

Background: Sec22 assembles into protein complexes to catalyze membrane fusion events in the early secretory pathway.
Results: A cysteine cross-linking approach to probe Sec22 arrangements in cellular membranes revealed efficient homodimer formation.
Conclusion: Sec22 homodimers are dynamic intermediates necessary for efficient intracellular transport.
Significance: First evidence for Sec22 homodimers and suggests Sec22 promotes assembly of higher-order fusion complexes.
Keywords: Endoplasmic Reticulum (ER), Golgi, Membrane Fusion, Membrane Trafficking, SNARE Proteins
Abstract
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein complexes play essential roles in catalyzing intracellular membrane fusion events although the assembly pathway and molecular arrangement of SNARE complexes in membrane fusion reactions are not well understood. Here we monitored interactions of the R-SNARE protein Sec22 through a cysteine scanning approach and detected efficient formation of cross-linked Sec22 homodimers in cellular membranes when cysteine residues were positioned in the SNARE motif or C terminus of the transmembrane domain. When specific Sec22 cysteine derivatives are present on both donor COPII vesicles and acceptor Golgi membranes, the formation of disulfide cross-links provide clear readouts on trans- and cis-SNARE arrangements during this fusion event. The Sec22 transmembrane domain was required for efficient homodimer formation and for membrane fusion suggesting a functional role for Sec22 homodimers. We propose that Sec22 homodimers promote assembly of higher-order SNARE complexes to catalyze membrane fusion. Sec22 is also reported to function in macroautophagy and in formation of endoplasmic reticulum-plasma membrane contact sites therefore homodimer assembly may regulate Sec22 activity across a range of cellular processes.
Introduction
Intracellular membrane fusion events in the eukaryotic secretory pathway depend on a family of membrane-bound SNARE proteins to catalyze the bilayer fusion stage (1, 2). The SNARE family is characterized by a conserved 60–70-amino acid heptad repeat region, termed SNARE motif, adjacent to a transmembrane or lipidic anchor. Several lines of evidence indicate that specific sets of SNARE proteins form stable complexes through assembly of their SNARE motifs into parallel four-helix coiled-coil bundles. During membrane fusion, the regulated assembly of SNARE complexes from opposed membranes in trans is thought to drive bilayer fusion. Post-membrane fusion SNARE complexes in cis are disassembled through an ATP- and NSF-dependent reaction that recycles the SNARE machinery for subsequent rounds of membrane fusion (3).
Crystallographic analyses of the synaptic and endosomal SNARE complexes has provided critical insight into the molecular architecture of this fusion machinery (4–6). These structures indicate that SNARE motifs are assembled into stable four-helix bundles with multiple layers of hydrophobic residues contributing to the stable core of the SNARE complex. An ionic layer of residues or “zero layer” lies near the center of the four-helix bundle and appears to be a conserved feature of several distinct SNARE complexes (4). This layer typically consists of one arginine residue contributed by a synaptobrevin-like protein, or R-SNARE, and three glutamine residues contributed from three Q-SNARE helices (7). Although a great deal is known about the structural requirements for SNARE complex assembly, SNARE protein dynamics in the context of membrane fusion events remain poorly understood. There are specific limitations in approaches to detect short-lived intermediates during SNARE-dependent membrane fusion and to monitor low affinity protein-protein or protein-lipid interactions that are likely to drive SNARE complex assembly (8). To address these issues we are developing new probes and in vitro assays to monitor SNARE-catalyzed fusion in the yeast ER2-Golgi transport stage.
Genetic, biochemical, and morphological studies in yeast indicate that Sec22 (an R-SNARE) plus Sed5, Bos1, and Bet1 (three Q-SNAREs) assemble into a SNARE complex and catalyze fusion of ER-derived vesicles with Golgi membranes (9). A cell-free ER-Golgi transport assay reconstituted with yeast components has provided a tractable model to dissect mechanisms of SNARE-dependent membrane fusion (10). We have generated a molecular model for the ER-Golgi SNARE complex based on known SNARE structures (11) and used this information to probe the fusion mechanism. Insertion of cysteine pairs into contact regions of an ER-Golgi SNARE complex allows us to monitor specific sites of interaction during fusion of ER-derived vesicles with Golgi membranes. A Bet1-Sec22 cross-linked heterodimer was detected when ER-derived vesicles bearing a specific cysteine derivative of Bet1 were fused with Golgi membranes containing a cysteine derivative of Sec22. Formation of this cross-linked heterodimer was temperature and time dependent and required the same components known to function in this fusion event. Moreover, the rate of heterodimer formation mirrored the rate of Golgi-specific carbohydrate modification of a secretory protein substrate used in this cell-free fusion assay to measure luminal content mixing (11).
In this study, we report that in addition to the formation of Sec22-Bet1 heterodimers, Sec22 homodimers were readily detected when cysteine derivatives of Sec22 were examined under oxidizing conditions. Sec22 disulfide cross-linked homodimers were efficiently produced when cysteine residues were inserted into specific positions of the SNARE motif or the C-terminal transmembrane segment. Our studies indicate that Sec22 homodimer assembly is dynamic, functionally important, and can be used to report on distinct topological stages of SNARE catalyzed fusion. We propose that Sec22 and possibly other R-SNARE proteins connect SNARE complexes into higher ordered arrangements for efficient bilayer fusion.
EXPERIMENTAL PROCEDURES
Plasmids and Plasmid Construction
A yeast expression vector for a C-terminally 3HA-tagged Sec22 (pRS313-SEC22–3HA) was constructed by first introducing a silent AflII restriction site at position +637 in the SEC22 ORF on pRS313-SEC22 (12). A 3HA fragment (including additional stop codons) was obtained by PCR amplification of pFa6–3HA-His3MX6 (13) using 5′ and 3′ primers containing AflII restriction sites. Following AflII digestion and ligation, clones were screened to identify proper directional orientation of the 3HA tag. Additionally, the SEC22–3HA region containing both 5′ and 3′ UTR was subcloned into pRS316 (14) via the NotI and SalI restriction sites (pRS316-SEC22–3HA).
To generate pRS316-BOS1, a ∼3.2-kb fragment containing the BOS1 coding sequence was obtained by KpnI and HindIII digestion of pAN105 (15). This fragment was gel purified and further digested with XmaI to yield a ∼1.7-kb BOS1-containing fragment. The KpnI-XmaI BOS1 fragment was then inserted into the KpnI and XmaI restriction sites of pRS316.
The transmembrane-swap construct pRS316-SEC22-BOS1TM(D153C) consisting of the Sec22 cytoplasmic region (amino acids 1–192) and the Bos1 transmembrane segment (amino acids 223–244) was obtained as follows: the DNA region coding for the Bos1 transmembrane segment was amplified from pRS316-BOS1. The corresponding PCR product (∼400 bp) was digested with BglII and KpnI and ligated into a ∼6 kb fragment derived from BclI, and KpnI digestion of pRS316-SEC22(D153C) (11).
Cysteine residues were introduced into Sec22 by site-directed mutagenesis using the QuikChange kit (Stratagene). A plasmid list is provided in Table 1 and sequences of oligonucleotide primers used in the construction of these plasmids are available upon request. All of the constructs were sequence verified by automated fluorescent sequencing using an Applied Biosystems 3730 DNA analyzer.
TABLE 1.
Plasmids used in this study
| Plasmid description | Source |
|---|---|
| pRS313-SEC22 | Ref. 12 |
| pRS313-SEC22-3HA | This study |
| pRS316-SEC22-3HA | This study |
| pRS313-SEC22(D153C) | Ref. 11 |
| pRS316-SEC22–3HA(D153C) | This study |
| pRS313-SEC22(K144C) | This study |
| pRS313-SEC22(I146C) | This study |
| pRS313-SEC22(S148C) | This study |
| pRS313-SEC22(S175C) | This study |
| pRS313-SEC22(L189C) | This study |
| pRS313-SEC22(A201C) | This study |
| pRS313-SEC22(L213C) | This study |
| pRS316-SEC22–3HA(L213C) | This study |
| pRS316-BOS1 | This study |
| pRS316-SEC22-BOS1TM(D153C) | This study |
Yeast Strains and Media
All Saccharomyces cerevisiae strains were derivatives of BY4742 (Invitrogen). Yeast strains CBY740 (Matα his3 leu2 ura3 lys2), CBY773 (Matα his3 leu2 ura3 lys2 sec22Δ::KAN), and CBY1584 (Matα his3 leu2 ura3 lys2 sec22Δ::KAN with pRS313-SEC22(D153C)) have been previously described (11). To generate strains expressing only Sec22-cysteine derivatives, corresponding plasmids were transformed into strain CBY773 using the lithium acetate method (16). For the preparation of semi-intact cells or microsomal membranes, strains were grown in selective medium (0.67% nitrogen base without amino acids, 2% dextrose, and required supplements) and then back-diluted and grown in rich medium (1% Bacto-yeast extract, 2% Bacto-peptone, and 2% dextrose) for several doublings until the A600 was between 0.6 and 0.8. Standard yeast protocols were used (17).
Antibodies and Immunoblotting
Antibodies directed against Sec22 (18), Sly1 (11, 19), Och1 and Erv41 (20), Yif1 (21), Bos1 (22), Sec61 (23), Sed5 (10), Gos1 (24), and α1,6-mannose linkages (25) have been described previously. Monoclonal anti-HA antibody was purchased from Sigma. Polyclonal anti-Cog3 antibodies were raised against a purified GST-Cog3 (residues 8–293) fusion protein expressed from pGEX-3X (GE Healthcare). Western blots were developed using the SuperSignal WestPico chemiluminescent substrate (Pierce Chemical) and developed on both films and with a UVP Bioimaging System.
In Vitro Budding and Transport Assays
Semi-intact cells from wild-type and Sec22 mutant strains were prepared as previously described (26). Microsomes were isolated to generate ER-derived vesicles for two-stage transport assays (27). Vesicle budding, tethering, and transport assays following [35S]glycopro-α-factor (gpαf) have been previously described (10, 25). Briefly, two-stage transport reactions were set-up such that ER-derived vesicles containing Sec22(D153C) or Sec22(L213C) were incubated with acceptor membranes expressing Sec22HA(D153C) or Sec22HA(L213C), respectively. The amount of α1,6-mannose modification of [35S]gpαf and the extent of formation of disulfide cross-linked Sec22p homodimers (22 × 22HA) were monitored in parallel reactions as described (11).
Oxidative Cross-linking of Cysteine-containing SNAREs
Disulfide cross-linking of SNAREs from semi-intact cells and two-stage transport reactions was performed as previously described (11). Briefly, cross-linking of cysteine-containing SNAREs within washed semi-intact cells was induced by the addition of freshly prepared Cu(1,10-phenanthroline)2SO4 (Cu2+/phen) to a final concentration of 200 μm for 15 min. The reactions were quenched with 5× SDS-PAGE non-reducing sample buffer containing excess N-ethylmaleimide to quench any remaining free sulfhydryls. After heating and brief centrifugation to pellet insoluble material, a portion of the sample was resolved by non-reducing SDS-PAGE, transferred onto nitrocellulose, and blotted with the indicated antibodies. Cross-linking of two-stage transport reactions was initiated by the addition of Cu2+/phen following the completion of the transport reaction. After quenching with N-ethylmaleimide, the reactions were centrifuged to pellet membranes. The membranes were solubilized in 2× non-reducing SDS-PAGE buffer, resolved by non-reducing SDS-PAGE, transferred onto nitrocellulose, and probed with the indicated antibodies. For densitometric analysis, bands on immunoblots were quantified using Labworks software package (UVP).
Immunoprecipitations
Native immunoprecipitations of Bos1 from detergent-solubilized membranes were performed as previously described (18) but with a few modifications. CBY740 containing pRS313-SEC22–3HA and CBY773 containing pRS313-SEC22–3HA(D153C) were used to prepare semi-intact cell membranes. Washed membranes were mock-treated or oxidized with 200 μm Cu2+/phen for 15 min and then quenched with excess NEM. Membranes were pelleted, resuspended, and then solubilized on ice in 0.1 ml of lysis buffer (25 mm HEPES, pH 7, 150 mm potassium acetate, 0.5 mm PMSF, and 1% Triton X-100) for 10 min. The solubilized extracts were centrifuged at 20,000 × g to pellet any insoluble material. The supernatant fraction was diluted 5-fold with immunoprecipitation buffer (25 mm HEPES, pH 7, 150 mm potassium acetate, and 0.1% Triton X-100) and mixed with anti-Bos1 antibodies cross-linked to protein A-beads. This mixture was incubated at 4 °C for 2 h, followed by multiple washings of the beads in immunoprecipitation buffer. The bound proteins were eluted from the beads by heating in SDS-PAGE sample buffer at 95 °C for 3 min. Proteins were resolved on polyacrylamide gels and transferred to nitrocellulose membranes for immunoblot.
Native immunoprecipitations of Sed5, Sly1, and Gos1 from detergent-solubilized membranes were performed using semi-intact cell membranes from CBY773 strains expressing Sec22HA or Sec22HA(D153C). Washed membranes were mock-treated or oxidized with 200 μm Cu2+/phen for 15 min and then quenched with excess NEM. Membranes were solubilized on ice in 0.45 ml of lysis buffer (15 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1 mm PMSF, and 1% Triton X-100) for 15 min. The solubilized extracts were centrifuged at 20,000 × g and 0.4 ml of the supernatant fraction was diluted with 0.7 ml of lysis buffer, then mixed with 1.5–2.0 μl of the indicated antiserum and protein A-magnetic beads (Pierce) at 10 μl of beads/μl of antiserum. After incubation at 4 °C overnight, beads were washed multiple times with cold immunoprecipitation buffer (15 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 1% Triton X-100). Bead-bound complexes were eluted by heating in SDS-PAGE sample buffer at 75 °C for 10 min. Eluted proteins were resolved on polyacrylamide gels and transferred to nitrocellulose membranes for immunoblot detection.
Subcellular Fractionation
Membrane fractions enriched in ER (P13) or Golgi (P100) were prepared from semi-intact cells (28). Approximately 4 A280 units of semi-intact cells were diluted to 1 ml with lysis buffer (0.4 m sorbitol, 150 mm KOAc, 20 mm HEPES, pH 7.0, 2 mm Mg(OAc)2, 1 mm PMSF) and lysed with a Dounce homogenizer (10 strokes) on ice. Unlysed cells were cleared by centrifuging the homogenate at 2500 × g for 10 min at 4 °C, and the supernatant fraction was centrifuged at 13,000 × g for 10 min at 4 °C to generate a p13 pellet (ER). The supernatant from the p13 fractionation was further centrifuged (100,000 × g for 15 min at 4 °C) to generate a p100 membrane pellet (Golgi). Both p13 and p100 pellets were solubilized in 30 μl of 5× sample buffer, resolved by SDS-PAGE, and analyzed by immunoblot.
RESULTS
Sec22 Forms Disulfide Cross-linked Homodimers between Cysteine Residues in SNARE Motifs
In our previous report (11), cysteine-disulfide cross-linking was used to monitor heteromeric interactions between adjacent proteins assembled into ER-Golgi SNARE complexes. Briefly, unique cysteine residues were positioned within the SNARE motifs of Bet1 and Sec22 such that under oxidizing conditions a disulfide cross-linked Bet1-Sec22 heterodimer formed that was resolved by non-reducing SDS-PAGE and detected by immunoblot. Interestingly, during our study of Bet1-Sec22 heterodimer formation, we detected a Cu2+/phen-dependent higher molecular weight protein species appearing in semi-intact cell membranes harboring certain Sec22-cysteine derivatives alone and not in membranes containing wild-type Sec22. In our current study we generated a series of cysteine replacements into the SNARE motif and transmembrane domain of Sec22 (Fig. 1) and performed experiments to identify the nature of these cross-linked species.
FIGURE 1.
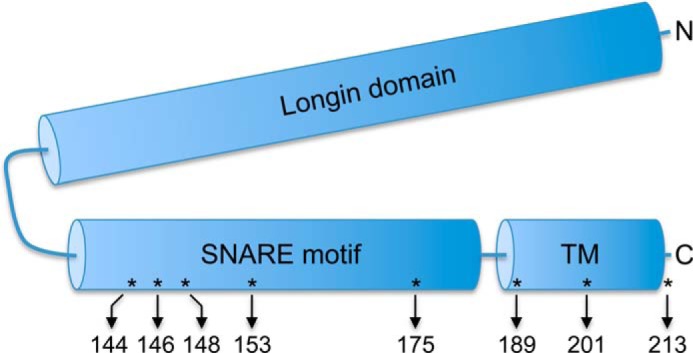
Position of cysteine substitutions in Sec22. Schematic representation of Sec22 showing the N-terminal longin domain (amino acids 1–124), SNARE motif (amino acids 132–186), and tail-anchor transmembrane (TM) domain (amino acids 189–212). The eight residues mutated to cysteines for this study are indicated by asterisks and the arrows show the relative amino acid positions.
As shown in Fig. 2, the addition of Cu2+/phen to membranes expressing Sec22-cysteine derivatives caused a decrease of Sec22 monomer (∼25-kDa; solid arrowhead) with a commensurate increase in a ∼50-kDa immunoreactive species (solid diamond). The extent of formation and the mobility of the 50-kDa immunoreactive species varied depending on the location of the engineered cysteine residues within the SNARE motif of Sec22. Under oxidizing conditions, we observed an ∼80% reduction in the amount of the Sec22 monomeric species when cysteines replaced specific residues to produce K144C, I146C, and D153C. However, considerably less formation of the 50-kDa immunoreactive species was observed when cysteines were introduced to generate S148C and S175C. Because the Sec22(D153C) variant yielded a high level of cross-linked adduct, we chose this mutant for further characterization.
FIGURE 2.

Sec22 SNARE motif cysteine-derivatives shift to higher molecular weight species under oxidizing conditions. Washed semi-intact cells expressing the indicated cysteine derivatives of Sec22 in CBY773 were incubated in the absence (−) or presence (+) of 0.2 mm Cu2+/phen as described under “Experimental Procedures.” After 15 min under oxidizing conditions, the reactions were terminated with sample buffer (containing NEM) and incubated at 70 °C for 5 min. Proteins were resolved by nonreducing SDS-PAGE (10%) and immunoblotted with anti-Sec22. A solid diamond indicates the position of the Sec22 cross-linked species, whereas a solid arrowhead indicates the position of wild-type Sec22.
We hypothesized that this oxidant-dependent 50-kDa species was most likely a Sec22-homodimer because the apparent molecular mass in non-reducing SDS-PAGE was approximately twice that of a Sec22-monomer. However, this molecular weight coincidence does not rule out the possibility that Sec22 was disulfide cross-linked to some unknown protein of a similar size. To test if Sec22 dimers were forming in semi-intact cell membranes, we constructed a CEN-based plasmid containing SEC22 fused in-frame with a triple-HA epitope at its C terminus. Expression of Sec22HA (∼31 kDa) in a sec22Δ strain was fully complemented when grown at 37 °C indicating the triple HA epitope did not interfere with biogenesis and function of Sec22 (data not shown). When Sec22HA(D153C) membranes were exposed to oxidizing conditions, a protein species (22HA × 22HA) having the expected mobility of a Sec22HA dimer was recognized by anti-Sec22 and anti-HA antibodies (Fig. 3, upper and lower panels, respectively). Furthermore, when Sec22(D153C) and Sec22HA(D153C) were co-expressed in the same strain, a new oxidant-dependent protein band (22 × 22HA) appeared on anti-Sec22 and anti-HA immunoblots that was not present in strains expressing only one of the SEC22 cysteine derivatives (Fig. 3). Additionally, because this new cross-linked band had a mobility between that of the two Sec22 homodimers, mixed Sec22-homodimers (22 × 22HA) must form that consist of Sec22(D153C) and Sec22HA(D153C). Variable amounts of unidentified cross-linked species were also observed in some experiments and are indicated by asterisks on Fig. 3. These products may result from cross-linking of Sec22(D153) to other cellular proteins. However, these unidentified products did not interfere with detection of cross-linked Sec22 homodimers.
FIGURE 3.

Sec22(D153C) and Sec22HA(D153C) form mixed homodimers when both derivatives are expressed in the same strain. Washed semi-intact cells expressing the indicated cysteine derivatives of Sec22 and Sec22HA in CBY773 were incubated in the absence (−) or presence (+) of 0.2 mm Cu2+/phen. After quenching reactions with NEM, proteins were resolved on non-reducing 10% SDS-PAGE and immunoblotted with anti-Sec22 or anti-HA. Positions for the Sec22-Sec22 homodimer (22 × 22), Sec22-Sec22HA mixed dimer (22 × 22HA), and Sec22HA-Sec22HA homodimer (22HA × 22HA) are indicated by arrows. Arrows also show the positions of monomeric Sec22 and Sec22HA. The asterisks indicate unidentified cross-linked products.
Sec22 Homodimers Are Associated with Bos1
Sec22 is known to assemble into ternary SNARE complexes with Bos1, Bet1, and Sed5 (22) that catalyze membrane fusion (29). Previous experiments demonstrated that immunoprecipitation of Bos1 from membrane-solubilized extracts efficiently recovered SNARE complexes containing Sec22 (18). To test if the Sec22 homodimer was incorporated into SNARE complexes or if homodimer assembly precluded association with other SNARE proteins, we used the Sec22HA(D153C) variant to capture Sec22 dimers and monitored their co-association with Bos1 in immunoprecipitation experiments.
As seen in Fig. 4, under native immunoprecipitation conditions Bos1 was efficiently recovered (∼50%) from detergent-solubilized extracts. The monomeric forms of both Sec22HA and Sec22HA(D153C) were efficiently co-immunoprecipitated with Bos1 in the absence or presence of Cu2+/phen-induced cross-links as expected. Importantly, after inducing disulfide cross-links into Sec22HA(D153C), ∼2% of Sec22HA(D153C) dimers immunoprecipitated with Bos1 indicating that Sec22 dimers can assemble into SNARE complexes with Bos1. These results demonstrate that Sec22 homodimer formation does not prevent stable association with other SNARE proteins.
FIGURE 4.

Sec22 homodimers are associated with Bos1. Washed semi-intact cells expressing Sec22HA in CBY740 and Sec22HA(D153C) in CBY773 were incubated in the absence (−) or presence (+) of 0.2 mm Cu2+/phen. After quenching reactions with NEM, membranes were solubilized with Triton X-100. Total solubilized extracts (T) and anti-Bos1 immunoprecipitates (IP) were resolved by non-reducing SDS-PAGE and immunoblotted for the indicated proteins. Total lanes represent 1/20 of the solubilized extracts loaded in the IP lanes. Yif1 is an integral membrane ER/Golgi protein and serves as a specificity control for the IP. Note recovery of the Sec22HA homodimer after cross-linking the D153C variant and IP with anti-Bos1 (far right lane).
To determine whether Sec22 homodimers assemble into complexes with other components of the ER/Golgi SNARE machinery, native immunoprecipitation experiments were conducted with anti-Sed5 and anti-Sly1 antibodies after inducing Sec22HA(D153C) cross-links. As shown in Fig. 5, Sec22HA homodimers were specifically detected in complex with the Q-SNARE protein Sed5 and the SM protein Sly1 that binds to Sed5. These results indicate that Sec22 homodimers are incorporated into ER-Golgi SNARE complexes with Bos1, Sed5, and Sly1. In addition, a fraction of Sec22 homodimers were detected in complex with Gos1 (Fig. 5C), a Q-SNARE protein that functions in intra-Golgi transport (30). This finding suggests that Sec22 homodimers may operate in other Sec22-dependent processes beyond fusion of ER-derived vesicles with Golgi membranes.
FIGURE 5.

Sec22 homodimers are associated with SNARE protein complexes. Washed semi-intact cells expressing Sec22HA in CBY773 and Sec22HA(D153C) in CBY773 were incubated in the absence (−) or presence (+) of 0.2 mm Cu2+/phen. Reactions were quenched with NEM and membranes were solubilized in Triton X-100 for immunoprecipitation of the target proteins Sed5, Sly1, or Gos1. Total solubilized extracts (T) and immunoprecipitates (IP) were resolved by non-reducing SDS-PAGE and immunoblotted for the indicated proteins. A, protein recovered in Sed5 immunoprecipitations. B, proteins recovered in Sly1 immunoprecipitations. C, proteins recovered in Gos1 immunoprecipitations. Total lanes represent 1/20 of the solubilized extracts loaded in the IP lanes. Erv41 serves as a specificity control and was not efficiently recovered in immunoprecipitation reactions. Note the Sec22HA dimer (22HA × 22HA) was recovered after cross-linking the D153C variant in Sed5, Sly1, and Gos1 immunoprecipitations.
Mixed Sec22(D153C) Cross-linked Homodimers Are Formed during in Vitro Two-stage Transport Assays
Having demonstrated the ability of Sec22 to form homodimers, we next investigated whether a Sec22-Sec22HA cross-linked species could be generated in cell-free transport assays as a reporter for membrane fusion. In two-stage transport reactions, COPII vesicles generated from Sec22(D153C) microsomes were incubated with Sec22HA(D153C) acceptor membranes in the presence of purified Uso1 and LMA1. At the end of the transport assay, parallel reactions were processed to measure Golgi-specific [35S]gpαf modification or were oxidized with Cu2+/phen to induce disulfide bond formation between Sec22(D153C)-Sec22HA(D153C) complexes and monitored on polyacrylamide gels. The inclusion of [35S]gpαf in the transport experiments provided an internal standard for vesicle fusion, and allowed us to compare the functional requirements for [35S]gpαf transport and mixed Sec22-homodimer (22 × 22HA) formation.
As shown in Fig. 6, the addition of Uso1 and LMA1 to the transport reactions stimulated [35S]gpαf modification reflecting increased fusion of vesicles with acceptor Golgi membranes. The addition of fusion factors to transport reactions also produced the ∼50-kDa Sec22-Sec22HA cross-linked species recognized by the anti-Sec22 antibody. Additional controls confirmed that the Sec22-Sec22HA cross-linked species was a direct result of transport. Under the conditions where reactions were carried out in the absence of fusion factors, or incubated on ice, only low levels of [35S]gpαf transport and cross-linked Sec22-Sec22HA were detected.
FIGURE 6.

Mixed Sec22-Sec22HA(D153C) homodimers form during in vitro vesicle fusion reactions. Two-stage transport reactions in which COPII vesicles containing [35S]gpαf were synthesized from microsomes expressing Sec22(D153C) in strain CBY1584 and mixed with Golgi acceptor membranes containing Sec22HA(D153C) in strain CBY773. Reactions were incubated at the indicated temperatures for 60 min in the presence of an ATP regeneration system and (+) or (−) the Uso1 and LMA1 fusion factors (Recon) to reconstitute transport. EGTA (3 mm) was added to reactions as indicated. The bar graph plots the amount of Golgi-modified [35S]gpαf to determine the transport percentage as a readout of vesicle fusion. Immunoblots of parallel reactions were probed with antibodies against Sec22 and Cog3 (loading control) to monitor the level of Sec22-Sec22HA dimer formation. For [35S]gpαf transport, data points are the average of duplicate reactions and the error bars represent the range.
Studies in mammalian cells have implicated Ca2+ in membrane fusion events from the ER-Golgi intermediate compartment (ERGIC) to the Golgi complex and for intra-Golgi transport (31–33). However, other SNARE-dependent intracellular fusion events in the early secretory pathway do not appear to require Ca2+ (33, 34). In yeast, addition of metal chelators such as EGTA or BAPTA inhibited apparent ER-Golgi transport as measured by Golgi-specific outer chain modification of [35S]gpαf (35, 36). However, for fusion of COPII vesicles with Golgi acceptor membranes, we demonstrated that formation of cross-linked Bet1-Sec22 complexes from opposed membranes was not inhibited by EGTA or BAPTA and instead our studies suggested that metal chelators deplete Golgi membranes of Mn2+ required for outer chain modification of [35S]gpαf by mannosyltranferase enzymes (11). To test if metal chelators influenced formation of the Sec22-Sec22HA cross-linked product when produced from fusion of Sec22(D153C) vesicles and Sec22HA(D153C) Golgi membranes, reactions were conducted in the presence of 3 mm EGTA (Fig. 6). Addition of EGTA inhibited Golgi-specific modification of [35S]gpαf to background levels as expected, whereas formation of disulfide cross-linked Sec22-Sec22HA was not affected. These collective results demonstrate that a specific Sec22-Sec22HA homodimer forms under conditions required for transport of [35S]gpαf and that homodimer assembly is not sensitive to metal chelation by EGTA.
Sec22 Homodimers Probed with Cysteine Residues Positioned in Transmembrane and Luminal Segments
SNARE motifs are structurally well characterized protein-protein interaction domains, however, far less is known regarding arrangements of SNARE protein transmembrane domains. It has been reported that SNARE protein transmembrane segments mediate homodimerization and promote efficient membrane fusion (37–39). Moreover, structural studies suggest that SNARE complex transmembrane domains align and extend through the membrane bilayer (6). To explore if Sec22 transmembrane domains are aligned in Sec22 homodimers, we introduced cysteines at positions L189C and A201C within the transmembrane segment (residues 187–112) and at position L213C, which is predicted to be just past the transmembrane domain and located in the lumen. Membranes were prepared from sec22Δ strains harboring CEN-based plasmids expressing these mutant proteins and disulfide cross-linking induced with Cu2+/phen. As observed in Fig. 7A, no cross-linked adduct was detected in WT and L189C membranes, whereas a minor product was detected in the A201C version. Strikingly, the L213C derivative generated an efficient cross-linked product of the size predicted for a Sec22 homodimer. To confirm the homodimer arrangement, Sec22(L213C) and Sec22HA(L213C) were coexpressed in the sec22Δ strain and membranes were treated with oxidant to induce disulfide cross-links. Immunoblots with anti-Sec22 and anti-HA antibodies (Fig. 7B) revealed cross-linked species of the expected sizes for Sec22 and Sec22HA homodimers as well as the mixed Sec22-Sec22HA homodimer of intermediate size. Based on these results, we conclude that Sec22 transmembrane segments are partially aligned in Sec22 homodimers. Moreover, efficient disulfide cross-linking of luminally positioned L213C residues provides a new tool to monitor fusion of luminal compartments.
FIGURE 7.

Cysteines positioned in the Sec22-transmembrane domain and luminal C terminus form disulfide cross-linked homodimers. Washed semi-intact cells expressing the indicated cysteine derivatives of (A) Sec22 or (B) Sec22 and Sec22HA in CBY773 were incubated in the absence (−) or presence (+) of 0.2 mm Cu2+/phen. After quenching reactions with NEM, proteins were resolved by non-reducing 10% SDS-PAGE and immunoblotted with anti-Sec22 or anti-HA. Arrows show the positions of monomeric and cross-linked forms of Sec22. The asterisk indicates a nonspecific cross-reactive species.
Mixed Sec22(L213C) Cross-linked Homodimers Are Formed in Two-stage Transport Assays
To determine whether formation of this mixed Sec22(L213C) homodimer could serve as an accurate readout for fusion of Sec22 containing compartments, we again performed two-stage transport assays that contained Sec22(L213C) COPII vesicles and Sec22HA(L213C) Golgi acceptor membranes (Fig. 8). Under conditions that reconstituted efficient transport of [35S]gpαf to Golgi membranes, Sec22(L213C)-Sec22HA(L213C) mixed homodimers were produced as observed for the Sec22(D153C) derivatives in Fig. 6. Addition of 3 mm EGTA to chelate divalent metals potently inhibited Golgi-specific modification of [35S]gpαf but did not diminish formation of mixed Sec22(L213C)-Sec22HA(L213C) cross-linked homodimers. These findings are consistent with our results using the D153C derivative and further confirm that Ca2+ is not required for fusion of COPII vesicles with Golgi membranes. Importantly, analysis of the luminally positioned Sec22(L213C) derivative excludes the possibility that disulfide cross-links formed between D153C residues within the Sec22 SNARE motif could reflect trans-SNARE complexes or hemi-fused membranes states.
FIGURE 8.

Formation of Sec22-Sec22-HA(L213C) homodimers in vesicle fusion reactions demonstrates luminal compartment mixing. Two-stage transport reactions in which COPII vesicles containing [35S]gpαf were synthesized from microsomes expressing Sec22(L213C) in CBY773 and mixed with Golgi acceptor membranes containing Sec22HA(L213C) expressed in CBY773. Reactions were incubated at the indicated temperatures for 60 min in the presence of an ATP regeneration system and (+) or (−) the LMA1 and Uso1 fusion factors (Recon) to reconstitute transport. EGTA (3 mm) was added to reactions as indicated. The bar graph plots the amount of Golgi-modified [35S]gpαf to reflect vesicle fusion. Immunoblots of parallel reactions were probed with antibodies against Sec22 and Sly1 (loading control) to monitor the level of Sec22-Sec22HA mixed dimer formation.
We also performed a kinetic analysis of mixed Sec22(L213C) homodimers that formed in reconstituted two-stage transport assays (Fig. 9). Here formation of the cross-linked Sec22 homodimer product and Golgi-specific modification of [35S]gpαf displayed time dependence with production of the disulfide cross-linked species detected as early as 5 min into the time course. Indeed, we observed that formation of mixed Sec22(L213C) cross-linked product was faster relative to Golgi modification of [35S]gpαf and suggests that outer-chain glycosylation of [35S]gpαf lags fusion of these Sec22 containing compartments.
FIGURE 9.

Time course of [35S]gpαf transport and Sec22(L213C)-Sec22HA(L213C) mixed dimer formation. Transport reactions were assembled with Sec22(L213C) vesicles containing [35S]gpαf and Sec22HA(L213C) acceptor membranes in the presence of fusion factors and an ATP regeneration system. The reactions were incubated at 23 °C for various times and then processed to determine the amount of Golgi-modified [35S]gpαf (closed circles) or Sec22-Sec22HA mixed dimer (closed squares). The amounts of α1,6-mannose precipitable [35S]gpαf and of mixed Sec22 dimer from the immunoblot (inset) were plotted as a percentage of maximum levels over time. Sly1 immunoblot serves as a loading control.
Role of the Sec22 Transmembrane Domain in Homodimer Formation
Our cross-linking analyses of the Sec22(A201C) and Sec22p(L213C) derivatives indicated that the transmembrane regions in Sec22 homodimers are closely aligned. To investigate whether the Sec22 transmembrane segment influences homodimer formation and membrane fusion, we generated a chimeric protein in which amino acid residues from the Bos1 transmembrane segment replaced the corresponding residues in Sec22. This chimera also included the D153C mutation to monitor formation of cross-linking between Sec22(D153C) and Sec22-Bos1TM(D153C). Initial characterization of this chimera (Fig. 10) shows that the protein is stably expressed but when exposed to Cu2+/phen, the membrane swapped variant displayed a greatly reduced level of disulfide cross-linking relative to the standard Sec22(D153C) derivative. Although cross-linked homodimer formation was reduced, a low level was reproducibly detected suggesting that the Sec22 transmembrane segment may stabilize homodimers but was not absolutely required for their assembly.
FIGURE 10.

Sec22-transmembrane domain influences dimerization efficiency. Washed semi-intact cells expressing the indicated cysteine-derivatives of Sec22 and Sec22-Bos1TM in CBY773 were incubated in the absence (−) or presence (+) of 0.2 mm Cu2+/phen for 15 min. After terminating the reactions with sample buffer containing NEM, proteins were resolved by non-reducing 10% SDS-PAGE and immunoblotted with anti-Sec22. Arrows shows the position of the cross-linked forms of Sec22 (22 × 22) and Sec22-Bos1TM (22 × 22TM).
To examine the biological consequences of reduced homodimer formation, we monitored the growth rate of cells expressing Sec22-Bos1TM(D153C) as the sole source of Sec22 and measured the efficiency of ER-Golgi transport in vitro using membranes prepared from these cells. In a serial dilution growth assay (Fig. 11A), both Sec22(D153C) and Sec22-Bos1TM(D153C) supported wild-type growth rates, whereas the sec22Δ strain was strongly thermosensitive. However, in vitro assays with membranes containing the Sec22-Bos1TM(D153C) chimera revealed a ∼35% reduction in COPII vesicle budding and a minimal affect on Uso1-dependent tethering relative to wild-type (Fig. 11B). Overall transport efficiency was reduced by ∼50% when compared with wild-type (Fig. 11C) indicating further defects in the vesicle fusion stage. Sec22 is known to function in both anterograde and retrograde fusion stages between the ER and Golgi (40) therefore partial loss of function with the Sec22-Bos1TM(D153C) chimera is likely to influence both ER and Golgi function. Although ER budding and vesicle fusion with Golgi membranes was compromised with the chimera, we note that the overall expression level and subcellular distribution of Sec22-Bos1TM(D153C) was not detectably altered compared with the wild-type protein (Fig. 11D). These findings show that the Sec22 transmembrane segment is necessary for efficient homodimer formation and for efficient ER-Golgi transport providing support for a model in which Sec22 homodimers are functionally significant in trafficking between ER and Golgi compartments.
FIGURE 11.

Sec22-Bos1TM membranes display modest defects in budding and transport. A, SEC22(D153C) and SEC22-BOS1TM(D153C) suppress sec22Δ temperature sensitivity. Serial dilutions of WT (CBY740), sec22Δ (CBY773), and sec22Δ (CBY773) expressing Sec22(D153C) or Sec22-Bos1TM(D153C) strains were spotted on YPD plates and incubated at 25 and 37 °C. B, membranes expressing Sec22-Bos1TM display budding defects in vitro. Washed semi-intact cell membranes were prepared from strains expressing Sec22(D153C) or Sec22-Bos1TM(D153C). Membranes were mock treated (NA), incubated with COPII proteins or COPII proteins plus Uso1 at 23 °C for 30 min, and freely diffusible vesicles were quantified to assess levels of vesicle budding and tethering. C, membranes expressing Sec22-Bos1TM(D153C) display in vitro transport defects. Washed semi-intact cell membranes were incubated with COPII, Uso1, and LMA1 proteins to reconstitute transport. Over a time course at 23 °C, the amount of Golgi-modified [35S]gpαf was measured to determine transport efficiency. Error bars represent the range of duplicate determinations. D, subcellular fractionation of lysed spheroplasts to enrich ER (P13) and Golgi (P100) membranes followed by immunoblot for Sec22 in addition to Sec61 (ER) and Och1 (Golgi) markers.
DISCUSSION
In this study we examined SNARE protein assemblies and topological arrangements that accompany fusion of COPII vesicles with Golgi membranes. A cysteine cross-linking analysis of Sec22 indicated that this R-SNARE protein forms homodimers that are efficiently captured when cysteines are positioned at specific locations within the SNARE motif and surprisingly in transmembrane or luminal regions. These distinct cysteine probes in Sec22 can be used in cell-free vesicle fusion assays to monitor formation of mixed homodimers in trans or in post-fusion cis arrangements. Our results show that fusion of COPII vesicles with Golgi membranes does not depend on Ca2+ because formation of mixed trans- and post-fusion cis-Sec22 homodimers was not inhibited by the chelating agent EGTA. Sec22 homodimers were also detected in association with other ER-Golgi and intra-Golgi Q-SNARE proteins suggesting that these SNARE complexes assemble into multimers to catalyze Sec22-dependent fusion processes. Finally, the native Sec22 transmembrane sequence was necessary for efficient homodimer formation and for efficient transport between ER and Golgi membranes indicating a physiological role for homodimer formation. The collective findings support a model in which higher-order oligomerization of ER-Golgi SNARE complexes drive efficient fusion of COPII vesicles with Golgi acceptor membranes.
Several lines of investigation have shown that specific SNARE proteins form homodimers and can assemble into higher-order arrangements. The R-SNARE synaptobrevin 2/VAMP2 has been shown through cross-linking and binding studies to form homodimers in vitro (37, 41, 42) and biomolecular fluorescence approaches indicate that VAMP2 molecules dimerize in cells (43). Moreover, these studies indicate that the VAMP2 transmembrane domain and adjacent residues near the cytosol-membrane interface are required for stable dimer formation (37, 42, 43). We similarly observed that the R-SNARE Sec22 assembles into homodimers and that its transmembrane domain is needed for efficient homodimer formation. Both monomeric and dimeric forms of Sec22 were detected in cellular membrane preparations suggesting that homodimer assembly is reversible and dynamic.
Although R-SNARE proteins appear to play a prominent role in multimerization, there is also evidence for Q-SNARE protein homodimer formation in studies on Vam3 (38) and Sso1 (39, 44). Again, the transmembrane domains of these Q-SNAREs are needed for homodimerization and for formation of supramolecular SNARE assemblies (39, 45). For other ER/Golgi SNARE proteins, we did not detect efficient formation of disulfide cross-linked homodimers when cysteine residues were inserted into Q-SNARE proteins Sed5 and Bet1, although this analysis was not exhaustive (data not shown). It is noteworthy that SNARE proteins are often detected in clusters or segregated into specific membrane domains and there are likely multiple mechanisms that could drive SNARE complexes into higher-order structures or clusters depending on their site of function or mode of regulation (46). Both protein-protein and protein-lipid interactions are reported to act synergistically in concentrating SNARE proteins into membrane fusion zones (47, 48).
The mechanisms by which supramolecular SNARE complex assemblies function in membrane fusion are not well understood. Estimates for how many SNARE complexes are needed for a single intracellular membrane fusion event range from one to nine (49–52). In vitro membrane fusion studies show that mutation of transmembrane residues needed for Vam3 SNARE homodimer formation inhibit the hemifusion to fusion transition, although fusion can still proceed under certain conditions (45). In vivo analyses of transmembrane domain mutations in syntaxin-1 and VAMP2 that interfere with oligomerization show moderate affects on neurotransmitter release but remain competent for membrane fusion (43, 53). Indeed, full replacement of SNARE protein transmembrane domains with lipid anchors in neuronal cells supports efficient synaptic vesicle fusion with only modest effects on fusion properties (54). Our findings on a transmembrane swapped form of Sec22 are in accord with these results. With diminished Sec22 homodimer assembly, no secretion or growth defects were observed in vivo although in vitro assays revealed decreased levels of fusion between COPII vesicles and Golgi acceptor membranes. These collective results suggest that transmembrane domain-dependent formation of SNARE homodimers is not required for membrane fusion events but somehow influences fusion efficiency. It is also possible that other protein or lipid components in SNARE-dependent fusion reactions promote higher-order assemblies in the absence of transmembrane domains. More recent work using minimally reconstituted fusion reactions where proteoliposome size can be controlled indicates that small vesicles fuse efficiently with single to low numbers of SNARE complexes, whereas dozens of SNARE complexes were necessary for fusion of large liposomes (55). Therefore variable levels of SNARE complex assemblies could be employed to fuse distinct intracellular membranes.
Our cross-linking results show that Ca2+ is not absolutely required for SNARE complex assembly or membrane fusion between COPII vesicles and Golgi acceptor membranes in cell-free transport reactions. Earlier studies on ER-Golgi transport had suggested a calcium requirement for the fusion stage (31, 36, 56) but our previous (11) and current work indicate that apparent transport inhibition by Ca2+ chelation is due to inhibition of the luminal glycosylation enzymes that report on fusion. Ca2+ clearly plays a critical role in triggered synchronous fusion of synaptic vesicles (57) but is not thought to be directly required for many intracellular membrane fusion reactions (58). However, there is ample evidence that Ca2+ levels play an important regulatory role in membrane fusion events through both in vivo and in vitro studies (58–60). Specific Ca2+ sensors and modes of regulation are still not well understood for membrane fusion. Therefore, use of disulfide cross-linkable probes in Sec22 and other SNARE proteins may provide robust assays for Ca2+-dependent regulation of membrane fusion in complex cell-free reactions.
Recent studies have revealed that Sec22 also functions in cellular processes beyond ER-Golgi transport. A subset of SNARE proteins that includes Sec22 is required in early stages of autophagy for autophagosome biogenesis in yeast (61). In animal cells, Sec22 has been shown to connect ER with the plasma membrane in neurons to support axonal and dendritic growth (62). Here ER-localized Sec22 assembles in trans with plasma membrane syntaxins and has a non-fusogenic role in lipid transfer between compartments. How Sec22 functions in autophagy and in generating ER-PM contact sites, or whether Sec22 homodimers are involved in these processes is not known. Use of cysteine probes in the Sec22 SNARE motif and transmembrane segment could provide powerful tools for dissection of these newly described cellular processes.
This work was supported, in whole or in part, by National Institutes of Health Grants GM52549 (to C. B.) and GM070096 (to J. J. F.).
- ER
- endoplasmic reticulum
- NEM
- N-ethylmaleimide
- Cu2+/phen
- Cu(1,10-phenanthroline)2SO4
- gpαf
- glycopro-α-factor
- BAPTA
- 1,2-bis(2-aminophenoxyl)ethane-N,N,N′,N′-tetraacetic acid.
REFERENCES
- 1. Söllner T., Bennett M. K., Whiteheart S. W., Scheller R. H., Rothman J. E. (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75, 409–418 [DOI] [PubMed] [Google Scholar]
- 2. Weber T., Zemelman B. V., McNew J. A., Westermann B., Gmachl M., Parlati F., Söllner T. H., Rothman J. E. (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 [DOI] [PubMed] [Google Scholar]
- 3. Südhof T. C., Rothman J. E. (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sutton R. B., Fasshauer D., Jahn R., Brunger A. T. (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4-Å resolution. Nature 395, 347–353 [DOI] [PubMed] [Google Scholar]
- 5. Antonin W., Fasshauer D., Becker S., Jahn R., Schneider T. R. (2002) Crystal structure of the endosomal SNARE complex reveals common structural principles of all SNAREs. Nat. Struct. Biol. 9, 107–111 [DOI] [PubMed] [Google Scholar]
- 6. Stein A., Weber G., Wahl M. C., Jahn R. (2009) Helical extension of the neuronal SNARE complex into the membrane. Nature 460, 525–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fasshauer D., Sutton R. B., Brunger A. T., Jahn R. (1998) Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. U.S.A. 95, 15781–15786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rizo J., Rosen M. K., Gardner K. H. (2012) Enlightening molecular mechanisms through study of protein interactions. J. Mol. Cell Biol. 4, 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barlowe C. K., Miller E. A. (2013) Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 193, 383–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao X., Ballew N., Barlowe C. (1998) Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO J. 17, 2156–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Flanagan J. J., Barlowe C. (2006) Cysteine-disulfide cross-linking to monitor SNARE complex assembly during endoplasmic reticulum-Golgi transport. J. Biol. Chem. 281, 2281–2288 [DOI] [PubMed] [Google Scholar]
- 12. Liu Y., Flanagan J. J., Barlowe C. (2004) Sec22p export from the endoplasmic reticulum is independent of SNARE pairing. J. Biol. Chem. 279, 27225–27232 [DOI] [PubMed] [Google Scholar]
- 13. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 14. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Newman A. P., Shim J., Ferro-Novick S. (1990) BET1, BOS1, and SEC22 are members of a group of interacting yeast genes required for transport from the endoplasmic reticulum to the Golgi complex. Mol. Cell. Biol. 10, 3405–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elble R. (1992) A simple and efficient procedure for transformation of yeasts. BioTechniques 13, 18–20 [PubMed] [Google Scholar]
- 17. Sherman F. (1991) Getting started with yeast. Methods Enzymol. 194, 3–21 [DOI] [PubMed] [Google Scholar]
- 18. Liu Y., Barlowe C. (2002) Analysis of Sec22p in endoplasmic reticulum/Golgi transport reveals cellular redundancy in SNARE protein function. Mol. Biol. Cell 13, 3314–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cao X., Barlowe C. (2000) Asymmetric requirements for a Rab GTPase and SNARE proteins in fusion of COPII vesicles with acceptor membranes. J. Cell Biol. 149, 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Otte S., Belden W. J., Heidtman M., Liu J., Jensen O. N., Barlowe C. (2001) Erv41p and Erv46p: new components of COPII vesicles involved in transport between the ER and Golgi complex. J. Cell Biol. 152, 503–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matern H., Yang X., Andrulis E., Sternglanz R., Trepte H. H., Gallwitz D. (2000) A novel Golgi membrane protein is part of a GTPase-binding protein complex involved in vesicle targeting. EMBO J. 19, 4485–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Søgaard M., Tani K., Ye R. R., Geromanos S., Tempst P., Kirchhausen T., Rothman J. E., Söllner T. (1994) A Rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell 78, 937–948 [DOI] [PubMed] [Google Scholar]
- 23. Stirling C. J., Rothblatt J., Hosobuchi M., Deshaies R., Schekman R. (1992) Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol. Biol. Cell 3, 129–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inadome H., Noda Y., Adachi H., Yoda K. (2005) Immunoisolaton of the yeast Golgi subcompartments and characterization of a novel membrane protein, Svp26, discovered in the Sed5-containing compartments. Mol. Cell. Biol. 25, 7696–7710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barlowe C. (1997) Coupled ER to Golgi transport reconstituted with purified cytosolic proteins. J. Cell Biol. 139, 1097–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baker D., Hicke L., Rexach M., Schleyer M., Schekman R. (1988) Reconstitution of SEC gene product-dependent intercompartmental protein transport. Cell 54, 335–344 [DOI] [PubMed] [Google Scholar]
- 27. Wuestehube L. J., Schekman R. W. (1992) Reconstitution of transport from endoplasmic reticulum to Golgi complex using endoplasmic reticulum-enriched membrane fraction from yeast. Methods Enzymol. 219, 124–136 [DOI] [PubMed] [Google Scholar]
- 28. Belden W. J., Barlowe C. (2001) Deletion of yeast p24 genes activates the unfolded protein response. Mol. Biol. Cell 12, 957–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parlati F., McNew J. A., Fukuda R., Miller R., Söllner T. H., Rothman J. E. (2000) Topological restriction of SNARE-dependent membrane fusion. Nature 407, 194–198 [DOI] [PubMed] [Google Scholar]
- 30. McNew J. A., Coe J. G., Søgaard M., Zemelman B. V., Wimmer C., Hong W., Söllner T. H. (1998) Gos1p, a Saccharomyces cerevisiae SNARE protein involved in Golgi transport. FEBS Lett. 435, 89–95 [DOI] [PubMed] [Google Scholar]
- 31. Beckers C. J., Balch W. E. (1989) Calcium and GTP: essential components in vesicular trafficking between the endoplasmic reticulum and Golgi apparatus. J. Cell Biol. 108, 1245–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Porat A., Elazar Z. (2000) Regulation of intra-Golgi membrane transport by calcium. J. Biol. Chem. 275, 29233–29237 [DOI] [PubMed] [Google Scholar]
- 33. Xu D., Hay J. C. (2004) Reconstitution of COPII vesicle fusion to generate a pre-Golgi intermediate compartment. J. Cell Biol. 167, 997–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J. L., Ahluwalia J. P., Stamnes M. (2002) Selective effects of calcium chelators on anterograde and retrograde protein transport in the cell. J. Biol. Chem. 277, 35682–35687 [DOI] [PubMed] [Google Scholar]
- 35. Rexach M. F., Schekman R. W. (1991) Distinct biochemical requirements for the budding, targeting, and fusion of ER-derived transport vesicles. J. Cell Biol. 114, 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lupashin V. V., Hamamoto S., Schekman R. W. (1996) Biochemical requirements for the targeting and fusion of ER-derived transport vesicles with purified yeast Golgi membranes. J. Cell Biol. 132, 277–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roy R., Laage R., Langosch D. (2004) Synaptobrevin transmembrane domain dimerization-revisited. Biochemistry 43, 4964–4970 [DOI] [PubMed] [Google Scholar]
- 38. Roy R., Peplowska K., Rohde J., Ungermann C., Langosch D. (2006) Role of the Vam3p transmembrane segment in homodimerization and SNARE complex formation. Biochemistry 45, 7654–7660 [DOI] [PubMed] [Google Scholar]
- 39. Lu X., Zhang Y., Shin Y. K. (2008) Supramolecular SNARE assembly precedes hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 15, 700–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Spang A., Schekman R. (1998) Reconstitution of retrograde transport from the Golgi to the ER in vitro. J. Cell Biol. 143, 589–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laage R., Langosch D. (1997) Dimerization of the synaptic vesicle protein synaptobrevin (vesicle-associated membrane protein) II depends on specific residues within the transmembrane segment. Eur. J. Biochem. 249, 540–546 [DOI] [PubMed] [Google Scholar]
- 42. Fdez E., Jowitt T. A., Wang M. C., Rajebhosale M., Foster K., Bella J., Baldock C., Woodman P. G., Hilfiker S. (2008) A role for soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex dimerization during neurosecretion. Mol. Biol. Cell 19, 3379–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fdez E., Martínez-Salvador M., Beard M., Woodman P., Hilfiker S. (2010) Transmembrane-domain determinants for SNARE-mediated membrane fusion. J. Cell Sci. 123, 2473–2480 [DOI] [PubMed] [Google Scholar]
- 44. Zhang Y., Shin Y. K. (2006) Transmembrane organization of yeast syntaxin-analogue Sso1p. Biochemistry 45, 4173–4181 [DOI] [PubMed] [Google Scholar]
- 45. Hofmann M. W., Peplowska K., Rohde J., Poschner B. C., Ungermann C., Langosch D. (2006) Self-interaction of a SNARE transmembrane domain promotes the hemifusion-to-fusion transition. J. Mol. Biol. 364, 1048–1060 [DOI] [PubMed] [Google Scholar]
- 46. van den Bogaart G., Lang T., Jahn R. (2013) Microdomains of SNARE proteins in the plasma membrane. Curr. Top. Membr. 72, 193–230 [DOI] [PubMed] [Google Scholar]
- 47. Fratti R. A., Jun Y., Merz A. J., Margolis N., Wickner W. (2004) Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J. Cell Biol. 167, 1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van den Bogaart G., Meyenberg K., Risselada H. J., Amin H., Willig K. I., Hubrich B. E., Dier M., Hell S. W., Grubmüller H., Diederichsen U., Jahn R. (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479, 552–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van den Bogaart G., Holt M. G., Bunt G., Riedel D., Wouters F. S., Jahn R. (2010) One SNARE complex is sufficient for membrane fusion. Nat. Struct. Mol. Biol. 17, 358–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sinha R., Ahmed S., Jahn R., Klingauf J. (2011) Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc. Natl. Acad. Sci. U.S.A. 108, 14318–14323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Domanska M. K., Kiessling V., Stein A., Fasshauer D., Tamm L. K. (2009) Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J. Biol. Chem. 284, 32158–32166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shi L., Shen Q. T., Kiel A., Wang J., Wang H. W., Melia T. J., Rothman J. E., Pincet F. (2012) SNARE proteins: one to fuse and three to keep the nascent fusion pore open. Science 335, 1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Han X., Wang C. T., Bai J., Chapman E. R., Jackson M. B. (2004) Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304, 289–292 [DOI] [PubMed] [Google Scholar]
- 54. Zhou P., Bacaj T., Yang X., Pang Z. P., Südhof T. C. (2013) Lipid-anchored SNAREs lacking transmembrane regions fully support membrane fusion during neurotransmitter release. Neuron 80, 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hernandez J. M., Kreutzberger A. J., Kiessling V., Tamm L. K., Jahn R. (2014) Variable cooperativity in SNARE-mediated membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 111, 12037–12042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Baker D., Wuestehube L., Schekman R., Botstein D., Segev N. (1990) GTP-binding Ypt1 protein and Ca2+ function independently in a cell-free protein transport reaction. Proc. Natl. Acad. Sci. U.S.A. 87, 355–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Südhof T. C. (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hay J. C. (2007) Calcium: a fundamental regulator of intracellular membrane fusion? EMBO Rep. 8, 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bentley M., Nycz D. C., Joglekar A., Fertschai I., Malli R., Graier W. F., Hay J. C. (2010) Vesicular calcium regulates coat retention, fusogenicity, and size of pre-Golgi intermediates. Mol. Biol. Cell 21, 1033–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parkinson K., Baines A. E., Keller T., Gruenheit N., Bragg L., North R. A., Thompson C. R. (2014) Calcium-dependent regulation of Rab activation and vesicle fusion by an intracellular P2X ion channel. Nat. Cell. Biol. 16, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nair U., Jotwani A., Geng J., Gammoh N., Richerson D., Yen W. L., Griffith J., Nag S., Wang K., Moss T., Baba M., McNew J. A., Jiang X., Reggiori F., Melia T. J., Klionsky D. J. (2011) SNARE proteins are required for macroautophagy. Cell 146, 290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Petkovic M., Jemaiel A., Daste F., Specht C. G., Izeddin I., Vorkel D., Verbavatz J. M., Darzacq X., Triller A., Pfenninger K. H., Tareste D., Jackson C. L., Galli T. (2014) The SNARE Sec22b has a non-fusogenic function in plasma membrane expansion. Nat. Cell Biol. 16, 434–444 [DOI] [PubMed] [Google Scholar]