

Background: HIV-1 Vpu displaces bone marrow stromal antigen 2 (BST2) from sites of viral assembly.
Results: A tryptophan residue near the C terminus of Vpu is required for this activity and interacts with the lipid bilayer.
Conclusion: Interaction of the C terminus of Vpu with the lipid bilayer helps HIV-1 escape restriction by BST2.
Significance: The topology of the cytoplasmic domain of Vpu with respect to the lipid bilayer is a key aspect of BST2 antagonism.
Keywords: Cell Biology, Cell Surface Protein, Human Immunodeficiency Virus (HIV), Innate Immunity, Membrane Trafficking, Nuclear Magnetic Resonance (NMR), Retrovirus, Tryptophan
Abstract
The restriction factor BST2 (tetherin) prevents the release of enveloped viruses from the host cell and is counteracted by HIV-1 Vpu. Vpu and BST2 interact directly via their transmembrane domains. This interaction enables Vpu to induce the surface down-regulation and the degradation of BST2, but neither of these activities fully accounts for the ability of Vpu to enhance virion release. During a study of naturally occurring Vpu proteins, we found that a tryptophan residue near the Vpu C terminus is particularly important for enhancing virion release. Vpu proteins with a W76G polymorphism degraded and down-regulated BST2 from the cell surface, yet they inefficiently stimulated virion release. Here we explore the mechanism of this anomaly. We find that Trp-76 is critical for the ability of Vpu to displace BST2 from sites of viral assembly in the plane of the plasma membrane. This effect does not appear to involve a general reorganization of the membrane microdomains associated with virion assembly, but rather is a specific effect of Vpu on BST2. Using NMR spectroscopy, we find that the cytoplasmic domain of Vpu and Trp-76 specifically interact with lipids. Moreover, paramagnetic relaxation enhancement studies show that Trp-76 inserts into the lipid. These data are consistent with a model whereby Trp-76 anchors the C terminus of the cytoplasmic tail of Vpu to the plasma membrane, enabling the movement of Vpu-bound BST2 away from viral assembly sites.
Introduction
HIV-1 counteracts the interferon-inducible restriction factor bone marrow stromal antigen 2 (BST2)5/tetherin with its accessory protein Vpu (1, 2). BST2 entraps lipid enveloped virions as they bud from the plasma membrane, preventing their release. Vpu, a single-pass type I transmembrane protein (3), binds directly to BST2, a single-pass type II transmembrane protein with a glycosylphosphatidylinositol anchor (4), via an anti-parallel interaction between the proteins' transmembrane domains (5–9). This interaction enables Vpu to decrease the concentration of BST2 on the plasma membrane and induce the degradation of BST2 by an endolysosomal mechanism (7, 8, 10–13).
However, the analysis of certain Vpu mutants reveals that neither the surface down-regulation nor the degradation of BST2 strictly correlates with the ability of Vpu to enhance virion release. For example, site-directed mutation of the β-TrCP-binding site, through which Vpu links its cellular targets to an E3 ubiquitin ligase complex, renders Vpu unable to degrade or down-regulate BST2 (2, 8, 10, 11, 14), yet such a Vpu mutant (Vpu-S52N,S56N) can partially enhance virion release (8, 11, 15, 16) compared with no Vpu (ΔVpu). The source of this residual activity has been attributed to the ability of Vpu to displace BST2 from virion-assembly sites in the plane of the membrane, an activity that has been roughly mapped to the C-terminal portion of the cytoplasmic domain of Vpu (16). During an analysis of naturally occurring Vpu proteins, we found the converse phenotype: the preserved ability to degrade and down-regulate BST2 yet impaired enhancement of virion release (17). This phenotype mapped to a W76G polymorphism, a mutation that presumably arises in infected hosts to provide escape from cytotoxic T lymphocytes. Site-directed mutagenesis confirmed that optimal enhancement of virion release depends upon Trp-76, which is near the C terminus of Vpu and is well conserved in certain HIV-1 clades (17). Substitution of a glycine for Trp-76 impairs virion release without affecting the ability of Vpu to co-localize with BST2, to down-regulate BST2 from the cell surface, or to reduce the steady-state amount of cellular BST2. Notably, the defect in virion release caused by the W76G substitution is additive with that caused by mutation of the β-TrCP-binding site (S52N,S56N), indicating that the effects of these mutations are mechanistically distinct.
How Trp-76 contributes to maximal virion release without affecting BST2 down-regulation or degradation is investigated here. Our central hypothesis was that Trp-76 is instrumental in the ability of Vpu to displace BST2 from sites of viral assembly along the plasma membrane (16). In support of this, we show using immunofluorescence microscopy that Trp-76 is required for the displacement of BST2 from viral assembly sites by Vpu. Because BST2 has been proposed to link lipid rafts to each other (4, 18) and to the actin cytoskeleton (19), we considered that Vpu might distort the membrane topology of viral assembly sites through its influence on BST2. Instead, we observed that cellular markers of lipid rafts and tetraspanin-enriched membrane domains (TEMs), proteins that define the assembly sites of HIV-1 virions (20–22), remained associated with Gag, the viral structural protein that drives viral assembly, despite the Vpu-induced redistribution of BST2. To elaborate the mechanism of the displacement effect, we show using NMR spectroscopy that the Vpu cytoplasmic domain interacts with lipids, and we support a model in which Trp-76 anchors the C-terminal region of the protein to the lipid bilayer. Although this interaction was enhanced in the presence of lipid head groups of net positive charge, phenylalanine did not substitute functionally for tryptophan at this position; this result suggests that the ability of tryptophan to bind lipid cannot be explained fully by a cation-π interaction but likely depends on the hydrophobic nature of the residue. Based on these data, we propose a refined topological model for the counteraction of BST2 by Vpu: tryptophan-mediated anchoring of the Vpu C terminus to the lipid bilayer enables Vpu to displace BST2 from virion-assembly sites.
EXPERIMENTAL PROCEDURES
Plasmids, Cells, and Reagents
Proviral plasmids pNL4-3, pNL4-3ΔVpu (pNL4–3/Udel (23)), pNL4-3-Vpu-S52N,S56N (24), pNL4-3-Vpu-W76G (17), pNL4-3-S52N,S56N + W76G (17), and pNL4-3-Vpu-A10F,A14F,A18F (25) have been described previously. Rev-dependent FLAG-tagged Vpu expression constructs for NL4-3-Vpu, Vpu-S52N,S56N, Vpu-W76G, Vpu-S52N,S56N, and Vpu-S52N,S56N + W76G were described previously (17). Expression plasmids encoding Vpu-W76F and Vpu-S52N,S56N + W76F with a C-terminal FLAG tag were constructed by site-directed mutagenesis of expression constructs for NL4-3-Vpu and Vpu-S52N,S56N, respectively, using a QuikChange II site-directed mutagenesis kit (Agilent Technologies). For NMR studies, DNA encoding the Vpu NL4-3 strain cytoplasmic domain (VpuCyto) sequence was inserted into pHLV Trp-leader (TrpΔLE) vector for expression as a TrpΔLE fusion. The methionine in VpuCyto was substituted with leucine to facilitate protein purification. Mutants W76G VpuCyto and W76F VpuCyto were prepared using the QuikChange Lightning Site-directed Mutagenesis Kit (Agilent Technologies) and primers were synthesized by Integrated DNA Technologies. HeLa P4-R5 cells (26–28) were acquired from the NIH AIDS Research and Reference Reagent program from Nathaniel Landau and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, penicillin/streptomycin, and 1 μg/ml of puromycin. The chelated manganese was prepared by adding excess manganese sulfate (MnSO4) to EDTA (0.5 m) in 20 mm HEPES, pH 7.3, buffer, and MnEDTA was precipitated overnight. The precipitated complex was collected by centrifugation, washed with methanol to remove excess amounts of MnSO4 and EDTA, and dried by lyophilization. The lipids 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-ethylphosphocholine (DMEPC), 1,2-dimyristoyl-3-trimethylammonium propane (DMTAP), 3β-[N-(N′,N′-dimethylaminoethane)carbamoyl]cholesterol hydrochloride (DC-cholesterol), and 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) were purchased from Avanti Polar Lipids, Inc. 5-Doxyl-stearic acid, free radical (5-DSA) and deuterium oxide (D2O) were obtained from Sigma.
Transfections
HeLa P4-R5 cells in 12-well plates were transfected with 600 ng of proviral plasmid or 600 ng of proviral mutant pNL4-3ΔVpu plus 500 ng of Rev-dependent Vpu-FLAG expression plasmid in triplicate using Lipofectamine 2000 (Life Technologies). Interferon-treated cells were cultured in 30 ng/ml of human interferon-α1 (Cell Signaling, 8927LC). Supernatants and cells were harvested the next day for p24 ELISA, immunoblot analysis, and staining for flow cytometry.
Virion Release Assays
Supernatants from provirus-transfected HeLa P4-R5 cells were collected, pelleted through a 20% sucrose cushion, then measured for Gag by p24 ELISA (Advanced Bioscience Laboratories) as previously described (2, 17).
Immunoblots
Immunoblot analysis for actin, BST2, and the FLAG epitope were performed as previously described (12, 25), developed using enhanced chemiluminescence and imaged using the ChemiDoc MP System (Bio-Rad). For PNGase F treatment, lysates in 1× SDS-PAGE buffer were diluted 2-fold with water, G7 Reaction Buffer (New England Biolabs), and Nonidet P-40, then treated with PNGase F (glycerol free, New England Biolabs, P0705S) for 2 h at 37 °C according to the manufacturer's instructions before immunoblotting.
Flow Cytometry
Transfected HeLa P4-R5 cells were stained with Alexa Fluor 647 anti-human CD317 (BST2, tetherin; RS38E (Biolegend)), or Alexa Fluor 647 mouse IgG1, κ isotype control, according to the manufacturer's instructions, fixed, and permeabilized using a BD Cytofix/Cytoperm kit according to the manufacturer's instructions, followed by staining with fluorescein isothiocyanate (FITC)-conjugated anti-HIV-1 p24 antibody (KC57; Beckman Coulter) as previously described (17). Surface expression of BST2 was analyzed using a BD Accuri C6 flow cytometer after gating on live p24-positive cells.
Immunofluorescence Microscopy
HeLa P4-R5 cells were plated at 120,000 per well on glass coverslips in a 24-well plate. The following day, they were transfected with 800 ng of proviral plasmid using Lipofectamine 2000 (Life Technologies). Four to six hours after transfection, the cell culture medium was changed to DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin. For studies of BST2 localization, 1000 units/ml of human interferon-α A/D (Sigma) was also added to the culture medium. For studies of Gag/BST2 and Gag/BST2/Vpu colocalization, 24 h post-transfection, the cells were fixed with 3% paraformaldehyde in PBS for 15 min at 4 °C, washed twice with PBS, then permeabilized with 0.2% Nonidet P-40 for 7 min at 4 °C. Fixed and permeabilized cells were washed twice with PBS, then blocked with 5% normal donkey serum and 3% BSA for 30 min at 4 °C. Cells were stained with rabbit anti-Vpu antiserum (from the NIH AIDS Research and Reference Reagent Program, contributed by Klaus Strebel) diluted 1:800 and/or mouse anti-BST2/HM1.24/CD317 antibody (Chugai Pharmaceutical Co., Kanagawa, Japan) diluted 1:250 for 30 min at 4 °C. Cells were washed three times with PBS, then incubated with Alexa Fluor 647-conjugated donkey anti-rabbit antibody and/or Rhodamine Red-X-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch) diluted 1:150 for 30 min at 4 °C, then washed four times with PBS. Cells were blocked with 5% mouse antiserum for 30 min at 4 °C, washed four times with PBS, then stained with FITC mouse anti-p24 (KC57) antibody (Beckman Coulter) diluted 1:150 for 30 min at 4 °C. The cells were washed 5 times with PBS, mounted, and imaged using a fluorescence microscope (Olympus) and SlideBook version 4.1 software (Intelligent Imaging Innovations, Denver, CO). For each field, a series of images along the z axis was collected, the images were deconvolved using the nearest-neighbor method, and a representative single image plane was chosen. Images were assembled using Adobe Photoshop software. For studies of Env and BST2 colocalization, cells were plated and transfected as above, then after 24 h blocked with 3% BSA, 5% donkey and 5% human serum in PBS for 30 min at 4 °C. Cells were stained with mouse anti-BST2/HM1.24/CD317 antibody (Chugai) diluted 1:250 and human anti-Env (2G12, from the NIH AIDS Research and Reference Reagent Program) diluted 1:150 for 30 min at 4 °C. Cells were washed three times with PBS, then stained with FITC-conjugated donkey anti-human antibody and Rhodamine Red-X-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch) diluted 1:150 for 30 min at 4 °C, then washed six times with PBS and fixed, mounted, and imaged as above. For studies of Env and membrane microdomain markers CD55, CD59, and CD81, after blocking with 3% BSA, 5% donkey and 5% human serum in PBS for 30 min at 4 °C, cells were stained with human anti-Env (2G12) diluted 1:150 and either mouse anti-CD55 (Biolegend) diluted 1:200, mouse anti-CD59 (Biolegend) diluted 1:200, or mouse anti-CD81 (Biolegend) diluted 1:80 for 30 min at 4 °C. Cells were washed three times with PBS, then stained with FITC-conjugated donkey anti-human antibody and Rhodamine Red-X-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch) diluted 1:150 for 30 min at 4 °C, then washed six times with PBS and fixed, mounted, and imaged as above. For studies of Gag and membrane microdomain markers CD55, CD59, and CD81, after blocking with 3% BSA and 5% donkey serum in PBS for 30 min at 4 °C, cells were stained with mouse anti-CD55 (Biolegend) diluted 1:200, mouse anti-CD59 (Biolegend) diluted 1:200, or mouse anti-CD81 (Biolegend) diluted 1:80 for 30 min at 4 °C. Cells were washed three times with PBS, then stained with Rhodamine Red-X-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch) diluted 1:150 for 30 min at 4 °C, then washed four times with PBS. Cells were fixed with 3% paraformaldehyde for 15 min at 4 °C, washed twice with PBS, then permeabilized with 0.2% Nonidet P-40 for 7 min at 4 °C. Cells were washed twice with PBS, blocked with 5% mouse antiserum for 30 min at 4 °C, washed four times with PBS, then stained with FITC mouse anti-p24 (KC57) antibody (Beckman Coulter) diluted 1:150 for 30 min at 4 °C. Cells were washed six times with PBS before imaging as above.
Protein Expression
The expression and purification of VpuCyto and mutants were performed essentially as previously described (30). The protein was overexpressed in BL21-CodonPlus competent cells (Agilent) in M9 medium for isotopically labeled samples. Purification was performed using nickel affinity chromatography, cyanogen bromide cleavage from the fusion protein, a second nickel affinity chromatography, and reverse phase HPLC. Milligram amounts of isotopically labeled protein were obtained for the NMR experiments.
Sample Preparation for NMR
Samples in micelles were prepared by dissolving purified and lyophilized protein powder of VpuCyto in an aqueous solution containing 100 mm DHPC with 10% (v/v) D2O with pH adjusted to 4.0. Samples for paramagnetic relaxation enhancement (PRE) measurements were prepared by adding either MnEDTA powder or 5-DSA powder to Vpu-containing DHPC in aqueous solution to a final concentration of 1 mm for MnEDTA and 0.5 mm for 5-DSA.
Samples with liposomes were prepared by mixing the trifluoroethanol-dissolved protein with chloroform-dissolved phospholipids, lipids, and/or cholesterol and were vaporated under a stream of nitrogen followed by lyophilization to generate proteoliposome films. These films were hydrated at 42 °C with aqueous buffer and vortexed to a final concentration of 0.1 mm protein and 1.8 mm lipid, and the pH of the samples was adjusted to 3.0.
Solution NMR Spectroscopy and Data Processing
The NMR experiments were performed on a 600 MHz Bruker AVANCE spectrometer equipped with a 5-mm triple resonance cryoprobe (1H, 13C, and 15N) and z axis pulsed gradient. All of the experiments were performed at 50 °C. 1H-15N heteronuclear single quantum correlation (HSQC) experiments were acquired with 2048 and 256 points in direct and indirect dimensions, respectively. Triple resonance HNCA, HNCACB, and HN(CO)CA experiments were performed on uniformly 13C- and 15N-labeled protein samples to obtain the backbone amide resonance assignments. The NMR data were processed using NMRPipe (31) and the figures were displayed using Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco). The resonance intensities and chemical shifts were extracted using Sparky. Chemical shift perturbations were calculated using the following equation.
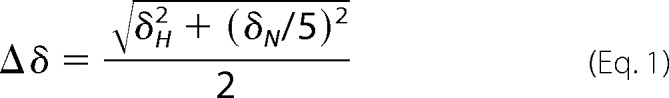 |
RESULTS
Mutation of Vpu Trp-76 to Glycine Specifically Impairs Virion Release in Interferon-treated Cells
We recently showed that mutation of Trp-76 near the C terminus of Vpu to glycine impairs the ability of Vpu to enhance virion release without affecting the surface down-regulation of BST2 (17). As BST2 is induced by interferon in response to HIV infection in vivo (32), we first evaluated the phenotypes of the Vpu-W76G mutant in the setting of the high levels of BST2 expression induced by α-interferon in vitro. HeLa P4-R5 cells were transfected in triplicate with NL4-3 proviral plasmids encoding no Vpu (NL4-3ΔVpu), wild-type Vpu, the β-TrCP-binding mutant Vpu-S52N,S56N, Vpu-W76G, and the combined mutant Vpu-S52N,S56N + W76G, and then treated (or not) with human interferon-α1 overnight. Viral supernatants were harvested and pelleted through a 20% sucrose cushion to isolate virion-associated Gag/p24 before analysis by p24 ELISA. Cells were collected and lysed for immunoblot as well as stained for surface BST2 and intracellular p24 before analysis by flow cytometry (FACS). Fig. 1A shows the results of ELISA for p24 released into the supernatants. Vpu-W76G was as impaired for virion release enhancement as Vpu-S52N,S56N, which cannot degrade BST2. The combined mutant Vpu-S52N,S56N + W76G was as impaired for virion release as the no Vpu mutant (ΔVpu), indicating that the S52N,S56N and W76G mutations have additive effects and that these regions of Vpu likely contribute to its function by distinct mechanisms. These relationships recapitulated our previous findings, but here they hold in the setting of the increased levels of BST2 induced by interferon. An immunoblot of the cell lysates is shown in Fig. 1B; the smears in the top panels are consistent with the reported heterogeneous glycosylation of BST2 in HeLa cells (12, 33, 34). In the bottom panels, the same lysates were treated with PNGase F, which cleaves N-linked glycans from BST2 and yielded discrete lower molecular weight bands. These immunoblots confirmed that BST2 expression was substantially increased with interferon treatment. They also show that Vpu-W76G induced nearly wild-type levels of BST2 degradation both in the presence or absence of interferon treatment (compare lane 3 to 7 (no IFN) and 4 to 8 (with IFN)). As expected, Vpu-S52N,S56N and Vpu-S52N,S56N + W76G did not degrade BST2 (compare lane 3 to 5 and 9 (no IFN); also lane 4 to 6 and 10 (+IFN)). The surface down-regulation of BST2 was likewise similar for wild-type and W76G mutant Vpu. Fig. 1C shows two-color FACS plots for BST2 surface staining (y axis) versus intracellular p24 expression (x axis) for these cells; the data indicate that the wild-type and the W76G mutant each down-regulate BST2, whereas the Vpu-S52N,S56N and Vpu-S52N,S56N + W76G mutants fail to down-regulate and cause a paradoxical increase in cell surface BST2. Fig. 1D shows the average MFI for surface BST2 in p24-positive (infected) cells measured in triplicate. In summary, in the presence or absence of interferon treatment, Vpu-W76G effectively down-regulated BST2, while Vpu-S52N,S56N and Vpu-S52N,S56N + W76G did not. Yet, Vpu-W76G was impaired for virion release.
FIGURE 1.

Characterization of the W76G mutation in interferon-treated cells expressing high levels of BST2. A, enhancement of HIV-1 virion release by “wild-type” Vpu and site-directed mutants (ΔVpu, S52N,S56N, W76G, and S52N,S56N + W76G). The concentration of pelletable p24 antigen in the culture supernatants in triplicate was measured by ELISA. Error bars represent the standard deviation. B, immunoblot analysis of cell lysates showing the induction of BST2 expression in HeLa-P4-R5 cells following treatment with interferon and degradation of BST2 by wild-type and W76G mutant Vpu. The top panels show untreated lysates, whereas the bottom panels show lysates that were treated with PNGase F. Actin is shown as a loading control. C, down-regulation of surface BST2 in cells transfected with proviral DNA harboring the indicated Vpu mutations in the absence (top panels) or presence (bottom) of interferon-α. Wild-type and W76G Vpu are able to down-regulate surface BST2, whereas the S52N,S56N and S52N,S56N + W76G mutants are not, regardless of interferon treatment. The two-color plots show BST2 expression (y axis) versus intracellular p24 expression (x axis). Results are representative of triplicates. D, bar graph of average MFI for surface BST2 in p24-positive cells. Error bars are the standard deviations of triplicates.
Trp-76 Is Required for the Displacement of BST2 from Sites of Viral Assembly
Because the W76G substitution did not markedly affect either the surface down-regulation or the degradation of BST2 by Vpu, but did impair the enhancement of virion release, we reasoned that this residue might play a role in the ability of Vpu to displace BST2 from sites of viral assembly within the plane of the plasma membrane (16). To test this, we transfected HeLa P4-R5 cells with HIV-1 (NL4-3) proviral plasmids encoding no Vpu (ΔVpu), wild-type, Vpu-S52N,S56N, Vpu-W76G, and Vpu-S52N,S56N + W76G and then treated the cells with human interferon-α overnight. Cells were fixed, permeabilized, and stained for BST2 and HIV Gag/p24, then analyzed by immunofluorescence microscopy as described under “Experimental Procedures.” Fig. 2A shows BST2 (in red), Gag (in green), and their overlap (in yellow) for cells transfected with the indicated Vpu-mutant proviral plasmids; the single image planes shown depict optical sections along the cover glass. In the absence of Vpu (ΔVpu), BST2 and Gag colocalized extensively as indicated by the substantial (yellow) overlap seen in the rightmost image. As reported previously (16), both wild-type Vpu and the β-TrCP-binding site mutant Vpu-S52N,S56N induced the displacement of BST2 from Gag at the plasma membrane. This is shown by the decrease in the yellow color in the merged image for wild-type (“WT”) and Vpu-S52N,S56N (“S52N,S56N”) compared with ΔVpu in Fig. 2A as well as the lower Pearson's correlation coefficients for the colocalization of BST2 and Gag in Fig. 2B. The effect of wild-type Vpu on the colocalization of BST2 and Gag is likely due both to surface down-regulation as well as the displacement of BST2 from Gag in the plane of the plasma membrane. In contrast, Vpu-S52N,S56N was unable to down-regulate BST2 (see Fig. 1, B–D), so that its ability to decrease the colocalization of BST2 and Gag in the plane of the plasma membrane is presumably due solely to the displacement effect. In the case of Vpu proteins with the W76G mutation, BST2 and Gag colocalized to a relatively greater extent; this was the case both when Vpu-W76G was compared with wild-type Vpu (p value 5.52 × 10−8, unpaired t test) and when the combination mutant Vpu-S52N,S56N + W76G was compared with Vpu-S52N,S56N (p value 2.22 × 10−14, unpaired t test). Moreover, when the Vpu-S52N,S56N + W76G mutations were combined, the coefficient of correlation between Gag and BST2 was the same as when no Vpu was expressed (p value 0.3407, unpaired t test). These data indicate that the colocalization of BST2 and Gag along the plasma membrane is inversely related to virion release (compare Fig. 1A with 2B). They also indicate that Trp-76 is critical to the ability of Vpu to displace BST2 from Gag within the plane of the membrane.
FIGURE 2.

Vpu displaces BST2 from Gag in a Trp-76-dependent manner. A, HeLa P4-R5 cells were transfected with proviral plasmids, either pNL4-3 (“WT”) or the indicated Vpu-mutants. The following day, the cells were fixed, permeabilized, and stained for p24 and BST2. Wide field images were acquired as a Z-series, and the plane along the cover glass that captures the cell surface is shown after deconvolution of the image using a “nearest neighbors” algorithm. The left column shows anti-BST2 staining; the middle column shows anti-Gag/p24; and the right column shows a merged image in which the overlapping Gag and BST2 appears yellow. Wild-type Vpu and the S52N,S56N mutant are each able to displace BST2 from Gag (compared with ΔVpu), and this activity is lost when Trp-76 is mutated to Gly. B, Pearson's correlation coefficients for the colocalization of BST2 and Gag along the plasma membrane in the presence of Vpu or mutants from the experiment shown in A. At least 25 cells were assessed for each condition, using the image plane adjacent to the cover glass. Error bars are the standard deviations. p values for unpaired two-tailed t tests were as noted on the graph and for ΔVpu versus WT, W76G, and S52N,S56N were <0.0001; for WT versus S52N,S56N, p = 0.0094; for WT versus W76G and S52N,S56N + W76G, p < 0.0001; and for W76G versus S52N,S56N and S52N,S56N + W76G, p < 0.0001.
To confirm that our staining for Gag was consistent with virion assembly sites and not nonspecific aggregates of Gag, we stained the surface of virus-producing cells for the HIV-1 envelope glycoprotein (Env). When the relationship between BST2 and sites of viral assembly stained for Env was analyzed, a pattern similar to that seen with Gag emerged (Fig. 3). For these experiments, HeLa P4-R5 cells were transfected with Vpu-mutant provirus as above, then the staining for Env and BST2 was performed to allow “co-patching” of the proteins as described under “Experimental Procedures.” In this technique, cells are labeled before chemical fixation at 4 °C with primary and fluorophore-labeled secondary antibodies to detect the proteins of interest. This allows antibody-mediated cross-linking and aggregation (co-patching) of the proteins, provided that they both initially reside in the same membrane microdomains. This approach yielded robust colocalization of BST2 and Env in the absence of Vpu. As shown in Fig. 3A, BST2 is in red, Env is in green, and regions of the plasma membrane where they overlap are in yellow in the merge column on the right. (Note that the single image planes shown here depict optical sections 1–3 μm above the cover glass, because the cell surface along the cover glass stained poorly without prior chemical fixation (data not shown).) Compared with provirus lacking vpu (ΔVpu), the colocalization of BST2 and Env was decreased by wild-type Vpu (“WT”) and Vpu-S52N,S56N (Fig. 3A), a conclusion supported by the lower Pearson's correlation coefficients for the colocalization of Env and BST2 (p value for ΔVpu versus WT: 3.79 × 10−13, for ΔVpu versus S52N,S56N: 5.93 × 10−8, unpaired t test) in Fig. 3B. Vpu-mutants W76G, S52N,S56N + W76G, and the transmembrane domain mutant A10F,A14F,A18F (which does not bind BST2) permitted co-patching of Env and BST2 to levels near that of ΔVpu (p values of 0.0810, 0.2486, and 0.7940, respectively, by unpaired t test), indicating that these mutants are impaired in their ability to displace BST2 from sites of viral assembly. For Vpu-A10F,A14F,A18F, this impairment presumably relates to its inability to bind BST2 (6).
FIGURE 3.

Staining for surface Env and BST2 in the presence of Vpu-mutants also reveals the displacement effect. A, HeLa P4-R5 cells were transfected with the indicated proviral Vpu-mutant plasmids and the next day stained for BST2 (in red) and Env (in green), then fixed and imaged. The merged image is shown on the right with BST2-Env overlap in yellow. Wild-type (WT) Vpu and the S52N,S56N mutant are each able to displace BST2 from Env (compared with ΔVpu), and this activity is lost for W76G and for the Vpu transmembrane domain mutant A10F,A14F,A18F, which is unable to interact with BST2. B, Pearson's correlation coefficients for the colocalization of BST2 and Env along the plasma membrane in the presence of Vpu or mutants from the experiment shown in A. At least 20 cells were assessed for each condition, using image planes 1–3 μm above the cover glass. Error bars are the standard deviations. p values for unpaired two-tailed t tests were as noted on the graph and for ΔVpu versus WT and S52N,S56N were <0.0001; for ΔVpu versus W76G, p = 0.0810; for WT versus W76G, S52N,S56N, S52N,S56N + W76G, and A10F,A14F,A18F, p < 0.0001; for W76G versus S52N,S56N, p < 0.0001; for W76G versus S52N,S56N + W76G, p = 0.0037; for W76G versus A10F,A14F,A18F, p = 0.0554; for S52N,S56N versus A10F,A14F,A18F, p < 0.0001; and for S52N,S56N + W76G versus A10F,A14F,A18F, p = 0.4075.
The most straightforward model for the Trp-76-dependent ability of Vpu to displace BST2 from sites of viral assembly involves a direct interaction between Vpu and BST2 at the plasma membrane. However, whether Vpu acts at the plasma membrane is somewhat controversial, with some data suggesting that the Vpu-BST2 interaction occurs within endosomes (10, 35) and/or the trans-Golgi network (7, 36–38), whereas other findings support a role for Vpu at the plasma membrane (8, 39). To investigate whether Vpu is positioned on or near the plasma membrane and to confirm that the displacement effect requires the interaction between the two proteins, we transfected HeLa P4-R5 cells with provirus harboring vpu mutants ΔVpu, Vpu-S52N,S56N, or the transmembrane domain mutant Vpu-A10F,A14F,A18F; the cells were stained for BST2, Gag, and Vpu, and then imaged the cells as described under “Experimental Procedures.” Fig. 4A shows expression of BST2 in red, Gag in green, and Vpu in blue with overlap shown in the merge column on the right. As in Fig. 2, the single image planes shown depict optical sections along the cover glass. Colocalization of BST2 and Vpu is shown in magenta, and as expected was less for Vpu-A10F,A14F,A18F, as these mutations disrupt the interaction between Vpu and BST2 (see Pearson's correlation coefficients for the colocalization of Vpu and BST2 in Fig. 4B). Consistent with the data of Fig. 2, the colocalization of BST2 and Gag (yellow in the merged image) was less for Vpu-S52N,S56N than for ΔVpu (Fig. 4, A and C), supporting the displacement effect. In contrast, Vpu-A10F,A14F,A18F was inactive; in its presence, BST2 and Gag colocalized to levels similar to those seen in the case of ΔVpu (see Pearson's correlation coefficients for the colocalization of Gag and BST2 in Fig. 4C). These imaging data are consistent with a location of Vpu on or near the plasma membrane, such that it can directly mediate the displacement effect. The data also confirm that interaction between the transmembrane domains of the proteins is required for Vpu to displace BST2 from viral assembly sites.
FIGURE 4.

The location of Vpu relative to BST2 and Gag reveals that Vpu is positioned to mediate the displacement effect. A, HeLa P4-R5 cells were transfected with HIV-1 (NL4-3) proviral plasmids encoding the indicated Vpu mutants, fixed, permeabilized, and stained for BST2 (first column, in red), Gag (second column, green), and Vpu (third column, blue). The merged image is shown on the right with BST2-Gag overlap in yellow, BST2-Vpu overlap in magenta, and overlap of all three in white. B, Pearson's correlation coefficients for the colocalization of Vpu mutants and BST2 from the experiment shown in A. At least 10 cells were assessed for each condition, using the image plane adjacent to the cover glass. Error bars are the standard deviations. The p value was calculated by unpaired two-tailed t test. C, Pearson's correlation coefficients for the colocalization of BST2 and Gag along the plasma membrane in the presence of Vpu or mutants from the experiment shown in A. At least 25 cells were assessed for each condition, using the image plane adjacent to the cover glass. Error bars are the standard deviations. The p values were calculated by unpaired two-tailed t test. The p value for ΔVpu versus A10F,A14F,A18F was 0.8311.
Vpu Does Not Reorganize Membrane Microdomains during the Displacement of BST2 from Sites of Viral Assembly
BST2 has been proposed to link lipid rafts to each other and to the actin cytoskeleton (19, 40), whereas HIV assembly sites are known to include both lipid rafts and TEMs (20–22). In light of this, we wondered whether the displacement of BST2 from viral assembly sites by Vpu might disrupt the organization of membrane microdomains at assembly sites. To test this hypothesis, HeLa P4-R5 cells were transfected with proviral plasmids lacking vpu (ΔVpu) or encoding the Vpu-S52N,S56N mutant, then stained for either Gag or Env and a lipid raft marker (CD55 or CD59) or the tetraspanin CD81 in a co-patching experiment as described under “Experimental Procedures.” Fig. 5A shows the merged images for Gag staining in green on the left and Env staining in green on the right as well as the indicated membrane microdomain markers in red. Colocalization is shown in yellow. The images on the left in Fig. 5A and the Pearson's correlation coefficients for the colocalization of Gag and the domain markers in Fig. 5B show that the Vpu-S52N,S56N mutant displaced neither CD55 nor CD81 from Gag. Instead, we observed a small but statistically significant increase in the colocalization between Gag and CD55 (p = 0.0442, unpaired t test), and Gag and CD81 (p = 0.0217, unpaired t test), the cause of which is currently obscure. Results for another lipid raft marker, CD59, were similar to CD55 (data not shown). The right column of Fig. 5A and the Pearson's correlation coefficients for the colocalization of Env and the domain markers in Fig. 5C show that the expression of Vpu-S52N,S56N had no effect on the colocalization of Env and CD55 (p = 0.4653, unpaired t test) or CD81 (p = 0.7983, unpaired t test). Overall, Vpu does not appear to substantially affect the organization of markers of membrane microdomains, either lipid raft or TEM, with respect to Gag and Env. Therefore, the displacement of BST2 from sites of viral assembly likely involves the direct and specific action of Vpu on BST2, and this does not induce a general reorganization of membrane microdomains.
FIGURE 5.

Vpu does not change the relationship of HIV assembly sites to markers of lipid rafts or tetraspanin-enriched microdomains. A, HeLa P4-R5 cells were transfected with either pNL4–3ΔVpu or pNL4–3-Vpu-S52N,S56N mutant proviral plasmids and stained for the indicated membrane microdomain marker: CD55 (top two rows) as a marker of lipid rafts and CD81 (bottom two rows) as a marker of tetraspanin-enriched microdomains (TEMs). In addition, cells were either then fixed, permeabilized, and stained for p24 (left column) or co-patched for Env (right column). For all panels, the domain marker (CD55 or CD81) is in red, Gag (left) or Env (right) are in green, and the overlap in yellow. B, Pearson's correlation coefficients for the colocalization of the indicated membrane microdomain markers and Gag along the plasma membrane in the presence or absence of Vpu-S52N,S56N. C, Pearson's correlation coefficients for the colocalization of the indicated membrane microdomain markers and co-patched Env in the presence or absence of Vpu-S52N,S56N. For the analysis in B and C at least 20 cells were assessed for each condition. Error bars are the standard deviations. p values were calculated by unpaired two-tailed t test.
Trp-76 but Not Gly-76 Interacts with Lipids
Tryptophans are known to anchor the transmembrane domains of proteins in membranes and typically occur at or near the non-polar/polar interface where the hydrophobic acyl chain meets the hydrophilic lipid head group. For example, Vpu contains such a conserved tryptophan near the C-terminal end of its transmembrane domain. Although membrane anchoring of cytoplasmic domains by tryptophan is apparently rare, we considered that such an anchoring activity of Trp-76 might explain its role in the BST2 displacement phenotype. To test this, we studied the interaction of the cytoplasmic domain of Vpu with lipids by NMR spectroscopy. For these studies, we expressed and purified uniformly 15N-labeled polypeptides corresponding to the wild-type Vpu cytoplasmic domain sequence (VpuCyto) and the tryptophan 76 to glycine mutant (W76G VpuCyto). A well resolved, fully assigned 1H-15N HSQC NMR spectrum for VpuCyto in aqueous solution was obtained (Fig. 6A, black contours). When DMPC liposomes were added to the aqueous solution, VpuCyto resonances line widths were broadened, resulting in reduced intensities. The reduced intensities indicate a change of reorientation rate due to the interaction of VpuCyto with the liposomes (Fig. 6B). In addition, chemical shift changes were observed for a number of resonances from VpuCyto residues; among them, Trp-76 was the most affected, suggesting that Trp-76 undergoes a significant environmental or conformational change upon addition of liposomes (Fig. 6C). When uniformly 15N-labeled W76G VpuCyto was dissolved in aqueous solution, the frequencies of most resonances were very similar to those of VpuCyto, suggesting a minimal perturbation in the structure upon mutation (Fig. 6D). When the DMPC liposomes were added to the aqueous solution containing W76G VpuCyto, smaller intensity changes were observed for resonances from W76G VpuCyto compared with wild-type VpuCyto (Fig. 6E). Moreover, no significant chemical shift changes were observed for Gly-76 (Fig. 6F). These data indicate that the cytoplasmic domain of Vpu interacts with lipids. The data also suggest that Trp-76 is an important determinant of that interaction and its molecular environment is altered upon lipid binding.
FIGURE 6.

NMR chemical shift perturbation shows that Trp-76 but not Gly-76 interacts with lipids. A, assigned 1H-15N HSQC NMR spectrum of uniformly labeled 15N-labeled VpuCyto peptide in H2O either with (blue) or without (black) DMPC liposomes. B, VpuCyto resonances relative intensity measured before and after addition of DMPC liposomes. C, chemical shift change induced in VpuCyto residues by addition of DMPC liposomes. D, assigned 1H-15N HSQC NMR spectrum of uniformly labeled 15N-labeled W76G VpuCyto peptide in H2O either with (blue) or without (black) DMPC liposomes. E, W76G VpuCyto resonances relative intensity measured before and after addition of DMPC liposomes. F, chemical shift change induced in W76G VpuCyto residues by addition of DMPC liposomes is shown to be much smaller than for Trp-76 (C).
Trp-76 Is Inserted in the Lipid Bilayer
Next we tested whether Trp-76 inserts into the lipid bilayer using PRE distance measurements. The PRE effects were measured by comparing the signal intensities with and without addition of either of two paramagnetic probes, MnEDTA and 5-DSA. MnEDTA is a bulky paramagnetic reagent restricted to the aqueous environment, whereas 5-DSA is restricted to the hydrophobic region of the lipid micelle as described in detail below. When MnEDTA was added to VpuCyto incorporated in DHPC micelles, signals from residues that are accessible to MnEDTA in solution are broadened and the corresponding intensities are lowered. In Fig. 7A, the largest intensity changes were observed among signals from residues in both the N- and C-terminal as well as the loop regions of VpuCyto, indicating that these regions are exposed to aqueous solvent and MnEDTA. However, resonance signals of Trp-76 amide and its indole in the C terminus were relatively unaffected, suggesting that Trp-76 was protected by insertion into the micelles (Fig. 7A). 5-DSA is a fatty acid that has a nitroxide functional group labeled at the fifth carbon position in the hydrophobic chain. When incorporated into the DHPC micelles, 5-DSA partitions to the hydrophobic region of the micelles, and the spin label is at the center of the micelle, as shown in the schematic in Fig. 7C. Consequently, 5-DSA can be used to detect the portion of the protein embedded in the micelles. For VpuCyto in DHPC micelles with 5-DSA, we observed that the residues in the membrane-proximal cytoplasmic domain helix were slightly more affected than the C-terminal region, but Trp-76 and especially its indole signals were the most broadened (Fig. 7B). Together, these PRE experiments using different lipid-partitioned paramagnetic probes suggest that Trp-76 embeds in the lipid micelles.
FIGURE 7.
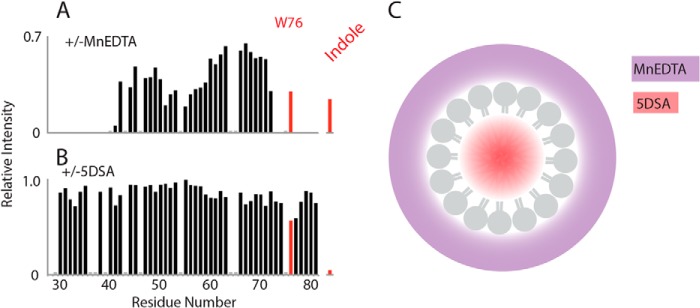
Trp-76 is inserted into micelles. Paramagnetic relaxation effect of MnEDTA and 5-DSA on VpuCyto in DHPC micelles characterized using differential signal intensities measured from 1H-15N HSQC spectra. A, relative intensity measured before and after addition of MnEDTA reveals that the Trp-76 amide and its indole are relatively unaffected compared with VpuCyto N-terminal, C-terminal, and loop regions, which are comparatively more exposed to aqueous solvent. B, relative intensity measured before and after addition of 5-DSA, demonstrating the largest effects on Trp-76 and its indole, which supports a structure where Trp-76 is embedded in the micelles. C, a schematic demonstrating the accessibility of MnEDTA (purple) and 5-DSA (red) in the DHPC micelles.
Trp-76 Can Interact with Positively Charged Lipids
The interaction of tryptophan with lipids can involve hydrophobic interactions as well as cation-π interactions between the aromatic ring and positively charged lipid head groups. To determine whether Vpu Trp-76 interacts with positively charged lipids, we compared the effects of positively charged and neutral lipids on VpuCyto resonances. When positively charged DMEPC liposomes (structure shown in Fig. 8G) were added to VpuCyto in aqueous medium, resonance intensities were further reduced as compared with the effect of adding neutral DMPC liposomes, especially for the second helix and the C terminus of the cytoplasmic domain. Moreover, the intensity for Trp-76 was drastically reduced upon addition of DMEPC liposomes (Fig. 8A). The same trend was observed for the chemical shift perturbation (Fig. 8B); Trp-76 was even more affected by positively charged lipids (DMEPC liposomes) than by neutral lipids (DMPC liposomes). Other positively charged lipids, specifically DMTAP and DMPC mixed with DC-cholesterol, were tested, and similar patterns of intensity changes and chemical shift perturbations were observed (Fig. 8, C–F). The structures of DMPC, DMEPC, DMTAP, and DC-cholesterol are shown in Fig. 8G. These data indicate an enhanced interaction of VpuCyto and Trp-76 with positively charged lipids relative to neutral lipids, and supports the notion that cation-π interactions might contribute to this interaction.
FIGURE 8.

Enhanced interaction of the Vpu cytoplasmic domain and Trp-76 with positively charged lipids. A, VpuCyto resonances relative intensity measured before and after addition of either DMPC liposomes (blue) or DMEPC liposomes (red). B, chemical shift change induced in VpuCyto residues by addition of either DMPC liposomes (blue) or DMEPC liposomes (red). C, relative intensity of VpuCyto resonances measured before and after addition of DMTAP liposomes. D, chemical shift change induced in VpuCyto residues by the addition of DMTAP liposomes. E, relative intensity of VpuCyto resonances measured before and after the addition of DMPC liposomes mixed with DC-cholesterol. F, chemical shift change induced in VpuCyto residues by the addition of DMPC liposomes mixed with DC-cholesterol. G, structures of DMPC, DMEPC, DMTAP, and DC-cholesterol.
Phenylalanine Cannot Substitute for the Activity of Trp-76
Because our NMR studies supported a possible role for cation-π interactions between the aromatic ring and positively charged lipid head groups during interaction of the Vpu cytoplasmic domain with membranes, we tested whether another aromatic amino acid, phenylalanine, could substitute functionally for Trp-76. To test whether Vpu-W76F could down-regulate BST2 from the cell surface, we transfected HeLa P4-R5 cells with proviral plasmids lacking vpu plus expression constructs encoding FLAG epitope-tagged wild-type NL4-3 Vpu, mutant Vpu-W76G or Vpu-W76F alone or in combination with the S52N,S56N mutations. As shown in the two-color FACS plots in Fig. 9A and the graph of average MFI for surface BST2 in p24-positive cells in Fig. 9B, Vpu-W76F was functional for surface down-regulation of BST2; its activity was indistinguishable from wild-type (Trp-76 Vpu) and W76G Vpu. The addition of the S52N,S56N mutations abrogated this activity. These results are shown quantitatively in Fig. 9C, where the BST2 down-regulation by each Vpu mutant is shown relative to that of wild-type Vpu. In contrast, the Vpu-W76F mutant did not support wild-type levels of virion release enhancement, as shown in Fig. 9D. Supernatant p24 released by provirus encoding Vpu-W76F was similar to that encoding Vpu-S52N,S56N, just under half that of wild-type NL4-3. Notably, Vpu-W76F was slightly more active for virion release enhancement than Vpu-W76G. Immunoblot for the FLAG epitope tag in Fig. 9E revealed no significant difference in the expression of the mutant Vpu proteins to account for their ability to enhance virion release.
FIGURE 9.

Phe-76 supports neither optimal virion release nor the binding of the C terminus of Vpu to lipids. A, two-color plots showing BST2 down-regulation (y axis) and p24 expression (x axis) in HeLa P4-R5 cells co-transfected with a proviral plasmid lacking Vpu (pNL4-3ΔVpu) and a Rev-dependent expression plasmid encoding the indicated Vpu mutants with a C-terminal FLAG epitope tag. Plots are representative of four experiments done in triplicate. B, bar graph of average MFI for surface BST2 in p24-positive cells. Error bars are the standard deviations of triplicates. C, quantification of relative levels of BST2 down-regulation by the Vpu mutants in A shows that Vpu-W76F is as functional for surface down-regulation of BST2 as WT and Vpu-W76G. Samples were gated on live p24-positive cells. The MFI of BST2 staining for each Vpu mutant was subtracted from that of the no-Vpu (ΔVpu) control, and this value was divided by the same calculation made for NL4-3 Vpu to derive the relative activity. Error bars are the standard deviations of triplicates. D, the concentration of pelletable p24 antigen in the culture supernatants from the cells shown in A was measured by ELISA. Vpu-W76F is impaired in its ability to enhance virion release compared with wild-type Vpu although not to the same extent as Vpu-W76G. Error bars represent the standard deviation of triplicates. E, immunoblot analysis of cell lysates for the FLAG epitope tag showing expression levels of the Vpu mutants from the cells in A. Actin is shown as a loading control. F, W76F VpuCyto resonances relative intensity measured before and after addition of DMPC liposomes. G, chemical shift change induced in W76F VpuCyto residues by addition of DMPC liposomes.
Similar to W76G (see Fig. 6, E and F), W76F VpuCyto showed no significant changes in its HSQC spectrum compared with wild-type VpuCyto. Also like W76G VpuCyto, both the intensity change (Fig. 9F) and the chemical shift perturbation data (Fig. 9G) indicate that Phe-76 and W76F VpuCyto do not interact with DMPC liposomes as strongly as wild-type VpuCyto. These results suggest that both the W76G and W76F mutations impair the protein-lipid interaction. The results also suggest that the interaction of Trp-76 with lipids is unlikely to solely depend on an aromatic side chain. Importantly, the functional data with regard to virion release are consistent with the NMR data: a phenylalanine (like a glycine) cannot support the function of Trp-76 with regard to enhancing virion release or interacting with the lipid bilayer.
DISCUSSION
The ability of Vpu to displace BST2 from sites of viral assembly in the plane of the plasma membrane appears to be an important aspect of the mechanism by which Vpu enhances virion release, distinct from both surface down-regulation and degradation of BST2. This activity is shown here to be a direct and specific effect of Vpu on BST2 localization relative to viral assembly sites (as defined by Gag and Env proteins), and is neither a consequence of a reorganization of the membrane microdomains (lipid rafts, TEMs) associated with viral assembly sites nor does it induce changes in those domains. This BST2-displacement activity of Vpu depends in large part on the C-terminal residue Trp-76, at least for clade B Vpu. This activity now explains the anomalous phenotype of the W76G polymorphism described in recent studies of naturally occurring Vpu proteins (17).
How does Trp-76 contribute to this displacement activity? Our NMR data obtained using recombinant protein associated with either DHPC micelles or with liposomes containing neutral or positively charged lipids support a direct interaction of Trp-76 with membrane phospholipids. Similar conclusions with regard to Trp-76 have been suggested previously based on chemical shift perturbation and PRE measurements on VpuCyto in DPC micelles (41). Based on these biochemical properties, we propose a modified model of the Vpu structure in which the extreme C terminus attaches to the membrane via insertion of Trp-76 into the lipid phase. This C-terminal attachment of Vpu to the membrane appears to be critical for activity, as neither the naturally occurring variant glycine nor our site-directed substitution of phenylalanine supported the membrane interaction or optimal virion release. Exactly how the C-terminal membrane attachment supports the ability of Vpu to displace BST2 from viral assembly sites remains to be elucidated. One possibility is that the C terminus of Vpu inserts into specific lipid domains that are distinct from viral assembly sites. The preference of Vpu for such sites and its direct interaction with BST2 would enable it to “drag” BST2 away from the sites of viral assembly. Another possibility is that the C-terminal membrane attachment correctly orients the cytoplasmic domain of Vpu such that it can interact with a cellular peripheral membrane or cytoskeletal protein, and this connection ultimately provides the movement of BST2 relative to viral assembly sites. Such a co-factor might, for example, be a clathrin adaptor, an hypothesis consistent with the role reported for the EXXXLV64 adaptor protein binding sequence in the displacement effect (16, 42). In either case, Vpu would presumably need to interact with BST2 at assembly sites initially, and then move together with BST2 away from those sites. In this regard, we speculate that Vpu might sample the C-terminal attached and unattached conformations, providing alternate binding (or not) to the membrane microdomains or cellular co-factors that support the displacement effect. This would allow Vpu to first bind BST2, and then direct it from the sites of viral assembly. A prior NMR study of the Vpu cytoplasmic domain supports this notion insofar as the C-terminal end of Vpu seems able to participate in more than one conformation: one in which Trp-76 is exposed to the aqueous phase and another in which it is embedded in the lipid phase (41).
Tryptophan residues in trans-membrane helices are commonly located at the boundary of the hydrocarbon core of the bilayer and the polar lipid head groups. Indeed, Vpu has such a tryptophan at the C-terminal end of its transmembrane domain, and this residue is strictly conserved among HIV-1 isolates (43, 44). In contrast, evidence for the involvement of membrane attachment via tryptophan such as we propose here is rare. We are aware of only two similar examples, vinculin and annexin V, neither of which are transmembrane proteins (29, 45). In the case of vinculin, like Vpu, the tryptophan occurs very near the C terminus of the protein and in an unfolded, flexible region. The membrane attachment sequence of vinculin, TPWYQ, is notable for a proline that precedes the Trp; this is also seen in the consensus sequence of clade B Vpu proteins, APWDV, but not in the case of annexin V, in which the key tryptophan is not located near the C terminus.
The mechanism by which tryptophan residues interact with biological membranes at the hydrophobic/hydrophilic interface seems to be multifaceted, potentially involving hydrophobic interactions with the hydrocarbon chain, cation-π interactions between the aromatic ring and positively charged lipid head groups, and hydrogen bonding between indole and lipid head groups. Here, our NMR data indicate that the cytoplasmic domain of Vpu interacted even more strongly with positively charged lipid than it did with neutral lipid. However, phenylalanine at position 76 supported neither the interaction with lipid nor the enhancement of virion release by Vpu. These results suggest that cation-π interactions, although they might contribute to the interaction, are apparently not sufficient for membrane insertion of the Vpu C terminus, and consequently, they are not sufficient to support enhancement of virion release. The interaction may be reinforced by hydrogen bonding between the tryptophan indole nitrogen and a lipid head group, which is not possible with phenylalanine. At least in the context of clade B Vpu proteins, tryptophan seems uniquely suited to this function. In this regard, however, we note that Trp-76, although conserved in clades B, D, and G, does not occur in Vpu proteins of other clades such as C, which is the most prevalent in the HIV-1 pandemic. The corresponding region of clade C Vpu typically contains the sequence RLLDV, and whether or not this sequence provides membrane insertion and/or the ability to displace BST2 from viral assembly sites remains to be determined.
Finally, we note that virion assembly sites are not random; in the case of HIV-1, they are enriched in markers of cholesterol-enriched lipid rafts and in tetraspanins. HIV-1 Gag specifically binds the acidic lipid phosphatidylinositol 4,5-bisphosphate, and its assembly appears to drive the coalescence of lipid rafts and TEMs (20–22). BST2 appears to associate both with lipid rafts and TEMs (40). Moreover, due to its unusual topology, an N-terminal transmembrane domain and a C-terminal glycosylphosphatidylinositol anchor, BST2 has been proposed to cross-link lipid rafts. Consequently, the displacement effect might well have disrupted the associations between membrane microdomains that accompany virion assembly. Surprisingly, we found no morphological evidence for this; Vpu displaced BST2 from both Gag and Env (viral assembly sites), whereas neither raft nor TEM markers were displaced from these sites. Our data suggest that the movement of BST2 relative to these sites by Vpu is specific, and it does not in turn cause a more general disruption of membrane topology.
In summary, the anomalous phenotype of the Vpu-W76G polymorphism, a defect in enhancing virion release despite preserved activity in down-regulating BST2 from the cell surface, can be explained by the key role of Trp-76 in the Vpu-mediated displacement of BST2 from virion assembly sites within the plane of the plasma membrane. This activity correlates with the ability of Trp-76 to mediate the attachment of the Vpu C terminus to the lipid bilayer in vitro, and it supports a modified view of the topology of Vpu with respect to the membrane. We speculate that membrane anchoring of the C terminus either drives a specific association of Vpu with lipid microdomains distinct from those where viral assembly occurs, or it orients the cytoplasmic domain of Vpu such that it can interact with an as yet unidentified cellular protein cofactor to mediate the displacement effect.
Acknowledgments
We utilized the Biotechnology BTRC for NMR Molecular Imaging of Proteins at the University of California, San Diego. We also thank Marissa Suarez for performing the p24 ELISA and other technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R21AI114397 (to J. G. and M. K. L.), R37AI081668 (to J. G.), and P41EB002031, RO1EB005161, RO1GM099986, and RO1GM066978 (to S. J. O.), The James B. Pendleton Charitable Trust and the University of California, San Diego, Center for AIDS Research (CFAR), NIH funded program Grant P30 AI036214, which is supported by the NIAID, NCI, NIMH, NIDA, NICHD, NHLBI, NIA, NIGMS, and NIDDK.
- BST2
- bone marrow stromal antigen 2
- β-TrCP
- β-transducin repeat containing protein
- TEMs
- tetraspanin-enriched membrane domains
- VpuCyto
- Vpu cytoplasmic domain
- DMPC
- 1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DMEPC
- 1,2-dimyristoyl-sn-glycero-3-ethylphosphocholine
- DHPC
- 1,2-dihexanoyl-sn-glycero-3-phosphocholine
- DMTAP
- 1,2-dimyristoyl-3-trimethylammonium propane
- DC-cholesterol
- 3β-[N-(N′,N′-dimethylaminoethane)carbamoyl]cholesterol hydrochloride
- 5-DSA
- 5-doxyl-stearic acid
- PRE
- paramagnetic relaxation enhancement
- HSQC
- heteronuclear single quantum correlation
- MFI
- mean fluorescent intensity
- PNGase F
- peptide:N-glycosidase F.
REFERENCES
- 1. Neil S. J., Zang T., Bieniasz P. D. (2008) Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430 [DOI] [PubMed] [Google Scholar]
- 2. Van Damme N., Goff D., Katsura C., Jorgenson R. L., Mitchell R., Johnson M. C., Stephens E. B., Guatelli J. (2008) The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3, 245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Park S. H., De Angelis A. A., Nevzorov A. A., Wu C. H., Opella S. J. (2006) Three-dimensional structure of the transmembrane domain of Vpu from HIV-1 in aligned phospholipid bicelles. Biophys. J. 91, 3032–3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kupzig S., Korolchuk V., Rollason R., Sugden A., Wilde A., Banting G. (2003) Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4, 694–709 [DOI] [PubMed] [Google Scholar]
- 5. McNatt M. W., Zang T., Hatziioannou T., Bartlett M., Fofana I. B., Johnson W. E., Neil S. J., Bieniasz P. D. (2009) Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 5, e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Skasko M., Wang Y., Tian Y., Tokarev A., Munguia J., Ruiz A., Stephens E. B., Opella S. J., Guatelli J. (2012) HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 287, 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dubé M., Roy B. B., Guiot-Guillain P., Binette J., Mercier J., Chiasson A., Cohen E. A. (2010) Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 6, e1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iwabu Y., Fujita H., Kinomoto M., Kaneko K., Ishizaka Y., Tanaka Y., Sata T., Tokunaga K. (2009) HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 284, 35060–35072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goffinet C., Allespach I., Homann S., Tervo H. M., Habermann A., Rupp D., Oberbremer L., Kern C., Tibroni N., Welsch S., Krijnse-Locker J., Banting G., Kräusslich H. G., Fackler O. T., Keppler O. T. (2009) HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5, 285–297 [DOI] [PubMed] [Google Scholar]
- 10. Mitchell R. S., Katsura C., Skasko M. A., Fitzpatrick K., Lau D., Ruiz A., Stephens E. B., Margottin-Goguet F., Benarous R., Guatelli J. C. (2009) Vpu antagonizes BST-2-mediated restriction of HIV-1 release via β-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5, e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Douglas J. L., Viswanathan K., McCarroll M. N., Gustin J. K., Früh K., Moses A. V. (2009) Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a βTrCP-dependent mechanism. J. Virol. 83, 7931–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tokarev A. A., Munguia J., Guatelli J. C. (2011) Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J. Virol. 85, 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Janvier K., Pelchen-Matthews A., Renaud J. B., Caillet M., Marsh M., Berlioz-Torrent C. (2011) The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 7, e1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mangeat B., Gers-Huber G., Lehmann M., Zufferey M., Luban J., Piguet V. (2009) HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its β-TrCP2-dependent degradation. PLoS Pathog. 5, e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miyagi E., Andrew A. J., Kao S., Strebel K. (2009) Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U.S.A. 106, 2868–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McNatt M. W., Zang T., Bieniasz P. D. (2013) Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 9, e1003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jafari M., Guatelli J., Lewinski M. K. (2014) Activities of transmitted/founder and chronic clade B HIV-1 Vpu and a C-terminal polymorphism specifically affecting virion release. J. Virol. 88, 5062–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rollason R., Korolchuk V., Hamilton C., Schu P., Banting G. (2007) Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J. Cell Sci. 120, 3850–3858 [DOI] [PubMed] [Google Scholar]
- 19. Rollason R., Korolchuk V., Hamilton C., Jepson M., Banting G. (2009) A CD317/tetherin-RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. J. Cell Biol. 184, 721–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nydegger S., Khurana S., Krementsov D. N., Foti M., Thali M. (2006) Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J. Cell Biol. 173, 795–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thali M. (2011) Tetraspanin functions during HIV-1 and influenza virus replication. Biochem. Soc. Trans. 39, 529–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krementsov D. N., Rassam P., Margeat E., Roy N. H., Schneider-Schaulies J., Milhiet P. E., Thali M. (2010) HIV-1 assembly differentially alters dynamics and partitioning of tetraspanins and raft components. Traffic 11, 1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klimkait T., Strebel K., Hoggan M. D., Martin M. A., Orenstein J. M. (1990) The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 64, 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schubert U., Bour S., Ferrer-Montiel A. V., Montal M., Maldarell F., Strebel K. (1996) The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 70, 809–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tokarev A., Suarez M., Kwan W., Fitzpatrick K., Singh R., Guatelli J. (2013) Stimulation of NF-κB activity by the HIV restriction factor BST2. J. Virol. 87, 2046–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deng H., Liu R., Ellmeier W., Choe S., Unutmaz D., Burkhart M., Di Marzio P., Marmon S., Sutton R. E., Hill C. M., Davis C. B., Peiper S. C., Schall T. J., Littman D. R., Landau N. R. (1996) Identification of a major co-receptor for primary isolates of HIV-1. Nature 381, 661–666 [DOI] [PubMed] [Google Scholar]
- 27. Morgenstern J. P., Land H. (1990) Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Charneau P., Mirambeau G., Roux P., Paulous S., Buc H., Clavel F. (1994) HIV-1 reverse transcription. A termination step at the center of the genome. J. Mol. Biol. 241, 651–662 [DOI] [PubMed] [Google Scholar]
- 29. Campos B., Mo Y. D., Mealy T. R., Li C. W., Swairjo M. A., Balch C., Head J. F., Retzinger G., Dedman J. R., Seaton B. A. (1998) Mutational and crystallographic analyses of interfacial residues in annexin V suggest direct interactions with phospholipid membrane components. Biochemistry 37, 8004–8010 [DOI] [PubMed] [Google Scholar]
- 30. Ma C., Marassi F. M., Jones D. H., Straus S. K., Bour S., Strebel K., Schubert U., Oblatt-Montal M., Montal M., Opella S. J. (2002) Expression, purification, and activities of full-length and truncated versions of the integral membrane protein Vpu from HIV-1. Protein Sci. 11, 546–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 32. Homann S., Smith D., Little S., Richman D., Guatelli J. (2011) Upregulation of BST-2/Tetherin by HIV infection in vivo. J. Virol. 85, 10659–10668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perez-Caballero D., Zang T., Ebrahimi A., McNatt M. W., Gregory D. A., Johnson M. C., Bieniasz P. D. (2009) Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139, 499–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kühl A., Banning C., Marzi A., Votteler J., Steffen I., Bertram S., Glowacka I., Konrad A., Stürzl M., Guo J. T., Schubert U., Feldmann H., Behrens G., Schindler M., Pöhlmann S. (2011) The Ebola virus glycoprotein and HIV-1 Vpu employ different strategies to counteract the antiviral factor tetherin. J. Infect Dis. 204, S850–S860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lau D., Kwan W., Guatelli J. (2011) Role of the endocytic pathway in the counteraction of BST-2 by human lentiviral pathogens. J. Virol. 85, 9834–9846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dubé M., Roy B. B., Guiot-Guillain P., Mercier J., Binette J., Leung G., Cohen E. A. (2009) Suppression of Tetherin-restricting activity upon human immunodeficiency virus type 1 particle release correlates with localization of Vpu in the trans-Golgi network. J. Virol. 83, 4574–4590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hauser H., Lopez L. A., Yang S. J., Oldenburg J. E., Exline C. M., Guatelli J. C., Cannon P. M. (2010) HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 7, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Petit S. J., Blondeau C., Towers G. J. (2011) Analysis of the human immunodeficiency virus type 1 M group Vpu domains involved in antagonizing tetherin. J. Gen. Virol. 92, 2937–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bour S., Perrin C., Strebel K. (1999) Cell surface CD4 inhibits HIV-1 particle release by interfering with Vpu activity. J. Biol. Chem. 274, 33800–33806 [DOI] [PubMed] [Google Scholar]
- 40. Billcliff P. G., Rollason R., Prior I., Owen D. M., Gaus K., Banting G. (2013) CD317/tetherin is an organiser of membrane microdomains. J. Cell Sci. 126, 1553–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wittlich M., Koenig B. W., Stoldt M., Schmidt H., Willbold D. (2009) NMR structural characterization of HIV-1 virus protein U cytoplasmic domain in the presence of dodecylphosphatidylcholine micelles. FEBS J. 276, 6560–6575 [DOI] [PubMed] [Google Scholar]
- 42. Jia X., Weber E., Tokarev A., Lewinski M., Rizk M., Suarez M., Guatelli J., Xiong Y. (2014) Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife 3, e02362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kobayashi T., Ode H., Yoshida T., Sato K., Gee P., Yamamoto S. P., Ebina H., Strebel K., Sato H., Koyanagi Y. (2011) Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 85, 932–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vigan R., Neil S. J. (2010) Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J. Virol. 84, 12958–12970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bakolitsa C., de Pereda J. M., Bagshaw C. R., Critchley D. R., Liddington R. C. (1999) Crystal structure of the vinculin tail suggests a pathway for activation. Cell 99, 603–613 [DOI] [PubMed] [Google Scholar]