

Background: The molecular basis of full-length heparin activation of Slit-Robo is poorly understood, despite its importance in nervous system development.
Results: Two separate binding sites of Robo1-full length heparin interaction were identified.
Conclusion: A model for heparin/heparan sulfate binding and activation of the Slit-Robo complex is proposed.
Significance: A previously unidentified Robo1 low affinity binding site for heparin may be required for signal transduction.
Keywords: Glycosaminoglycan, Heparin, Heparin-binding Protein, Mass Spectrometry (MS), Structural Biology, Hydroxyl Radical Protein Footprinting, Protein-Carbohydrate Interaction
Abstract
Interaction of transmembrane receptors of the Robo family and the secreted protein Slit provides important signals in the development of the central nervous system and regulation of axonal midline crossing. Heparan sulfate, a sulfated linear polysaccharide modified in a complex variety of ways, serves as an essential co-receptor in Slit-Robo signaling. Previous studies have shown that closely related heparin octasaccharides bind to Drosophila Robo directly, and surface plasmon resonance analysis revealed that Robo1 binds more tightly to full-length unfractionated heparin. For the first time, we utilized electron transfer dissociation-based high spatial resolution hydroxyl radical protein footprinting to identify two separate binding sites for heparin interaction with Robo1: one binding site at the previously identified site for heparin dp8 and a second binding site at the N terminus of Robo1 that is disordered in the x-ray crystal structure. Mutagenesis of the identified N-terminal binding site exhibited a decrease in binding affinity as measured by surface plasmon resonance and heparin affinity chromatography. Footprinting also indicated that heparin binding induces a minor change in the conformation and/or dynamics of the Ig2 domain, but no major conformational changes were detected. These results indicate a second low affinity binding site in the Robo-Slit complex as well as suggesting the role of the Ig2 domain of Robo1 in heparin-mediated signal transduction. This study also marks the first use of electron transfer dissociation-based high spatial resolution hydroxyl radical protein footprinting, which shows great utility for the characterization of protein-carbohydrate complexes.
Introduction
During neuronal development, growing axons are initially attracted to the midline by following specific pathways and encountering a number of guidance cues in their extracellular environment (1). A ligand-receptor system known to be very important in axon guidance involves the interaction of Slit and Robo at the CNS midline (2). Members of the Roundabout (Robo) family of proteins are single-pass transmembrane cell adhesion molecules that are highly conserved across many species, from Caenorhabditis elegans to humans (3). Four Robo proteins (Robo1, Robo2, Robo3/Rig-1, and Robo4/magic Roundabout) have been identified in mammals (4, 5). Robo1–3 are expressed on the surface of growing axons (1), whereas Robo4 is specifically expressed in vascular endothelial cells and hematopoietic stem cells (6). In all identified Robo receptors except for vertebrate Robo4, the Ig1 and Ig2 domains have been evolutionarily conserved and are crucial for forming the Robo-Slit protein-protein complex. Slit-Robo interactions are required in the development of the central nervous system and regulation of axonal midline crossing as well as influencing dendrite repulsion and self-avoidance (7). Slit-Robo signaling is also essential for many neurodevelopmental processes, including formation of the olfactory tract, the optic nerve, and motor axon fasciculation (8, 9). In addition, Slit-Robo signaling contributes to cell migration (10), leukocyte chemotaxis (11), and the development of other tissues, such as the lung, kidney, muscle, and breast (12, 13). Mutations in Robo genes have been linked to multiple human disorders, such as dysplasia (14).
Previous studies have shown that heparan sulfate is required for Slit-Robo signaling (15, 16) and acts at least in part by promoting the formation of a ternary Slit-Robo-HS signaling complex (16–18). Heparan sulfate (HS)2 is a member of the glycosaminoglycan (GAG) family, consisting of linear polysaccharides bearing repeating disaccharide units of (-4GlcNAcα1,4GlcUAβ1-) that are modified by variable epimerization of the uronic acid, possible N-sulfation or deacetylation of the GlcNAc and O-sulfation of both monosaccharides. Heparin is a closely related GAG typically stored in the secretory vesicles of mast cells that tends to be more heavily sulfated. HS and heparin chains are attached to extracellular matrix proteins to form HS proteoglycans (19, 20). HS-protein interactions have been shown to modulate protein activity by various mechanisms, such as stabilizing proteins at their sites of production and in the matrix for future mobilization, protecting proteins against degradation, binding ligands to other receptors, and regulating enzyme activity (20). GAG-protein interactions are essential to many biological processes, including those involving cell-cell communication, cell migration, and stabilization of cellular structure (20). However, the large size, heterogeneous structure, and inherent flexibility of GAG chains make the detailed structural interrogation of protein-GAG interactions difficult, particularly for full-length GAG chains. Previous studies have shown that heparin octasaccharides bind to Drosophila Robo directly, and the heparin dp8 binding sites have been identified by mutagenesis and partial direct crystallographic observation of a bound heparin oligosaccharide (21). These observations indicate that HS/heparin plays a direct and essential role in Slit-Robo signaling. However, these studies of Robo1-heparin binding are all performed on relatively small oligosaccharides. Previous surface plasmon resonance (SPR) analysis of Robo1-heparin interactions revealed that heparin dp8 bound to Robo1 with considerably lower affinity than full-length unfractionated heparin (22). Additionally, heparin dp8 SPR binding curves fit a single state binding model, whereas full-length heparin-Robo1 SPR binding curves fit a two-state binding model, including a fast binding event similar to the single state model for heparin dp8 and an additional slow binding event that is not observed with the dp8 heparin ligands (22). These data indicate that the dp8 ligand does not fully engage the Robo1 binding site, and a structural characterization of the Robo1-unfractionated heparin complex is required to understand the interaction with extended HS/heparin ligands. However, unfractionated heparin is a polydisperse mixture of large, flexible chains (∼10–40 kDa) with a variety of modification patterns. Standard high resolution structural technologies are unable to handle such a diverse mixture of highly dynamic complexes.
Hydroxyl radical protein footprinting (HRPF) coupled with mass spectrometry (MS) has become an increasingly popular structural analysis technique to probe molecular interaction interfaces, given its fast time frame for protein modification, high labeling efficiency, and stability of labeled analyte (23). HRPF oxidizes amino acid side chains at a rate based on a combination of the inherent reactivity and the accessibility to the hydroxyl radical diffusing in solution. This rate dependence on solvent accessibility makes HRPF a very promising technique to probe intact protein structure (24), protein conformational change (25), protein-protein interactions (26), and protein-ligand binding surfaces in solution (27). Generally, in an HRPF experiment, the protein of interest is exposed to diffusing hydroxyl radicals, and the radicals covalently react with the solvent-accessible side chains of residues (28). Certain methods of radical generation for HRPF, including fast photochemical oxidation of proteins (FPOP) (29) and electron accelerator radiolysis (30), are able to complete the labeling process faster than the protein can undergo labeling-induced conformational changes, ensuring that the HRPF is not probing an artifactual structure. The stable modifications to the protein side chains are analyzed by liquid chromatography-mass spectrometry (LC-MS), relatively quantifying modified and unmodified peptides, and the MS signal of the oxidized version(s) of each peptide is compared with the MS signal of the unoxidized version of the same peptide to quantify oxidation. By comparing the same protein under two different states (e.g. ligand-bound versus ligand-free), changes in solvent accessibility caused by the structural differences are detected. Traditional HRPF quantitation was performed on the peptide level, allowing for structural information. If multiple amino acids on the same peptide were oxidized (a common occurrence in HRPF), this method yielded structural information with a spatial resolution limited by the size of the peptide. Although such information is useful for monitoring large scale protein conformational changes or large protein-protein interaction interfaces, it is much less useful for studying smaller protein-ligand interaction interfaces. High spatial resolution HRPF necessitates not only the identification of the sites of oxidation on a residue level but also the quantitation of the level of oxidation at each site within the peptide, providing structural information with higher spatial resolution and more accuracy. Moreover, because HRPF is measuring the changes in the solvent accessibility of the protein, it can be carried out in the presence of a complex mixture of ligands, only some of which bind tightly to the protein.
In our previous work, we have successfully demonstrated the accuracy of electron transfer dissociation (ETD)-based methods for quantifying multiple adjacent sites of isomeric oxidation products as well as the general applicability of ETD-based dissociation to quantify oxidized peptide isomers with multiple sites of oxidation on both a series of synthetic peptide oxidation isomers and peptides from Robo1 Ig1-2 protein generated by HRPF (31). Here, this technology is applied to characterizing the interaction of Robo1 Ig1-2 with unfractionated heparin. We have identified basic residues in and around the previously identified site for high affinity heparin binding (21) as well as a previously unidentified second binding site near the N terminus of Robo1 that is evolutionarily conserved and influences heparin binding as determined by site-directed mutagenesis, SPR, and heparin affinity chromatography. We have also identified residues in the hydrophobic core of the Ig2 domain that show altered solvent accessibility upon binding of unfractionated heparin, suggesting a heparin binding-induced conformational change in the Ig2 domain that may be involved in heparin-induced signal transduction. We propose a model of Robo-Slit-heparin/HS binding and signaling involving the previously identified high affinity heparin binding site and a second low affinity binding site near the N terminus that is present in the Slit-Robo complex, which is important for triggering the conformational changes required for signal transduction. This work also marks the first determination of protein-GAG interaction by high resolution HRPF, providing binding interface information down to the amino acid residue level in some cases.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human Robo1 (Ig domains 1 and 2) was generated as a soluble secreted fusion protein in HEK293 suspension cultures as described previously (22). Briefly, the Robo1 protein coding region was designed and synthesized for in-frame insertion into the pGEn2 mammalian expression vector (32) as a fusion protein expression product. The vector encodes a 25-amino acid signal sequence that is followed by a His8 tag, AviTag, and the “superfolder” GFP coding region. The synthetic Robo1 cassette was subcloned downstream of the vector sequence as an EcoRI-HindIII restriction fragment containing an N-terminal 7-amino acid recognition sequence of the tobacco etch virus (TEV) protease and residues 58–266 of human Robo1 (UniProt Q9Y6N7). For generation of mutants that eliminate the primary heparin binding site (Mutant I, Arg136 → Ala, Lys137 → Ala) and the secondary heparin binding site (Mutant II, Arg62 → A, Arg69 → Ala), the wild type Robo1 expression construct was subjected to site-directed mutagenesis using the QuikChangeTM mutagenesis kit (Stratagene, La Jolla, CA). The wild type recombinant product was expressed in suspension culture mutant HEK293S GnTI− cells (ATCC catalogue no. CRL-3022) by transient transfection as a polyethyleneimine-DNA complex as described previously (33). Following expression for 6 days, the cultures were harvested and clarified by centrifugation at 3500 rpm for 15 min, and the recombinant product was purified by nickel-nitrilotriacetic acid Superflow (Qiagen, Valencia, CA) chromatography as described (22). The eluted GFP-Robo1 fusion protein was concentrated to ∼1 mg/ml by ultrafiltration with a 10-kDa molecular mass cut-off membrane (Millipore, Billerica, MA), and tags and glycans were excised by treatment with recombinant TEV protease and endoglycosidase F1 and further purified as described (33). For surface plasmon resonance binding studies, wild type and mutant forms of Robo1 were expressed in wild type HEK293 cells (FreeStyle 293-F cells, Invitrogen) and purified by nickel-nitrilotriacetic acid chromatography and subjected to TEV protease cleavage without the addition of endoglycosidase F1 and further purified as described (33). The protein preparation was concentrated by ultrafiltration to ∼3 ml and was further purified on a Superdex 75 column (GE Healthcare) preconditioned with a buffer containing 137 mm NaCl, 2.7 mm KCl, 10 mm NaH2PO4, 2 mm KH2PO4, pH 7.4. Peak fractions of Robo1 were collected and concentrated. Later MS analysis indicated that the final expressed products are lacking the C-terminal lysine, resulting in an expressed protein of residues 58–265.
HRPF of Proteins
HRPF of Robo1 with and without a heparin ligand was performed by FPOP as described previously (29, 34). Robo1 (20 μm) and unfractionated heparin (estimated at 40 μm based on average mass of heparin chain) were mixed together and incubated for 1 h. Both Robo1 alone and Robo1 with heparin were combined with glutamine and nanopure water to give a final protein concentration of 20 μm Robo1, ∼40 μm unfractionated heparin in a 20-μl final volume. Glutamine was added as a radical scavenger at a concentration of 20 mm. Angiotensin peptide (10 μm) was added to each protein solution as a radical dosimeter to monitor the available radical dose in each protein solution. Concentrated hydrogen peroxide (1 m) was added to each replicate of a triplicate set to give a final concentration of 100 mm just prior to flow through the laser beam path for oxidative modification. The protein samples were loaded into a 100-μl syringe and introduced via a syringe pump coupled to the 100-μm inner diameter fused silica tubing. Samples flowed through the beam path of an EX100 KrF excimer laser at 248 nm with a laser power of 50 mJ/pulse (GAM Laser Inc., Orlando, FL). The flow rate of the syringe pump was set at 12.19 μl/min, with the laser pulse repetition rate set such that each segment of protein sample was irradiated with a single ∼20-ns UV pulse with a 10% unirradiated buffer region between irradiated segments to help account for sample diffusion and laminar flow (29, 35). Samples were collected in a microcentrifuge tube containing methionine amide (0.5 μg/μl) and catalase (0.5 μg/μl) with sodium phosphate (50 mm, pH 7.4) to immediately quench oxidation by destroying excess hydrogen peroxide and other reactive oxygen species. In addition, control samples were made with hydrogen peroxide under the same conditions, but they were not subjected to laser irradiation.
Following irradiation, each sample was lyophilized and resuspended in 20 μl of 50 mm sodium phosphate. Samples were reduced with 5 mm DTT and incubated at 65 °C for 30 min. After cooling to room temperature, samples were digested overnight with one of three proteases: trypsin at 37 °C at an enzyme/protein ratio of 1:20 or Lys-C or Glu-C at a ratio of 1:100. The digested samples were stored at −20 °C for LC-MS/MS analysis.
LC-MS/MS Acquisition and Data Analysis
Samples were analyzed on a LTQ Orbitrap XL (Thermo Fisher Scientific) controlled by Xcalibur version 2.0.7 software (Thermo Fisher Scientific). Samples were loaded on a 75-μm (inner diameter, 105 mm) C18 fritless reverse phase column (packed in house, YMC GEL ODS-AQ120 ÅS-5, Waters) by nitrogen pressure bomb. Samples were eluted directly into the nanospray source of an LTQ Orbitrap XL with a 160-min linear gradient consisting of 5–100% acetonitrile with a constant concentration of 0.1% formic acid over 100 min at a flow rate of 250 nl/min. The spray voltage was set to 2.0 kV, and the temperature of the heated capillary was set to 200 °C. Full MS scans were acquired from m/z 200 to 2000 at a nominal resolution of 60,000 for ions of m/z 400 with the Fourier transform mass spectrometer. To obtain MS/MS data of oxidized peptides, ions were fragmented by ETD with the corresponding parent ion mass list. ETD-based precursor activation was carried out for 100 ms, including charge state-dependent supplemental activation. Precursor ions were isolated with a width of 3 m/z units.
Unoxidized Robo1 controls, oxidized Robo1, and oxidized Robo1-unfractionated heparin complex peptide sequences were identified using MASCOT version 2.2.2 (Matrix Science, London, UK) and ByOnic version 1.2-250 (Protein Metrics, San Carlos, CA). All tandem mass spectra assignments and sites of oxidation were verified manually due to peptide modification complexity. The tryptic and Lys-C-digested peptides and corresponding oxidation products were identified from the LC-MS runs manually to calculate the average oxidation events per peptide in the sample. Peptide level quantitation of the average oxidation events per peptide is calculated by summing the ion intensities of all of the oxidized peptide masses multiplied by the number of oxidation events required for the mass shift (e.g. one event for +16, two events for +32) and then dividing by the sum of the ion intensities of all unoxidized and oxidized peptide masses,
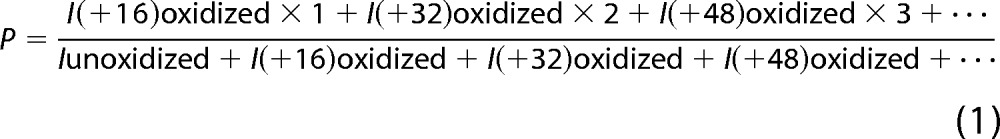 |
where P denotes the average oxidation events per peptide and I represents the intensity of each of oxidized and unoxidized peptide forms.
Residue level quantitation was calculated by the fragment ion intensities from ETD to determine oxidation extent at specific residue sites based on our previous study (31) and a similar approach reported by Jumper et al. (36) for carbene-based footprinting. Briefly, oxidized peptide with oxidation at multiple sites can generate both oxidized and unoxidized sequence ions in its MS/MS spectrum. The fractional oxidation of a given sequence ion is defined as the ratio of the oxidized sequence ion intensity to the sum of the intensity of the corresponding oxidized and unoxidized sequence ion,
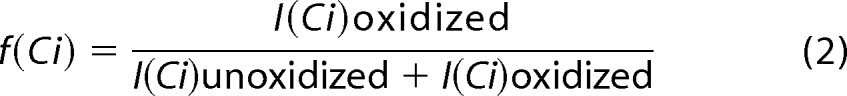 |
where f(Ci) denotes the fractional oxidization of sequence ion i (e.g. oxidized c3 ions generated by ETD), and I(Ci) denotes the intensity of the oxidized and unoxidized ion of interest (e.g. oxidized c3 ion plus unoxidized c3 ion). The absolute level of oxidation for a given amino acid residue i is based on both the average oxidation event of peptide and the fractional oxidation of the corresponding sequence ions, as shown in Equation 3,
where P is the average oxidation events/peptide as derived from Equation 1, and the term in square brackets is the fractional oxidation difference of two adjacent sequence ions, Ci and Ci (−1). Multiplying the average number of oxidations/peptide by the fraction of that oxidation that occurs on a given amino acid residue yields the average oxidation events per residue. For ETD MS/MS spectra where ETD fragmentation ions are not adjacent in sequence, fractional oxidation for multiple contiguous residues within the peptide can be calculated by using non-adjacent ETD fragments in Equation 3, resulting in a spatial resolution of HRPF that is limited by the distance between ETD fragmentation sites.
Dynamic Light Scattering
A DynaPro NanoStar system (Wyatt Technology Corp., Santa Barbara, CA) was used to conduct all dynamic light scattering (DLS) measurements. Laser wavelength was 825 nm. Samples of different concentration were held at 25 °C by a temperature-controlled sample holder and allowed to equilibrate for 120 s prior to analysis. Each size measurement was determined from 20 scans, 5 s per scan. All DLS data were collected and analyzed using DYNAMIC V6TM software, version 6.3.01. Hydrodynamic radius calculations based on the Robo1 XRC structure were performed using WINHYDROPRO10 (37).
Heparin Affinity Chromatography
TEV protease-digested, purified Robo1 proteins devoid of the GFP fusion tags (500 μg each of wild type, mutant I, or mutant II Robo1 protein) were injected onto a 5-ml HiTrap Heparin HP column (GE Healthcare) equilibrated in 0.05 m Tris-HCl, pH 7.5. Bound proteins were eluted with a linear NaCl gradient of 0–1 m NaCl over 5 column volumes at a flow rate of 1 ml/min.
Surface Plasmon Resonance
The biotinylated heparin was prepared by reaction of sulfo-N-hydroxysuccinimide long chain biotin (Thermo Fisher Scientific) with free amino groups of unsubstituted glucosamine residues in the polysaccharide chain following a published procedure (38). The SPR analysis was conducted using a BIAcore 3000 (GE Healthcare) SPR instrument. The biotinylated heparin was immobilized to a streptavidin chip (GE Healthcare,) based on the manufacturer's protocol. In brief, a 20-μl solution of the heparin-biotin conjugate (0.1 mg/ml) in HBS-EP running buffer was injected over flow cell 2 of the streptavidin chip at a flow rate of 10 μl/min. The successful immobilization of heparin was confirmed by the observation of a 100–200 resonance unit (RU) increase in the sensor chip. The control flow cell (flow cell 1) was prepared by a 1-min injection with saturated biotin. To measure the interaction between heparin and Robo1, protein samples were diluted in HBS-EP buffer (0.01 m HEPES, 0.15 m NaCl, 3 mm EDTA, 0.005% surfactant P20, pH 7.4). Different dilutions of protein samples were injected at a flow rate of 50 μl/min. At the end of the sample injection, the same buffer was flowed over the sensor surface to facilitate dissociation. After a 2-min dissociation time, the sensor surface was regenerated by injecting with 50 μl of 0.25% SDS to obtain a fully regenerated surface. The response was monitored as a function of time (sensorgram) at 25 °C. Data were analyzed using BIAevaluate version 4.0.1 using a 1:1 Langmuir model (corrected for bulk signal); Robo1 (A) binds to heparin (B) to form complex (AB),
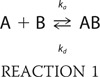 |
where ka is the apparent association rate constant (m−1 s−1) and kd is the apparent dissociation rate constant (s−1). The apparent dissociation equilibrium constant (KD) is kd/ka.
RESULTS
The interaction of Robo1 and heparin previously described by SPR analysis suggests that Robo1 binds more tightly to full-length unfractionated heparin, and the binding event with unfractionated heparin contains a slow component that is not detected in the Robo1-heparin dp8 interaction (22). To investigate the binding site between Robo1 and unfractionated heparin at a high spatial resolution, Robo1 and Robo1-heparin samples were aliquoted into three replicates, and each sample was oxidized for HRPF by FPOP. As the protein is exposed to the short burst of hydroxyl radicals, the radicals react with the amino acid side chains with an apparent rate that is a function of the amino acid sequence, which is constant between samples, and the exposure of the side chain to the solvent, which changes at the binding interface as the heparin binds to the protein, as well as changing in the vicinity of any heparin binding-induced conformational changes. Immediately after FPOP oxidation, the samples were quenched to eliminate excess hydrogen peroxide and any other long lived secondary oxidants, digested with multiple enzymes to achieve full sequence coverage of the protein, and analyzed by LC-MS/MS.
High Spatial Resolution HRPF of Robo1-Heparin Complex
The Robo1 protein analyzed in this study consists of the two domains Ig1-2 with 209 amino acids and an N-glycan site (Fig. 1). To study the interaction of Robo1 with heparin, a Robo-heparin complex was prepared by mixing Robo1 with unfractionated heparin at an approximate 1:2 molar ratio. The samples were oxidized under the same experimental conditions with a peptide radical dosimeter used to ensure that all samples received the same amount of available hydroxyl radical. Peptide level quantitation of oxidation for Robo1-heparin was performed and compared with the extent of oxidation of Robo1 alone (Fig. 2). Chromatography of the oxidized residues was intentionally performed so that peptide oxidation isomers did not resolve. A residue level quantitation was employed by measuring the intensity of unoxidized and oxidized c/z ions generated by ETD and calculating the relative average oxidation for each residue as described under “Experimental Procedures.” In order to determine the statistical difference in the amount of oxidation for these residues, two-tailed independent Student's t tests were used for statistical analysis, with an α of 0.05 selected for statistical significance. Residues exhibiting statistically significant differences upon heparin binding are shown in Table 1. The amounts of oxidation of Robo1 and Robo1-heparin and protection for these residues are also shown. As can be seen in Table 1, residues Arg62, Phe66-Pro67-Pro68, Arg69, Ile70-Val71, Ser80-Lys81, and Ala166-Ile167-Leu168-Arg169 have heparin-bound oxidation levels less than 50% of the Robo1 without heparin, indicating strong protection upon heparin binding. Phe129-Leu130-Arg131, Cys147, Met180, and Met189 have heparin-bound oxidation levels greater than 50% of Robo1 without heparin, although statistically significant protection is observed.
FIGURE 1.

Amino acid sequence of Robo1 Ig1-2 domain. Identified oxidized amino acids are colored red. The underline indicates the N-glycan site. Unoxidized residues in the Ig1 domain are colored green; unoxidized residues in the Ig2 domain are colored blue.
FIGURE 2.

Hydroxyl radical protein footprinting of Robo1-heparin. Extent of oxidation of Robo1 with unfractionated heparin (gray bars) and without unfractionated heparin (black bars) at the peptide level is shown. Error bars, S.D. from a triplicate set of HRPF analyses.
TABLE 1.
Independent two-tailed Student's t test of HRPF between Robo1 and Robo1-heparin
The oxidation of Robo1 and Robo1-heparin at residue level are shown as the average number of oxidation events per residue. S.D. values were calculated from triplicate samples.
| Protected residues | Average oxidation events/residue Robo1 ± S.D. | Average oxidation events/residue Robo1-heparin ± S.D. | p value | Percentage protection upon heparin binding |
|---|---|---|---|---|
| % | ||||
| Arg62 | 0.198 ± 0.035 | 0.048 ± 0.023 | 0.02 | 75.8 |
| Phe66-Pro67-Pro68 | 0.185 ± 0.011 | 0.040 ± 0.014 | <0.01 | 78.4 |
| Arg69 | 0.012 ± 0.003 | 0.003 ± 0.001 | 0.03 | 75.0 |
| Ile70-Val71 | 0.056 ± 0.001 | 0.015 ± 0.005 | <0.01 | 73.2 |
| Ser80-Lys81 | 0.062 ± 0.016 | 0.016 ± 0.003 | 0.04 | 74.2 |
| Phe129-Leu130-Arg131 | 0.433 ± 0.024 | 0.364 ± 0.002 | <0.01 | 15.9 |
| Cys147 | 0.582 ± 0.063 | 0.399 ± 0.028 | 0.03 | 31.4 |
| Ala166-Ile167-Leu168-Arg169 | 0.057 ± 0.001 | 0.028 ± 0.003 | <0.01 | 50.9 |
| Met180 | 1.078 ± 0.028 | 0.726 ± 0.021 | <0.01 | 32.6 |
| Met189 | 0.967 ± 0.030 | 0.683 ± 0.020 | 0.01 | 29.4 |
Identification of Heparin Binding Sites
Heparin-protein binding interactions are most often mediated by interactions between basic residues and the negative charges present on heparin. Previous mutagenesis studies of Robo1 of Drosophila melanogaster found four basic residues that affect elution from a heparin column: Lys81, Arg131, Arg136, and Lys137 (21). These residues are conserved within human Robo1. To evaluate the definition of the binding site for full-length heparin, we examined the residue level HRPF protection information at or around basic residues in the structure. Residue level quantitation by ETD for peptide 61–69 was employed to determine the oxidation extent at specific residue sites (Fig. 3a). An example product ion ETD spectrum for singly oxidized peptide 61–69 is shown in Fig. 4. Residues Leu61, Arg62, Phe66-Pro67-Pro68, and Arg69 all show significant protection upon binding to heparin. The average number of oxidation events per residue for Arg62 decreases from 0.198 to 0.048. Residues Phe66-Pro67-Pro68 are not typical for a binding site for heparin because heparin interacts with positively charged amino acids in most GAG-protein interaction systems. These residues, which show decreased oxidation from 0.185 to 0.040, are adjacent to the basic residue Arg69 shown in Fig. 3b, which is also a minor oxidation target that shows a large oxidation decrease from 0.012 to 0.003, which is a 75% reduction in oxidation and is statistically significant. Protection of the Phe66-Pro67-Pro68 region and the Ile70-Val71 region before and after the Arg69 site (discussed below) confirms the protection observed in the Arg69 residue through binding to heparin. These residues were not previously detected as heparin-binding and are distal to the binding site identified previously (21).
FIGURE 3.
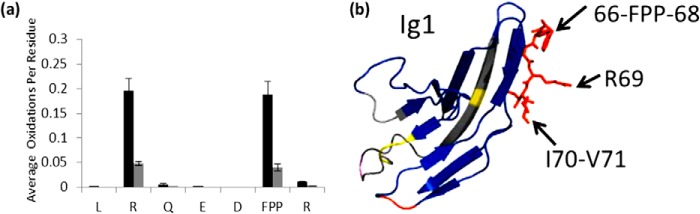
Residues involved in binding with unfractionated heparin. a, extent of oxidation of Robo1 alone (black bars) compared with heparin-bound Robo1 (gray bars) at the residue level. Error bars, S.D. from a triplicate set of experiments. b, protected residues Phe66-Pro67-Pro68, Ile70, and Val71 surround protected basic residue Arg69 in three-dimensional space, suggesting that Arg69 interacts with heparin, which shields neighboring residues. Protected residue Arg62 is not shown in the crystal structure.
FIGURE 4.

Representative ETD spectrum of singly oxidized peptide LRQEDFFPR for Robo1 alone (a) and Robo1 with heparin (b). The asterisks indicate the product ions that are oxidized. The ETD spectrum shows unoxidized and oxidized c ions as pairs from c2 to c8, whereas unoxidized and oxidized c1, c6, and c7 ions are absent in the spectrum. Oxidized z3 and unoxidized and oxidized z4–z7 ions generated by ETD are also shown in the spectrum. The intensities of c ions are preferred to calculate for quantitation because the interference between unoxidized y ions and oxidized z ions prevents accurate quantification in mass spectrometers with lower resolution, as demonstrated by our previous work.
Protection was also observed in and around the previously identified heparin binding site. Residues Phe129-Leu130-Arg131 show a decreased amount of oxidation from 0.433 to 0.364 (Fig. 5a). Previous mutagenesis and x-ray crystallography studies have indicated that Arg131 is involved in heparin interaction (21), which is strongly supported by our high spatial resolution HRPF data (Fig. 5b). Because we do not have sufficient ETD fragment ions to differentiate oxidation of these 3 amino acids, we must quantitate oxidation of this three amino acid stretch as a whole. Taken together, these 3 amino acids exhibit only modest protection upon heparin binding. However, of the 3 amino acids in this stretch, Phe129 is substantially more chemically reactive and has not been previously implicated in the heparin binding interface. All other factors being equal, the majority of oxidation of this 3-amino acid stretch would be expected to occur on Phe129. If the solvent accessibility of Phe129 is not substantially impacted by heparin binding, even strong protection from heparin binding at Arg131 would be overshadowed by the greater reactivity and negligible protection of Phe129 upon heparin binding.
FIGURE 5.
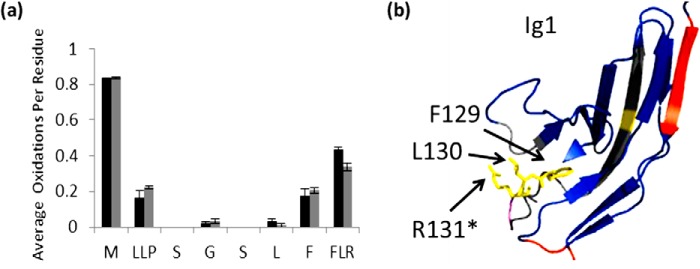
Residues involved in binding with unfractionated heparin. a, extent of oxidation of Robo1 alone (black bars) compared with heparin-bound Robo1 (gray bars) at the residue level. Error bars, S.D. from a triplicate set of experiments. b, structure highlighting Phe129-Leu130-Arg131 are shown in yellow. An asterisk indicates the residue involved in a heparin dp8 binding reported previously (21).
Residue level oxidations for peptide 70–81 are shown in Fig. 6a. The amount of oxidation for residues Ile70-Val71 in peptide 70–81 decreases from 0.056 to 0.015. As mentioned previously, these residues are directly adjacent to Arg69, which is heavily protected upon heparin binding. In addition to protection of Ile70-Val71, peptide 70–81 also shows strong protection of Ser80-Lys81 upon heparin binding. Lys81 was shown to bind with heparin in a previous study (21), which we can confirm by high spatial resolution HRPF. The N-glycosylated peptide 151–169 shows either no oxidation or no protection on its residues except for the C terminus (Fig. 6b). Protected residues Ala166-Ile167-Leu168-Arg169 in peptide 151–169 are at a basic patch of the protein adjacent to residues previously shown to be involved in heparin dp8 binding (Fig. 6c). We have identified significant protection of these residues upon binding to unfractionated heparin, revealing Arg169 to be involved in the extended binding site, along with the previously identified residues involved in dp8 binding (Lys81, Arg131, Arg136, and Lys137) (21). The latter two residues were not identified by HRPF because they were not oxidized under ligand-free or ligand-bound conditions, although they were reported to be involved in binding with heparin. Our inability to detect significant oxidation of these residues in either the free or bound state made us unable to probe these residues directly. No basic residues were found to be protected by heparin binding within the Ig2 domain.
FIGURE 6.
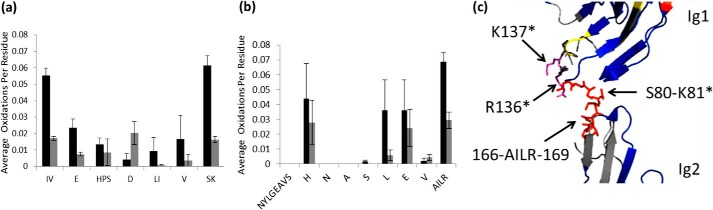
Residues identified as binding sites around basic patch. Error bars, S.D. from a triplicate set of experiments. a, residue level extent of oxidation for peptide 70–81 of Robo1 (black bars) and Robo1-heparin (gray bars). b, residue level extent of oxidation for peptide 151–169 of Robo1 (black bars) and Robo1-heparin (gray bars). c, structure shows the protected residues Ser80-Lys81 and Ala166-Ile167-Leu168-Arg169 (red) are at a basic patch with basic residues in magenta unidentified by HRPF. Asterisks indicate the residue identified as interacting with heparin dp8 previously (21).
Changes of Protection of Non-binding Residues upon Heparin Binding
A significant decrease of oxidation was also shown in residues Cys147, Met180, and Met189 (Fig. 7a). These residues were the sole sites of oxidation in peptides 138–150 and 174–195 with a decrease in oxidation from 0.582 to 0.400 for peptide 138–150 and from 1.121 to 0.761 for peptide 174–195 (Fig. 2). These residues are all highly reactive to hydroxyl radicals. Previous studies have indicated that residues with high inherent reactivities exhibit drastic changes to their HRPF oxidation level in response to relatively modest changes in the average solvent-accessible surface area (27); therefore, although the protection observed in these three residues is statistically significant, the fact that only these three highly reactive non-binding residues were observed to be modestly protected by heparin binding suggests that the changes in structure and/or dynamics they reflect are relatively modest. Larger conformational changes would be expected to result in measurable changes in the oxidation of a wider variety of less reactive amino acids.
FIGURE 7.
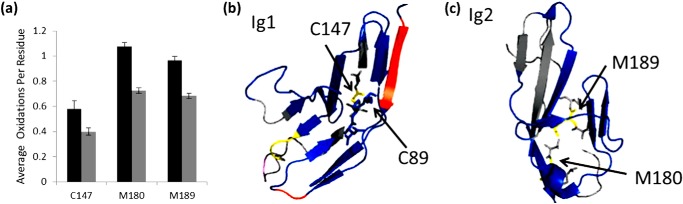
Non-binding residues show significant protections. a, residue level extent of oxidation of Robo1 alone (black bars) compared with heparin-bound Robo1 (gray bars) for residues Cys147, Met180, and Met189. Error bars, S.D. from a triplicate set of experiments. b, structural model showing Cys147 (yellow) involved in the disulfide bond with Cys89 in Ig1 domain. c, hydrophobic residues Met180 and Met189 are shown in yellow in the Ig2 domain of Robo1.
Cys147 is involved in the core disulfide bond of the Ig1 domain (Fig. 7b) and is fully protected from solvent in the static x-ray crystal structure (39). Oxidation of this cysteine in the heparin-free form despite its full burial in the x-ray structure may indicate dynamics in the Ig1 domain in solution. The modest decrease of oxidation of this highly reactive residue upon heparin binding may indicate a possible modest stabilization of the folded structure of the Ig1 domain upon ligand binding. Alternatively, heparin interactions could induce a modest conformational change not captured in the x-ray crystal structure upon binding of dp8. The change in the structure and/or dynamics of the Ig1 domain must be minor to only cause measurable changes in the HRPF footprint for Cys147 and not for other residues we measured as unchanged. It should also be noted that the disulfide partner of Cys147 (Cys89) remains almost wholly protected in the heparin-bound and heparin-free form and is unaffected by heparin binding. Exposure and labeling of only one-half of a disulfide bond has been reported previously for lysozyme, where it indicated exposure of one sulfur of the disulfide bond to solvent (40).
Met180 and Met189 are hydrophobic residues in and around the core of the Ig2 domain (Fig. 7c) and, similar to Cys147, are highly reactive with hydroxyl radicals. Protection of these residues in the Ig2 domain distal from the extended heparin binding site suggests that this protection is probably due to either a heparin-induced multimerization event or a binding-induced change in conformation and/or dynamics of the Ig2 domain. Previous studies showed no evidence of Robo1 multimerization upon heparin binding, and DLS of Robo1 at a concentration 5 times greater than that used for HRPF also showed that Robo1 exists as a monomer. DLS of the Robo1-heparin complex was inconclusive due to the polydisperse nature of the ligand but indicated that oligomerization beyond a dimer was unlikely (Table 2). Additionally, the lack of protection of any less reactive residues in the Ig2 domain suggests that no stable multimer exists; otherwise, we would expect to see widespread protection along the monomer-monomer interface(s). A very modest change in conformation and/or dynamics of Robo1 upon binding to heparin would be consistent with prior SPR data that indicated a two-state binding model for Robo1 interaction with unfractionated heparin (22), and with the very limited changes in HRPF oxidation observed in Ig2 here.
TABLE 2.
Dynamic light scattering results for Robo1 and unfractionated heparin
| Sample | Ra |
|---|---|
| nm | |
| 0.1 mm Robo1 | 2.7 |
| 1 mm heparin | 2.4 |
| 0.5 mm Robo1 + 1 mm heparin | 3.7 |
a The hydrodynamic radius for the monomer of Robo1 as calculated from the x-ray crystal structure (Protein Data Bank code 2V9Q) is 2.85 nm.
Heparin Binding of Site-directed Mutants
The HRPF data indicate protection of two basic, conserved residues at the N terminus of the Robo1 Ig1 domain. In order to determine whether these two residues are involved in binding to heparin rather than protected due to a binding-induced structural event, site-directed point mutations were made and expressed as described under “Experimental Procedures.” Mutant I consists of a double mutation at the previously identified binding site near the Ig1-Ig2 domain interface, transforming two known heparin binding basic residues to alanine (Arg136 → Ala, Lys137 → Ala). This double mutant in the Drosophila Robo1 homolog (K122A/K123A) was previously shown to strongly decrease binding to a heparin column (21). Mutant II consists of a double mutation converting both N-terminal basic residues identified as protected by heparin binding to alanine (Arg62 → Ala, Arg69 → Ala).
The effect of the mutations on heparin binding was tested by heparin affinity chromatography, with the results shown in Fig. 8. Mutant I shows a large decrease in the NaCl concentration required for elution from the column, as reported previously (21). Mutant II shows a much smaller, but reproducible, decrease in the NaCl concentration required for elution. These results suggest that the N-terminal binding site identified by HRPF is directly involved in heparin binding but influences the overall affinity of Robo1 to heparin less than the previously identified binding site near the Ig1-Ig2 interface.
FIGURE 8.
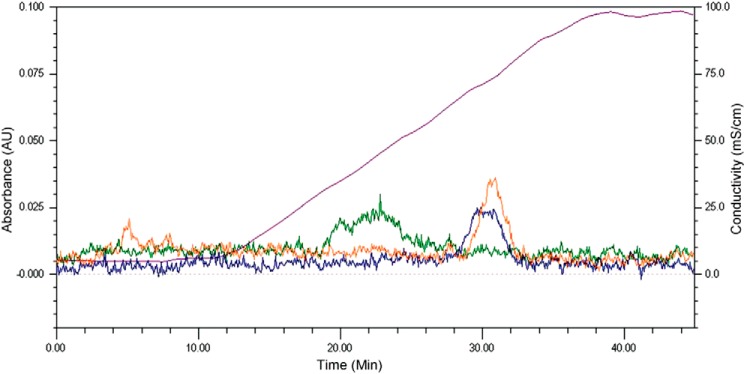
Heparin affinity chromatography of Robo1 site-directed mutants. The chromatogram is a representative replicate; the analyses were performed in duplicate, and the elution profiles were reproducible. Orange, WT Robo1; green, Mutant I (R136A/K137A); blue, Mutant II (R62A/R69A). The purple curve shows the conductivity of the elution buffer (millisiemens/cm) due to the increasing NaCl concentration, with the scale shown on the right y axis.
These Robo1 mutants were also tested for binding to heparin by SPR using immobilized heparin. Sensorgrams of the Robo1 (WT), Mutant I, and Mutant II interactions with the immobilized heparin chip are shown in Fig. 9, A–C, respectively. Each sensorgram was globally fitted using the 1:1 Langmuir model to generate the binding kinetics data. Although this kinetic model is a simplification of the interactions of Robo1 with the complex mixture of heparin sequences immobilized on the SPR chip, the 1:1 Langmuir model fit the binding data of both mutants very well and allows us to make comparisons of apparent binding rate constants. The 1:1 Langmuir model does not fit the WT data quite as well, as reported previously (22); however, fitting of the WT data to a two-state model did not result in a substantial enough improvement to warrant use of this model, possibly due to the use of a slower association period than reported previously (data not shown). We retained the 1:1 Langmuir model for all sensorgrams to facilitate direct comparisons of the data. The SPR data show that the interaction of both mutants with heparin displayed a slower apparent on-rate (ka), a faster apparent off-rate (kd), and consequently a reduced apparent affinity (higher KD) to heparin as compared with the Robo1 WT. The SPR data suggest that both binding sites contribute to heparin binding affinity. Mutant I resulted in a 35-fold increase in the apparent KD for heparin interaction, whereas Mutant II resulted in a 25-fold increase. (Table 3). Given the complexity of the Robo1-heparin interaction and the presence of multiple diverse binding sequences in unknown abundances, it is difficult to quantitatively determine exact binding affinities for each of the two binding sites discussed here. However, the heparin affinity column and SPR data both indicate a role for the N-terminal basic residues identified by HRPF in binding to heparin.
FIGURE 9.
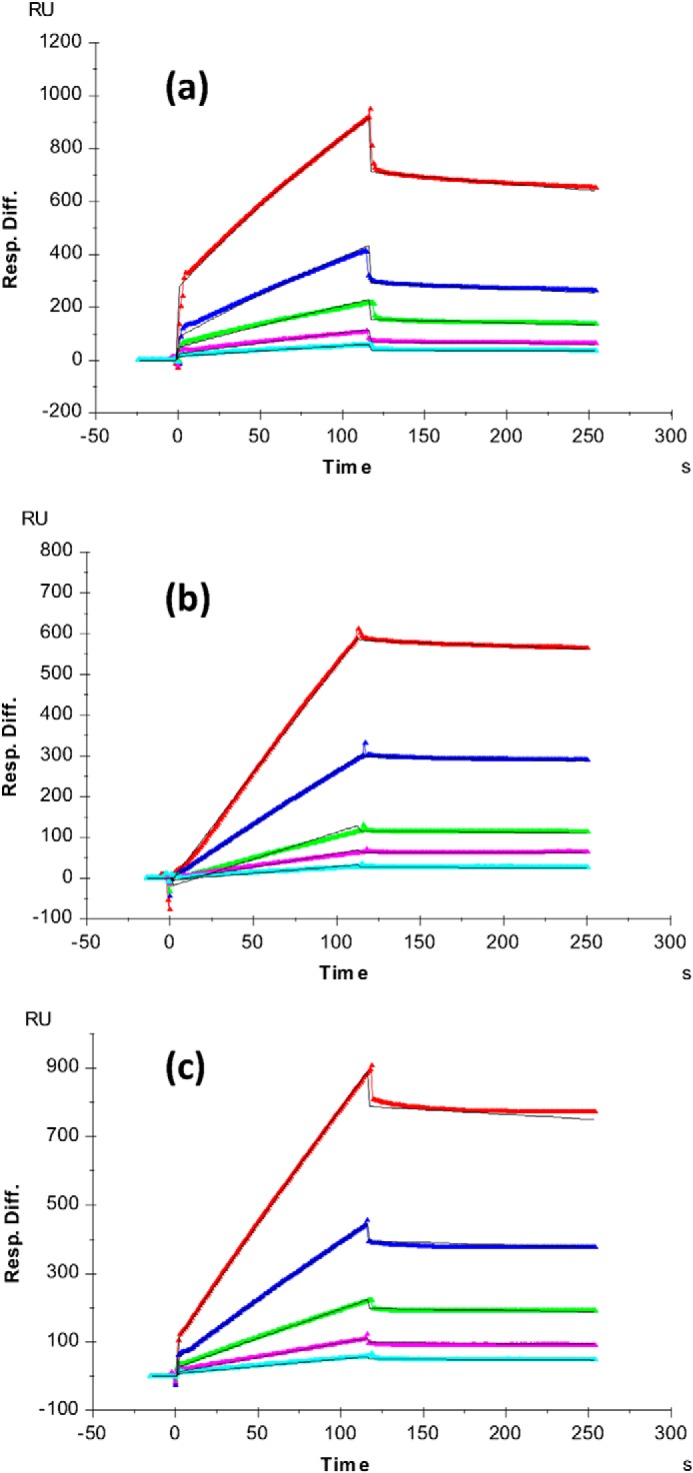
SPR sensorgrams of Robo1-heparin interaction. a, Robo1 WT/heparin; b, Mutant I (R136A/K137A)/heparin; c, Mutant II (R62A/R69A)/heparin. Concentrations of Robo1 (from top to bottom) are as follows: 1000 nm (red), 500 nm (blue), 250 nm (green), 125 nm (magenta), and 63 nm (cyan). The black curves are the fitting curves from a 1:1 Langmuir model from BIAevaluate version 4.0.1. Resp. Diff., response difference.
TABLE 3.
Summary of kinetic data of Robo1-heparin interactions
The data in parentheses are the S.D. values from global fitting of five different concentration injections.
| Interaction | ka | kd | KD |
|---|---|---|---|
| m−1 s−1 | s−1 | m | |
| Robo1 WT/heparin | 2.8 × 103 (±2.9 × 102) | 8.6 × 10−4 (±5.1 ×10−5) | 3.1 × 10−7 |
| Robo1 Mutant I/heparin | 31 (±2) | 3.4 × 10−4 (±3 ×10−5) | 1.1 × 10−5 |
| Robo1 Mutant II/heparin | 44 (±1.4) | 3.4 × 10−4 (±1.5 × 10−5) | 7.8 × 10−6 |
DISCUSSION
We have carried out a high spatial resolution hydroxyl radical protein footprinting analysis to study the structure of the complex of Robo1 and unfractionated heparin. These HRPF results revealed heparin-induced protection at and around the previously identified site of heparin dp8 interaction as well as a second binding site containing basic residues near the N terminus of the Ig1 domain. No protection of basic residues in the Ig2 domain was observed, indicating that heparin does not interact directly with the Ig2 domain. This is consistent with previous studies suggesting no direct interaction between heparin and the Ig2 domain (21).
Based on the results of our HRPF analysis, combined with the previously reported x-ray crystal structures of the human Slit2-Robo1 complex (39), mutagenesis studies of heparin binding to Drosophila Robo1 (21) and human Slit2 (18), and SPR studies of Robo1-heparin/HS interaction (22), we propose a model for Slit2-Robo1-heparin/HS ternary complex formation and signaling shown in Fig. 10. In this model, the initial interaction between Robo1 and heparin/HS occurs at what we propose to be the high affinity binding site centered around the previously identified residues in the Robo1 Ig1 domain (Lys81, Arg131, Arg136, Lys137, and Arg169) and the Slit2 D2 domain (Arg465, Arg466, Lys470, Arg471, Lys476, and Lys479), based on our heparin affinity column and SPR data for Mutants I and II. This initial binding event serves to stabilize the Robo1-Slit2 ternary complex and stabilizes the complex with the long heparin/HS chain. A separate binding motif on the heparin-HS chain then interacts with the novel binding site on Robo1 that we report here (Arg62 and Arg69) as well as with conserved basic residues spatially adjacent in Slit2 (Arg310, Lys328, Lys329, Arg331, and Arg332). Based on the SPR and heparin affinity column data, we propose that this N-terminal binding site is lower affinity and serves to enhance and stabilize the overall ternary complex.
FIGURE 10.

A model of the mechanism of Slit2-Robo1-heparin interactions. The Slit2-Robo1 complex has two binding sites for heparin: the previously identified high affinity binding site near the Ig1-Ig2 interface of Robo1 (magenta) and a novel low affinity binding site located near the disordered N terminus of Robo1 as well as within adjacent conserved basic residues in Slit2 (cyan). Full-length heparin/HS binds first to the high affinity binding site, which then allows for binding of a separate portion of the heparin/HS chain to the low affinity binding site. The binding to the low affinity binding site prompts conformational changes required for signal transduction. This model was generated using the x-ray crystal structure of the second LRR domain of human Slit2 in complex with the Ig1 domain of human Robo1 (Protein Data Bank code 2V9T). The heparin tetrasaccharide and the Ig2 domain of Drosophila Robo1 were aligned and joined from the x-ray crystal structure of dRobo1 bound to heparin dp8 (Protein Data Bank code 2VRA).
In addition to the basic residues in the Ig1 domain, HRPF also found modest heparin-induced protection at Cys147 involved in the core disulfide bond of the Ig1 domain. Interestingly, Cys147 exhibits protection upon binding, but its disulfide partner Cys89 remains strongly protected in both the heparin-free and heparin-bound states. Because the reactive atom in cysteine is the sulfur, this suggests that only the Cys147 side of the disulfide bond is exposed to any substantial degree in heparin-free Robo1. The first β-strand of the Ig1 domain sits atop this disulfide bond, and this strand and the preceding flexible region contain the two residues making up the N-terminal binding site for heparin. We hypothesize that the N-terminal tail and the first β-strand of the Robo1 Ig1 domain are inherently disordered, and heparin binding to the low affinity binding site consisting of Arg62 and Arg69 stabilizes the β-strand, protecting the Cys147 side of the disulfide bond from solvent. Mutagenesis studies would not observe a strong effect on heparin binding upon mutagenesis of this low affinity site (18, 21) because elution from the heparin binding column would be dictated primarily by the affinity of the high affinity binding site. Examination of the low affinity binding site within the Robo1 structure indicates that, in the unbound state, the side chain orientation of the Arg62 and Arg69 residues of Robo1 are not in a geometry conducive to formation of a shared binding site between Robo1 and Slit2 (Fig. 10). However, our evidence suggests that this region may be flexible in the unbound state, allowing for an induced fit binding of the heparin/HS polysaccharide.
Our results also show a modest protection of hydrophobic residues Met180 and Met189 in the Ig2 domain upon binding to unfractionated heparin (Fig. 7). Met180 is only partially protected from solvent by residues involved in the small C-terminal α-helix of the Ig2 domain and is heavily oxidized in the absence of heparin. In the static x-ray crystal structure, Met189 is highly protected from solvent, yet we detect heavy oxidation of this residue as well. These results suggest that Met189 may be somewhat more solvent-exposed in the absence of heparin than is indicated by the crystal structure. Met189 is protected from solvent largely by the β-strand containing Met180, suggesting that this strand may be dynamic in solution in the heparin-free state, exposing both Met180 and Met189.
Previous SPR studies performed on Robo1 Ig1-2 interacting with unfractionated heparin indicated a two-state binding mechanism, including a slow event upon binding to unfractionated heparin that was not present upon binding to heparin octasaccharides (22). Although SPR data presented here were not able to conclusively verify the two-state binding model, the fit of the WT Robo1 to a standard 1:1 Langmuir binding model was not as good as the fit of either Mutant I or Mutant II, indicating that the WT Robo1 may have a more complex binding mechanism than either mutant. Our binding model contains a second, low affinity binding site for full-length heparin/HS chains. Our model suggests that the heparin/HS chain loops back and interacts with a distal binding site near the N terminus of the Robo1 Ig1 domain and potentially the Slit2 D2 domain, an interaction that would not be present for shorter heparin/HS chains. We additionally find evidence for a modest change in the conformation and/or dynamics of the Ig2 domain upon heparin binding to the Ig1 domain but no evidence for a large conformational change under these conditions. We hypothesize that this change in conformation/dynamics may be due to heparin/HS binding at the low affinity binding site, which would explain why previous SPR data found a two-state mechanism of binding only for full-length heparin, not for shorter oligosaccharides (22). The model also provides a testable hypothesis for the mechanism of Slit-Robo-heparin/HS interaction in signal transduction.
Acknowledgments
We thank Professor Lianchun Wang for providing porcine intestinal heparin. We thank Dr. Chau-wen Chou of the Proteomic and Mass Spectrometry Facility at the University of Georgia for the mass spectral data acquisition assistance as well as Professor Lance Wells and Rob Bridger. We also thank Dr. Gregory Wylie and Professor James Prestegard for assistance with dynamic light scattering.
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Grant P41GM103390 as part of the Research Resource for Integrated Glycotechnology.
- HS
- heparan sulfate
- GAG
- glycosaminoglycan
- SPR
- surface plasmon resonance
- HRPF
- hydroxyl radical protein footprinting
- FPOP
- fast photochemical oxidation of proteins
- ETD
- electron transfer dissociation
- TEV
- tobacco etch virus
- DLS
- dynamic light scattering
- RU
- resonance units.
REFERENCES
- 1. Garbe D. S., Bashaw G. J. (2004) Axon guidance at the midline: from mutants to mechanisms. Crit. Rev. Biochem. Mol. Biol. 39, 319–341 [DOI] [PubMed] [Google Scholar]
- 2. Dickson B. J., Gilestro G. F. (2006) Regulation of commissural axon pathfinding by slit and its robo receptors. Annu. Rev. Cell Dev. Biol. 22, 651–675 [DOI] [PubMed] [Google Scholar]
- 3. Evans T. A., Bashaw G. J. (2012) Slit/Robo-mediated axon guidance in Tribolium and Drosophila: divergent genetic programs build insect nervous systems. Dev. Biol. 363, 266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kidd T., Brose K., Mitchell K. J., Fetter R. D., Tessier-Lavigne M., Goodman C. S., Tear G. (1998) Roundabout controls axon crossing of the CNS midline and defines a novel subfamily of evolutionarily conserved guidance receptors. Cell 92, 205–215 [DOI] [PubMed] [Google Scholar]
- 5. Huminiecki L., Gorn M., Suchting S., Poulsom R., Bicknell R. (2002) Magic roundabout is a new member of the roundabout receptor family that is endothelial specific and expressed at sites of active angiogenesis. Genomics 79, 547–552 [DOI] [PubMed] [Google Scholar]
- 6. Carmeliet P., Tessier-Lavigne M. (2005) Common mechanisms of nerve and blood vessel wiring. Nature 436, 193–200 [DOI] [PubMed] [Google Scholar]
- 7. Gibson D. A., Tymanskyj S., Yuan R. C., Leung H. C., Lefebvre J. L., Sanes J. R., Chédotal A., Ma L. (2014) Dendrite self-avoidance requires cell-autonomous slit/robo signaling in cerebellar purkinje cells. Neuron 81, 1040–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li H. S., Chen J. H., Wu W., Fagaly T., Zhou L., Yuan W., Dupuis S., Jiang Z. H., Nash W., Gick C., Ornitz D. M., Wu J. Y., Rao Y. (1999) Vertebrate slit, a secreted ligand for the transmembrane protein roundabout, is a repellent for olfactory bulb axons. Cell 96, 807–818 [DOI] [PubMed] [Google Scholar]
- 9. Fricke C., Lee J. S., Geiger-Rudolph S., Bonhoeffer F., Chien C. B. (2001) astray, a zebrafish roundabout homolog required for retinal axon guidance. Science 292, 507–510 [DOI] [PubMed] [Google Scholar]
- 10. Wang B., Xiao Y., Ding B. B., Zhang N., Yuan X. B., Gui L., Qian K. X., Duan S., Chen Z., Rao Y., Geng J. G. (2003) Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell 4, 19–29 [DOI] [PubMed] [Google Scholar]
- 11. Wu J. Y., Feng L., Park H. T., Havlioglu N., Wen L., Tang H., Bacon K. B., Jiang Z. H., Zhang X. C., Rao Y. (2001) The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature 410, 948–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Englund C., Steneberg P., Falileeva L., Xylourgidis N., Samakovlis C. (2002) Attractive and repulsive functions of Slit are mediated by different receptors in the Drosophila trachea. Development 129, 4941–4951 [DOI] [PubMed] [Google Scholar]
- 13. Kramer S. G., Kidd T., Simpson J. H., Goodman C. S. (2001) Switching repulsion to attraction: Changing responses to slit during transition in mesoderm migration. Science 292, 737–740 [DOI] [PubMed] [Google Scholar]
- 14. Jen J. C., Chan W. M., Bosley T. M., Wan J., Carr J. R., Rüb U., Shattuck D., Salamon G., Kudo L. C., Ou J., Lin D. D., Salih M. A. M., Kansu T., al Dhalaan H., Al Zayed Z., MacDonald D. B., Stigsby B., Plaitakis A., Dretakis E. K., Gottlob I., Pieh C., Traboulsi E. I., Wang Q., Wang L., Andrews C., Yamada K., Demer J. L., Karim S., Alger J. R., Geschwind D. H., Deller T., Sicotte N. L., Nelson S. F., Baloh R. W., Engle E. C. (2004) Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science 304, 1509–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu H. (2001) Cell-surface heparan sulfate is involved in the repulsive guidance activities of Slit2 protein. Nat. Neurosci. 4, 695–701 [DOI] [PubMed] [Google Scholar]
- 16. Inatani M., Irie F., Plump A. S., Tessier-Lavigne M., Yamaguchi Y. (2003) Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science 302, 1044–1046 [DOI] [PubMed] [Google Scholar]
- 17. Esko J. D., Lindahl U. (2001) Molecular diversity of heparan sulfate. J. Clin. Invest. 108, 169–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hussain S. A., Piper M., Fukuhara N., Strochlic L., Cho G., Howitt J. A., Ahmed Y., Powell A. K., Turnbull J. E., Holt C. E., Hohenester E. (2006) A molecular mechanism for the heparan sulfate dependence of Slit-Robo signaling. J. Biol. Chem. 281, 39693–39698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Capila I., Linhardt R. J. (2002) Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 41, 391–412 [DOI] [PubMed] [Google Scholar]
- 20. Bishop J. R., Schuksz M., Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 21. Fukuhara N., Howitt J. A., Hussain S. A., Hohenester E. (2008) Structural and functional analysis of Slit and heparin binding to immunoglobulin-like domains 1 and 2 of Drosophila Robo. J. Biol. Chem. 283, 16226–16234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang F., Moniz H. A., Walcott B., Moremen K. W., Linhardt R. J., Wang L. (2013) Characterization of the interaction between Robo1 and heparin and other glycosaminoglycans. Biochimie 95, 2345–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu G., Chance M. R. (2007) Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 107, 3514–3543 [DOI] [PubMed] [Google Scholar]
- 24. Aye T. T., Low T. Y., Sze S. K. (2005) Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry. Anal. Chem. 77, 5814–5822 [DOI] [PubMed] [Google Scholar]
- 25. Chance M. R. (2001) Unfolding of apomyoglobin examined by synchrotron footprinting. Biochem. Biophys. Res. Commun. 287, 614–621 [DOI] [PubMed] [Google Scholar]
- 26. Goldsmith S. C., Guan J. Q., Almo S., Chance M. (2001) Synchrotron protein footprinting: A technique to investigate protein-protein interactions. J. Biomol. Struct. Dyn. 19, 405–418 [DOI] [PubMed] [Google Scholar]
- 27. Charvátová O., Foley B. L., Bern M. W., Sharp J. S., Orlando R., Woods R. J. (2008) Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. J. Am. Soc. Mass. Spectrom. 19, 1692–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maleknia S. D., Brenowitz M., Chance M. R. (1999) Millisecond radiolytic modification of peptides by synchrotron x-rays identified by mass spectrometry. Anal. Chem. 71, 3965–3973 [DOI] [PubMed] [Google Scholar]
- 29. Gau B. C., Sharp J. S., Rempel D. L., Gross M. L. (2009) Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 81, 6563–6571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Watson C., Janik I., Zhuang T., Charvátová O., Woods R. J., Sharp J. S. (2009) Pulsed electron beam water radiolysis for submicrosecond hydroxyl radical protein footprinting. Anal. Chem. 81, 2496–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li X., Li Z., Xie B., Sharp J. S. (2013) Improved identification and relative quantification of sites of peptide and protein oxidation for hydroxyl radical footprinting. J. Am. Soc. Mass Spectrom. 24, 1767–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barb A. W., Meng L., Gao Z., Johnson R. W., Moremen K. W., Prestegard J. H. (2012) NMR characterization of immunoglobulin G Fc glycan motion on enzymatic sialylation. Biochemistry 51, 4618–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meng L., Forouhar F., Thieker D., Gao Z., Ramiah A., Moniz H., Xiang Y., Seetharaman J., Milaninia S., Su M., Bridger R., Veillon L., Azadi P., Kornhaber G., Wells L., Montelione G. T., Woods R. J., Tong L., Moremen K. W. (2013) Enzymatic basis for N-glycan sialylation: structure of rat α2,6-sialyltransferase (ST6GAL1) reveals conserved and unique features for glycan sialylation. J. Biol. Chem. 288, 34680–34698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hambly D. M., Gross M. L. (2005) Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass. Spectrom. 16, 2057–2063 [DOI] [PubMed] [Google Scholar]
- 35. Konermann L., Stocks B. B., Czarny T. (2010) Laminar flow effects during laser-induced oxidative labeling for protein structural studies by mass spectrometry. Anal. Chem. 82, 6667–6674 [DOI] [PubMed] [Google Scholar]
- 36. Jumper C. C., Bomgarden R., Rogers J., Etienne C., Schriemer D. C. (2012) High-resolution mapping of carbene-based protein footprints. Anal. Chem. 84, 4411–4418 [DOI] [PubMed] [Google Scholar]
- 37. Ortega A., Amorós D., García de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hernaiz M., Liu J., Rosenberg R. D., Linhardt R. J. (2000) Enzymatic modification of heparan sulfate on a biochip promotes its interaction with antithrombin III. Biochem. Biophys. Res. Commun. 276, 292–297 [DOI] [PubMed] [Google Scholar]
- 39. Morlot C., Thielens N. M., Ravelli R. B. G., Hemrika W., Romijn R. A., Gros P., Cusack S., McCarthy A. A. (2007) Structural insights into the Slit-Robo complex. Proc. Natl. Acad. Sci. U.S.A. 104, 14923–14928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharp J. S., Becker J. M., Hettich R. L. (2004) Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal. Chem. 76, 672–683 [DOI] [PubMed] [Google Scholar]