 and 7412, have been validated in previous studies (14). As seen in Fig. 2A, SALL4 in HL60 or NB4 cells was not significantly knocked down by Dox-mediated scramble shRNA, whereas 7410 expression substantially repressed SALL4 levels by up to ∼90%, which is closely correlated with Dox dosages. 7412 showed similar effects, and the expression levels of housekeeping genes were not affected by either shRNA (data not shown). Therefore, this shRNA-mediated SALL4 knockdown was specific. In addition, Dox-mediated SALL4 knockdown was confirmed in another AML cell line, TEX (ATRA-resistant), and two primarily isolated patient samples by Western blotting. In the three tested cell lines, an ∼85%, ∼83% and ∼78% reduction of SALL4 levels (qRT-PCR evaluation) caused approximately ∼7.7-, ∼6.9-, and ∼3.5-fold up-regulation of CD11b-expressing cells, respectively (Fig. 2B). Moreover, the SALL4 reduction-induced differentiation was accompanied by a corresponding increase in apoptotic marker annexin-V levels (Fig. 2C and data not shown), which is consistent with our previous findings using retrovirus-mediated SALL4 knockdown. Next we explored the therapeutic potential of whether SALL4 knockdown and ATRA may synergistically induce AML differentiation. We studied both ATRA-responsive and ATRA-resistant AML cell lines as well as primarily isolated patient samples. ATRA-resistant TEX cells mimic the features of primary AML and of leukemia-initiating cells and are more than 90% CD34 positive (20). These cells were infected with TRIPZ-SALL4shRNA lentivirus and selected with puromycin. In an initial study using HL60 cells, cotreatment of ATRA and Dox for 3 days already resulted in an increasing number of “differentiated” cells, as distinguished by their elongated, spindle, or irregular polygon shapes in culture (Fig. 2A). Further flow cytometry assays confirmed that, in these cells as well as other AML cell types, cotreatment of ATRA and Dox fundamentally increased the CD11b-expressing myeloid cells that were significantly higher than either treatment alone (Fig. 2, B and C). In double drug-treated cells, there were also markedly increased annexin-V expression levels, assumingly because of the direct apoptosis-inducing effect of SALL4 knockdown and enhanced post-maturation cell death.
FIGURE 2.

SALL4 knockdown enhances ATRA-mediated differentiation. A, HL60 cells infected with TripZ shRNA 7410 virus were treated with either ATRA (1 μm), Dox (1 μg/ml), or both for 60 h. Dox-induced TripZ-TurboRFP expression in cells was monitored, and representative pictures were taken using the EVOS fl microscope (×20). Dox-induced SALL4 knockdown in HL60 and NB4 cells was determined by qRT-PCR normalized to GAPDH levels. Error bars represent mean ± S.D. of three independent experiments. Shown is a Western blot analysis revealing SALL4 depletion after 3 days of Dox (1.2 μg/ml) induction in the TEX cell line (representing at least three repeats) and patient-derived primary AML cells. Ctrl, control. B, flow cytometry assays were performed for the indicated cell lines after ATRA induced cell differentiation with or without Dox (shown as micrograms per milliliter)-mediated SALL4 knockdown. Error bars represent mean ± S.D. of four independent experiments. C, representative flow cytometry assays showing CD11b and annexin-V expression levels in differentially treated cell lines or primary patient (AML-M2) blast samples. D, the cell morphology of the indicated cells was analyzed by May-Grünwald-Giemsa staining after treatment with the indicated drugs for 60 h. Pictures were taken through a Nikon Eclipse 90i microscope (×60). E, HL60 and NB4 cells were treated with the indicated drugs and analyzed with a superoxide anion production kit. The kinetic curve shows luminescence changes when measured with a SpectraMax® M3 microplate reader. F, ATRA and SALL4 knockdown suppress AML tumor growth in vivo. Drug treatments were started 10 days after tumor cell injection. Palpable tumors were present for the established tumor model before initiating drug treatment. Dox (added to 250 ml of drinking water at a final concentration of 1 mg/ml), ATRA (15 mg/kg), or vehicle (30 ml of dimethyl sulfoxide and 70 ml of water) were injected intraperitoneally twice per day for 1 week, followed by once a day for 1 week and every other day for the third week. Dox was fed to mice for the entire 3 weeks. Top panel, the results are representative of the samples in each group. Bottom panel, tumor volume growth curves are expressed as the mean ± S.E. (n = 4 per group). Data were analyzed by Student's t test.
We next performed Wright-Giemsa staining to verify the microscopic and flow cytometry findings. Compared with dimethyl sulfoxide-treated controls, ATRA-treated cells had modestly decreased nucleus to cytoplasmic ratios, whereas ATRA and Dox double-treated cells had a more mature morphology, with an increased cytoplasmic to nuclear ratio and obvious nuclear segmentation (Fig. 2D). In parallel with the morphologic findings, cell myeloid maturation was also analyzed by measurement of superoxide anion status. One feature of myeloid maturation is the production of superoxide anion used by the mature myeloid cell to kill ingested microorganisms (23). As seen in Fig. 2E, in ATRA- or Dox-treated HL60 cells, there was enhanced superoxide production compared with dimethyl sulfoxide controls (up to 93-fold and 3.5-fold, respectively), whereas cotreatment of ATRA and Dox in these cells further increased superoxide anion levels up to 400-fold. The significantly enhanced superoxide anion production by double-drug treatment was also observed in NB4 cells. Taken together, the results from the studies mentioned above demonstrate that SALL4 knockdown and ATRA synergistically induce AML myeloid differentiation.
To further validate the enhanced anti-AML effects in vivo, we examined leukemic cell growth in a xenograft mouse model. HL60 cells infected with vector control or TripZ-SALL4 shRNA lentivirus were injected into the flanks of nonobese diabetic/SCID mice. Compared with the dimethyl sulfoxide control, ATRA or Dox administration resulted in a moderate to remarkable tumor inhibition (∼22% reduction for ATRA and 53% reduction for Dox), whereas simultaneous treatment of both drugs further inhibited tumor growth by up to 79% (Fig. 2F, ATRA+Dox versus ATRA or Dox group, p < 0.01). Notably, there was no remarkable progressive weight loss observed in either mono or double drug-treated groups during the experiment. Therefore, ATRA and SALL4 inhibition not only induce AML differentiation and maturation in culture but also synergistically impair tumor growth in vivo.
SALL4 Affects the Expression Levels of ATRA Target Genes and Physically Interacts with RARα
Having confirmed the synergism of SALL4 reduction and ATRA in impairing AML activity, we sought to explore the underlying molecular mechanisms. We started with an ATRA target gene, RARβ (which contains the RA-responsive element), and tested its promoter activity using HEK293 cells that were transfected with RARβ-Luc plasmid and TripZ-SALL4 shRNA. As seen in Fig. 3A, ATRA administration to these cells increased RARβ promoter activity about 4.1-fold. However, when cells were cotreated with Dox to knock down endogenous SALL4 expression, RARβ promoter activity was further increased 1.5- to 2-fold, depending on the Dox dosage. Therefore, these data suggest that SALL4 blocks ATRA activity in regulating its target gene expression. To verify this notion, we checked a group of well studied ATRA target genes in HL60 cells (19, 24). Interestingly, although ATRA administration for 36 h dramatically induced mRNA expression levels of RARβ, CYP26, and ALOX5AP, Dox-mediated SALL4 knockdown also resulted in a significant up-regulation of these gene expressions in a dose-dependent fashion (Fig. 3B). In addition, the myeloid regulators ID2 and C/EBPα (CCAAT/enhancer binding protein, α), which were also identified as SALL4-regulated genes (21, 25), were also up-regulated. Furthermore, the increased expression of these genes was further enhanced when cotreated with ATRA at different doses (Fig. 3B). These data suggest that SALL4 knockdown enhances ATRA-mediated AML differentiation, at least in part, via ATRA target pathways. Interestingly, ATRA also directly down-regulated SALL4 expression in a time-dependent fashion, as demonstrated by both qRT-PCR and Western blot assays (Figs. 3B and 5A). We have demonstrated previously that SALL4 in hematopoietic precursor cells represses important differentiation and apoptosis pathways. From these studies, it is likely that ATRA also affects AML phenotypes by modulating important stem cell factors such as SALL4.
FIGURE 3.

SALL4 affects ATRA target gene expression. A, in HEK293 cells, the RARβ promoter activity under different treatments was measured 24 h after plasmid transfection and Dox administration (shown as micrograms per milliliter). Relative luciferase units (RLU) are expressed as the mean ± S.E. (n = 3). Ctrl, control. B, the effects of SALL4 knockdown on the expression of ATRA-target genes. HL60 cells were treated with the indicated drugs for 30 h. The relative mRNA levels of the indicated genes were determined by qRT-PCR and quantified on the basis of three independent experiments. Error bars denote S.D. *, p < 0.05; **, p < 0.01 versus ATRA only.
FIGURE 5.

ATRA dissociates SALL4 and LSD1 from RARα. A, HL60 cells were treated with 1 μm ATRA for 24 and 48 h. Then, cell lysates were either Western-blotted (WB, top panel, 20 μg input in each lane) or immunoprecipitated with SALL4 and RARa antibody (bottom panel, 200 μg input in each IP), respectively, and Western-blotted with indicated antibodies. Representative results are shown, and experiments were repeated independently at least three times. IB, immunoblot. B, ChIP analysis. HL60 cells were treated with the indicated drugs for 60 h. Chromatin was extracted and immunoprecipitated with the indicated antibodies, followed by DNA elution, PCR, and agarose gel electrophoresis to analyze the tested RA-responsive element regions or identified SALL4 binding sequences. Band intensities from the agarose gel were quantified and normalized with input chromatin. Values are averages of four separate experiments. *, p < 0.05; **, p < 0.01.
ATRA exerts its effects through specific nuclear receptors (RARα and RXRs). Studies of the molecular mechanism that blocks differentiation in non-APL AML cells suggest a diminished expression of RARα, and RARα restoration can induce differentiation of human AML cells (26). On the basis of the abovementioned ATRA target gene expression findings and the fact that either RARα or SALL4 associates with important epigenetic factors such as DNA methyltransferases, lysine-specific demethylase 1 (LSD1), and histone deacetylases, we next explored the potential association of SALL4 with RARα. Initially utilizing HEK293 cells overexpressing FLAG-RARα and HA-tagged SALL4 isoforms or different deletion mutants, we observed that SALL4 and RARα physically interact by co-IP) assays (Fig. 4A). In a separate study, we further detected a physical interaction of endogenous SALL4 and RARα in HL60 cells (Fig. 4B). In addition to the co-IP experiments, we also performed immunofluorescence staining for SALL4 and RARα, and, as shown in Fig. 4C, their proteins are clearly colocalized in HL60 cell nuclei.
FIGURE 4.

SALL4 interacts with RARα. A, SALL4 isoforms and mutant SALL4 deletions are shown schematically (left panel). Hemagglutinin-tagged SALL4 isoforms or mutants were transfected into HEK293 cells. IP was performed using the Dynabeads protein G kit and immunoblotted with anti-RARα antibody (right panel). B, HL60 whole cell lysates were immunoprecipitated and tested with the indicated antibodies (Ab). IgG was used as a control. IB, immunoblot. C, the localization and expression of SALL4 and RARα proteins in HL60 cells. The arrowheads indicate colocalization of the two proteins in the nucleus. The results were detected by immunofluorescence staining, and representative images were taken using a Nikon Eclipse 90i microscope (×60).
ATRA Leads to Dissociation of SALL4 and Epigenetic Corepressors from RARα
SALL4 has been shown to dynamically recruit epigenetic modifiers to target genes depending on the cell context. Because SALL4 directly interacts with RARα, we deduce that RARα, SALL4, and its associated epifactors may form a complex, whereas ATRA treatment will alter the complex structure and affect target gene expression. To test this probability, we first performed a series of co-IP assays and examined each protein interaction in HL60 cells in the presence or absence of ATRA treatment. LSD1 is included in this study because it has been shown to contribute to SALL4-repressive effects, and inhibition of LSD1 enhances ATRA-mediated AML differentiation. As shown in Fig. 5A, following treatment of ATRA for 24–48 h, SALL4 and LSD1 were increasingly dissociated from RARα, as judged by the relative density of the Western bands normalized to an internal control. To further confirm that RARα, SALL4, and LSD1 bind dynamically to the same ATRA-target gene promoters and that SALL4 and LSD1 dissociate from these genes following ATRA treatment, we next performed ChIP assays. As seen in Fig. 5B, endogenous SALL4 and LSD1 were recruited to the RARβ, ID2, and CYP26 promoters in the absence of ATRA (lane 1, second and third panels). However, when treated with ATRA, SALL4 and LSD1 were released from the same promoter regions (Fig. 5B, lanes 1 and 2, the second and third panels). Because LSD1 is able to convert dimethylated histone H3 at lysine 4 (H3K4) to mono- and unmethylated H3K4, we then examined the H3K4 methylation status of the tested promoters. In the ATRA- or Dox-treated AML samples, corresponding to decreased LSD1 and SALL4 binding at the tested promoters, there were increased H3K4 dimethylation levels, albeit to different extents (Fig. 5B, lanes 1∼3, fourth panel). Interestingly, in ATRA and Dox double-treated samples, the dimethylated H3K4 levels at these promoters were increased further (Fig. 5B, lane 4, fourth panel). Therefore, these data demonstrate that SALL4 forms a complex with RARα and LSD1 on the same ATRA target genes to modulate their expression, whereas treatment of ATRA and SALL4 knockdown stimulates active histone modifications and up-regulates downstream differentiation gene expression.
The Anti-leukemia Effect of ATRA and SALL4 Knockdown Is Enhanced Further by Coinhibition of LSD1
In a couple of recent studies, it has been shown that inhibition of LSD1 by either gene-specific knockdown or using enzymatic inhibitors such as TCP can reactivate ATRA sensitivity in ATRA-resistant AMLs and diminish the engraftment of primary AML samples in vivo (20, 27). Because LSD1 acts as a coregulator for SALL4-repressive functions, we compared the anti-AML effects of ATRA/TCP cotreatment with that of ATRA plus SALL4 knockdown and then investigated the overall effect of coinhibiting SALL4, LSD1, plus ATRA treatment in inducing AML differentiation and apoptosis. We tested the same AML cell lines and primary AML patient samples as we did previously. The AML differentiation and apoptosis-inducing effects of ATRA/TCP cotreatment versus ATRA plus SALL4 knockdown are generally comparable in most cases, whereas the latter treatment tends to induce more severe apoptosis. Interestingly, compared with mono and dual combination (ATRA+Dox) treatments, triple drug (ATRA+Dox+TCP) treatment caused further increased cellular differentiation, as judged by myeloid marker expression levels, cell morphological appearances, and their superoxide anion production status. In addition, the triple combination inhibited the proliferation of HL60 leukemic xenografts by nearly 91% compared with the control-treated groups in vivo (Fig. 6). Again, none of the treated mice showed signs of illness or significant weight loss during the experiments.
FIGURE 6.

The synergistic effects of ATRA and SALL4 knockdown are further enhanced by coinhibition of LSD1. These experiments were conducted in parallel with mono or double drug treatments (the results are shown in Fig. 2), and the results can therefore be compared. A, HL60 and TEX cells were treated with ATRA+Dox+TCP for 60 h, and CD11b levels were detected by flow cytometry assays. B, representative flow cytometry profiles showing the CD11b and annexin-V expression levels in primary AML cells obtained from patient AML1. C, cell morphology of the indicated cells by May-Grünwald-Giemsa staining was evaluated after ATRA+Dox+TCP treatments. Representative images were taken using a Nikon Eclipse 90i microscope (×60). D, dose-response (for ATRA) and time course studies showing superoxide anion production in ATRA+Dox+TCP-treated HL60 cells. Ctrl, control. E, triple drug treatment significantly decreased tumor growth in xenograft studies. Tumor volume growth curves are expressed as the mean ± S.E. (n = 3 in the control group, n = 4 per other group). **, p < 0.01 versus A+T or A+D group. A, ATRA; T, TCA; D, Dox.
DISCUSSION
The use of ATRA has introduced the concept of differentiation therapy. However, ATRA has been largely ineffective in non-APL subtypes of AML. Therefore, it is important to identify the cellular signaling pathways that are aberrantly activated and act to inhibit myeloid differentiation. We have shown previously that SALL4 plays a key role in maintaining leukemic cell proliferation. Here we addressed its role in modulating leukemic differentiation and its therapeutic potential in ATRA-based differentiation therapy.
In APL, the PML-RARα fusion protein causes a strong dominant-negative effect, resulting in a blockade of RARα-induced differentiation. The RARα signaling pathway, which is important for myeloid differentiation, can also be inhibited by other binding partners in non-APL AMLs. Our data support that SALL4 overexpression in leukemia interferes with RA-RARα signaling, causing inhibition of differentiation and, therefore, contributing to the leukemic phenotype. For therapeutic potential studies, we first show that SALL4 expression levels tightly associate with ATRA effects (Figs. 1 and 2). When SALL4 is up-regulated, the anti-AML effects of ATRA were largely blocked, whereas a reduction of endogenous SALL4 substantially improves these ATRA effects. In fact, we also show that ATRA directly down-regulates SALL4 expression by both Western blotting and mRNA quantitation in dose-response and time course studies, suggesting that SALL4 is a downstream target of ATRA (Figs. 3 and 5). This assumption is supported by a recent study using embryonal day 8.5 mouse embryos. When treated with RA, SALL4 was identified as one of the most severely down-regulated genes in the presomitic mesoderm (28). In our ChIP assays, ATRA treatment also led to decreased SALL4 binding to target genes and dissociation of SALL4 and epigenetic corepressors from RARα (Fig. 5). Therefore, ATRA treatment may also affect AML phenotypes by modulating important stem cell factors such as SALL4.
In addressing the molecular mechanisms of the SALL4 inhibitory effect on ATRA activity, we found that SALL4 forms a complex with RARα and LSD1 but dissociated from this complex in the presence of ATRA treatment. In the absence of an agonist such as ATRA, RA receptors bind to RXR and recruit the nuclear receptor corepressor or silencing mediator for retinoid and thyroid hormone receptors along with epifactors such as histone deacetylases or DNA methyltransferases to repress gene expression (29, 30). A recent study suggests that LSD1 also contributes to RARα-mediated transcription via repressive histone H3 methylation (31). Because SALL4 directly interacts with RARα and also dynamically recruits these epifactors, it would inhibit AML differentiation via suppressing the RARα pathway. At physiological concentrations (10−9∼10−8 m), ATRA binds to the RARα-RXR heterodimer and induces dissociation of corepressors interchangeably with association of the coactivator complex, including CBP/p300, causing chromatin decondensation and activation of transcription (29). It is likely that sustained expression of repressive oncoproteins (such as SALL4) in leukemia may interfere with this switch and block differentiation gene transcription. As shown in Fig. 4, although the corepressor complex containing SALL4 and LSD1 gradually dissociates from RARα on the RA-responsive element, SALL4 remains localized on the tested promoters, even with pharmacological doses of ATRA.
SALL4 is expressed in both APL and non-APL AMLs. In exploring how SALL4 interferes differentially with ATRA-mediated pathways in these cells, we noticed that, although in NB4 (AML-M3) cells ATRA down-regulates SALL4 expression rapidly to a background level, in HL60 (AML-M2) cells, this ATRA-mediated SALL4 reduction was slower and moderate (Fig. 7A). The next question will be how SALL4 is eventually degraded or recycled via unknown pathways in these different cell types. On the other hand, although SALL4 protein interacts with RARα (∼52 kDa) in both cell types, we failed to detect an interaction of SALL4 with the PML-RARα fusion protein (∼130 kDa) in NB4 APL cells (Fig. 7B). This raises the possibility that, by avoiding interference of repressive factors such as SALL4, the PML-RARα protein in APL may allow “easier” access and binding of ATRA and, consequently, a more efficient switch from corepressor to coactivator interaction, which eventually leads to clinically meaningful significance (Fig. 7C). However, further extensive mechanistic studies are needed to address this assumption.
FIGURE 7.
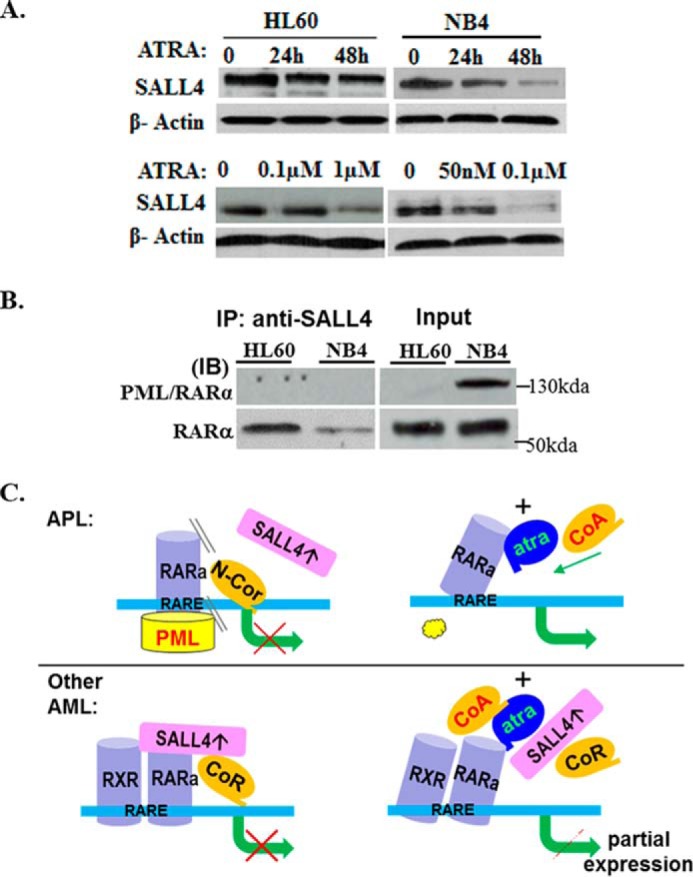
SALL4 differentially interferes with the ATRA-RA pathways in APL and non-APL AML cells. A, comparison of ALL4 protein expression in HL60 and NB4 cells after ATRA treatment at different time points or with different dosages by Western blotting. β-Actin was used as an internal loading control. B, co-IP experiments to detect potential SALL4 and PML-RARα interaction in NB4 cells. HL60 and NB4 cell nuclear extracts (200 μg) were immunoprecipitated with IP-grade SALL4 antibody and Western-blotted with anti-PML/RARα and anti-RARα antibodies, respectively. The experiments were repeated at least twice, and similar results were obtained. IB, immunoblot. C, a tentative model for the molecular interaction of SALL4 with RARα/RXR and PML-RARα in APL and non-APL AMLs. See text for details.
In testing the therapeutic potentials, we examined the anti-AML effects by combined treatments of ATRA plus coinhibition of SALL4 and LSD1. It is worthwhile, however, to note that, except for LSD1, coinhibition of other epifactors (such as DNA methyltransferase and histone deacetylase) should also generate better anti-AML outcomes because these factors are binding partners of SALL4 and key components of the nuclear receptor corepressor complex. The efficacy of each different combination in inducing AML differentiation and apoptosis will be addressed in further functional and mechanistic studies.
In conclusion, we discovered an unknown aspect of SALL4 in leukemia. The overexpression of SALL4 in AML patients might be a critical event of the multistep process of leukemogenesis, and sustained expression of SALL4 plays an important role in modulating leukemia phenotypes. This will have potential clinical significance in combating non-APL AMLs and also other leukemia and cancers.
Acknowledgments
We thank Dr. Luciano Croce for the RARβ-Luc plasmid and Dr. John Dick and Cheryl Szombati at the University Health Network for preparing the TEX cell line.
Note Added in Proof
The data shown in Fig. 5A were not correct in the version of this article that was published on March 3, 2015 as a Paper in Press. Specifically, the panels shown for RARα antibody probing of RARα and SALL4 immunoprecipitates were reversed. This error has been corrected. In addition, Lai-Han Leung was originally listed as Elaine Leung in the version of the article published on March 3, 2015 as a Paper in Press.
This work was supported by American Cancer Society Research Scholar Grant RSG-12-216-01-LIB (to J. Y.) and New York State Stem Cell Science Grants C0281148 (to Y. M.) and C026716 (to the Stony Brook Stem Cell Center).
- AML
- acute myeloid leukemia
- ATRA
- All-trans retinoic acid
- APL
- acute promyelocytic leukemia
- PML
- promyelocytic leukemia
- RARα
- RA receptor α
- RXR
- retinoid X receptor
- qRT-PCR
- quantitative RT-PCR
- TCP
- tranylcypromine
- Dox
- doxycycline
- IP
- immunoprecipitation
- H3K4
- histone H3 at lysine 4.
REFERENCES
- 1. Kohlhase J., Schuh R., Dowe G., Kühnlein R. P., Jäckle H., Schroeder B., Schulz-Schaeffer W., Kretzschmar H. A., Köhler A., Müller U., Raab-Vetter M., Burkhardt E., Engel W., Stick R. (1996) Isolation, characterization, and organ-specific expression of two novel human zinc finger genes related to the Drosophila gene spalt. Genomics 38, 291–298 [DOI] [PubMed] [Google Scholar]
- 2. Al-Baradie R., Yamada K., St Hilaire C., Chan W. M., Andrews C., McIntosh N., Nakano M., Martonyi E. J., Raymond W. R., Okumura S., Okihiro M. M., Engle E. C. (2002) Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 71, 1195–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang J., Chai L., Fowles T. C., Alipio Z., Xu D., Fink L. M., Ward D. C., Ma Y. (2008) Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 105, 19756–19761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu Q., Chen X., Zhang J., Loh Y. H., Low T. Y., Zhang W., Zhang W., Sze S. K., Lim B., Ng H. H. (2006) Sall4 interacts with Nanog and co-occupies Nanog genomic sites in embryonic stem cells. J. Biol. Chem. 281, 24090–24094 [DOI] [PubMed] [Google Scholar]
- 5. Lim C. Y., Tam W. L., Zhang J., Ang H. S., Jia H., Lipovich L., Ng H. H., Wei C. L., Sung W. K., Robson P., Yang H., Lim B. (2008) Sall4 regulates distinct transcription circuitries in different blastocyst-derived stem cell lineages. Cell Stem Cell 3, 543–554 [DOI] [PubMed] [Google Scholar]
- 6. Yang J., Aguila J. R., Alipio Z., Lai R., Fink L. M., Ma Y. (2011) Enhanced self-renewal of hematopoietic stem/progenitor cells mediated by the stem cell gene Sall4. J. Hematol. Oncol. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang J., Liao W., Ma Y. (2012) Role of SALL4 in hematopoiesis. Curr. Opin. Hematol. 19, 287–291 [DOI] [PubMed] [Google Scholar]
- 8. Aguila J. R., Liao W., Yang J., Avila C., Hagag N., Senzel L., Ma Y. (2011) SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood 118, 576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang X., Yuan X., Zhu W., Qian H., Xu W. (2015) SALL4: An emerging cancer biomarker and target. Cancer Lett. 357, 55–62 [DOI] [PubMed] [Google Scholar]
- 10. Jeong H. W., Cui W., Yang Y., Lu J., He J., Li A., Song D., Guo Y., Liu B. H., Chai L. (2011) SALL4, a stem cell factor, affects the side population by regulation of the ATP-binding cassette drug transport genes. PloS ONE 6, e18372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma Y., Cui W., Yang J., Qu J., Di C., Amin H. M., Lai R., Ritz J., Krause D. S., Chai L. (2006) SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood 108, 2726–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oikawa T., Kamiya A., Kakinuma S., Zeniya M., Nishinakamura R., Tajiri H., Nakauchi H. (2009) Sall4 regulates cell fate decision in fetal hepatic stem/progenitor cells. Gastroenterology 136, 1000–1011 [DOI] [PubMed] [Google Scholar]
- 13. Zeng S. S., Yamashita T., Kondo M., Nio K., Hayashi T., Hara Y., Nomura Y., Yoshida M., Hayashi T., Oishi N., Ikeda H., Honda M., Kaneko S. (2014) The transcription factor SALL4 regulates stemness of EpCAM-positive hepatocellular carcinoma. J. Hepatol. 60, 127–134 [DOI] [PubMed] [Google Scholar]
- 14. Yang J., Chai L., Gao C., Fowles T. C., Alipio Z., Dang H., Xu D., Fink L. M., Ward D. C., Ma Y. (2008) SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood 112, 805–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown G., Hughes P. (2012) Retinoid differentiation therapy for common types of acute myeloid leukemia. Leuk. Res. Treatment 939021, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ozpolat B., Lopez-Berestein G., Adamson P., Fu C. J., Williams A. H. (2003) Pharmacokinetics of intravenously administered liposomal all-trans-retinoic acid (ATRA) and orally administered ATRA in healthy volunteers. J. Pharm. Pharm. Sci. 6, 292–301 [PubMed] [Google Scholar]
- 17. Launay S., Giannì M., Diomede L., Machesky L. M., Enouf J., Papp B. (2003) Enhancement of ATRA-induced cell differentiation by inhibition of calcium accumulation into the endoplasmic reticulum: cross-talk between RAR α and calcium-dependent signaling. Blood 101, 3220–3228 [DOI] [PubMed] [Google Scholar]
- 18. Yang J., Corsello T. R., Ma Y. (2012) Stem cell gene SALL4 suppresses transcription through recruitment of DNA methyltransferases. J. Biol. Chem. 287, 1996–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uribesalgo I., Buschbeck M., Gutiérrez A., Teichmann S., Demajo S., Kuebler B., Nomdedéu J. F., Martín-Caballero J., Roma G., Benitah S. A., Di Croce L. (2011) E-box-independent regulation of transcription and differentiation by MYC. Nat. Cell Biol. 13, 1443–1449 [DOI] [PubMed] [Google Scholar]
- 20. Schenk T., Chen W. C., Göllner S., Howell L., Jin L., Hebestreit K., Klein H. U., Popescu A. C., Burnett A., Mills K., Casero R. A., Jr., Marton L., Woster P., Minden M. D., Dugas M., Wang J. C., Dick J. E., Müller-Tidow C., Petrie K., Zelent A. (2012) Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu L., Souto J., Liao W., Jiang Y., Li Y., Nishinakamura R., Huang S., Rosengart T., Yang V. W., Schuster M., Ma Y., Yang J. (2013) Histone lysine-specific demethylase 1 (LSD1) protein is involved in Sal-like protein 4 (SALL4)-mediated transcriptional repression in hematopoietic stem cells. J. Biol. Chem. 288, 34719–34728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lubbert M., Kuendgen A. (2014) Combining DNA methyltransferase and histone deacetylase inhibition to treat acute myeloid leukemia/myelodysplastic syndrome: achievements and challenges. Cancer 121, 498–501 [DOI] [PubMed] [Google Scholar]
- 23. Stegmaier K., Corsello S. M., Ross K. N., Wong J. S., Deangelo D. J., Golub T. R. (2005) Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood 106, 2841–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones J. E., Wang L., Kropf P. L., Duan R., Johnson D. E. (2009) Src family kinase gene targets during myeloid differentiation: identification of the EGR-1 gene as a direct target. Leukemia 23, 1933–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nigten J., Breems-de Ridder M. C., Erpelinck-Verschueren C. A., Nikoloski G., van der Reijden B. A., van Wageningen S., van Hennik P. B., de Witte T., Löwenberg B., Jansen J. H. (2005) ID1 and ID2 are retinoic acid responsive genes and induce a G0/G1 accumulation in acute promyelocytic leukemia cells. Leukemia 19, 799–805 [DOI] [PubMed] [Google Scholar]
- 26. Glasow A., Prodromou N., Xu K., von Lindern M., Zelent A. (2005) Retinoids and myelomonocytic growth factors cooperatively activate RARA and induce human myeloid leukemia cell differentiation via MAP kinase pathways. Blood 105, 341–349 [DOI] [PubMed] [Google Scholar]
- 27. Sakamoto K., Imamura T., Yano M., Yoshida H., Fujiki A., Hirashima Y., Hosoi H. (2014) Sensitivity of MLL-rearranged AML cells to all-trans retinoic acid is associated with the level of H3K4me2 in the RARα promoter region. Blood Cancer J. 4, e205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Savory J. G., Edey C., Hess B., Mears A. J., Lohnes D. (2014) Identification of novel retinoic acid target genes. Dev. Biol. 395, 199–208 [DOI] [PubMed] [Google Scholar]
- 29. Altucci L., Gronemeyer H. (2001) The promise of retinoids to fight against cancer. Nat. Rev. Cancer 1, 181–193 [DOI] [PubMed] [Google Scholar]
- 30. Klein E. S., Wang J. W., Khalifa B., Gavigan S. A., Chandraratna R. A. (2000) Recruitment of nuclear receptor corepressor and coactivator to the retinoic acid receptor by retinoid ligands: influence of DNA-heterodimer interactions. J. Biol. Chem. 275, 19401–19408 [DOI] [PubMed] [Google Scholar]
- 31. Lee S. W., Cho Y. S., Na J. M., Park U. H., Kang M., Kim E. J., Um S. J. (2010) ASXL1 represses retinoic acid receptor-mediated transcription through associating with HP1 and LSD1. J. Biol. Chem. 285, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]