

Background: N-Acetylmuramic acid α-1-phosphate (MurNAc-α1-P) uridylyltransferase MurU contributes to a shortcut in Pseudomonas putida cell wall biosynthesis accounting for intrinsic fosfomycin resistance.
Results: This work provides the first structural analysis of a MurNAc-α1-P uridylyltransferase.
Conclusion: MurNAc-α1-P and UTP are coordinated at distinct sites in the active center.
Significance: The presented structure represents a “minimal nucleotidyltransferase domain” and may provide structural guidelines for inhibitor design.
Keywords: Bacteria, Cell Wall, Drug Resistance, Enzyme Structure, Structural Biology, N-Acetylmuramic Acid α-1-Phosphate, UDP-MurNAc, Fosfomycin, Peptidoglycan Biosynthesis
Abstract
The N-acetylmuramic acid α-1-phosphate (MurNAc-α1-P) uridylyltransferase MurU catalyzes the synthesis of uridine diphosphate (UDP)-MurNAc, a crucial precursor of the bacterial peptidoglycan cell wall. MurU is part of a recently identified cell wall recycling pathway in Gram-negative bacteria that bypasses the general de novo biosynthesis of UDP-MurNAc and contributes to high intrinsic resistance to the antibiotic fosfomycin, which targets UDP-MurNAc de novo biosynthesis. To provide insights into substrate binding and specificity, we solved crystal structures of MurU of Pseudomonas putida in native and ligand-bound states at high resolution. With the help of these structures, critical enzyme-substrate interactions were identified that enable tight binding of MurNAc-α1-P to the active site of MurU. The MurU structures define a “minimal domain” required for general nucleotidyltransferase activity. They furthermore provide a structural basis for the chemical design of inhibitors of MurU that could serve as novel drugs in combination therapy against multidrug-resistant Gram-negative pathogens.
Introduction
The bacterial peptidoglycan is a large, meshlike macromolecule consisting of polysaccharide chains of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc)4 links that are connected via short oligopeptide bridges (1). The biosynthesis of peptidoglycan is highly conserved and essential for bacteria, and enzymes within this synthetic pathway therefore represent attractive targets for antibacterial drug design. The first committed precursor molecule in this pathway is UDP-MurNAc, which is synthesized in the cytosol from phosphoenolpyruvate and UDP-GlcNAc in two steps that are catalyzed by the enzymes MurA and MurB. The naturally produced phosphoenolpyruvate analog fosfomycin ((1R,2S)-1,2-epoxypropylphosphonic acid) irreversibly inhibits MurA, resulting in a reduced pool of UDP-MurNAc, decelerated biosynthesis of peptidoglycan, and eventually cell lysis (2). Many bacteria, particularly Gram-negative organisms such as Pseudomonas aeruginosa, are characterized by a high intrinsic resistance to fosfomycin, which limits its use for the treatment of infections (3). In the bacterial life cycle, the peptidoglycan polymer is constantly degraded and resynthesized, and bacteria are able to reuse a large proportion of their peptidoglycan in a process called cell wall recycling (4–6). We have recently discovered a connection between cell wall recycling and intrinsic fosfomycin resistance (7, 8).
In Escherichia coli, peptidoglycan catabolites are imported into the cytoplasm (6, 9) where they are further degraded by the etherase MurQ, a distinctive enzyme of the E. coli peptidoglycan recycling pathway with orthologs in many other organisms (10, 11). MurQ cleaves the d-lactic acid substituent of 6-OH phosphorylated MurNAc, thereby merging the catabolism of the two cell wall sugars at the stage of the common metabolite GlcNAc-6-P that is subsequently converted to glucosamine-6-P by the deacetylase NagA (6, 10, 12). Recent studies revealed that the occurrence of cell wall recycling enzymes differs among bacteria. Specifically, all pseudomonades and many other Gram-negative bacteria, including important pathogens, lack an ortholog of MurQ. These bacteria instead possess a shortcut from MurNAc-6-P to UDP-MurNAc that has been described recently for Pseudomonas putida KT2440 and P. aeruginosa PA01/PA14 (7, 8). This pathway bypasses the de novo biosynthesis of UDP-MurNAc and thus endows the bacterium with an intrinsic resistance against the antibiotic fosfomycin. Three novel, sequentially acting enzymes were identified to account for this shortcut: a MurNAc-6-P phosphatase, the kinase AmgK that rephosphorylates the resulting MurNAc at its C1 hydroxyl group, and the nucleotidyltransferase MurU that generates UDP-MurNAc from UTP and MurNAc-α1-P.
MurU belongs to the enzyme family of sugar nucleotidyltransferases (SNTs; also referred to as sugar nucleotide pyrophosphorylases, EC 2.7.7.x) whose members share a similar domain organization but do not necessarily display high sequence identity (13). MurU orthologs from Pseudomonas species, however, display sequence identities of over 70%, arguing for a general relevance of the insights gained from MurU of P. putida for these organisms. Moreover, previous studies revealed functional orthologs of P. putida MurU in P. aeruginosa PAO1 (70.8% amino acid sequence identity), Neisseria meningitidis MC58 (56.2% identity), and in Caulobacter crescentus CB15 (34.3% identity) (8).
Mutations or deletions of amgK and/or murU rendered the Pseudomonas strains more sensitive to fosfomycin (7, 8), which brings the shortcut from MurNAc-6-P to UDP-MurNAc into a pharmacological focus of interest. Accounting for the recent rise of multidrug-resistant bacteria, fosfomycin, because of its low toxicity and low cross-resistance with other antibiotics, has regained significant therapeutic relevance especially in combination therapy with other antibiotics (14). The structural analysis of MurU is of particular interest in this context because this enzyme is highly selective for MurNAc-α1-P (8), and its crystal structure might thus help to elucidate some key features for this selectivity and possibly provide a rationale for designing inhibitory drugs specifically targeting MurU and the above described shortcut in cell wall recycling.
Although several SNTs have been structurally characterized, there is no structure available of an SNT that specifically accepts MurNAc-α1-P as its sugar substrate (13). To advance the understanding of how SNTs use MurNAc-α1-P, we have solved crystal structures of native MurU and MurU enzyme-substrate complexes. Comparisons of these structures with the structures of related SNTs provide insights into the narrow substrate specificity of the enzyme, reveal mechanistic details, and establish a platform for the design of inhibitors.
EXPERIMENTAL PROCEDURES
Cloning of murU, Protein Expression, and Purification
murU of P. putida was cloned in vector pET29b (Novagen), heterologously overexpressed in E. coli as a C-terminally His6-tagged MurU fusion protein, and purified by nickel affinity and gel filtration chromatography as described previously (8). The purified protein (yield ∼20 mg/liter of culture) was concentrated to 5–10 mg/ml and could be stored for several weeks at −20 °C in gel filtration buffer (30 mm Tris-HCl, pH 7.6, 50 mm NaCl) without significant loss of enzyme activity.
HPLC Analysis of Purified MurU
High performance liquid chromatography (HPLC; Dionex BioLC) with a size exclusion column (BioSep-SEC-S3000, 600 × 7, 800 mm, 5-μm particle size; Phenomenex, Aschaffenburg) was used at a flow rate of 1 ml/min (buffer, 20 mm Na2HPO4, 500 mm NaCl, pH 7.4) to assess the association state of native MurU. Standard proteins used for gel filtration separation by HPLC were albumin (molecular mass, 66 kDa), chymotrypsinogen (25.6 kDa), and ribonuclease (13.7 kDa). The theoretical molecular mass of the His6-tagged version of MurU is 24,879 Da. Analytical gel filtration of MurU by HPLC revealed an apparent molecular weight of a monomer, indicating that the physiologically active moiety of MurU is the monomer (see Fig. 1A).
FIGURE 1.

A, HPLC analysis confirms a monomeric state of MurU. Retention times obtained from size exclusion chromatography for the standard proteins albumin (66 kDa), chymotrypsinogen (25.6 kDa), and ribonuclease (13.7 kDa) (filled dots) were plotted against their molecular mass. The retention time for MurU and its corresponding apparent molecular mass (18.87 min and 27.5 kDa) are indicated by an asterisk. B, NTP substrate specificity assay using radiolabeled MurNAc α-1-[32P]phosphate. C, overall structure of a ternary complex of MurU bound to its substrate MurNAc-α1-P and the substrate analog UppNHp. Ligand positions are based on two different crystals that were each soaked with MurNAc-α1-P and UppNHp or UpNHpp an MgCl2, respectively (see Table 1). Substrates are depicted as sticks and colored according to the atom type (oxygens in red, nitrogens in blue, carbons in yellow, and phosphorus in orange). The protein is shown in schematic representation with the colors highlighting the subdomains referred to in the text (blue, C-terminal helix; orange, sugar-binding domain; marine blue, nucleotidyltransferase signature motif). Secondary structure elements are numbered from the N to C terminus. Bottom, schematic representation of the reaction catalyzed by MurU.
Enzymatic Preparation of MurNAc-α1-P
The enzymatic preparation of the MurU substrate MurNAc-α1-P was conducted as described (8), and the concentration of MurNAc-α1-P in preparations was determined by assaying the phosphate release with malachite green upon treatment with alkaline phosphatase according to previously established protocols (15).
Enzyme Activity Assay
The malachite green assay also served to evaluate the enzymatic activity of purified MurU. No MgCl2 had to be added to yield active enzyme; however, addition of EDTA (1 mm) led to complete loss of MurU activity, indicating the requirement of divalent cations, e.g. Mg2+, which has been shown previously for other nucleotidyltransferases (13).
Substrate Specificity Assays
The nucleotidyl triphosphate specificity of MurU was analyzed using 5 μl of radiolabeled AmgK reaction product (MurNAc α-1-[32P]phosphate) in Tris-HCl buffer, pH 7.6 containing 0.25 unit of bakers' yeast inorganic pyrophosphate (Sigma-Aldrich) to drive the transferase reaction toward completion and a 50 mm concentration of either UTP, ATP, CTP, GTP, or TTP nucleotidyl triphosphate (Sigma-Aldrich) in 25-μl reaction volumes at 37 °C. The nucleotidyl transfer reactions were started in parallel by the addition of 1 μg of purified MurU, and 5 μl of the reaction mixtures were spotted immediately (0 min) and after 180 min of incubation on a TLC plate. Reaction products were separated in basic solvent with n-butyl alcohol, methanol, 25% (w/v) ammonium hydroxide, water (5:4:2:1). The radioactive products were detected using a Typhoon Trio+ biomolecular imager (GE Healthcare). The 32P-radiolabled substrate MurNAc-α1-P was prepared by adding 50 mm MurNAc to a reaction mixture containing 100 mm Tris-HCl, pH 7.6, 100 mm ATP, pH 7.6, 10 mm MgCl2, 140 kBq of [γ-32P]ATP in a total volume of 100 μl. The reaction was started by the addition of 25 μg of purified AmgK and incubated for 3 h at 25 °C. MurNAc-α1-P but not GlcNAc-α1-P or Glc-α1-P was the only accepted sugar phosphate substrate that yielded UDP-MurNAc along with UTP (8). The presence of 0, 0.5, 1, 5, and 10 mm GlcNAc-α1-P in the same setup did not have an effect on product formation. MurU also displays narrow specificity for UTP, and no other tested nucleotide triphosphate (ATP, CTP, TTP, or GTP) served as substrate (Fig. 1B).
Protein Crystallization
MurU crystallized in space group P 6122 with one monomer in the asymmetric unit and a solvent content of the crystals of around 50% (16). Crystals were grown using the sitting drop vapor diffusion method at 20 °C. In 96-well crystallization plates, 300 nl of protein solution (7.5–10.5 mg/ml) were mixed with 300 nl of a well solution that contained 0.05–0.1 m buffer (MES, pH 6 or 6.5; HEPES, pH 7; or Tris, pH 8.5) and 1.0–1.5 m (NH4)2SO4.
For experimental phasing crystals were soaked with 250 mm NH4I for 10 min. Enzyme-substrate complexes were formed by soaking crystals with (i) 1.25–2.5 mm MurNAc-α1-P and 3.0–3.5 mm β,γ non-hydrolyzable UTP analog UppNHp (Jena Bioscience) for 75 min as well as with (ii) 1.25–2.5 mm MurNAc-α1-P, 2.8–3.7 mm α,β non-hydrolyzable UTP analog UpNHpp (Jena Bioscience), and 20 mm MgCl2 for 2 h. Crystals were transferred into a cryoprotecting solution containing 0.8–3.3 m (NH4)2SO4 as well as 5–25% (v/v) glycerol and flash frozen in liquid nitrogen prior to data collection.
X-ray Diffraction
All data were collected at 100 K on PILATUS® detectors at the synchrotron radiation beamline ID14-4 of the European Synchrotron Radiation Facility in Grenoble, France as well as at beamlines X06SA and X06DA of the Swiss Light Source in Villigen, Switzerland. Diffraction patterns of the non-liganded enzyme as well as of the enzyme-ligand complexes were generated using an x-ray wavelength of 1.0 Å, whereas diffraction data of the NH4I-soaked crystals for experimental phasing were obtained at a wavelength of 1.7 Å.
Structure Determination and Model Building
Indexing, integrating, and scaling of diffraction data were done with the XDS software package (17). Experimental phases for the non-liganded enzyme data were determined in a single anomalous dispersion experiment (18, 19), exploiting the anomalous signal of iodine in NH4I-soaked crystals at a wavelength of 1.7 Å. Initial phases were derived by determining the substructure of four iodine atoms per asymmetric unit with the help of the program suite SHELXC/D/E (20) and further extended and refined with SHARP/autoSHARP (21) and PHENIX (22). Molecular replacement for all ligand-bound structures was done with Phaser (23) using the structure of the native enzyme as a search model. Model building, refinement, and validation was performed with Coot (24), PHENIX, the CCP4 suite (25), the Protein Data Bank data validation and deposition portal, and the MolProbity web page (26). To remove model bias, simulated annealing was performed for all structures, and simulated annealing omit density maps were calculated for the ligand-bound structures using phenix.refine. The TLSMD (Translation Libration Screw Motion Determination) web server (27) was used for TLS parameterization of the protein. Coordinate and parameter files for MurNAc-α1-P, UpNHpp, and UppNHp were generated using phenix.elbow. Ligands were built manually according to the difference and simulated annealing omit density maps using Coot, and their coordinates were further refined with PHENIX.
The final structures contain all residues with the exception of some poorly ordered segments between Gly134 and His136 and around Ala154 in the native and complex 1 structures (Table 1) as well as the C-terminal His6 tag (in all structures). Structure figures were generated with PyMOL (28), and electrostatic potentials were calculated with PyMOL-implementations (29). For structural comparison, structural models were superimposed using the alignment algorithm implemented in PyMOL with complex 1 as the reference structure. For the native structure 177 atoms and for complex 2 175 atoms were aligned with a root mean square deviation of 0.179 and 0.144 Å, respectively.
TABLE 1.
Statistics of x-ray data collection and structure refinement
Values in parentheses refer to the highest resolution shell. SAD, single anomalous dispersion; r.m.s., root means square; CC, correlation coefficient; ESRF, European Synchrotron Radiation Facility; SLS, Swiss Light Source.
| Parameter | Native data set (Protein Data Bank code 4Y7T) | NH4I-soaked for SAD phasing | MurNAc-α1-P + UppNHp, complex 1 (Protein Data Bank code 4Y7V) | Mg2+ + MurNAc-α1-P + UpNHpp, complex 2 (Protein Data Bank code 4Y7U) |
|---|---|---|---|---|
| Data collection | ||||
| Beamline | ID14-4, ESRF | X06DA, SLS PXIII | X06DA, SLS PXIII | X06DA, SLS PXIII |
| Wavelength (Å) | 1.0 | 1.7 | 1.0 | 1.0 |
| Space group | P 6122 | P 6122 | P 6122 | P 6122 |
| Cell dimensions | ||||
| a, b, c (Å) | 72.61, 72.61, 158.47 | 72.71, 72.71, 159.01 | 72.35, 72.35, 162.48 | 72.75, 72.75, 163.13 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 |
| Resolution (Å) | 50.0-1.8 (1.9-1.8) | 30.0-2.4 (2.4-2.4) | 50.0-1.8 (2.1-1.8) | 50.0-1.7 (2.0-1.7) |
| |I/σ| | 13.3 (2.0) | 44.8 (9.8) | 27.1 (2.0) | 30.7 (2.1) |
| |I/σ| of anomalous signal | 1.8 (0.84) | |||
| CC1/2 (%) | 99.9 (88.9) | 100.0 (98.4) | 100.0 (80.6) | 100.0 (84.2) |
| Rmeas (%) | 22.4 (804.6) | 7.9 (52.3) | 9.3 (236.9) | 6.4 (200.2) |
| Rpim (%) | 4.1 (193.3) | 1.2 (8.3) | 1.5 (38.0) | 1.0 (33.3) |
| Completeness (%) | 99.9 (99.6) | 99.9 (100.0) | 99.9 (100.0) | 100.0 (100.0) |
| Measured reflections | 704,884 | 741,478 | 916,020 | 1,044,585 |
| Unique reflections | 23,701 | 18,399 | 24,133 | 28,942 |
| Redundancy | 29.7 | 40,3 | 38.0 | 36.1 |
| Wilson B (Å2) | 44.2 | 43,6 | 47.1 | 46.7 |
| Refinement | ||||
| Resolution (Å) | 40.4-1.8 | 49.6-1.8 | 49.8-1.7 | |
| Rwork/Rfree (%) | 21.1/24.5 | 21.5/25.4 | 22.8/26.4 | |
| No. of atoms | ||||
| Protein | 1,683 | 1,652 | 1,702 | |
| H2O | 80 | 67 | 54 | |
| Ligands: | ||||
| MurNAc/-1-P | 41 | 24a | ||
| UxxP/UDP | 14 | 29a | ||
| SO42− | 20 | 5 | 5 | |
| Mg2+/Na+/NH4+ | 2 | |||
| Glycerol | 42 | 28 | 28 | |
| Total | 1,783 | 1,807 | 1,791 | |
| B-factors (Å2) | ||||
| Protein | 46.68 | 51.84 | 55.95 | |
| H2O | 49.96 | 56.86 | 53.13 | |
| Ligands | ||||
| MurNAc/-1-P | 48.71 | 68.05 | ||
| UxxP/UDP | 86.05 | 106.52 | ||
| Mg2+/Na+/NH4+ | 6.95 | |||
| SO42− | 88.38 | 94.51 | 105.33 | |
| Glycerol | 48.38 | 57.29 | 71.61 | |
| r.m.s.a deviations | ||||
| Bond length (Å) | 0.015 | 0.007 | 0.008 | |
| Bond angles (°) | 1.526 | 1.074 | 1.192 | |
| Ramachandran plot | ||||
| Favored regions (%) | 94.17 | 98.17 | 93.04 | |
| Allowed regions (%) | 4.93 | 1.83 | 6.09 | |
| Outliers (%) | 0.90 | 0.00 | 0.87 | |
a Ligands refined without hydrogens.
RESULTS
Overall Structure and Substrate-binding Region of MurU
The structure of MurU was determined at 1.8-Å resolution using single anomalous dispersion (Table 1). The polypeptide chain folds into a three-dimensional structure similar to those observed in other SNTs with a large N-terminal “core” domain that contains the catalytically active center, a dinucleotide-binding region showing a Rossmann fold, and a C-terminal auxiliary domain (13). The core domain of MurU adopts an α/β-sandwich fold with a central, mixed, seven-stranded β-sheet flanked on both sides by a total of eight α-helices (Fig. 1). The C-terminal auxiliary domain essentially consists of a single, long α-helix (α9) that projects from the core domain.
MurNAc-α1-P, but not the related GlcNAc-α1-P or Glc-α1-P compound, can function as sugar phosphate substrate for MurU, yielding UDP-MurNAc along with UTP (8). Moreover, the presence of GlcNAc-α1-P did not have an inhibitory effect on product formation in an inhibition assay (data not shown). MurU also displays narrow specificity for UTP as no other tested nucleotide triphosphate (ATP, CTP, TTP, or GTP) is accepted as a substrate according to a 32P radioassay (Fig. 1B). With the aim of understanding how MurU binds and interacts with its substrates and catalyzes the uridyl transfer reaction, we solved structures of MurU in complex with (i) MurNAc-α1-P and the β,γ non-hydrolyzable UTP analog UppNHp (complex 1 in Table 1) and with (ii) MurNAc-α1-P, the α,β non-hydrolyzable UTP analog UpNHpp and 20 mm MgCl2 (complex 2 in Table 1). The catalytically active site is located at a solvent-exposed cleft whose bottom is formed by one edge of the central β-sheet and at two of its sides by helices α5 and α8 as well as the N-terminal amino acids 1–13. This N-terminal section, which also comprises strand β1, is widely conserved among SNTs and carries the canonical signature motif GXGXR (Gly9–Arg13 in MurU) that has been referred to previously (13). The third side of the substrate-binding cleft is flanked by a subdomain that is inserted between strands β5 and β6 of the central β-sheet of MurU. This subdomain consists of a three-stranded antiparallel β-sheet (strands β8–β10) that is connected to β5 by a highly flexible loop that was not fully visible in any of our crystal structures (Fig. 1). Functionally, this subdomain is involved in the coordination of MurNAc-α1-P (or generally the sugar moiety in structurally related enzymes) and can be referred to as the sugar-binding domain (see also Ref. 30).
The MurNAc-α1-P-binding Site of MurU
The data sets for complex 1 and complex 2 revealed very clear, unbiased difference electron density (mFo − DFc) for MurNAc-α1-P, and thus this ligand could be modeled with high confidence (Fig. 2). The MurNAc-α1-P sugar ring assumes a 4C1 chair conformation in all our MurU structures. MurNAc-α1-P forms hydrogen bonds to the side chain carboxylates of Asp205 and Asp140 as well as polar contacts to the side chain of Asn105 (Fig. 2A). Furthermore, there is a partial polar interaction to the backbone carbonyl of Phe160 (for the sake of clarity, it is not explicitly shown in Fig. 2). The side chains of Phe141 and Phe160 provide a hydrophobic patch that accommodates the hydrophobic portion of the MurNAc-α1-P lactyl moiety and the sugar ring. The side chains of Leu181 and Leu185, which are also facing the lactyl-CH3, also contribute to this interaction. The carboxyl group of the lactyl moiety is accommodated in the vicinity of Lys180 in a solvent-filled groove displaying a predominantly positive surface potential (Fig. 2C). The structures of the two complexes also contain a bound sulfate ion that was likely incorporated from the crystallization solution. The sulfate binds in the vicinity of the MurNAc-α1-P lactyl group (Fig. 2). The sulfate ions have elevated temperature factors (B-factors) in all structures, indicating low occupancy and/or high mobility.
FIGURE 2.
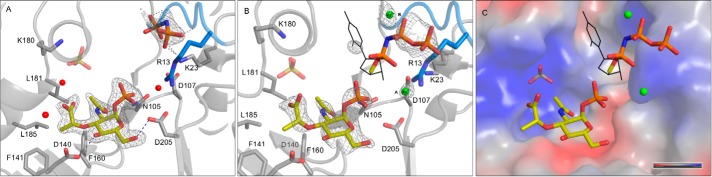
A and B, crystal structures of MurU soaked with MurNAc-α1-P + UppNHp (A; complex 1) and MurNAc-α1-P + UppNHp and MgCl2 (B; complex 2) with omit difference maps (mFo − DFc) for the ligands contoured at 2.5 σ. Parts of the substrates for which omit difference electron density could be obtained are depicted as sticks, Mg2+ ions are depicted as green spheres, and those parts of UpNHpp for which no or only 2mFo − DFc density could be obtained are represented as black lines. The SNT signature motif is highlighted in marine blue. Side chains that are involved in ligand coordination are represented as sticks. Ligands and side chains are colored by atom type (oxygens in red, nitrogens in blue, phosphorus in orange, sulfur in gold, magnesium in green, and carbons in the colors used for the domains in Fig. 1. C, surface representation of the substrate-binding site colored according to the surface potential (gradient from blue (positive) to red (negative)). Note that the parts of UpNHpp for which no reliable density could be obtained were modeled in the most likely possible conformation by relying on geometrical restraints and the clear electron density for the pyrophosphate part as an anchor point.
The UTP-binding Region of MurU
In contrast to MurNAc-α1-P, the densities obtained for the uridyl substrates were much weaker in both complexes and did not allow for a confident placing of the uridyl ring. Nevertheless the “pNHp” portion of UppNHp (Fig. 2A) or the pyrophosphate of UpNHpp (Fig. 2B), respectively, could be clearly localized. They are being coordinated by backbone nitrogens of residues 10–12 of the nucleotidyltransferase signature motif as well as the side chain of Lys23 (Fig. 2, A and B). The positive surface potential of this motif is significantly enhanced by the side chain of Arg13 and helps compensate the negative charge of the pyrophosphate (Fig. 2C). The slightly different orientation that is adopted by the pNHp in comparison with the α-phosphate of UpNHpp can be attributed to the different geometrical environment of nitrogen compared with oxygen in the respective ligands.
As the uridyl moieties were not visible in either structure, we modeled their approximate location and orientation based on the visible portion of the nucleotides and geometrical restraints (Fig. 2, B and C, black tracings). Independently performed molecular dynamics simulations (data not shown) arrived at a conformation of the uridyl moiety that almost coincided with the orientations modeled, suggesting that the depicted conformation might be a good approximation of the structure that UTP actually adopts when bound to MurU. However, it would not be valid to deduce any specific interactions with the protein from these models.
In the non-liganded structure of MurU, a SO42− is coordinated instead of the pyrophosphate at the respective site. However, in contrast to the native data set, the densities in the complexes are larger, and their elongated shape cannot be explained by a spherical sulfate ion, thus clearly arguing for the presence of the pyrophosphate moiety of the non-hydrolyzable substrate UpNHpp or the pyrophosphate mimic pNHp, respectively, being retained at the active site.
Two Magnesium Ions Are Likely Part of the Active Site
An EDTA-induced enzymatic inactivation indicated that MurU requires Mg2+ or equivalent divalent cations as a cofactor to achieve its full enzymatic activity (8). We therefore also included 20 mm MgCl2 in the soaking solution when solving the structure of complex 2. An unbiased difference electron density map (mFo − DFc) revealed two spherical features near the α-phosphate of MurNAc-α1-P and the pyrophosphate moiety of UpNHpp that could result either from water molecules or from an ion of similar size (Fig. 2, A and B). However, in MurU, MurNAc-α1-P binding was obviously independent of the presence or absence of Mg2+ in the soaking solution, indicating that the Mg2+ is not required for substrate binding per se. To justify the modeling of Mg2+ in our structure, we analyzed the coordination environment at the putative cation positions (which will be termed position A/AMg2+ and position B/BMg2+ hereafter). At position A, the side chains of Asp205 and Asp107 (Fig. 2B) as well as the backbone nitrogen of Gly207 (not explicitly shown in Fig. 2B) would stabilize a putative AMg2+. A Mg2+ in position B in contrast would not be coordinated by any amino acids of MurU but to the phosphates of the uridyl substrate. A similar coordination of divalent cations in an SNT has been described previously (31). Moreover, we performed molecular dynamics simulations that also predicted the presence of Mg2+ at the MurU active site. With the crystal structure as a starting model, the positions and radii of putative metal ions could be calculated and optimized by keeping only the protein atoms close to their crystallographic positions through the application of a high force field constant on the protein coordinates (data not shown). Although it was not suitable to distinguish among Mg2+, NH4+, or Na+, the ion positions predicted in this molecular dynamics approach were quite robust in different simulation scenarios and coincided nicely with the ion positions deduced from the x-ray data. Taken together, it seems reasonable and plausible to model Mg2+ ions at the two catalytically relevant positions A and B.
Structural Changes upon Ligand Binding
The comparison of the complex 1 and 2 structures with the non-liganded enzyme reveals no large domain rearrangements upon ligand binding. In particular, there is no significant movement of the C-terminal helix relative to the N-terminal core domain, which is in contrast to what has been observed for the larger C-terminal domains of other SNTs (32). However, there are some distinct side-chain reorientations of residues involved in MurNAc-α1-P coordination upon substrate binding (Fig. 3). The side chain of Asp140 is rotated by almost 90° and reoriented toward the sugar substrate, thereby facilitating hydrogen bonding to C4-OH of MurNAc-α1-P (Fig. 3). Interestingly, this side chain has rather low B-factors in both its substrate-bound and its non-liganded state, indicating that the described movement is a switch between two distinct states. Another, more subtle reorientation takes place at the conserved Arg13 of the signature motif. Here, we observe a slight reorientation of the Arg13 guanidinium group toward the pyrophosphate moiety of UpNHpp (Fig. 3) or UppNHp, respectively. Other residues involved in substrate binding, such as Asp205, Phe106, and Asn105, remain essentially unchanged upon substrate binding (Fig. 3). It appears that MurU provides a preformed binding pocket for MurNAc-α1-P that requires no large domain rearrangements for substrate binding.
FIGURE 3.
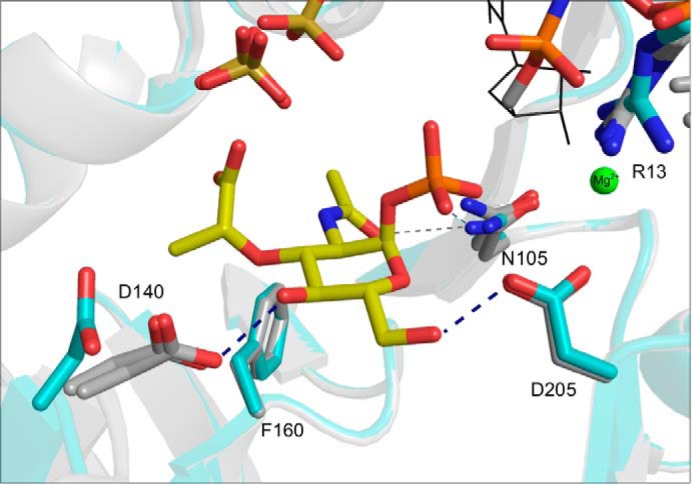
Superposition of complex structures 1 and 2 (gray) with the native structure (cyan). The MurNAc-α1-P, UppNHp, and Mg2+ ions (representation and coloring as in Fig. 2) are shown together with a sulfate ion that is seen in all three structures. Ligands and side chains involved in ligand coordination are represented as sticks and colored by atom type (see legend to Fig. 2). Note that Asp140 is rotated by about 90° toward the C4-hydroxyl group of MurNAc-α1-P in both ligand-bound structures.
MurU Defines a “Minimal Functional Domain” for Sugar Nucleotidyltransferase Activity
A Dali (33) search revealed significant structural similarities among MurU and several other SNTs. Of those, the glucose-1-phosphate thymidylyltransferase RmlA from P. aeruginosa (Protein Data Bank code 1G1L (30)), the mannose-1-phosphate guanylyltransferase GMPase of Thermotoga maritima (Protein Data Bank code 2X5Z (32)), and the N-terminal SNT domain of bifunctional GlcNAc-1-P uridylyltransferase GlmU from E. coli (Protein Data Bank code 1FWY (34)) were chosen for structural comparison with MurU. This selection was predominantly made to cover a range of substrates that, taken together, feature the specificity requirements of MurU as closely and all-encompassing as possible and thus provide a helpful guideline to integrate the structural analysis of MurU into a broader context of SNTs. The comparison of the overall structures of P. putida MurU (MurUP.p.), P. aeruginosa RmlA (RmlAP.a.), E. coli GlmU (GlmUE.c.), and T. maritima GMPase (GMPaseT.m.) clearly visualizes the common domain organization of SNTs (Fig. 4). All four enzyme folds can be subdivided into an N-terminal and a C-terminal domain with the N-terminal domain housing the sugar nucleotidyltransferase activity. The N-terminal domains of the enzymes are similar in structure as they all feature a Rossmann fold containing nucleotide binding motifs and a central seven-stranded β-sheet with an additional “sugar-binding” domain inserted between strands β5 and β6. The C-terminal domain of MurU, however, is strikingly different from that of RmlAP.a., GlmUE.c., and GMPaseT.m.. Although in the latter three enzymes the C-terminal domain is rather large and projects quite prominently from the N-terminal nucleotidyltransferase domain, the corresponding region of MurU is small, essentially consisting of a single α-helix. The C-terminal domain is involved in the regulation of enzymatic activity in several SNTs, for example by mediating oligomerization and/or by binding allosteric inhibitors (13), and in some cases, it may even influence substrate specificity (32). By contrast, MurU is a monofunctional enzyme that, according to gel filtration profiles, is monomeric in solution (Fig. 1A).
FIGURE 4.

Combined schematic representations of the MurU structure (same orientation as Fig. 1) as well as those of the structurally related Glc-1-P thymidylyltransferase RmlA (P. aeruginosa; Protein Data Bank code 1G1L), GlcNAc uridylyltransferase GlmU (E. coli; Protein Data Bank code 1FWY), and Man-1-P guanylyltransferase GMPase (T. maritima; Protein Data Bank code 2X5Z) structures. All four proteins are shown with a semitransparent surface. Of the multimeric enzymes RmlAP.a., GlmUE.c., and GMPaseT.m. only one monomer is depicted. The coloring highlights similarities in domain organization: blue, C-terminal domain; orange, sugar-binding domain; marine blue, SNT signature motif. Note that, despite the obvious structural similarities, the sequence identities of these enzymes to MurU are quite low (RmlA; 22.6%; GlmU, 16.6%; GMPase, 18.7%).
The role of the truncated C-terminal domain in MurU is unclear. No significant reorientation of the C-terminal helix α9 is observed upon substrate binding, and no residues from α9 are involved in substrate binding or the coordination of the catalytically relevant Mg2+ ions (Figs. 2 and 3). Although the C-terminal domain has some regulatory functions in related enzymes, MurU does not seem to require an “elaborate” C terminus for enzymatic activity. This defines the N-terminal core domain as a minimal functional domain required for MurNAc-α1-P uridylyltransferase activity.
DISCUSSION
We have solved structures of MurU of P. putida in native and two ligand-bound states at high resolution. Taken together, these structures allow for the identification of critical structural differences to other, related SNTs and may help explaining the remarkably narrow specificity of MurU for MurNAc-α1-P. They furthermore provide a structural basis for suggesting a catalytic mechanism.
MurU binds its substrate MurNAc-α1-P through numerous polar and hydrophobic interactions, and specificity for the MurNAc group over related compounds such as GlcNAc or GalNAc is in part achieved through contacts that specifically target the unique lactyl group of the sugar. The observation that GlcNAc-α1-P does not act as a competitive inhibitor for MurU moreover indicates that this compound does not bind to the enzyme in accordance with the specific contacts made by the substrate MurNAc-α1-P that cannot be made by GlcNAc-α1-P.
Although the location and orientation of the uridyl ring of the second substrate remain somewhat speculative as we do not observe density for it, the pyrophosphate portions of UpNHpp and the pNHp of UppNHp are clearly visible and allow placement of at least this part of the nucleotide with confidence. The pyrophosphate mimics one of the products of the uridyl transfer reaction and is tightly coordinated by residues of the SNT signature motif. Magnesium ions or other divalent cations have been found to be essential for or to enhance the enzymatic activities of other SNTs (5, 13, 31, 35), and it is likely that MurU likewise relies on bound magnesium ions to perform the nucleotidyl transfer reaction. The narrow specificity for UTP does not necessarily imply a high binding affinity for the nucleotide. However, on the basis of our structural data, it would not be valid to draw conclusions on the UTP binding mode of MurU.
In comparison with the functionally related SNTs RmlAP.a., GlmUE.c., and GMPaseT.m. (Fig. 4), the substrate-binding cleft of MurU is significantly more solvent-exposed. Regarding this fact and considering the distinct binding mode for MurNAc-α1-P, we consider it likely that MurNAc-α1-P binds first to the enzyme in a preformed binding pocket followed by Mg2+-coordinated UTP. After uridyl transfer, the remaining pyrophosphate together with the Mg2+ would then be released immediately after the reaction followed by the product UDP-MurNAc.
Extensive studies of GlmU orthologs of various organisms have shown that the phosphoryl transfer reaction involves nucleophilic activation of the phosphoryl oxygen of the sugar substrate, which in turn attacks the α-phosphate of the NTP substrate, resulting in the formation of the NDP sugar product and a concomitant release of pyrophosphate. This has been proposed to occur in a SN2 reaction for GlmU with the reaction proceeding through a pentavalent phosphorane involving two catalytically relevant Mg2+ ions with distinct functions (31). One Mg2+ (AMg2+) enables nucleophile activation and substrate coordination and is coordinated by distinct active site residues, whereas a second Mg2+ (BMg2+) contributes to the stabilization of the transition state of the reaction. Because of its specific requirements for coordination geometry, BMg2+ promotes product formation by changing its coordination state and thus destabilizing the scissile bond (31). According to our structural data and the likely locations of the magnesium ions, a similar mechanism seems also plausible for MurU (Fig. 5). Thereby the extensive coordination of the leaving group of the UMP transfer would result in a stabilization of the reaction product, which in turn destabilizes the scissile bond between Pα and Pβ of UTP and thus renders it susceptible to a nucleophilic attack by the phosphate group of MurNAc-α1-P. Such an attack might well be facilitated by the AMg2+ ion in position A in analogy to what has been described for GlmUMtb. However, referring to our structure, it is possible that residue Asp205 not only coordinates the Mg2+ ion but that this interaction moreover enables the Asp205 carboxyl group to catalytically abstract a proton of the MurNAc-α1-P phosphate group that in turn initiates the nucleophilic attack (Fig. 5).
FIGURE 5.

The reacting substrates and putative catalytic and coordinating residues are depicted together with the catalytically relevant Mg2+ ions. The dashed lines indicate polar contacts with the numbers giving the respective interatomic distances in Å.
Altering the nucleotide selectivity of SNTs through engineering of the substrate-binding site is a field of particular interest because with the biosynthesis of NDP sugars SNTs provide precursors for a large range of bioglycosylation pathways. Chemoenzymatic approaches for in vitro glycorandomization with a focus on engineering the sugar phosphate selectivity were introduced more than 10 years ago (36, 37). In 2007, Moretti and Thorson (38) were able to also enhance the latent NTP substrate flexibility of the thymidylyltransferase RmlA, and in 2011, Moretti et al. (39) were able to expand its promiscuity via directed evolution based on x-ray structural data. We think that MurU forms a promising target for such an approach.
The recent rediscovery of fosfomycin in antibiotic combination therapy against multidrug-resistant bacteria has identified intrinsic fosfomycin resistance as a remarkable clinical problem (14). MurU significantly contributes to this resistance, thus making a specific inhibitor for this enzyme a desirable objective. Because MurU is highly specific for its sugar substrate (8), the MurNAc-α1-P binding pocket might be a promising target for structure-guided drug design. Although this pocket is shallow and solvent-accessible, it still contains features that could be exploited, such as the hydrophobic pocket that accommodates the methyl group of the lactyl moiety of MurNAc-α1-P or the charged surface that faces the carboxylate group of MurNAc-α1-P. The design of an initial inhibitor should therefore mimic this lactyl pattern and perhaps involve optimization of contacts that favor its binding over the actual substrate.
The atomic coordinates and structure factors (codes 4Y7T, 4Y7V, and 4Y7U) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MurNAc
- N-acetylmuramic acid
- MurNAc-α1-P
- N-acetylmuramic acid α-1-phosphate
- 6-P
- 6-phosphate
- SNT
- sugar nucleotidyltransferase
- UppNHp
- uridine 5′-[(β,γ)-imido]triphosphate
- UpNHpp
- uridine 5′-[(α,β)-imido]triphosphate.
REFERENCES
- 1. Litzinger S., Mayer C. (2010) in Prokaryotic Cell Wall Compounds (König H., Claus H., Varma A., eds) pp. 3–52, Springer Verlag, Berlin [Google Scholar]
- 2. Kahan F. M., Kahan J. S., Cassidy P. J., Kropp H. (1974) The mechanism of action of fosfomycin (phosphonomycin). Ann. N. Y. Acad. Sci. 10, 364–386 [DOI] [PubMed] [Google Scholar]
- 3. Cox G., Wright G. D. (2013) Intrinsic antibiotic resistance: mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 303, 287–292 [DOI] [PubMed] [Google Scholar]
- 4. Mayer C. (2012) Bacterial cell wall recycling. in Encyclopedia of Life Science, John Wiley and Sons, Ltd., Chichester, UK [Google Scholar]
- 5. Johnson J. W., Fisher J. F., Mobashery S. (2013) Bacterial cell-wall recycling. Ann. N. Y. Acad. Sci. 1277, 54–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park J. T., Uehara T. (2008) How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol. Mol. Biol. Rev. 72, 211–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borisova M., Gisin J., Mayer C. (2014) Blocking peptidoglycan recycling in Pseudomonas aeruginosa attenuates intrinsic resistance to fosfomycin. Microb. Drug Resist. 20, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gisin J., Schneider A., Nägele B., Borisova M., Mayer C. (2013) A cell wall recycling shortcut that bypasses peptidoglycan de novo biosynthesis. Nat. Chem. Biol. 9, 491–493 [DOI] [PubMed] [Google Scholar]
- 9. Dahl U., Jaeger T., Nguyen B. T., Sattler J. M., Mayer C. (2004) Identification of a phosphotransferase system of Escherichia coli required for growth on N-acetylmuramic acid. J. Bacteriol. 186, 2385–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jaeger T., Mayer C. (2008) N-Acetylmuramic acid 6-phosphate lyases (MurNAc etherases): role in cell wall metabolism, distribution, structure, and mechanism. Cell. Mol. Life Sci. 65, 928–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jaeger T., Arsic M., Mayer C. (2005) Scission of the lactyl ether bond of N-acetylmuramic acid by Escherichia coli “etherase.” J. Biol. Chem. 280, 30100–30106 [DOI] [PubMed] [Google Scholar]
- 12. Hadi T., Dahl U., Mayer C., Tanner M. E. (2008) Mechanistic studies on N-acetylmuramic acid 6-phosphate hydrolase (MurQ): an etherase involved in peptidoglycan recycling. Biochemistry 47, 11547–11558 [DOI] [PubMed] [Google Scholar]
- 13. Singh S., Phillips G. N., Jr., Thorson J. S. (2012) The structural biology of enzymes involved in natural product glycosylation. Nat. Prod. Rep. 29, 1201–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Michalopoulos A. S., Livaditis I. G., Gougoutas V. (2011) The revival of fosfomycin. Int. J. Infect. Dis. 15, e732–e739 [DOI] [PubMed] [Google Scholar]
- 15. Baykov A. A., Evtushenko O. A., Avaeva S. M. (1988) A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal. Biochem. 171, 266–270 [DOI] [PubMed] [Google Scholar]
- 16. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 17. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abendroth J., Gardberg A. S., Robinson J. I., Christensen J. S., Staker B. L., Myler P. J., Stewart L. J., Edwards T. E. (2011) SAD phasing using iodide ions in a high-throughput structural genomics environment. J. Struct. Funct. Genomics 12, 83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dauter M., Dauter Z. (2007) Phase determination using halide ions. Methods Mol. Biol. 364, 149–158 [DOI] [PubMed] [Google Scholar]
- 20. Sheldrick G. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vonrhein C., Blanc E., Roversi P., Bricogne G. (2007) Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 [DOI] [PubMed] [Google Scholar]
- 22. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Painter J., Merritt E. A. (2006) TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111 [Google Scholar]
- 28. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC, New York [Google Scholar]
- 29. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blankenfeldt W., Asuncion M., Lam J. S., Naismith J. H. (2000) The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). EMBO J. 19, 6652–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jagtap P. K., Verma S. K., Vithani N., Bais V. S., Prakash B. (2013) Crystal structures identify an atypical two-metal-ion mechanism for uridyltransfer in GlmU: its significance to sugar nucleotidyl transferases. J. Mol. Biol. 425, 1745–1759 [DOI] [PubMed] [Google Scholar]
- 32. Pelissier M. C., Lesley S. A., Kuhn P., Bourne Y. (2010) Structural insights into the catalytic mechanism of bacterial guanosine-diphospho-d-mannose pyrophosphorylase and its regulation by divalent ions. J. Biol. Chem. 285, 27468–27476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown K., Pompeo F., Dixon S., Mengin-Lecreulx D., Cambillau C., Bourne Y. (1999) Crystal structure of the bifunctional N-acetylglucosamine 1-phosphate uridyltransferase from Escherichia coli: a paradigm for the related pyrophosphorylase superfamily. EMBO J. 18, 4096–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maruyama D., Nishitani Y., Nonaka T., Kita A., Fukami T. A., Mio T., Yamada-Okabe H., Yamada-Okabe T., Miki K. (2007) Crystal structure of uridine-diphospho-N-acetylglucosamine pyrophosphorylase from Candida albicans and catalytic reaction mechanism. J. Biol. Chem. 282, 17221–17230 [DOI] [PubMed] [Google Scholar]
- 36. Barton W. A., Biggins J. B., Jiang J., Thorson J. S., Nikolov D. B. (2002) Expanding pyrimidine diphosphosugar libraries via structure-based nucleotidylyltransferase engineering. Proc. Natl. Acad. Sci. U.S.A. 99, 13397–13402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barton W. A., Lesniak J., Biggins J. B., Jeffrey P. D., Jiang J., Rajashankar K. R., Thorson J. S., Nikolov D. B. (2001) Structure, mechanism and engineering of a nucleotidylyltransferase as a first step toward glycorandomization. Nat. Struct. Biol. 8, 545–551 [DOI] [PubMed] [Google Scholar]
- 38. Moretti R., Thorson J. S. (2007) Enhancing the latent nucleotide triphosphate flexibility of the glucose-1-phosphate thymidylyltransferase RmlA. J. Biol. Chem. 282, 16942–16947 [DOI] [PubMed] [Google Scholar]
- 39. Moretti R., Chang A., Peltier-Pain P., Bingman C. A., Phillips G. N., Jr., Thorson J. S. (2011) Expanding the nucleotide and sugar 1-phosphate promiscuity of nucleotidyltransferase RmlA via directed evolution. J. Biol. Chem. 286, 13235–13243 [DOI] [PMC free article] [PubMed] [Google Scholar]