

Background: Salivary glands exhibit a HCO3−-dependent fluid secretion mechanism.
Results: Ae4 and Ae2 are Cl−/HCO3− exchangers in mouse submandibular acinar cells; however, only Ae4 gene disruption inhibits fluid secretion.
Conclusion: Ae4 mediates HCO3−-dependent Cl− uptake that is crucial for fluid secretion.
Significance: We show for the first time that the Ae4 Cl−/HCO3− exchanger plays a critical role in the transcellular movement of Cl− in secretory acinar cells.
Keywords: Bicarbonate, Chloride Transport, Epithelium, Exchanger, Secretion, Salivary Gland
Abstract
Transcellular Cl− movement across acinar cells is the rate-limiting step for salivary gland fluid secretion. Basolateral Nkcc1 Na+-K+-2Cl− cotransporters play a critical role in fluid secretion by promoting the intracellular accumulation of Cl− above its equilibrium potential. However, salivation is only partially abolished in the absence of Nkcc1 cotransporter activity, suggesting that another Cl− uptake pathway concentrates Cl− ions in acinar cells. To identify alternative molecular mechanisms, we studied mice lacking Ae2 and Ae4 Cl−/HCO3− exchangers. We found that salivation stimulated by muscarinic and β-adrenergic receptor agonists was normal in the submandibular glands of Ae2−/− mice. In contrast, saliva secretion was reduced by 35% in Ae4−/− mice. The decrease in salivation was not related to loss of Na+-K+-2Cl− cotransporter or Na+/H+ exchanger activity in Ae4−/− mice but correlated with reduced Cl− uptake during β-adrenergic receptor activation of cAMP signaling. Direct measurements of Cl−/HCO3− exchanger activity revealed that HCO3−-dependent Cl− uptake was reduced in the acinar cells of Ae2−/− and Ae4−/− mice. Moreover, Cl−/HCO3− exchanger activity was nearly abolished in double Ae4/Ae2 knock-out mice, suggesting that most of the Cl−/HCO3− exchanger activity in submandibular acinar cells depends on Ae2 and Ae4 expression. In conclusion, both Ae2 and Ae4 anion exchangers are functionally expressed in submandibular acinar cells; however, only Ae4 expression appears to be important for cAMP-dependent regulation of fluid secretion.
Introduction
Fluid and electrolyte transport is essential to the physiology of secretory epithelia. Many diseases are related to dysfunction of the fluid secretion process, such as cystic fibrosis, pancreatitis, and Sjögren syndrome (1–5). Secretory epithelia share a basic molecular transport process originally proposed as a “pump leak” or “two-membrane” mechanism (i.e. secretory epithelial cells exhibit an active pump-mediated ion transport pathway targeted to one membrane that works in series with a passive ion transport pathway in the opposite membrane) (6–8). Stimulated fluid secretion in salivary gland acinar cells is dependent on a pump leak mechanism to drive unidirectional transcellular Cl− movement. This process requires the intracellular Cl− concentration to be above its electrochemical equilibrium so that Cl− can be secreted across the apical membrane. Cl− secretion is accompanied by paracellular Na+ and osmotic driven water movement (9, 10). Targeted disruption of Nkcc1 produces a dramatic 65% reduction in saliva secretion by the mouse parotid gland, demonstrating that Na+-K+-2Cl− cotransporter 1 (Nkcc1) is the main mechanism for concentrating Cl− in acinar cells (11). A parallel HCO3−-dependent Cl− uptake mechanism appears to be responsible for much of the remaining Nkcc1-independent fluid secretion (12–14). The current fluid secretion model proposes that this secondary Nkcc1-independent Cl− uptake pathway is a basolateral Cl−/HCO3− exchanger. Consistent with this model, there is an approximately 35% reduction in muscarinic receptor-stimulated saliva secretion in the absence of HCO3− (12–14).
The Ae2 (Slc4a2) anion exchanger has been localized to the plasma membrane in mouse parotid and sublingual acinar cells, and it has been suggested that this exchanger is responsible for HCO3−-dependent fluid secretion in salivary glands (12). Ae2 is one of 10 members of the Slc4a family (Slc4a1–5 and Slc4a7–11). The designation Slc4a6 was later withdrawn because Slc4a6 and Slc4a7 represent the same gene product. Slc4a1–3 (Ae1–3, respectively) are Na+-independent Cl−/HCO3− exchangers, whereas Slc4a4, -5, -7, -8, and -10 (NBCe1, NBCe2, NBCn1, NDCBE, and NBCn2, respectively) are Na+-coupled HCO3− transporters. The function of Slc4a11 (BTR1) is unknown, but it does not appear to transport HCO3−. The HCO3− transport mechanism for Ae4 (Slc4a9) remains controversial. It has been suggested that Ae4 might mediate Cl−/HCO3− exchange or Na+-HCO3− cotransport (15–17). Ae4 was localized to the basolateral membrane of mouse submandibular gland duct cells, but the expression and function of Ae4 has not been reported in salivary gland acinar cells (18).
It has been proposed that the Ae2 Cl−/HCO3− exchanger is involved in saliva secretion (12, 19). However, the role of Ae2 has not been directly assessed in salivary glands because systemic Ae2−/− mice die prior to adulthood due to severe acidosis (20). The present study demonstrates that saliva secretion stimulated by a physiological combination of muscarinic and β-adrenergic receptor agonists was reduced by 35% in Ae4−/− mice but was not changed in a conditional knock-out mouse model lacking Ae2 in acinar cells (Ae2−/− mice). These results indicate that Ae4 is a major contributor to fluid secretion by mouse submandibular salivary glands. The secretory dysfunction observed in Ae4−/− mice correlated with decreased Cl−-concentrating capacity of acinar cells during β-adrenergic receptor activation of cAMP signaling.
EXPERIMENTAL PROCEDURES
Materials and Animals
All reagents were purchased from Sigma-Aldrich unless otherwise indicated. C57Bl/6 and BS/129Svj mice were used as controls when appropriate. Gene targeting and genotyping protocols for Ae2fl/fl (C57Bl/6 background), Ae4−/− (BS/129Svj background), and Aqp5-Cre (ACID) mice were as described previously (21–23). Acinus-specific Ae2−/− mice (Ae2fl/fl-ACID-Cre) were generated using the Cre/LoxP system by crossing Ae2fl/fl mice with Aqp5-Cre mice. Cre-recombinase expression is under control of the aquaporin 5 gene promoter in Aqp5-Cre (ACID) mice (23). Because aquaporin 5 is exclusively expressed in the acinar cells of mouse salivary glands (24, 25), this system permitted the ablation of Ae2 exchangers only in acinar cells. Littermate Ae2+/flCre+/−, Ae2fl/flCre−/−, and Ae2+/+Cre+/− mice were used as controls. Ae4−/−-Ae2−/− double knock-out mice were generated by crossing Ae4−/− with Ae2fl/fl-ACID-Cre mice, and littermate Ae4+/−-Ae2+/flCre−/− and Ae4+/+-Ae2+/flCre+/− mice were used as controls. Genotyping was performed by PCR from tail biopsies. All animals were housed in microisolator cages with ad libitum access to laboratory chow and water with a 12-h light/dark cycle. Experiments were performed on female and male animals aged between 2 and 4 months. All animal procedures were approved by the NIDCR, National Institutes of Health, Animal Care and Use Committee (ASP 13-686).
Ex Vivo Submandibular Gland Perfusion
Ex vivo submandibular gland perfusion was performed essentially as reported previously (26). Briefly, the gland was transferred to a perfusion chamber, where the common carotid was cannulated (31-gauge) and perfused with a HCO3−-containing, high Cl− solution (B+; see below). Salivation was stimulated by perfusion with the cholinergic receptor agonist carbachol (CCh3; 0.3 μm) plus the β-adrenergic receptor agonist isoproterenol (IPR; 5 μm). The progression of saliva within the capillary tube was recorded every minute. Experiments were performed at 37 °C.
Acinar Cell Preparation
Submandibular gland acinar cells were isolated by enzyme digestion as reported previously with minor modifications (26). Briefly, dissected glands were finely minced and digested in Eagle's minimum essential medium containing 0.015% trypsin for 10 min at 37 °C and then centrifuged. The cell pellet was rinsed with Eagle's minimum essential medium containing 0.2% trypsin inhibitor and then digested for 25 min in the presence of 0.5 mg/ml collagenase 2 (Worthington). After centrifugation, the cell pellet was further digested with collagenase for another 25 min and finally resuspended in Eagle's minimum essential medium. Cells were continuously gassed with a mixture of 95% O2, 5% CO2.
Intracellular Cl− and pH Measurements
During the second collagenase incubation, acinar cells were loaded with 5 mm 6-methoxy-N-(3-sulfopropyl) quinolinium (SPQ) for 25 min or 2 μm 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (BCECF-AM) for 15 min. Acinar cell clumps were seeded in a perfusion chamber maintained at 37 °C and mounted on the stage of an inverted microscope (Nikon, Eclipse TE 300) equipped with a Polychrome IV imaging system coupled to a high speed digital camera (TILL Photonics, Inc). SPQ fluorescence was excited at 340 nm, and emissions were collected at 510 nm. BCECF fluorescence was excited at 440 and 490 nm, and emissions were collected at 530 nm. The resting intracellular pH was determined using the high K+/nigericin protocol as described previously (27). The resting intracellular Cl− concentration was measured using the nigericin-tributyltin protocol essentially as described previously (28). Briefly, cells were exposed to calibration solutions containing 20, 50, and 80 mm Cl−. The calibration solution was as follows: 20 mm KCl, 120 mm potassium gluconate, 5 mm glucose, 10 mm HEPES, 0.005 mm nigericin, and 0.01 mm tributyltin (pH 7.4). The Cl− was changed from 20 to 80 by replacement of potassium gluconate with KCl.
50 μm bumetanide, 50 μm T16Ainh-A01 (Tocris Bioscience), 30 μm ethoxyzolamide (EZA), and 10 μm ethyl-isopropyl amiloride were used in some experiments to inhibit Na+/K+/2Cl− cotransporters, Ca2+-dependent Cl− channels, carbonic anhydrases, and Na+/H+ exchangers, respectively. The Cl− uptake experiment included three stages: 1) intracellular Cl− depletion by exposing the cells to a low Cl− (4 mm) bath solution; 2) inhibition of Nkcc1 cotransporter and Tmem16A (transmembrane member 16A) channel by exposure to 50 μm bumetanide and 50 μm T16Ainh-A01, respectively, in the low Cl− bath solution; and 3) Cl− uptake by re-exposure to a high Cl− bath solution in the presence of bumetanide and T16Ainh-A01, inhibitors included to block the two main Cl− movement pathways in acinar cells, Nkcc1 and Tmem16A, respectively (11, 29). Under these conditions, the HCO3−-dependent Cl− uptake should be independent of Nkcc1 and Tmem16A. Inhibitors were not included in control experiments.
Bath solutions were as follows: HCO3−-containing, high Cl− solution (B+): 4.3 mm KCl, 120 mm NaCl, 25 mm NaHCO3, 5 mm glucose, 10 mm HEPES, 1 mm CaCl2, and 1 mm MgCl2; HCO3−-free, high Cl− solution (B−): 4.3 mm KCl, 145 mm NaCl, 5 mm glucose, 10 mm HEPES, 1 mm CaCl2, and 1 mm MgCl2, pH 7.4; HCO3−-containing, low Cl− solution: 4.3 mm potassium gluconate, 120 mm sodium gluconate, 25 mm NaHCO3, 5 mm glucose, 10 mm HEPES, 1 mm CaCl2, and 1 mm MgCl2, pH 7.4; HCO3−-free, low Cl− solution: 4.3 mm potassium gluconate, 145 mm sodium gluconate, 5 mm glucose, 10 mm HEPES, 1 mm CaCl2, and 1 mm MgCl2, pH 7.4; HCO3−-containing, Na+-free solution: same composition as B+ solution but with replacement of NaCl by N-methyl-d-glucamine-Cl and NaHCO3 by choline-HCO3; HCO3−-containing, Na+-free, low Cl− solution: same composition as HCO3−-containing, low Cl− solution but with replacement of NaCl by N-methyl-d-glucamine-glutamate and NaHCO3 by choline-HCO3. HCO3−-containing solutions were gassed with 95% O2, 5% CO2 for at least 30 min. HCO3−-free solutions were gassed with 100% O2.
HCO3− in ex Vivo Saliva
Ex vivo saliva samples obtained in response to CCh+ IPR were collected and stored at −86 °C. The concentration of HCO3− in saliva was determined colorimetrically as described by the manufacturer (Diazyme Laboratories, Poway, CA). The amount of HCO3− secreted in saliva (expressed as microequivalents/10 min) was calculated by dividing the [HCO3−] by the amount of saliva collected during 10 min of stimulation.
Statistical Analysis
Results are presented as normalized data for SPQ fluorescence, where the starting fluorescence (F0) is divided by the fluorescence over time (F0/F), except when the intracellular Cl− concentration was determined. Normalized data from BCECF fluorescence (490/440 nm ratio) is obtained by dividing the fluorescence over time by starting fluorescence (F/F0), except when the intracellular pH was determined. Results are given as means ± S.E. Statistical significance was determined using Student's t test or analysis of variance followed by Bonferroni's post hoc test using Origin version 7.0 Software (OriginLab Corp., Northampton, MA). p < 0.05 was considered statistically significant. All experiments were performed using separate preparations from at least three different mice for each condition. n refers to the number of experiments performed.
RESULTS
Submandibular Gland Fluid Secretion and Cl− Uptake Capacity Are Decreased in Ae4−/− Mice
Saliva secretion is reduced 60–70% in mice lacking Nkcc1 Na+-K+-2Cl− cotransporter activity (11), suggesting that there is another basolateral Cl− uptake mechanism in salivary gland acinar cells. It has been proposed that the residual secretory response in Nkcc1−/− mice is driven by the basolateral Ae2 anion exchanger, Cl− uptake pathway (11, 12). Additionally, Ae4 has been detected in salivary gland duct cells (18); however, acinar expression has not been reported to date. To investigate whether Ae4 and/or Ae2 contribute to salivation, ex vivo submandibular gland secretions were collected from conditional Ae2−/− and systemic Ae4−/− mice. The flow rate and total volume of secreted saliva were measured in response to stimulation with 0.3 μm CCh plus 5 μm IPR (30, 31). The main Cl− efflux pathway in mouse salivary gland acinar cells is the apical Ca2+-activated Tmem16A Cl− channel (29), whereas Cl− uptake by basolateral Nkcc1 Na+-K+-2Cl− cotransporters (11) is up-regulated by cAMP and Ca2+ signaling (32–34). Therefore, stimulation of muscarinic (CCh) and β-adrenergic (IPR) signaling pathways elevates intracellular Ca2+ and cAMP, respectively, and induces maximal and sustained salivation by activation of the main Cl− uptake and exit pathways.
Saliva collected from ex vivo submandibular glands suggests that Ae4 expression is important for salivation (Fig. 1A). The amount of saliva secreted by Ae4−/− mice during the initial 2–3 min of stimulation was comparable with that secreted by control littermates. In contrast, there was a sustained decrease in flow rate at later time points (after 3 min), producing less than half the amount of saliva in Ae4−/− mice. The total amount of saliva collected during 10 min of stimulation was 35 ± 4.7% less than secreted by Ae4 WT mice (Fig. 1A, inset). Unexpectedly, the kinetics of salivary flow and the total saliva collected were essentially identical in Ae2−/− and control mice (Fig. 1, B and inset to B). Note that the kinetics of saliva secretion appear to be somewhat different in control (and Ae2−/−) mice from Ae4 wild type mice. In both Ae2−/− and their control mice, the saliva secretion rate during the initial 2–3 min of stimulation gradually decreased to a sustained level. In contrast, the secretion rate is relatively constant during the entire 10-min stimulation period in Ae4 wild type mice (Fig. 1A). Therefore, data analyses were performed comparing knock-out mice with their respective controls. This difference in kinetics is probably due to subtle differences in the secretory machinery between the Ae4 and Ae2 mouse strains (BS/129Svj and C57Bl/6, respectively). Regardless, the total volume of saliva secreted is comparable in Ae4 wild type and Ae2 control mice (Fig. 1, A (inset) and B (inset)), suggesting that the differences in the kinetics of fluid secretion during the first 2–3 min do not significantly influence the sustained secretion or the total amount of saliva secreted.
FIGURE 1.

Agonist-induced fluid secretion and changes in intracellular [Cl−] in Ae4−/− and Ae2−/− mice. A and B, ex vivo submandibular glands were perfused with a physiological solution containing 0.3 μm CCh and 5 μm IPR to stimulate secretion. A, flow rate (μl/min) and total saliva (μl/10 min, inset), were diminished in Ae4−/− compared with WT (gray and open symbols, respectively). Inset, comparison of total saliva secreted. *, p < 0.05, Student's t test (Ae4 WT, n = 6; Ae4−/−, n = 6). B, flow rate (μl/min) and total saliva (μl/10 min; inset), were essentially identical in Ae2−/− and control mice (control, n = 6; Ae2−/−, n = 6). C and D, acinar cells were loaded with SPQ to determine the intracellular [Cl−] and were stimulated with 0.3 μm CCh and 5 μm IPR during the indicated time. An initial Cl− exit (ΔF) was followed by Cl− reuptake. Cl− reuptake was slower in Ae4−/− (gray symbols) compared with Ae4 WT (open symbols). D, Cl− exit and Cl− reuptake were not altered in Ae2−/− compared with control mice. The initial Cl− exit (ΔF) was calculated from region indicated by brackets from individual experiments in C and D. The initial Cl− uptake rate was calculated using the slope obtained by linear regression analysis from the region indicated by dashed red lines from individual experiments in C and D. Representative experiments are shown in C and D. A summary of the initial Cl− exit and initial Cl− uptake rates is shown in Table 1. Error bars, S.E.
Cl−-dependent fluid secretion requires the intracellular Cl− concentration ([Cl−]i) to be greater than its electrochemical equilibrium potential. Consistent with the [Cl−]i being important for regulating Cl−-dependent fluid secretion, acinar cells from Nkcc1−/− mice exhibit a decreased resting [Cl−]i (11). To evaluate whether Ae4 or Ae2 might also be important for concentrating the [Cl−]i, the Cl−-sensitive fluorescent indicator SPQ was used to directly measure resting [Cl−]i (11, 29, 35, 36). We found that the resting [Cl−]i was significantly lower in Ae4−/− mice (27 ± 3.1% less), but [Cl−]i was not different in Ae2−/− compared with their controls (Table 1). Taken together, these results suggest that 1) the decrease in stimulated fluid secretion by Ae4−/− mice might be due to decreased Cl− uptake and [Cl−]i, and 2) Ae2 expression is not required to set the resting intracellular [Cl−], but this does not rule out the possibility that Ae2 is important for Cl− uptake during stimulation. To more directly test these different possibilities, we measured the intracellular [Cl−] changes induced by CCh plus IPR stimulation under physiological conditions. Fig. 1C shows that stimulation evoked a biphasic [Cl−]i response in both Ae4−/− and WT mice acinar cells (i.e. a rapid Cl− loss (ΔF, brackets), consistent with Cl− efflux through Tmem16A (Ano1 (anoctamine 1)) Cl− channels (29), followed by Cl− uptake). The calculated initial rate of Cl− uptake (compare dashed red lines) is clearly slower in Ae4−/− mice (Fig. 1C). A summary of the initial rates of Cl− uptake showed a 55.4 ± 4.5% lower rate in Ae4−/− mice (Table 1, Cl− uptake rate, CCh + IPR). In contrast, the Cl− uptake rates during CCh plus IPR stimulation were not significantly different in Ae2−/− and control mice (Fig. 1D and Table 1).
TABLE 1.
Calculated parameters from intracellular [Cl−] and pH measurements
Resting intracellular [Cl−] and pH were calculated from experiments performed in isolated acinar cells exposed to physiological HCO3−-containing solutions. Resting intracellular [Cl−] was significantly lower in Ae4−/− compared with WT mice and was not altered in Ae2−/− compared with control mice. Resting intracellular pH was unchanged in Ae4−/− and Ae2−/− mice compared with their controls. The magnitude of the initial Cl− exit (ΔF) and initial Cl− uptake rates were calculated from experiments like those shown in Fig. 1, C and D. Experiments were performed in isolated acinar cells bathed by physiological HCO3−-containing solutions and stimulated with only CCh, only IPR, or CCh + IPR. Initial Cl− exit in cells stimulated with only CCh or CCh + IPR was not different in Ae4−/− and Ae2−/− mice compared with their controls. In cells stimulated with CCh + IPR, the Cl− uptake rate was significantly slower in Ae4−/− compared with WT mice and was not altered in Ae2−/− compared with control mice. In cells stimulated only with CCh, the Cl− uptake rate was not different in Ae4−/− and Ae2−/− mice compared with their controls. In cells stimulated only with IPR, the Cl− uptake rate was lower in Ae4−/− compared with WT mice. The Cl− uptake rate was not different in Ae2−/− compared with controls.
| Ae4 WT | Ae4−/− | Ae2 control | Ae2−/− | |
|---|---|---|---|---|
| Resting Cl−i (mm) | 50.10 ± 1.50 | 36.50 ± 1.60a | 53.40 ± 1.80 | 54.50 ± 1.80 |
| (n = 7) | (n = 6) | (n = 6) | (n = 6) | |
| Resting pHi | 6.91 ± 0.07 | 6.89 ± 0.02 | 6.87 ± 0.01 | 6.95 ± 0.05 |
| (n = 4) | (n = 4) | (n = 4) | (n = 6) | |
| ΔF Cl− exit (CCh + IPR) | −0.21 ± 0.01 | −0.18 ± 0.02 | −0.16 ± 0.02 | −0.15 ± 0.01 |
| (n = 10) | (n = 9) | (n = 13) | (n = 18) | |
| ΔF Cl− exit (CCh) | −0.18 ± 0.03 | −0.15 ± 0.01 | −0.14 ± 0.02 | −0.17 ± 0.02 |
| (n = 7) | (n = 6) | (n = 8) | (n = 9) | |
| Cl− uptake rate (10−3 s−1) (CCh + IPR) | 2.02 ± 0.10 | 0.90 ± 0.09b | 2.11 ± 0.50 | 2.39 ± 0.20 |
| (n = 10) | (n = 9) | (n = 13) | (n = 18) | |
| Cl− uptake rate (10−3 s−1) (CCh) | 2.18 ± 0.20 | 2.30 ± 0.10 | 2.20 ± 0.20 | 2.30 ± 0.20 |
| (n = 7) | (n = 6) | (n = 8) | (n = 9) | |
| Cl− uptake rate (10−3 s−1) (IPR) | 0.40 ± 0.07 | 0.20 ± 0.03c | 0.70 ± 0.10 | 0.58 ± 0.05 |
| (n = 4) | (n = 7) | (n = 7) | (n = 6) |
a p < 0.001, Student's t test.
b p < 0.001, Student's t test.
c p < 0.05, Student's t test.
These results imply that Ae4, but not Ae2, is critical for Cl−-dependent fluid secretion and, thus, challenge the hypothesis that Ae2 is an important HCO3−-dependent Cl− uptake pathway. Another possibility is that Ae4 might be involved in acinar luminal HCO3− efflux to promote HCO3−-dependent fluid secretion. To evaluate this possibility, we calculated the amount of HCO3− ions secreted in saliva in response to CCh + IPR. We found no differences in the amounts of HCO3− ions secreted (microequivalents/10 min of stimulation) by WT and Ae4−/− glands: 5.3 ± 1.4 (n = 7) versus 7.1 ± 0.8 (n = 7), WT versus Ae4−/−, respectively (p = 0.28, Student's t test). Similarly, Ae2−/− glands secreted similar amounts of HCO3− (microequivalents/10 min of stimulation) in response to CCh + IPR: 4.0 ± 0.4 (n = 5) versus 6.3 ± 1.8 (n = 6), control versus Ae2−/− glands, respectively (p = 0.28, Student's t test).
However, the above results could at least partially reflect compensation by another Cl− uptake mechanism. For example, decreased salivation by Nhe1−/− mice is accompanied by an increase in Nkcc1 mRNA, suggesting a partial compensation by overexpression of Nkcc1 (37). Because Nkcc1 is a major Cl− uptake pathway in salivary glands, we investigated whether cotransporter activity might be altered in Ae2−/− or Ae4−/− mice. Nkcc1-dependent Cl− uptake was monitored in SPQ-loaded acinar cells as described under “Experimental Procedures.” In order to isolate Nkcc1 activity, experiments were performed using HCO3−-free solutions (B−) supplemented with 30 μm ethoxyzolamide + 50 μm T16Ainh-A01 to inhibit Cl−/HCO3− exchangers, carbonic anhydrases, and Ca2+-dependent Cl− channels, such as Tmem16A, respectively. Representative experiments shown in Fig. 2A reveal that Nkcc1 activity was identical in Ae4−/− and in Ae2−/− mice compared with their littermate control mice, ruling out a change in Nkcc1 functional activity in the knock-out mice (results summarized in Fig. 2B). Cl− uptake was virtually absent in the presence of bumetanide (95.6 ± 0.9% reduction), indicating that the response was totally dependent on Nkcc1 (Fig. 2, A and B, black symbols). The Cl− uptake rates calculated for the different genotypes of control mice (see “Experimental Procedures”) were not significantly different and thus are shown as a single mean (Fig. 2B, open bar).
FIGURE 2.

Na+-K+-2Cl− cotransporter and Na+/H+ exchanger activities in Ae2−/− and Ae4−/− mouse acinar cells. Cl− uptake experiments were performed in SPQ-loaded acinar cells from Ae2−/− and Ae4−/− mice. HCO3−-free solutions (B−) containing 30 μm ethoxyzolamide (EZA) and 50 μm T16Ainh-A01 were used. A, after intracellular Cl− was depleted in a low Cl− bath solution, Cl− uptake was initiated by the addition of high Cl− in Ae4−/− (light gray symbols), Ae2−/− (dark gray symbols), and control mice (open symbols). After the initial intracellular Cl− depletion of control cells, 50 μm bumetanide (Bum) was included to inhibit Nkcc1 Na+-K+-2Cl− cotransporter activity (black symbols). B, summary of the initial Cl− uptake rates calculated from experiments like those shown in A from the regions indicated with the dashed red lines (control, n = 16; Ae4−/−, n = 8; Ae2−/−, n = 10; control + bumetanide, n = 9). Analysis of variance followed by Bonferroni's post hoc test was performed. a, significant difference from control, Ae4−/−, and Ae2−/− relative to control + bumetanide (p < 0.001). C, the intracellular pH was measured in BCECF-AM-loaded acinar cells stimulated with 0.3 μm CCh and 5 μm IPR during the indicated time. An alkalinization was recorded in Ae4−/− (light gray symbols), Ae2−/− (dark gray symbols), and control mice (open symbols). The initial alkalinization was changed to an acidification when cells were exposed to an Na+/H+ exchange inhibitor (10 μm (EIPA), black symbols, n = 7). D, summary of the initial alkalinization rates calculated from experiments like those shown in C from the regions indicated with the dashed red line. Control, n = 13; Ae4−/−, n = 8; Ae2−/−, n = 6. Results are given as means ± S.E. (error bars). Data from Ae4 WT and Ae2 control mice are shown as a single bar in B and D because they were not statistically different. Representative experiments are shown in A and C.
It has been demonstrated that the main Cl− exit pathway in salivary glands acinar cells is the Tmem16A Cl− channel. During CCh stimulation, the initial Cl− exit recorded in SPQ-loaded acinar cells was absent in Tmem16A−/− mice (29). Here, we measured the initial Cl− exit rates from experiments like those shown in Figs. 1, C and D (ΔF, brackets). We found no differences in the initial Cl− exit rates in Ae4−/− and Ae2−/− mice, compared with their respective controls (Table 1, ΔF Cl− exit, CCh + IPR), suggesting that Tmem16A channel activity is not changed by loss of Ae4 or Ae2 expression.
HCO3−-dependent saliva secretion depends on an intracellular source of HCO3− to maintain the activity of Cl−/HCO3− exchangers. Nhe1−/− mice exhibit hyposalivation, suggesting that Nhe1 Na+/H+ exchangers play a role in setting the intracellular pH (HCO3− concentration) and, consequently, HCO3−-dependent saliva secretion (37). Here, we measured the intracellular pH in acinar cells stimulated with CCh plus IPR to test whether the Nhe exchanger activity is altered in Ae4−/− and Ae2−/− mice. Fig. 2C shows that stimulation evoked an intracellular alkalinization but was an acidification in the presence of a Na+/H+ exchange inhibitor (EIPA; black symbols), suggesting that the stimulation-induced alkalinization depends on Nhe Na+/H+ exchanger activation. The Nhe-dependent alkalinization rate was not different in Ae4−/− and Ae2−/− compared with their controls, indicating that Nhe exchanger activity is the same in these knock-out mice. Fig. 2D shows a summary of the initial alkalinization rates calculated from the dashed red lines in Fig. 2C. The alkalinization rates for the different control genotypes were not significantly different, and they are shown as a single mean (Fig. 2D, open bar). Additionally, we measured the resting intracellular pH and found no differences in Ae4−/− and Ae2−/− acinar cells compared with their controls (Table 1). These results suggest that the primary intracellular pH regulatory mechanisms are not affected in Ae4−/− and Ae2−/− mice.
HCO3−-dependent Cl− Uptake Is Decreased in the Acinar Cells of Ae2−/− and Ae4−/− Mice
The current salivary gland fluid secretion model includes a basolateral Cl−/HCO3− exchanger as part of the Cl− uptake process necessary to drive luminal Cl− secretion in acinar cells (12, 14, 38). However, the molecular nature of the HCO3−-dependent Cl− uptake activity is unknown. Results presented in Fig. 1, A–D, suggest that Ae4 may play a critical role in Cl−-dependent saliva secretion by acinar cells. To investigate these observations further, we directly tested for Cl−/HCO3− anion exchanger activity in acinar cells loaded with the Cl−-sensitive fluorescent dye SPQ. A Cl− depletion and reuptake strategy was used to isolate Cl−/HCO3− exchanger activity (see Fig. 3A and “Experimental Procedures” for details). Cl− uptake rapidly occurred following the addition of a high Cl− bath solution to Cl−-depleted acinar cells in control experiments (Control, no inhibitors; Fig. 3B, open symbols). A significantly slower Cl− uptake was observed when Nkcc1 Na+-K+-2Cl− cotransporter and Tmem16A (Ano1) Cl− channel inhibitors were present (gray symbols). Importantly, the inhibitor-resistant Cl− uptake in acinar cells was completely abolished in the absence of HCO3− (black symbols). Thus, this experimental approach isolates the Cl−/HCO3− exchange activity as HCO3−-dependent, inhibitor-resistant Cl− uptake. Fig. 3C summarizes the Cl− uptake rates calculated from experiments like those shown in Fig. 3B. Inhibition of Nkcc1 and Tmem16A reduced the Cl− uptake rate 74 ± 2.6% (Control versus Inh), whereas HCO3− removal further reduced the Cl− uptake rate to 96.3 ± 1.4% of control (Inh B−). These results are consistent with the Na+-K+-2Cl−cotransporter Nkcc1 being the primary Cl−-concentrating pathway and with Cl−/HCO3− exchange playing a smaller, but significant, role in Cl− uptake by submandibular gland acinar cells. These results agree with a previous study in mouse parotid glands, where a similar inhibition of Cl− uptake was reported in acinar cells from Nkcc1−/− mice or cells treated with the Na+-K+-2Cl− cotransporter inhibitor bumetanide (11).
FIGURE 3.
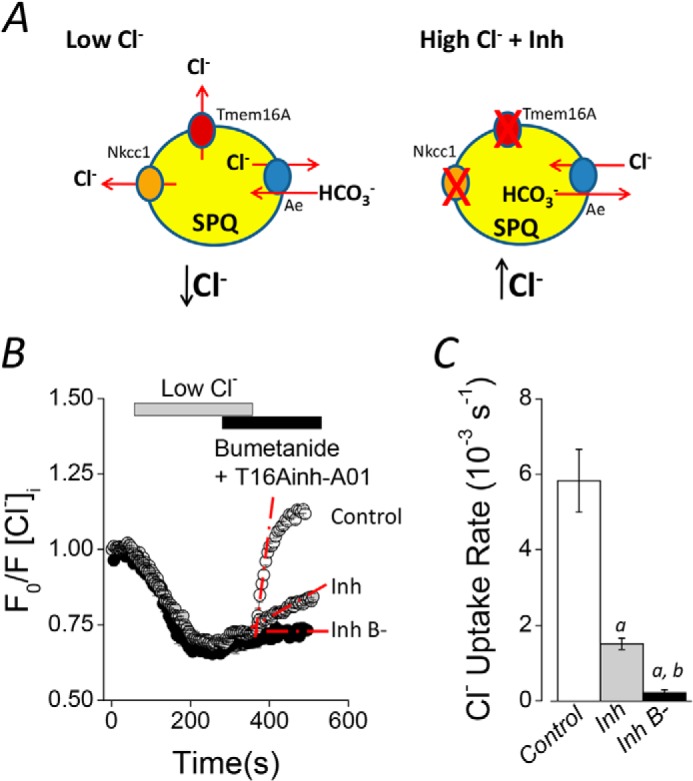
Cl−/HCO3− exchanger activity in mouse acinar cells. A, experimental strategy to isolate the Cl−/HCO3− exchanger activity in SPQ-loaded acinar cells. Intracellular Cl− depletion was produced by shifting to a low Cl− concentration bath solution (left side, Low Cl−). After Cl− depletion, 50 μm bumetanide and 50 μm T16Ainh-A01 were included in the bath solution to inhibit Nkcc1 Na+/K+/2Cl− cotransporter and Tmem16A Cl− channel activities, respectively. Cl− reuptake was initiated by shifting to high Cl− concentration bath solution in the continued presence of bumetanide and T16Ainh-A01 (right side, High Cl− + Inh). B, experiments containing HCO3− (25 mm) in the absence of inhibitors (Control; open symbols), including inhibitors (Inh; gray symbols), or in HCO3−-free solutions, including bumetanide, T16Ainh-A01, and 30 μm ethoxyzolamide (Inh B−; data replotted from Fig. 2A for comparison). C, summary of the initial rates of Cl− uptake for experiments like those shown in B. The rate was calculated using the slope obtained by linear regressions from the region indicated by dashed red lines from individual experiments (control, n = 10; inhibitors, n = 7; HCO3−-free solutions, n = 9). Analysis of variance followed by Bonferroni's post hoc test was performed. a and b, significant difference from control and inhibitor experiments, respectively (p < 0.001). All results are given as means ± S.E. (error bars). Representative experiments are shown in B.
To determine whether a member of the Slc4a gene family is involved in Cl−/HCO3− exchange-dependent Cl− uptake in submandibular acinar cells, we measured intracellular Cl− in cells from Ae2−/− and Ae4−/− mice. Fig. 4A shows that Cl− uptake in response to an inwardly directed Cl− chemical gradient was significantly slower in cells from Ae4−/− (light gray symbols) and Ae2−/− (dark gray symbols) mice than in control cells (open symbols). The Cl− uptake rates for the different control mouse genotypes were not different and thus are shown as a single mean (Fig. 4B, open bar). The rate of Cl− uptake was significantly slower in Ae4−/− and Ae2−/− acinar cells (38.2 ± 6.6 and 55.1 ± 3.4%, respectively) compared with that observed in control acinar cells. Moreover, Cl− uptake under HCO3−-free conditions demonstrated that the remaining activity in both Ae4−/− and Ae2−/− mice was totally dependent on HCO3− (Fig. 4, C and D). Consequently, the remaining HCO3−-dependent Cl− uptake activity in Ae4−/− mice is consistent with the presence of Ae2-mediated Cl−/HCO3− exchange activity, whereas the presence of Ae4-mediated Cl−/HCO3− exchange activity can explain the remaining Cl− uptake in Ae2−/− mice. We also found that much of the remaining anion exchanger activity in Ae2−/− acinar cells was Na+-dependent in the presence of bumetanide and T16Ainh-A01. The Cl− reuptake rate (10−3 s−1) was dramatically reduced by a HCO3−-containing, Na+-free solution: 1.35 ± 0.16 (n = 7) versus 0.51 ± 0.09 (n = 7), Ae2−/− Na+-containing versus Ae2−/− Na+-free solutions, respectively; p = 0.001, Student's t test), suggesting that Ae4 is Na+-dependent.
FIGURE 4.

Altered Cl− uptake in Ae4−/− and Ae2−/− mouse acinar cells. Cl− uptake experiments were performed as described in the legend to Fig. 3 in HCO3−-containing solutions (B+). Cl− reuptake is shown as normalized data. A, shifting to a high Cl− bath solution elicited Cl− uptake in Cl−-depleted acinar cells of control mice (open symbols). Cl− uptake was slower in Ae4−/− and Ae2−/− (light gray and dark gray symbols, respectively). B, summary of the initial rates of Cl− uptake for experiments like those shown in A. Initial rates were calculated as in Fig. 3. Data from Ae4 WT and Ae2 control mice are shown as a single bar because they were not statistically different (Ae4 WT, n = 7; Ae4−/−, n = 8; Ae2 control, n = 8; Ae2−/−, n = 10). Analysis of variance followed by Bonferroni's post hoc test was performed. a, different from control (p < 0.01); b, different from Ae4−/− (p < 0.05). C, Cl− uptake was absent in HCO3−-free solutions (B−) containing 30 μm EZA when cells were shifted from a low to a high Cl− bath solution in Ae4−/− mice (gray symbols) and Ae2−/− (open symbols). Data from experiments performed in HCO3−-containing solutions (B+) are replotted from A for comparison (dashed line, Ae4−/−; dotted line, Ae2−/−). D, summary of the initial rates of Cl− uptake (Ae4−/− B− (gray bar), n = 5; Ae2−/− B− (open bar), n = 3). Data from Ae4−/− and Ae2−/− in B+ solutions are replotted for comparison (dashed and dotted bars, respectively). **, p < 0.001, Student's t test. All results are given as means ± S.E. (error bars). Representative experiments are shown in A and C.
Although the above results are consistent with the functional expression of both Ae2 and Ae4 in acinar cells, the expression of other anion exchangers of the Slc4a or Slc26a families cannot be ruled out. To investigate this point in more detail, we measured the Cl−/HCO3− exchanger activity in double Ae4−/−-Ae2−/− knock-out mice. Fig. 5A demonstrates that Cl− uptake was severely reduced in double knock-out mice. A summary of the results (Fig. 5B) from experiments like those shown in Fig. 5A shows that the Cl− uptake rate was reduced by 87.7 ± 3.1% in double knockouts, consistent with Ae4 and Ae2 being responsible for nearly all of the Cl−/HCO3− exchanger activity in acinar cells.
FIGURE 5.
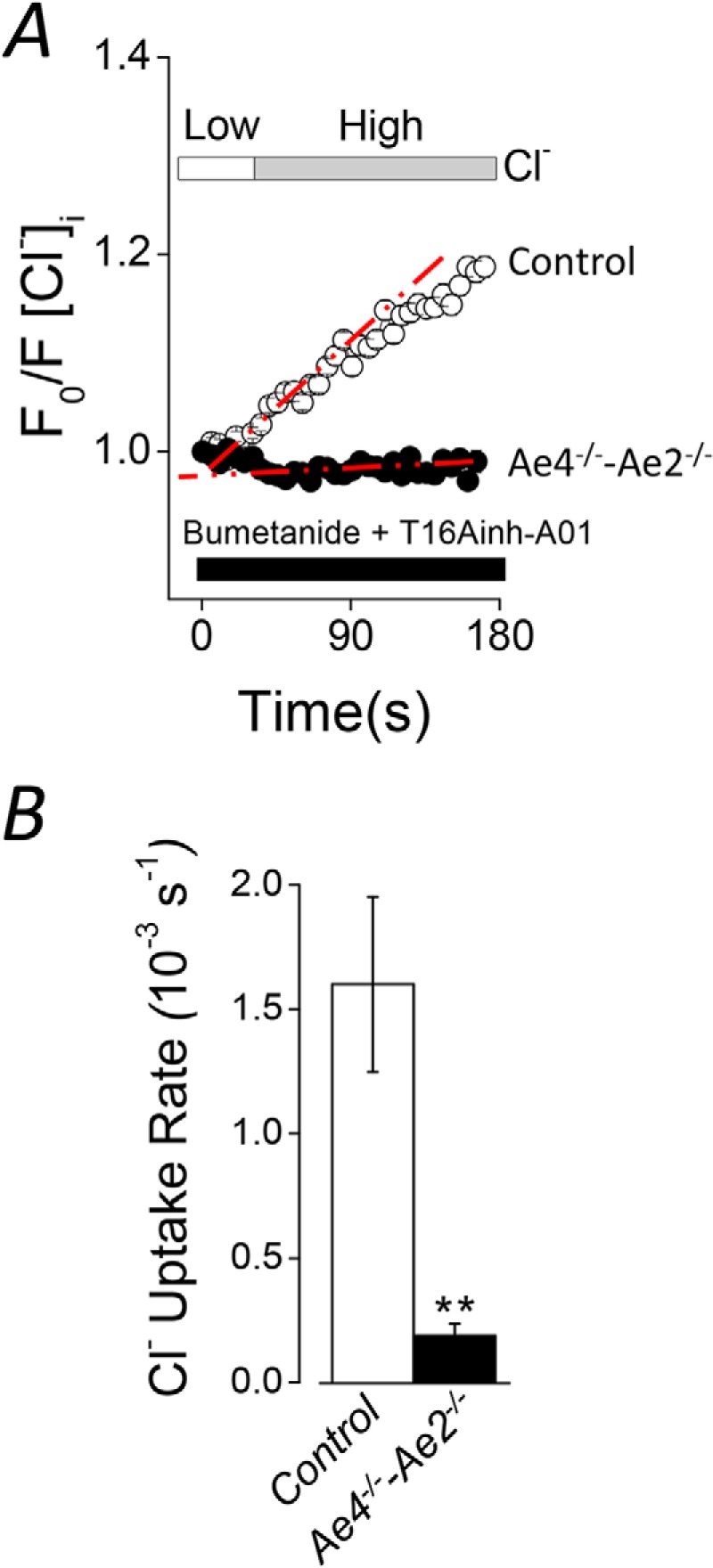
Severely reduced Cl− uptake in double Ae4−/−-Ae2−/− mouse acinar cells. Cl− uptake experiments were performed as described in the legend to Fig. 3 in HCO3−-containing solutions (B+). Cl− reuptake is shown as normalized data. A, Cl− uptake was severely reduced in double Ae4−/−-Ae2−/− (black symbols) compared with control mice (open symbols). B, summary of the initial Cl− uptake rates calculated from the regions indicated with the dashed red lines (Ae4-Ae2 control, n = 8; Ae4−/−-Ae2−/−, n = 6). **, p < 0.001, Student's t test. All results are given as means ± S.E. (error bars). Representative experiments are shown in A.
Taken together, the above results demonstrate that both Ae2 and Ae4 are co-expressed in mouse submandibular acinar cells and are able to drive HCO3−-dependent Cl− uptake (Figs. 4 and 5). However, only Ae4 activity appears to be important for stimulated fluid secretion (Fig. 1). One plausible explanation for this apparent discrepancy is different modes of regulation by CCh and IPR. We speculated that the increase in CCh-induced intracellular Ca2+ and IPR-stimulated cAMP levels could exert specific and possibly opposite effects on Ae4 and Ae2 activities. To investigate this hypothesis, we measured [Cl−]i changes in acinar cells stimulated with only CCh or only IPR. Experiments were performed as described in the legend to Fig. 1, C and D. We found that the initial Cl− exit rates during muscarinic versus muscarinic + β-adrenergic stimulation were not different in Ae4−/− and Ae2−/− mice compared with their controls (Table 1, ΔF Cl− exit, CCh), suggesting that Tmem16A Cl− channel activity was not different in controls, Ae4−/−, and Ae2−/− mice. Moreover, the initial Cl− reuptake rates during only CCh stimulation were not different in Ae2−/− or Ae4−/− mice compared with their controls (Table 1, Cl− uptake rate, CCh). We also measured [Cl−]i changes in response to only β-adrenergic receptor stimulation and found that the initial Cl− exit was absent, suggesting negligible Tmem16A Cl− channel activity under these conditions. However, a relatively slow net Cl− uptake was present during IPR stimulation. This β-adrenergic-induced Cl− uptake was significantly slower in Ae4−/− acinar cells but was not different in Ae2−/− compared with control cells (Table 1, Cl− uptake rate, IPR). These results suggest that the decreased Cl− uptake observed in the presence of only IPR (Table 1, Cl− uptake rate, IPR) or a combination of CCh + IPR in Ae4−/− mice (Fig. 1C) may be dependent on β-adrenergic receptor activation of intracellular cAMP signaling pathways. To more directly address this possibility, reuptake after Cl− depletion experiments like those shown in Figs. 3–5 were performed. A nearly 3-fold increase in the Cl− uptake rate (10−3 s−1) was observed upon β-adrenergic receptor stimulation in control acinar cells: 1.56 ± 0.19 (n = 10) versus 4.33 ± 0.91 (n = 13), without versus plus IPR, respectively; p = 0.016, Student's t test. In contrast, β-adrenergic receptor stimulation had no effect on the Cl− reuptake rate (10−3 s−1) in A4−/− acinar cells: 0.95 ± 0.10 (n = 8) versus 1.08 ± 0.09 (n = 9), without versus plus IPR, respectively; p = 0.366, Student's t test.
DISCUSSION
The electroneutral movement of Na+, K+, and Cl− ions by the Nkcc1 Na+-K+-2Cl− cotransporter drives the accumulation of intracellular Cl− in most secretory epithelia (11, 39–41). However, pharmacological block of the basolateral Na+-K+-2Cl− cotransporter by loop diuretics like bumetanide only partially inhibits fluid secretion, suggesting that Nkcc1 is not the only Cl− uptake pathway involved in salivation. Indeed, disruption of Nkcc1 expression in mouse parotid glands confirmed that a considerable residual saliva secretion remains in the absence of Na+-K+-2Cl− cotransporter activity. It has been proposed that this secondary Cl− uptake pathway depends on Cl−/HCO3− exchanger activity in secretory epithelial cells (39, 42–46). Although the molecular identity of the anion exchanger in salivary gland acinar cells is unknown, this HCO3−-dependent mechanism appears to play an important role for Cl− uptake during prolonged muscarinic stimulation (9, 11, 14, 47). It has been proposed that the ubiquitously expressed, basolateral Ae2 Cl−/HCO3− exchanger may contribute to the Nkcc1-independent fluid secretion in several secretory epithelial cells, including salivary glands (11, 12, 48). Several of these studies used pharmacological approaches to investigate anion exchanger-mediated Cl− uptake. However, specific inhibitors of Slc4 family members have not been reported. Stilbene derivatives, such as DIDS (4,4′-diisothiocyanostilbene-2,2′-disulfonic acid), SITS (4-acetamido-4′-isothiocyanatostilbene-2,2′-disulfonic acid), or DNDS (4,4′-dinitrostilbene-2,2′-disulfonic acid), inhibit Cl−/HCO3− exchangers but also block Cl− channels, Na+-HCO3− cotransporters, and Na+-driven Cl−/HCO3− exchangers (16, 49). Moreover, it has been reported that stilbenes inhibit Na+-K+-ATPases and adenylyl cyclases (50–53). Therefore, conclusions derived from studies using stilbene derivatives should be interpreted with caution.
In the present study, we used a non-pharmacological, genetic ablation strategy to directly investigate the contribution of Ae4 and Ae2 to stimulated fluid secretion in submandibular glands. Systemic Ae4−/− mice have no gross phenotypic abnormalities and develop normally. Functional evaluation of these mice suggests that Ae4 expression is critical for NaCl transport in renal intercalated cells (54), whereas Ae4 appears not to be important for HCO3− transport in the duodenum (22). Systemic deletion of Ae2 is lethal (20), making it impossible to investigate the function of Ae2 exchangers in the adult salivary gland. Consequently, we generated an acinus-specific, conditional Ae2−/− mouse (Ae2fl/fl-ACID-Cre mice) to study Ae2 function in submandibular salivary glands. Ex vivo experiments show that Ae4−/− mice secreted about 35% less total saliva in response to a combination of muscarinic and β-adrenergic receptor agonists, whereas saliva secretion was unaffected in Ae2−/− mice. The simplest interpretation of these results is that Ae2 contributes little, if any, to stimulated fluid secretion, whereas Ae4 expression plays a critical role in salivation under physiological conditions.
To further test this hypothesis, we measured the [Cl−]i in acinar cells. The current secretion model predicts that the [Cl−]i is lower and the Cl− uptake rate is slower in Ae4−/− mice, whereas the [Cl−]i will be essentially normal in Ae2−/− mice. Indeed, the resting [Cl−]i was reduced, and Cl− uptake was significantly slower in the acinar cells of Ae4−/− mice during stimulation with CCh and IPR, consistent with an important role for Ae4 in Cl−-dependent secretion. Moreover, the resting [Cl−]i and stimulated uptake of Cl− was unchanged in Ae2−/− mice, suggesting that the primary Cl− uptake mechanism is not altered in Ae2−/− mice. However, we cannot rule out the possibility that the functional activity of other key ion transport pathways involved in the fluid secretion process may be altered in an attempt to compensate for loss of Ae4 and/or Ae2 Cl−/HCO3− exchanger activity. The bumetanide-sensitive, cAMP-stimulated short circuit current (Isc) was increased in colonic mucosa from Ae2−/− animals, consistent with increased Nkcc1 activity in these mice (46). Moreover, decreased salivation in Nhe1−/− mice may be partially compensated for by increased expression of Nkcc1 (37). However, we found that Nkcc1 activity was unchanged in Ae4−/− and Ae2−/− mice, demonstrating that up-regulation of Nkcc1 activity does not compensate for loss of Ae4 or Ae2 expression.
Alternatively, the initial rapid Cl− exit stimulated by muscarinic receptor activation is mediated by Tmem16A (Ano1) channels (29). Thus, changes in Tmem16A Cl− channel activity in Ae4−/− or Ae2−/− mice might be expected to alter stimulation-induced Cl− efflux. However, the magnitude and the rate of Cl− exit induced in acinar cells by only CCh or CCh plus IPR stimulation were not different in Ae4−/− and Ae2−/− mice, demonstrating that Tmem16A channel activity is unchanged. Another possible compensatory mechanism could include Nhe Na+/H+ exchangers, which regulate HCO3−-dependent saliva secretion by a mechanism that involves a functional interplay with Cl−/HCO3− exchangers (38). We found that the stimulation-induced, Nhe-mediated alkalinization was not altered in Ae4−/− and Ae2−/− mice, suggesting that Nhe exchanger activity is comparable in these mice. These latter results also imply that carbonic anhydrase activity is unchanged in Ae4−/− and Ae2−/− mice. Carbonic anhydrases appear to be important modulators of Cl−/HCO3− exchanger activity in several cell types, including salivary gland acinar cells (12, 48). Moreover, the resting intracellular pH was not different in Ae4−/− and Ae2−/− mice (Table 1). Although pH experiments do not directly measure carbonic anhydrase activity, the lack of an effect on the resting intracellular pH or on the intracellular pH during stimulation in Ae4−/− and Ae2−/− mice suggests that carbonic anhydrase activity is not significantly affected in these animals.
The hyposalivation observed in Ae4−/− mice is consistent with the Ae4 anion exchanger playing a major role in HCO3−-dependent, Cl−-driven salivation. However, such organ level studies do not directly address which anion exchangers are functionally expressed in submandibular acinar cells. Our results verified that the major Cl− uptake pathway in submandibular acinar cells is the bumetanide-sensitive Na+-K+-2Cl− cotransporter (Fig. 3), consistent with previous reports of the Nkcc1 activity in parotid acinar cells (9, 11). We also recorded a significant bumetanide-resistant Cl− uptake pathway, which was totally dependent on HCO3−. This observation is in agreement with the expression of Cl−/HCO3− exchangers in submandibular acinar cells. Slc4a and Slc26a gene families encode anion exchangers that mediate Cl−/HCO3− exchanger in several tissues (15, 16). Of these, Ae2 (Slc4a2) and Ae4 (Slc4a9) are expressed in salivary glands (11, 12, 18, 48). Ae2 is a widely expressed electroneutral Cl−/HCO3− exchanger, whereas Ae4 has a more restricted tissue distribution, and its transport mechanism remains controversial (16, 18, 54–57). Here, we investigated the functional expression of Ae2 and Ae4 in submandibular acinar cells from acinus-specific Ae2−/− and systemic Ae4−/− mice.
Our results demonstrate that nearly all of the Cl−/HCO3− exchanger activity in submandibular acinar cells is dependent on Ae4 and Ae2 expression. The HCO3−-dependent secretion model presumes that anion exchangers are localized to the basolateral membrane of acinar cells. Ae2 is expressed in the basolateral membrane in acinar cells (12); however, Ae4 had not been previously detected in salivary gland acinar cells. Antibodies to Ae4 exchanger protein failed to show specific labeling in submandibular glands using tissue from Ae4−/− mice as negative controls. Our functional data argue for basolateral localization of Ae4 in acinar cells. Nevertheless, we cannot rule out the possibility that Ae4 is in the apical membrane, where it may be functionally coupled to apical Cl− channels to mediate fluid secretion by a mechanism similar to that described in the pancreatic ducts (5).
Our results clearly demonstrate that Ae4 and Ae2 are both functionally expressed in submandibular acinar cells; however, only Ae4 appears to significantly support fluid secretion during combined muscarinic and β-adrenergic receptor stimulation. Dual stimulation is observed in vivo in glands that are innervated by both the parasympathetic and sympathetic systems. It is worth noting that the Cl− chemical gradient favors its uptake by an electroneutral Cl−/HCO3− exchanger; therefore, it is not clear why Ae2 does not appear to be very important for Cl− uptake-dependent fluid secretion. One conceivable explanation is differential regulation of Ae4 and/or Ae2 by intracellular factors triggered by muscarinic and β-adrenergic stimulation. Indeed, in contrast to the increased Cl− uptake induced by only IPR or CCh plus IPR, the rate of Cl− uptake stimulated with only the muscarinic agonist CCh was not significantly different in Ae4−/− mice. Moreover, Cl− uptake stimulated by CCh, IPR, or CCh plus IPR is essentially identical in Ae2−/− mice and their controls (Table 1). These unexpected results reveal a unique regulation of the fluid secretion process in submandibular acinar cells that relies on the expression of the Ae4 anion exchanger and strongly suggests that Ae4 is activated by cAMP. We also found that Ae4 activity depends on Na+. The Na+ dependence of Ae4 can be explained by either an allosteric modulation of Ae4-mediated Cl−/HCO3− exchange by Na+ or the Ae4 exchanger transports Na+, Cl−, and HCO3−. However, these hypotheses require more investigation to unravel the complexity of the Ae4-mediated ion transport process.
The present study clearly demonstrates that Ae4 mediates Cl−/HCO3− exchange in mouse submandibular acinar cells and that its activity is important for the secretion of saliva when stimulated by a combination of muscarinic and β-adrenergic receptor agonists. Ae4-mediated activity requires β-adrenergic signaling, suggesting a fluid secretory mechanism dependent on cAMP, whereas the Ae2 anion exchanger does not appear to play a significant part in this process. The underlying mechanism appears to be most important during sustained stimulation (i.e. after 2–3 min of stimulation). Stimulation causes a dramatic increase in Na+/H+ exchanger activity, which increases the intracellular pH and HCO3− concentration (see Fig. 2C and Refs. 58 and 59). It is important to note that the Na+/H+ exchanger-induced alkalization has a slow time course that is consistent with the time delay noted for the sustained decrease in salivation observed in Ae4−/− mice. This increase in [HCO3−] would enhance Cl− uptake via Cl−/HCO3− exchange, in accordance with a role for Ae4 as a major Cl− uptake mechanism during sustained secretion.
Acknowledgments
We thank Drs. Z. Borok, E. D. Crandall, and U. P. Flodby for kindly providing the Aqp5-Cre (ACID) mice.
This work was supported, in whole or in part, by the National Institutes of Health (NIH), NIDCR, Division of Intramural Research (to the Secretory Mechanisms and Dysfunction Section) and NIH Grant DK050594 (to G. E. S.).
- CCh
- carbachol
- IPR
- isoproterenol
- SPQ
- 6-methoxy-N-(3-sulfopropyl) quinolinium
- BCECF-AM
- 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester
- EZA
- 6-ethoxyzolamide.
REFERENCES
- 1. Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073–1080 [DOI] [PubMed] [Google Scholar]
- 2. Quinton P. M. (1999) Physiological basis of cystic fibrosis: a historical perspective. Physiol. Rev. 79, S3–S22 [DOI] [PubMed] [Google Scholar]
- 3. Lee M. G., Muallem S. (2008) Pancreatitis: the neglected duct. Gut 57, 1037–1039 [DOI] [PubMed] [Google Scholar]
- 4. Atkinson J. C., Wu A. J. (1994) Salivary gland dysfunction: causes, symptoms, treatment. J. Am. Dent. Assoc. 125, 409–416 [DOI] [PubMed] [Google Scholar]
- 5. Lee M. G., Ohana E., Park H. W., Yang D., Muallem S. (2012) Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol. Rev. 92, 39–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koefoed-Johnsen V., Ussing H. H. (1958) The nature of the frog skin potential. Acta Physiol. Scand. 42, 298–308 [DOI] [PubMed] [Google Scholar]
- 7. Reuss L. (2001) Ussing's two-membrane hypothesis: the model and half a century of progress. J. Membr. Biol. 184, 211–217 [DOI] [PubMed] [Google Scholar]
- 8. Catalán M. A., Peña-Munzenmayer G., Melvin J. E. (2014) Ca2+-dependent K+ channels in exocrine salivary glands. Cell Calcium 55, 362–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melvin J. E., Turner R. J. (1992) Cl− fluxes related to fluid secretion by the rat parotid: involvement of Cl−-HCO3− exchange. Am. J. Physiol. 262, G393–G398 [DOI] [PubMed] [Google Scholar]
- 10. Nauntofte B. (1992) Regulation of electrolyte and fluid secretion in salivary acinar cells. Am. J. Physiol. 263, G823–G837 [DOI] [PubMed] [Google Scholar]
- 11. Evans R. L., Park K., Turner R. J., Watson G. E., Nguyen H. V., Dennett M. R., Hand A. R., Flagella M., Shull G. E., Melvin J. E. (2000) Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J. Biol. Chem. 275, 26720–26726 [DOI] [PubMed] [Google Scholar]
- 12. Nguyen H. V., Stuart-Tilley A., Alper S. L., Melvin J. E. (2004) Cl−/HCO3− exchange is acetazolamide sensitive and activated by a muscarinic receptor-induced [Ca2+]i increase in salivary acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G312–G320 [DOI] [PubMed] [Google Scholar]
- 13. Case R. M., Conigrave A. D., Favaloro E. J., Novak I., Thompson C. H., Young J. A. (1982) The role of buffer anions and protons in secretion by the rabbit mandibular salivary gland. J. Physiol. 322, 273–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pirani D., Evans L. A., Cook D. I., Young J. A. (1987) Intracellular pH in the rat mandibular salivary gland: the role of Na-H and Cl-HCO3 antiports in secretion. Pflugers Arch. 408, 178–184 [DOI] [PubMed] [Google Scholar]
- 15. Pushkin A., Kurtz I. (2006) SLC4 base (HCO3−, CO32−) transporters: classification, function, structure, genetic diseases, and knockout models. Am. J. Physiol. Renal Physiol. 290, F580–F599 [DOI] [PubMed] [Google Scholar]
- 16. Romero M. F., Chen A. P., Parker M. D., Boron W. F. (2013) The SLC4 family of bicarbonate (HCO3−) transporters. Mol. Aspects Med. 34, 159–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parker M. D., Boron W. F. (2013) The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol. Rev. 93, 803–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ko S. B., Luo X., Hager H., Rojek A., Choi J. Y., Licht C., Suzuki M., Muallem S., Nielsen S., Ishibashi K. (2002) AE4 is a DIDS-sensitive Cl−/HCO3− exchanger in the basolateral membrane of the renal CCD and the SMG duct. Am. J. Physiol. Cell Physiol. 283, C1206–C1218 [DOI] [PubMed] [Google Scholar]
- 19. Melvin J. E., Yule D., Shuttleworth T., Begenisich T. (2005) Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu. Rev. Physiol. 67, 445–469 [DOI] [PubMed] [Google Scholar]
- 20. Gawenis L. R., Ledoussal C., Judd L. M., Prasad V., Alper S. L., Stuart-Tilley A., Woo A. L., Grisham C., Sanford L. P., Doetschman T., Miller M. L., Shull G. E. (2004) Mice with a targeted disruption of the AE2 Cl−/HCO3− exchanger are achlorhydric. J. Biol. Chem. 279, 30531–30539 [DOI] [PubMed] [Google Scholar]
- 21. Coury F., Zenger S., Stewart A. K., Stephens S., Neff L., Tsang K., Shull G. E., Alper S. L., Baron R., Aliprantis A. O. (2013) SLC4A2-mediated Cl−/HCO3− exchange activity is essential for calpain-dependent regulation of the actin cytoskeleton in osteoclasts. Proc. Natl. Acad. Sci. U.S.A. 110, 2163–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Simpson J. E., Schweinfest C. W., Shull G. E., Gawenis L. R., Walker N. M., Boyle K. T., Soleimani M., Clarke L. L. (2007) PAT-1 (Slc26a6) is the predominant apical membrane Cl−/HCO3− exchanger in the upper villous epithelium of the murine duodenum. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1079–G1088 [DOI] [PubMed] [Google Scholar]
- 23. Flodby P., Borok Z., Banfalvi A., Zhou B., Gao D., Minoo P., Ann D. K., Morrisey E. E., Crandall E. D. (2010) Directed expression of Cre in alveolar epithelial type 1 cells. Am. J. Respir. Cell Mol. Biol. 43, 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krane C. M., Melvin J. E., Nguyen H. V., Richardson L., Towne J. E., Doetschman T., Menon A. G. (2001) Salivary acinar cells from aquaporin 5-deficient mice have decreased membrane water permeability and altered cell volume regulation. J. Biol. Chem. 276, 23413–23420 [DOI] [PubMed] [Google Scholar]
- 25. Ma T., Song Y., Gillespie A., Carlson E. J., Epstein C. J., Verkman A. S. (1999) Defective secretion of saliva in transgenic mice lacking aquaporin-5 water channels. J. Biol. Chem. 274, 20071–20074 [DOI] [PubMed] [Google Scholar]
- 26. Nakamoto T., Brown D. A., Catalán M. A., Gonzalez-Begne M., Romanenko V. G., Melvin J. E. (2009) Purinergic P2X7 receptors mediate ATP-induced saliva secretion by the mouse submandibular gland. J. Biol. Chem. 284, 4815–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomas J. A., Buchsbaum R. N., Zimniak A., Racker E. (1979) Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18, 2210–2218 [DOI] [PubMed] [Google Scholar]
- 28. Nakamoto T., Srivastava A., Romanenko V. G., Ovitt C. E., Perez-Cornejo P., Arreola J., Begenisich T., Melvin J. E. (2007) Functional and molecular characterization of the fluid secretion mechanism in human parotid acinar cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R2380–R2390 [DOI] [PubMed] [Google Scholar]
- 29. Romanenko V. G., Catalán M. A., Brown D. A., Putzier I., Hartzell H. C., Marmorstein A. D., Gonzalez-Begne M., Rock J. R., Harfe B. D., Melvin J. E. (2010) Tmem16A encodes the Ca2+-activated Cl− channel in mouse submandibular salivary gland acinar cells. J. Biol. Chem. 285, 12990–13001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Romanenko V. G., Nakamoto T., Catalán M. A., Gonzalez-Begne M., Schwartz G. J., Jaramillo Y., Sepúlveda F. V., Figueroa C. D., Melvin J. E. (2008) Clcn2 encodes the hyperpolarization-activated chloride channel in the ducts of mouse salivary glands. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G1058–G1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Catalán M. A., Nakamoto T., Gonzalez-Begne M., Camden J. M., Wall S. M., Clarke L. L., Melvin J. E. (2010) Cftr and ENaC ion channels mediate NaCl absorption in the mouse submandibular gland. J. Physiol. 588, 713–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paulais M., Turner R. J. (1992) Beta-adrenergic upregulation of the Na+-K+-2Cl− cotransporter in rat parotid acinar cells. J. Clin. Invest. 89, 1142–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reynolds A., Parris A., Evans L. A., Lindqvist S., Sharp P., Lewis M., Tighe R., Williams M. R. (2007) Dynamic and differential regulation of NKCC1 by calcium and cAMP in the native human colonic epithelium. J. Physiol. 582, 507–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Evans R. L., Turner R. J. (1997) Upregulation of Na+-K+-2Cl− cotransporter activity in rat parotid acinar cells by muscarinic stimulation. J. Physiol. 499, 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sekler I., Kobayashi S., Kopito R. R. (1996) A cluster of cytoplasmic histidine residues specifies pH dependence of the AE2 plasma membrane anion exchanger. Cell 86, 929–935 [DOI] [PubMed] [Google Scholar]
- 36. Chao A. C., Dix J. A., Sellers M. C., Verkman A. S. (1989) Fluorescence measurement of chloride transport in monolayer cultured cells. Mechanisms of chloride transport in fibroblasts. Biophys. J. 56, 1071–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park K., Evans R. L., Watson G. E., Nehrke K., Richardson L., Bell S. M., Schultheis P. J., Hand A. R., Shull G. E., Melvin J. E. (2001) Defective fluid secretion and NaCl absorption in the parotid glands of Na+/H+ exchanger-deficient mice. J. Biol. Chem. 276, 27042–27050 [DOI] [PubMed] [Google Scholar]
- 38. Park K., Evans R. L., Melvin J. E. (2000) Functional roles of Na+/H+ exchanger isoforms in saliva secretion. J. Korean Med. Sci. 15, S5–S6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Flagella M., Clarke L. L., Miller M. L., Erway L. C., Giannella R. A., Andringa A., Gawenis L. R., Kramer J., Duffy J. J., Doetschman T., Lorenz J. N., Yamoah E. N., Cardell E. L., Shull G. E. (1999) Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 274, 26946–26955 [DOI] [PubMed] [Google Scholar]
- 40. Delpire E., Lu J., England R., Dull C., Thorne T. (1999) Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 22, 192–195 [DOI] [PubMed] [Google Scholar]
- 41. Haas M., Forbush B., 3rd. (2000) The Na-K-Cl cotransporter of secretory epithelia. Annu. Rev. Physiol. 62, 515–534 [DOI] [PubMed] [Google Scholar]
- 42. Grubb B. R., Lee E., Pace A. J., Koller B. H., Boucher R. C. (2000) Intestinal ion transport in NKCC1-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 279, G707–G718 [DOI] [PubMed] [Google Scholar]
- 43. Grubb B. R., Pace A. J., Lee E., Koller B. H., Boucher R. C. (2001) Alterations in airway ion transport in NKCC1-deficient mice. Am. J. Physiol. Cell Physiol. 281, C615–C623 [DOI] [PubMed] [Google Scholar]
- 44. Seidler U., Bachmann O., Jacob P., Christiani S., Blumenstein I., Rossmann H. (2001) Na+/HCO3− cotransport in normal and cystic fibrosis intestine. JOP 2, 247–256 [PubMed] [Google Scholar]
- 45. Walker N. M., Flagella M., Gawenis L. R., Shull G. E., Clarke L. L. (2002) An alternate pathway of cAMP-stimulated Cl secretion across the NKCC1-null murine duodenum. Gastroenterology 123, 531–541 [DOI] [PubMed] [Google Scholar]
- 46. Gawenis L. R., Bradford E. M., Alper S. L., Prasad V., Shull G. E. (2010) AE2 Cl−/HCO3− exchanger is required for normal cAMP-stimulated anion secretion in murine proximal colon. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G493–G503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Novak I., Young J. A. (1986) Two independent anion transport systems in rabbit mandibular salivary glands. Pflugers Arch. 407, 649–656 [DOI] [PubMed] [Google Scholar]
- 48. Gonzalez-Begne M., Nakamoto T., Nguyen H. V., Stewart A. K., Alper S. L., Melvin J. E. (2007) Enhanced formation of a HCO3− transport metabolon in exocrine cells of Nhe1−/− mice. J. Biol. Chem. 282, 35125–35132 [DOI] [PubMed] [Google Scholar]
- 49. Suzuki M., Morita T., Iwamoto T. (2006) Diversity of Cl− channels. Cell. Mol. Life Sci. 63, 12–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Faelli A., Tosco M., Orsenigo M. N., Esposito G. (1984) Effects of the stilbene derivatives SITS and DIDS on intestinal ATPase activities. Pharmacol. Res. Commun. 16, 339–350 [DOI] [PubMed] [Google Scholar]
- 51. Vega F. V., Cabero J. L., Mårdh S. (1988) Inhibition of H,K-ATPase and Na,K-ATPase by DIDS, a disulphonic stilbene derivative. Acta Physiol. Scand. 134, 543–547 [DOI] [PubMed] [Google Scholar]
- 52. Shahidullah M., Wei G., Delamere N. A. (2013) DIDS inhibits Na-K-ATPase activity in porcine nonpigmented ciliary epithelial cells by a Src family kinase-dependent mechanism. Am. J. Physiol. Cell Physiol. 305, C492–C501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kleinboelting S., Diaz A., Moniot S., van den Heuvel J., Weyand M., Levin L. R., Buck J., Steegborn C. (2014) Crystal structures of human soluble adenylyl cyclase reveal mechanisms of catalysis and of its activation through bicarbonate. Proc. Natl. Acad. Sci. U.S.A. 111, 3727–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chambrey R., Kurth I., Peti-Peterdi J., Houillier P., Purkerson J. M., Leviel F., Hentschke M., Zdebik A. A., Schwartz G. J., Hübner C. A., Eladari D. (2013) Renal intercalated cells are rather energized by a proton than a sodium pump. Proc. Natl. Acad. Sci. U.S.A. 110, 7928–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tsuganezawa H., Kobayashi K., Iyori M., Araki T., Koizumi A., Watanabe S., Kaneko A., Fukao T., Monkawa T., Yoshida T., Kim D. K., Kanai Y., Endou H., Hayashi M., Saruta T. (2001) A new member of the HCO3− transporter superfamily is an apical anion exchanger of β-intercalated cells in the kidney. J. Biol. Chem. 276, 8180–8189 [DOI] [PubMed] [Google Scholar]
- 56. Xu J., Barone S., Petrovic S., Wang Z., Seidler U., Riederer B., Ramaswamy K., Dudeja P. K., Shull G. E., Soleimani M. (2003) Identification of an apical Cl−/HCO3− exchanger in gastric surface mucous and duodenal villus cells. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G1225–G1234 [DOI] [PubMed] [Google Scholar]
- 57. Parker M. D., Boron W. F., Tanner M. J. A. (2002) Characterization of human “AE4” as an electroneutral, sodium-dependent bicarbonate transporter. FASEB J. 16, A796 [Google Scholar]
- 58. Evans R. L., Bell S. M., Schultheis P. J., Shull G. E., Melvin J. E. (1999) Targeted disruption of the Nhe1 gene prevents muscarinic agonist-induced up-regulation of Na+/H+ exchange in mouse parotid acinar cells. J. Biol. Chem. 274, 29025–29030 [DOI] [PubMed] [Google Scholar]
- 59. Nguyen H. V., Shull G. E., Melvin J. E. (2000) Muscarinic receptor-induced acidification in sublingual mucous acinar cells: loss of pH recovery in Na+-H+ exchanger-1 deficient mice. J. Physiol. 523, 139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]