

Background: Amyloids made of apolipoprotein A-I (apoA-I) contribute to the growth of the atherosclerotic plaques.
Results: ApoA-I methionine oxidation by physiological levels of myeloperoxidase induces amyloid formation.
Conclusion: Myeloperoxidase-mediated oxidation not only impairs the physiological functions of apoA-I but also promotes protein loss in form of amyloids.
Significance: Our findings identify the physiological mechanism transforming wild-type apoA-I into an amyloidogenic protein.
Keywords: Amyloid, Apolipoprotein, Atherosclerosis, Fibril, Myeloperoxidase, 2D (Two-dimensional) Infrared Spectroscopy, Amyloidosis, Apolipoprotein A-I, Oxidation
Abstract
High plasma levels of apolipoprotein A-I (apoA-I) correlate with cardiovascular health, whereas dysfunctional apoA-I is a cause of atherosclerosis. In the atherosclerotic plaques, amyloid deposition increases with aging. Notably, apoA-I is the main component of these amyloids. Recent studies identified high levels of oxidized lipid-free apoA-I in atherosclerotic plaques. Likely, myeloperoxidase (MPO) secreted by activated macrophages in atherosclerotic lesions is the promoter of such apoA-I oxidation. We hypothesized that apoA-I oxidation by MPO levels similar to those present in the artery walls in atherosclerosis can promote apoA-I structural changes and amyloid fibril formation. ApoA-I was exposed to exhaustive chemical (H2O2) oxidation or physiological levels of enzymatic (MPO) oxidation and incubated at 37 °C and pH 6.0 to induce fibril formation. Both chemically and enzymatically oxidized apoA-I produced fibrillar amyloids after a few hours of incubation. The amyloid fibrils were composed of full-length apoA-I with differential oxidation of the three methionines. Met to Leu apoA-I variants were used to establish the predominant role of oxidation of Met-86 and Met-148 in the fibril formation process. Importantly, a small amount of preformed apoA-I fibrils was able to seed amyloid formation in oxidized apoA-I at pH 7.0. In contrast to hereditary amyloidosis, wherein specific mutations of apoA-I cause protein destabilization and amyloid deposition, oxidative conditions similar to those promoted by local inflammation in atherosclerosis are sufficient to transform full-length wild-type apoA-I into an amyloidogenic protein. Thus, MPO-mediated oxidation may be implicated in the mechanism that leads to amyloid deposition in the atherosclerotic plaques in vivo.
Introduction
Low incidence of cardiovascular disease associates with high plasma levels of apolipoprotein A-I (apoA-I). In contrast, apoA-I dysfunction promotes atherosclerosis (1). Myeloperoxidase (MPO)2-mediated oxidation is one foremost mechanism that generates dysfunctional apoA-I (2, 3). ATP binding cassette transporter A1-dependent cholesterol release from cells (4–6) and lechitin:cholesterol acyltransferase activation (7), two central physiological functions of apoA-I, are impacted by MPO-mediated modifications of apoA-I. Importantly, activated macrophages in the atherosclerotic lesions secrete high levels of MPO that target apoA-I locally and reduce its anti-atherogenic function where it is mostly needed (3, 6, 8). Recent studies demonstrated a 100-fold increase in the amount of lipid-free apoA-I in atherosclerotic arteries compared with normal arteries (9). Notably, this apoA-I is lipid-free rather than HDL-associated (9), heavily oxidized (10–12), and cross-linked (9). These findings suggest that oxidized apoA-I not only lacks its primary physiological functions but also accumulates in the atherosclerotic plaques.
A different dysfunctional fate for apoA-I is represented by protein aggregation and formation of amyloid fibrils. At least 19 apoA-I variants have been described as amyloidogenic (hereditary apoA-I amyloidoses) (13–15). The manifestation of the amyloid disease and the target organs (e.g. heart, arteries, skin, larynx, testes, and kidneys) vary with the nature and position of the mutation (16–18). In the majority of the cases of hereditary apoA-I amyloidosis in which the composition of the amyloid fibrils has been investigated, N-terminal fragments of apoA-I have been identified as the main protein component of the fibrils (16). However, a recent study based on laser microdissection and tandem mass spectrometry proteomics revealed for the first time that full-length mutant apoA-I is the primary protein component of amyloid material extracted from patients with four different amyloidogenic apoA-I mutations (15), confirming previous reports on the presence of full-length apoA-I in amyloid fibrils (19, 20).
But the amyloidogenic potential of apoA-I extends beyond hereditary apoA-I amyloidosis. Primarily thanks to the work of Westermark et al. (19, 22, 25–29), a novel scenario is emerging in which wild-type apoA-I is the main component of amyloid fibrils isolated from, so far, lung nodules (21), knee joints (22), peripheral neural tissues (sciatic nerve) (23), aortic valves (24), aorta atherosclerotic plaques (25, 26), and peripheral atherosclerotic plaques (27). Interestingly, in some cases, not only N-terminal fragments but full-length wild-type apoA-I was found in the amyloid fibrils extracted from clinical samples (23).
The high incidence of amyloid fibrils associated with aortic intima and with atherosclerotic lesions indicates that amyloid deposition in the intima may contribute to atherosclerosis progression (25, 26, 28, 29); however, the extent of this contribution is yet to be demonstrated (27). The identification of apoA-I as the main component of atherosclerosis-associated amyloids suggests a link between local apoA-I oxidation and increased amyloidogenic potential of oxidized wild-type apoA-I. MPO could be the primary mediator of this oxidation (6, 8, 11, 12). Unfortunately, the oxidation state of apoA-I in amyloid clinical samples composed of wild-type apoA-I (full-length or fragments) is largely unknown (23), and this causal link remains hypothetical. However Wong et al. (30) recently reported that extensive in vitro apoA-I Met oxidation by H2O2 promotes an amyloidogenic phenotype. Prompted by this observation and the ability of MPO to facilitate Met oxidation in vivo (12), we hypothesized that MPO-mediated oxidation of apoA-I can produce protein variants that are structurally destabilized and amyloidogenic.
To test this hypothesis, we exposed plasma apoA-I to physiological levels of MPO (in the MPO-H2O2-Cl− system). To induce fibrils formation, oxidized apoA-I was incubated at 37 °C and pH 6.0, as previously described by Wong et al. (30). Solid material generated after incubation exhibited the hallmarks of amyloid fibrils: binding to Thioflavin T and Congo red, an increase in β-structure, reactivity toward the anti-amyloid fibril-specific antibody OC, and fibrillar morphology when observed by electron microscopy. ApoA-I variants with Met to Leu substitutions were used to investigate the role of each of the three methionines in the structural destabilization process, leading to amyloid fibril formation. Oxidation of Met-148 and Met-86 resulted in increased amyloidogenic traits, whereas oxidation of Met-112 did not contribute to the process of amyloid formation. Our results support the hypothesis that oxidation of apoA-I by MPO levels similar to those found in atherosclerotic lesions is sufficient to render apoA-I amyloidogenic and identify a potential mechanistic link between local inflammation and amyloid deposition in atherosclerosis.
EXPERIMENTAL PROCEDURES
Materials
Sequencing grade endoproteinase Glu-C and modified trypsin were purchased from Promega. MPO from human polymorphonuclear leukocytes was obtained from Calbiochem. Tobacco etch virus protease was produced as described in Tubb et al. (31). Ultrapure grade Thioflavin T (ThT) and Congo red (CR) were purchased from AnaSpec. The anti-amyloid fibrils OC antibody was obtained from EMD Millipore. CPC Scientific synthesized the isotopically labeled peptides. Except where indicated, all other materials were purchased from Sigma.
Plasma ApoA-I
Blood of healthy volunteers was obtained from Research Blood Components, LLC. The blood was donated to the blood bank according to the rules of the institutional review board and with written consent from the donors. HDL was isolated from fresh EDTA-treated plasma from a single donor by density gradient ultracentrifugation in the 1.063–1.21 g/ml density range (32), dialyzed against 10 mm sodium phosphate, pH 7.5, degassed, and stored in the dark at 4 °C. ApoA-I was isolated and purified from HDL solutions as described (33). Purified apoA-I was refolded from 6 m guanidine chloride by extensive dialysis against oxidation buffer (OB, 10 mm sodium phosphate, 100 μm diethylene triamine pentaacetic acid, 100 mm NaCl, pH 7.5). Protein purity was confirmed by SDS-PAGE before freezing the samples for storage. Protein concentration was determined by the BCA assay (Pierce) and spectrophotometrically (ϵ280 = 1.13 (mg/ml)−1cm−1 (34)).
Production of Recombinant Proteins
Single amino acid mutations were introduced into the DNA sequence of wild-type human apoA-I by primer-directed PCR mutagenesis. A His6 tag and the cleavage site for the tobacco etch virus protease were also engineered at the protein N terminus as described (31). Multiple Met to Leu and Trp to Phe mutations (35) were generated one by one using the mutated DNA as template for the subsequent primer-directed PCR reactions. The mutated DNA was subcloned into the pET-20b bacterial expression vector (Novagen), and complete DNA sequences were confirmed by sequencing. ApoA-I proteins were expressed in BL21 Escherichia coli (Invitrogen) and purified by nickel affinity chromatography as described (31). The N-terminal His tag was then removed by cleavage with tobacco etch virus protease followed by purification by Ni-affinity chromatography to capture the cleaved tag fragments and the tobacco etch virus protease (31) and yield the mature target protein. Protein purity was confirmed by SDS-PAGE. Protein preparations were dialyzed against OB and stored frozen until use.
Protein Oxidation
Plasma apoA-I was oxidized using two different systems. Chemical oxidation was performed in OB with excess of H2O2 and overnight incubation at 37 °C. H2O2-low-apoA-I and H2O2-high-apoA-I samples were oxidized with 750:1 and 1000:1 mol/mol H2O2:apoA-I ratios, respectively. The concentration of H2O2 was determined spectrophotometrically (ϵ240 = 39.4 m−1 cm−1) (36). These conditions are known to oxidize the three Mets of apoA-I with no significant modifications of other residues (30). Enzymatic oxidation by MPO was performed in OB in the presence of the MPO-H2O2-Cl− system for 90 min at 37 °C. The MPO concentration was 60 nm, and H2O2:apoA-I molar ratios were 2:1 and 3:1 for MPO-2:1-H2O2-apoA-I and MPO-3:1-H2O2-apoA-I, respectively. Enzymatic oxidation was terminated by the addition of a 10-fold molar excess (relative to H2O2) of l-methionine. After oxidation, the protein samples were extensively dialyzed against fibrillation buffer (FB, 10 mm sodium phosphate, pH 6.0) for 2 days with two buffer exchanges. Control samples (Ctrl) were incubated under the same conditions as for chemical oxidation (overnight at 37 °C in OB) but in the absence of H2O2 and then treated according to the same procedure used for oxidized samples. Quantification of Met oxidation by liquid chromatography-electrospray ionization mass spectrometry (LC-MS) is reported in Table 1. When apoA-I was incubated with a 3:1 molar excess of H2O2, as in the MPO-3:1-H2O2-apoA-I system but in the absence of MPO, Met oxidation was negligible compared with intact apoA-I and ≤ 5%.
TABLE 1.
Met oxidation was quantified by LC-MS as described under “Experimental Procedures”
The percentage of oxidation of single Met residues is reported as the mean and S.E. n is the number of independent experiments as defined under “Experimental Procedures.” N/A, non-applicable for the specific protein variant.
| Sample name | Oxidation System | H2O2:apoA-I (mol:mol) | Met(O)86 (n) | Met(O)112 (n) | Met(O)148 (n) |
|---|---|---|---|---|---|
| Mean (%) ± S.E. | Mean (%) ± S.E. | Mean (%) ± S.E. | |||
| Intact plasma apoA-I | 1.3 ± 0.5 (n = 3) | 2.3 ± 1.0 (n = 3) | 2.7 ± 0.2 (n = 3) | ||
| Ctrl plasma apoA-I | None | 0:1 | 1.6 ± 0.8 (n = 6) | 2.7 ± 0.5 (n = 7) | 1.6 ± 0.3 (n = 7) |
| MPO-2:1-H2O2-apoA-I | MPO-H2O2-Cl− | 2:1 | 41.6 ± 5.9 (n = 5) | 76.0 ± 4.1 (n = 5) | 57.8 ± 4.6 (n = 5) |
| MPO-3:1-H2O2-apoA-I | MPO-H2O2-Cl− | 3:1 | 59.9 ± 5.0 (n = 4) | 96.6 ± 1.4 (n = 4) | 86.6 ± 4.0 (n = 4) |
| H2O2-low-apoA-I | H2O2 | 750:1 | 67.5 ± 5.6 (n = 4) | 99.8 ± 0.2 (n = 4) | 94.5 ± 3.1 (n = 4) |
| H2O2-high-apoA-I | H2O2 | 1000:1 | 93.0 ± 2.5 (n = 4) | 100.0 ± 0.0 (n = 4) | 98.1 ± 1.3 (n = 4) |
| Ctrl rapoA-I | 0.4 ± 0.0 (n = 3) | 2.5 ± 0.4 (n = 6) | 4.2 ± 1.0 (n = 6) | ||
| H2O2-rapoA-I | H2O2 | 750:1 | 70.7 ± 5.2 (n = 5) | 98.9 ± 0.5 (n = 5) | 86.3 ± 2.2 (n = 5) |
| H2O2-M86L-rapoA-I | H2O2 | 750:1 | N/A | 99.9 ± 0.1 (n = 3) | 94.5 ± 2.2 (n = 3) |
| H2O2-M112L-rapoA-I | H2O2 | 750:1 | 70.9 ± 3.3 (n = 3) | N/A | 86.9 ± 1.9 (n = 3) |
| H2O2-M148L-rapoA-I | H2O2 | 750:1 | 37.9 ± 2.8 (n = 6) | 99.9 ± 0.1 (n = 3) | N/A |
Amyloid Fibril Formation
Oxidized apoA-I samples in FB were diluted to 0.8 mg/ml (28.4 μm) in sterile 1.5-ml microcentrifuge tubes and incubated at 37 °C with continuous vortexing. At the indicated time points, sample aliquots were withdrawn for analysis. Sodium azide significantly reduced the kinetics of fibril formation compared with samples incubated under similar conditions but in the absence of the bacteriostatic, so this chemical was omitted from fibril formation incubations. To minimize the chance of sample contamination and bacterial growth, sterile material was used for aliquot withdrawal.
Fluorescence Spectroscopy
Other laboratories reported that ThT interferes with the kinetics of amyloid fibril formation in real-time continuous assays in which ThT is added to the aggregating solution (37). In agreement with these findings, the kinetics of amyloid formation by oxidized apoA-I was significantly slower when the protein was incubated in the presence of ThT. To prevent any ThT interference with the fibrillation process, protein incubation was performed in the absence of the dye. ThT fluorescence emission spectra were collected by single time point dilutions in which a small sample aliquot was withdrawn from the incubation mix and added to a 500 μm ThT stock solution (in 50 mm sodium phosphate, pH 7.4). The protein sample and the ThT stock solution were combined in a 3.0 mm MicroQuartz fluorometer cell (Starna) to produce 140 μl of 18 μm ThT and 0.08 mg/ml apoA-I (2.84 μm) in 50 mm sodium phosphate, pH 7.4. ThT fluorescence was measured on a Horiba Jobin-Yvon fluoromax-4 spectrofluorometer using an excitation wavelength of 450 nm and 2.5 and 5.0-nm slit widths for the excitation and emission monochronomators, respectively. For each sample, three ThT emission spectra were collected between 460 and 550 nm with 1-nm scanning increments and averaged.
Far UV Circular Dichroism (CD) Spectroscopy
Far UV CD spectra (185–250 nm) were collected on an AVIV 400 spectropolarimeter. For CD analysis, protein samples before and after fibrillation were diluted in FB to ∼20 μg/ml. The CD data were normalized to final protein concentrations and reported as molar residue ellipticity, [Θ]. Isolation of solid material after incubation of samples under fibrillation conditions was achieved by spinning the samples at 10,000 × g for 20 min. Clear supernatants were removed and stored at 4 °C for further analysis. Solid pellets were washed two times by resuspending the material in FB, pelleting again, and discarding the supernatant. The final pellets were resuspended in a volume of FB equal to the original supernatant solution. For CD analysis, the final fibril suspensions were diluted with FB to ∼20 μg/ml based on the protein concentration of the original sample before fibrillation. CD analysis of turbid samples may be affected by light scattering. To ensure that the spectral shape of resuspended pellets was not an artifact due to light scattering, CD spectra of freshly diluted samples were recorded in the 0.01–0.1 mg/ml concentration range. The overall spectral shape was reproducible for all the concentrations, thus excluding significant aberrations produced by light scattering.
Fourier Transform Infrared (FTIR) Spectroscopy
FTIR spectra were recorded on a Direct Detect™ spectrometer (EMD Millipore). The Direct Detect™ utilizes a membrane technology optimized for the detection and quantification of proteins. The protein samples, 2 μl, were applied directly to a card-based hydrophilic polytetrafluoroethylene membrane; the membrane was thoroughly dried within the instrument before an IR spectrum was collected between 4000 and 800 cm−1. The solvent background was subtracted in all the spectra reported.
Isolation of solid material from samples incubated under fibrillation conditions was performed as described for CD spectroscopy. The final pellets were resuspended in a volume of FB equal to the original supernatant solution. A 2-μl drop of sample suspension was deposited onto the membrane and dried before FTIR analysis.
Two-dimensional Infrared (IR) Spectroscopy
Two-dimensional IR spectra were collected with an ultrafast laser system in pump-probe geometry with a pulse shaper. For sample preparation, a 2-μl drop of liquid sample in aqueous buffer was deposited on a CaF2 window and dried under nitrogen flow. H/D exchange was achieved by rehydrating the sample by adding 2 μl of D2O and drying under nitrogen flow. The rehydrating/drying steps were repeated five times for each sample to ensure complete deuteration of the solvent without changing the buffer composition. To verify that the protein or fibril structure was not altered by such rehydrating/drying treatments, after fibrillation, samples were deposited on a Direct Detect™ polytetrafluoroethylene membrane and exposed to similar rehydrating/drying cycles. No significant changes in the FTIR spectra of samples before and after rehydrating/drying treatments were observed (data not shown). For two-dimensional IR analysis, solid material from samples incubated under fibrillation conditions was harvested as described for CD spectroscopy. The solid pellets were washed 3 times with D2O and resuspended in D2O before depositing a 2-μl drop onto the CaF2 window. As the solid material was already washed with D2O, only two H/D exchange steps were performed on the deposited sample. To ensure an equal path length between samples, the sample cell was assembled by placing a 56-μm spacer between two CaF2 windows. By using a pulse shaper to acquire the two-dimensional IR data, one full two-dimensional IR spectrum can be collected in less than a minute. The spectra reported are the average of 15–20-min acquisitions.
Electron Microscopy (EM)
After fibrillation, protein samples were diluted to 0.08 mg/ml before depositing a 4-μl sample drop on a grid and staining with phosphotungstate as described in Mehta et al. (38). EM micrographs were collected using a CM12 transmission electron microscope (Philips Electron Optics) equipped with a Teitz 1K x 1K CCD camera (TVIPS; Gauting Germany). The average fibril width in each micrograph was estimated by measuring the width of the fibril in 40 different points along the fibril length.
Congo Red Binding
CR binding was detected spectrophotometrically according to the method of Klunk et al. (39). Briefly, CR (1:5, mol of protein:mol of CR) was added to the protein samples, and the absorbance spectrum (425–650 nm) of the mixture was collected on a Shimadzu UV-1800 spectrophotometer. Isolation of solid material from samples incubated under fibrillation conditions was performed as described for CD spectroscopy. The final pellets were resuspended in a volume of FB equal to the original supernatant solution.
Immunoblotting with the Anti-amyloid Fibrils OC Antibody
Dot-blot analysis was performed as described (40). At selected times during fibrillation, 20 μl of incubated mixture were deposited on a nitrocellulose membrane and air-dried. At the end of the experiment, the protein load of the dots was qualitatively evaluated by Ponceau S staining. The protein stain was then washed away from the membrane with TBS-T (0.05% Tween 20 in 50 mm Tris-HCl, 150 mm NaCl, pH 7.4), and the membrane was blocked with 5% nonfat milk in TBS-T. Incubation with OC antibody (41) (1:1000 dilution) was performed overnight at 4 °C in TBS-T in the presence of 5 mg/ml BSA. HRP-conjugated secondary antibody was used for detection with ECL substrate.
Mass Spectrometry
Quantitative analysis of apoA-I Met oxidation was performed after proteolysis with modified trypsin or endoproteinase GluC by LC-MS (36). Briefly, to adjust the pH within the 7.5–8.5 range, 20 μg of protein in 8.2 mm Tris-HCl, 150 mm NaCl and 0.1 mm EDTA was buffered with 0.1 m ammonium bicarbonate or 0.2 m ammonium bicarbonate, 5 mm EDTA for trypsin or endoproteinase GluC digestion, respectively, and incubated at 37 °C with the protease (enzyme substrate ratio 1:32, w/w) in the presence of 10% acetonitrile. After a 16–18-h incubation, new enzyme was added at the same ratio, and the incubation was continued for another 8–10 h. The reactions were stopped by adding 10% TFA to lower the pH to 2–3. Under such conditions, >95% of the Met-112- and Met-148-containing peptides in the trypsin digest were 108WQEEMELYR and 141LSPLGEEMR. Similarly, 86MSKDLEE accounted for >95% of the Met-86-containing peptides produced by digestion with endoproteinase GluC. These three peptides were used for oxidation quantification.
Peptide analysis was performed by RP chromatography (HPLC system 2695, Waters Associates, Milford, MA) on a Protein and Peptide C18 column (25 cm × 2 mm, Vydac) interfaced with a quadrupole mass spectrometer Quattro LC (Waters Associates, Milford, MA) in electrospray ionization mode. For analysis, 80 μl of digest containing 200 pmol of protein was injected in the column. After a 3-min wash at a flow rate of 0.2 ml/min, the column was developed with a 60-min gradient from 0 to 40% acetonitrile in the presence of 0.1% formic acid. Data were collected in the continuous mode and analyzed with Mass-Lynx v3.3. Integrated extracted ion current (iXIC) of the +2 ions of the 108WQEEMELYR and 141LSPLGEEMR tryptic peptides were used to calculate the extent of Met oxidation using the following formula: oxidation = (R × oxidized peptide-iXIC)/((R × oxidized peptide-iXIC) + non-oxidized-peptide-iXIC), where R is a constant corresponding to the ratio of the slopes of the plots of iXIC in function of peptide concentration for non-oxidized and oxidized peptides. The ratios were determined in a series of runs with isotopically labeled synthetic peptides of known concentration. The dynamic range for the +2 ions of the tryptic peptides was linear from 1 to at least 200 pmol/80 μl. Oxidation of Met-86 was quantified in a similar way from the iXIC of the +1 ion of the 86MSKDLEE peptide, as the plot of the +2 ion iXIC in function of the peptide concentration was not linear. The dynamic range for this peptide extended from 2 to at least 250 pmol/80 μl.
Denaturing Gel Electrophoresis (SDS-PAGE)
4–20% Tris-glycine gels (Novex™, Invitrogen) were loaded with 3 μg of protein per lane in the presence of SDS and β-mercaptoethanol and run at 125 V for 2 h in SDS-Tris-glycine buffer. Gels were stained with GelCode™ blue stain reagent (Thermo Scientific).
Statistical Analysis
Experiments were replicated by independently executing the process of plasma apoA-I or recombinant apoA-I preparation, oxidation, LC-MS quantification, fibrillation, and sample analysis. Statistical analyses were conducted using SAS 9.3 (SAS Institute Inc., Cary, NC). Means and S.E. were calculated for all variables. Differences in kinetics of amyloid fibril formation (t½) between groups of apoA-I variants were assessed using the appropriate analysis of variance model. If a significant overall difference was found, the Tukey-Kramer's post hoc method for multiple comparisons was employed to determine which groups differed. A significance level of 0.05 was used for all statistical tests.
RESULTS
ApoA-I Oxidation
To investigate whether levels of MPO-mediated apoA-I oxidation similar to those encountered in atherosclerotic arteries are sufficient to promote apoA-I amyloid fibril formation (2), we incubated human plasma purified apoA-I in the presence of the MPO-H2O2-Cl− system. To replicate (patho)physiologically plausible conditions in vitro, we used low H2O2 to apoA-I molar ratios (3:1 and 2:1) (10). Under these oxidative conditions a fraction of Met residues is transformed to methylsulfoxide (Met(O)), whereas modifications of Tyr and Trp residues are negligible (10, 36). As a positive control, apoA-I was oxidized by chemical reaction with a large molar excess of H2O2. It has been previously demonstrated that in these non-enzymatic conditions, the three apoA-I Met residues are exhaustively oxidized with no significant modifications of other residues and that the oxidized protein becomes amyloidogenic (30).
A few conclusions can be derived from the analysis of the Met oxidation levels obtained with the different oxidation systems (Table 1). First, natural Met oxidation levels in plasma apoA-I from healthy individuals were very low (<3%), with no significant differences among the three Met residues. Second, chemical oxidation by a large molar excess (1000:1) of H2O2 achieves high levels of Met oxidation, with Met-86 only slightly less affected (∼93% versus 98–100% of Met-148 and Met-112, respectively). However, at a lower excess of H2O2 (750:1), oxidation of Met-86 was significantly reduced (∼67%) compared with the other two residues (≥94%). These results suggest that in lipid-free apoA-I, the native protein folding partially shields Met-86 from H2O2 oxidation, whereas Met-112 and Met-148 are more readily accessible to the oxidant.
In the presence of the MPO-H2O2-Cl− system, the levels of Met oxidation were strictly dependent on the H2O2 concentration in the incubation mix. At a 3:1 molar excess of H2O2 (MPO-3:1-H2O2-apoA-I), a hierarchy in the susceptibility to oxidation of the three Met residues was clearly illustrated (Table 1): Met-112 > Met-148 > Met-86. In the presence of a lower (2:1) molar excess of H2O2 (MPO-2:1-H2O2-apoA-I), Met oxidation was significantly reduced, but the trend in susceptibility to oxidation was unchanged. Notably, no signs of protein degradation were detectable by SDS-PAGE in any of the oxidized proteins independently from the oxidation system or H2O2 concentrations used Fig. 1.
FIGURE 1.
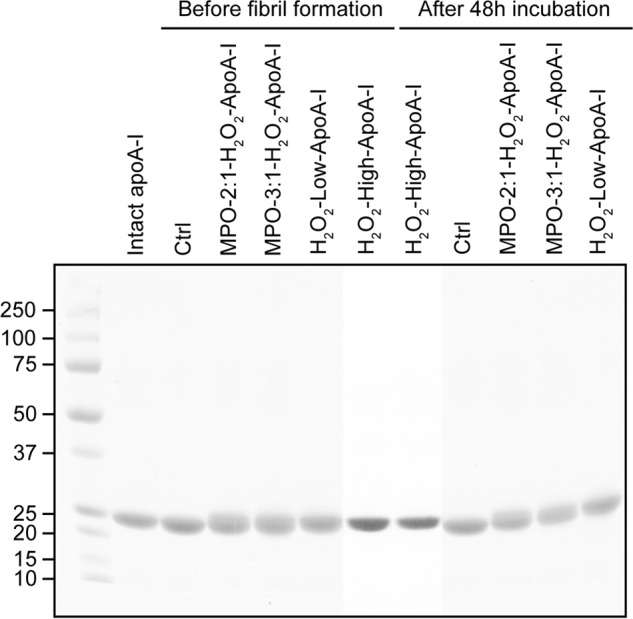
SDS-PAGE analysis of apoA-I samples after oxidation and after 48 h under fibrillation conditions. The figure represents a composite image in which lanes from two electrophoretic runs were combined by alignment of the Mr markers (Precision Plus protein standards, Bio-Rad).
Kinetics of Amyloid Fibril Formation
Oxidized and control apoA-I proteins were incubated under fibrillation conditions (pH 6.0, 37 °C, with continuous vortexing), and the ability of the protein samples to increase the fluorescence of the ThT dye was measured over time. Binding of the ThT aromatic moieties to the β-strands that are characteristic of amyloid fibril structures produces an increase in ThT fluorescence intensity that can be used as a means to detect amyloid fibril formation.
Time-dependent generation of amyloid fibrils in a sample is illustrated by the increase in ThT fluorescence; Fig. 2A. The intensity of ThT fluorescence emission at the maximal emission wavelength was plotted versus time in Fig. 2B. Upon incubation under fibrillation conditions, control apoA-I remained clear for up to 1 week, and no significant change in ThT fluorescence was detected. In contrast, the H2O2-high-apoA-I sample became hazy readily, and ThT fluorescence increased within the first 20 min of incubation. Haziness and ThT fluorescence increased exponentially and reached a steady state at ∼10 h of incubation. The extremely short lag phase indicates that, in these in vitro conditions, formation of fibrillar oligomers occurs rapidly during the very initial phase of fibrillation and that likely, early aggregates are capable of seeding further amyloid formation.
FIGURE 2.

Kinetics of amyloid fibril formation by ThT fluorescence. At the indicated time points, an aliquot of the mixture incubated under fibrillation conditions was added to a ThT stock solution, and the fluorescence emission spectrum of the sample was recorded as described under “Experimental Procedures”. Panel A, representative ThT fluorescence emission spectra time course of H2O2-high-apoA-I. Panel B, comparison of the fibrillation kinetics of H2O2-high-apoA-I (red), MPO-3:1-H2O2-apoA-I (blue), and MPO-2:1-H2O2-apoA-I (green). Reported values are the means of the intensities of ThT fluorescence emission at the maximal emission wavelength. Error bars are S.E. for 3–5 independent experiments, as defined under “Experimental Procedures.” ApoA-I samples (black) were incubated under the same oxidation conditions as the H2O2-oxidized samples but in the absence of H2O2. Solid lines are the fit of mean experimental values by exponential or logarithmic curves, as indicated in the figure. a.u., arbitrary units.
Notably, amyloid fibril formation in MPO-3:1-H2O2-apoA-I progressed with kinetics similar to those of H2O2-high-apoA-I, despite the significantly lower Met(O) levels (Table 1). The levels of Met(O) in MPO-3:1-H2O2-apoA-I were closer to those of MPO-2:1-H2O2-apoA-I than to the oxidation levels of H2O2-high-apoA-I. For this reason the significantly lower rate of amyloid fibril formation by MPO-2:1-H2O2-apoA-I was somehow unexpected. These results indicate that a relatively small reduction in total Met oxidation levels (Table 1) or in the oxidation level of a specific Met residue in MPO-2:1-H2O2-apoA-I was sufficient to slow down amyloid fibril formation. The largest difference in Met oxidation between MPO-2:1-H2O2-apoA-I and MPO-3:1-H2O2-apoA-I was observed for Met-148 (∼58% versus ∼87%, respectively) compared with ∼20% reduction for Met-86 and Met-112. These data suggest that the oxidation state of Met-148 may be critical for promoting amyloid formation. Remarkably, in all protein samples, no degradation occurred upon incubation under fibrillation conditions (Fig. 1). This integrity was maintained up to 2 weeks of incubation (data not shown).
Structural Changes upon Amyloid Fibril Formation
Although apoA-I amyloids extracted from patients with hereditary apoA-I amyloidosis retain some α-helical structure, β-structures are the dominant stabilizing elements in the fibrils (42). To investigate whether amyloid formation in vitro by oxidized apoA-I is also accompanied by a significant increase in β-structure, protein samples were analyzed by CD and FTIR spectroscopies before and after fibrillation.
The far-UV CD spectrum of intact apoA-I featured the two minima typical of α-helical structures (208 and 222 nm, black arrows, Fig. 3). Either chemical or enzymatic oxidation of apoA-I resulted in <10% loss of overall secondary structure, with no change in spectral shape (Fig. 3, A and B). However, after incubation under fibrillation conditions for 24 h, the CD spectrum of solid material isolated from H2O2-high-apoA-I was substantially different from the spectrum of H2O2-high-apoA-I before fibrillation (Fig. 3A). The minimum at 222 nm was red-shifted to ∼225 nm, and the intensity of the negative band at 208 nm was significantly reduced. These new features are consistent with a substantial increase in β-structures in the solid fraction of the sample (43). Remarkably, solid material isolated from MPO-2:1-H2O2-apoA-I displayed similar CD spectral features (Fig. 3B).
FIGURE 3.

Far-UV CD analysis. Far-UV CD spectra (185–250 nm) of intact apoA-I (black solid lines), oxidized samples before fibrillation (gray solid lines), and isolated solid material from samples after 24 h of incubation under fibrillation conditions (open dots). Panel A, H2O2-high-apoA-I. Panel B, MPO-2:1-H2O2-apoA-I.
FTIR analysis confirmed and expanded the conclusions derived from CD spectroscopy. The FTIR spectra reported in Fig. 4 span the region of the amide I band, where IR signals from backbone carbonyl stretch modes correlate with the protein secondary structure. A peak in the 1613–1643 cm−1 frequency range of the amide I region corresponds to β-sheet-rich structures, whereas a peak at ∼1650 cm−1 represents α-helical and random coil structures (44). The presence of only one defined peak at ∼1655 cm−1 in the amide I region of the FTIR spectrum of control apoA-I (Fig. 4A, Ctrl) indicates that the secondary structure of the original protein is predominantly α-helical and random-coil, with minimal contribution from β-structures. No significant changes in secondary structure were observed in any of the oxidized proteins (Fig. 4A). However, upon incubation under fibrillation conditions, the appearance of a new band at ∼1622 cm−1 in the oxidized proteins (Fig. 4B) was consistent with the formation of a significant amount of β-structures. Although the change occurred in all oxidized samples, the amount of β-structure after 48 h of incubation correlated with the levels of Met oxidation, i.e. the intensity of the peak at 1622 cm−1 was higher in MPO-3:1-H2O2-apoA-I than in MPO-2:1-H2O2-apoA-I. Notably, no further increase in intensity at 1622 cm−1 was detectable in the H2O2-oxidized apoA-I samples.
FIGURE 4.
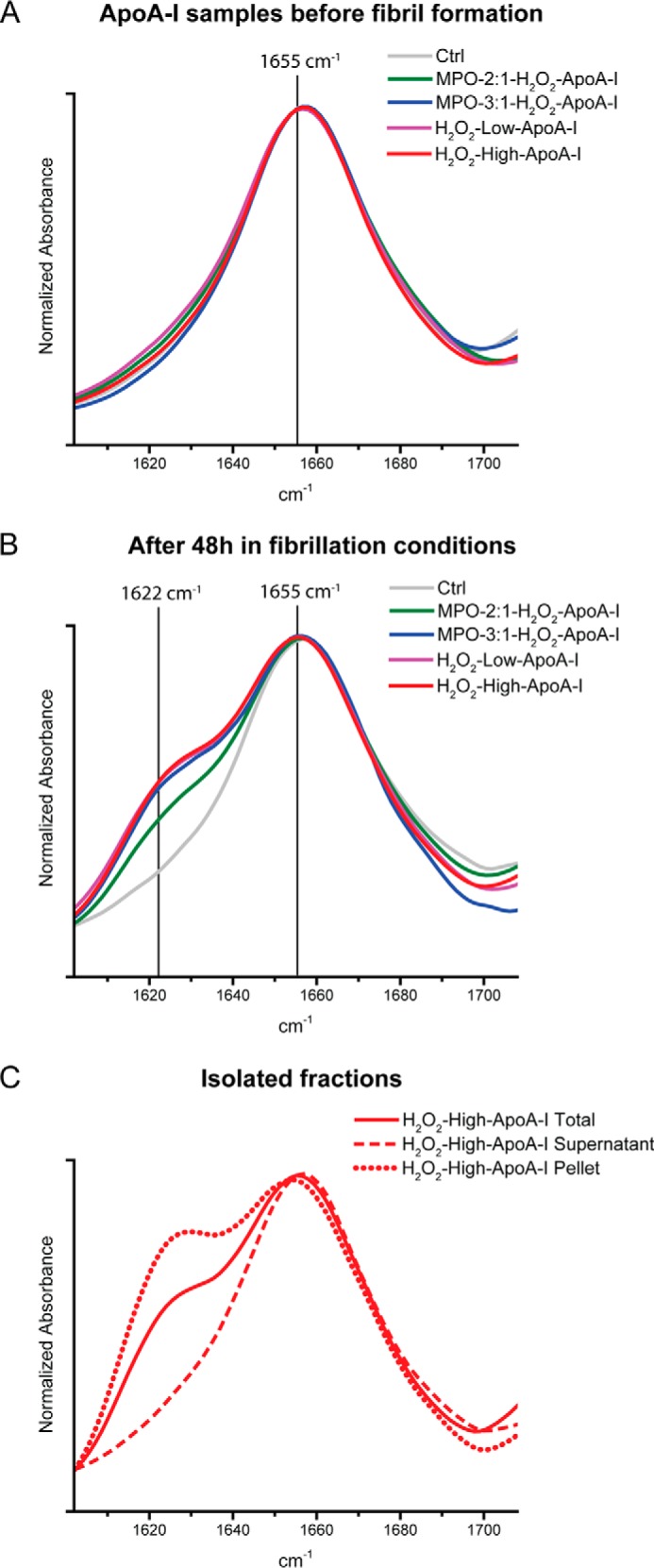
FTIR spectra in the amide I region (1602–1708 cm−1) of apoA-I samples. Control and oxidized samples before fibrillation (panel A) and after 48 h under fibrillation conditions (panel B) are shown. Panel C, FTIR analysis of supernatant and solid material isolated from H2O2-high-apoA-I.
To evaluate the distribution of secondary structural elements between the soluble fraction and the solid material generated in H2O2-high-apoA-I after 48 h of incubation under fibrillation conditions, the solid material was separated from the clear supernatant solution as described under “Experimental Procedures.” FTIR analysis indicated that after aggregation, the residual soluble fraction of the sample was exclusively composed of α-helical/random-coil structures (Fig. 4C). Furthermore, the solid material did not consist of pure β-structures but was a mixture of β-sheet and α-helical/random-coil elements (Fig. 4C). Overall, the CD and FTIR analyses confirmed that all the aggregated material was enriched in β-structures and that the oxidation promoted by physiological levels of MPO is sufficient to trigger formation of β-structure rich amyloid fibrils.
Two-dimensional IR Spectroscopy Analysis
To strengthen the spectroscopic conclusion from CD and FTIR data that amyloid β-sheets are present in our samples, we collected two-dimensional IR spectra. Due to the physics of two-dimensional IR spectroscopy and the nature of amyloid β-sheets, the signal strength of amyloid β-sheets is amplified in two-dimensional IR spectra, making two-dimensional IR a very sensitive diagnostic technique for studying amyloid fiber formation. This technique has been used to probe the dynamics of amyloid fibril formation in human islet amyloid polypeptide (45, 46), amyloid β peptides (47), γD-crystallin (48), and other proteins. Two-dimensional IR spectra of apoA-I are reported in Fig. 5. The pair of positive (yellow-orange) and negative (green-blue) peaks in the amide I band region of the two-dimensional IR spectra originates from fundamental (ν = 1 → 0) and overtone (ν = 1 → 2) transitions, respectively, both of which were probed by the two-dimensional IR pulse. The overtone and the fundamental peaks are centered at different probe frequency due to the anharmonicity of the potential energy surface. The diagonal slices through the fundamental peaks (shown under each two-dimensional IR spectrum) reveal similar features as observed by FTIR.
FIGURE 5.

Two-dimensional IR analysis. Two-dimensional IR spectra and diagonal slices (below each panel) of oxidized apoA-I samples before fibril formation (panels A and D), after 48 h under fibrillation conditions (B and E), and of isolated solid material (panels C and F). H2O2-low-apoA-I (panel A–C) and MPO-2:1-H2O2-apoA-I (panels D–F) are shown.
In the two-dimensional IR spectrum of H2O2-low-apoA-I (Fig. 5A), the broad peak centered at ωpump = 1650 cm−1 indicates that despite almost complete Met oxidation, before incubation under fibrillation conditions H2O2-low-apoA-I was still largely composed of α-helices and random coil motifs, in accordance with the CD and FTIR results (Figs. 3 and 4). Completely new spectral features occurred in the sample after 48 h incubation under fibrillation conditions (Fig. 5B). The new intense and sharp peak at ∼1620 cm−1 indicates formation of amyloid β-sheets. The line-width and anharmonic shift of this feature are also consistent with amyloid fiber formation and comparable to what observed in previously studied amyloid fibrils (45–48). Notably, a similar enrichment in β-sheet structure was observed in MPO-2:1-H2O2-apoA-I incubated under the same conditions (Fig. 5, D and E).
In FTIR spectroscopy the signal intensity scales as |μ|2 (with μ = transition dipole moment), whereas in two-dimensional IR the intensity scales as |μ|4. As highly ordered β-sheets have a large transition dipole moment, their two-dimensional IR signal intensity is greatly enhanced. Thus, the fact that the 1620 cm−1 peak that was a minor contributor in the FTIR spectrum is the dominant feature of the two-dimensional IR spectrum indicates that the transition dipole is enhanced at least three times, which is a definitive signature of amyloid formation. The intensity at ∼1650 cm−1 (Fig. 5, B and E) indicates that α-helices and random-coil motifs are also present, in agreement with the FTIR results. Both two-dimensional IR spectra of solid material isolated from MPO-2:1-H2O2-apoA-I (Fig. 5F) and H2O2-low-apoA-I (Fig. 5C) were consistent with the structural elements expected for amyloid fibrils. No large differences between the two spectra were observed, suggesting that the structure of the amyloid fibrils is independent of the oxidation method.
Morphology of Amyloid Fibrils
Samples incubated under fibrillation conditions were imaged by electron microscopy as described under “Experimental Procedures.” Amyloid fibers are well known to form polymorphs in which the same protein can generate multiple fibrillar structures. In H2O2-oxidized apoA-I, convoluted fibrillar tangles as well as more defined individual threads and rod structures formed within the same sample. Representative images of such morphologies are reported in Fig. 6, A, B, and C. These polymorphs coexisted in samples from different plasma apoA-I preparations after incubation under fibrillation conditions as short as 3 h. Although the relative frequency of the different fibril morphologies varied in different fibrillation experiments, no enrichment of a specific structure versus the others was observed for longer incubation times. In all morphologies, the fibril width averaged from 6 to 10 nm. Rod structures were ∼200-nm long (Fig. 6C), and individual thread morphologies extended up to ∼400 nm (Fig. 6B). Fibril size and morphologies were comparable with those of amyloid fibrils previously reported for oxidized apoA-I (30) and amyloidogenic mutants of apoA-I (49). In some cases spherical features were observable in a few areas of the grid (e.g. Fig. 6C). These structures were not reproducible and did not correlate with the fibrillation experimental conditions (e.g. incubation time, vortexing speed, oxidation system). Therefore, for the scope of this paper, we will not attribute these morphologies to specific molecular species involved in the process of fibril formation.
FIGURE 6.

EM analysis. Representative EM images of the three fibrillar morphologies found in H2O2-oxidized apoA-I samples. Convoluted fibrillar tangles (A), individual threads (B), and rods (C). The three morphologies often coexisted in the same sample. Similar morphologies were observed in MPO-oxidized samples. D, thread-like structure in MPO-3:1-H2O2-oxidized apoA-I. Negative staining EM micrographs were obtained after 24 h incubation under fibrillation conditions. Mean width of amyloid fibrils in each micrograph and S.E. are reported.
Notably, although the total amount of fibrillar structures in MPO-oxidized apoA-I was lower than in H2O2-oxidized apoA-I samples incubated under the same conditions, MPO-oxidized apoA-I produced fibrils of similar morphologies and size; one thread-like structure is reported in Fig. 6D. This imaging study indicates that MPO-oxidized apoA-I can yield amyloid fibrils with morphological features similar to those produced by amyloidogenic apoA-I variants and other amyloidogenic proteins.
Congo Red Binding and Immunoblotting with the Anti-amyloid Fibril-specific OC Antibody
To further confirm the amyloid nature of the material produced by oxidized apoA-I, we measured CR binding and reactivity toward a specific antibody against amyloid fibrils (OC). The spectrophotometric assay described by Klunk et al. (39) was used for detecting binding of the CR dye to amyloid material (Fig. 7A). A red shift of ∼25 nm in the absorbance maximum of CR that was added to our samples after 24 h of incubation under fibrillation conditions clearly indicates that CR binds to the aggregated material produced by either chemically or enzymatically oxidized apoA-I.
FIGURE 7.
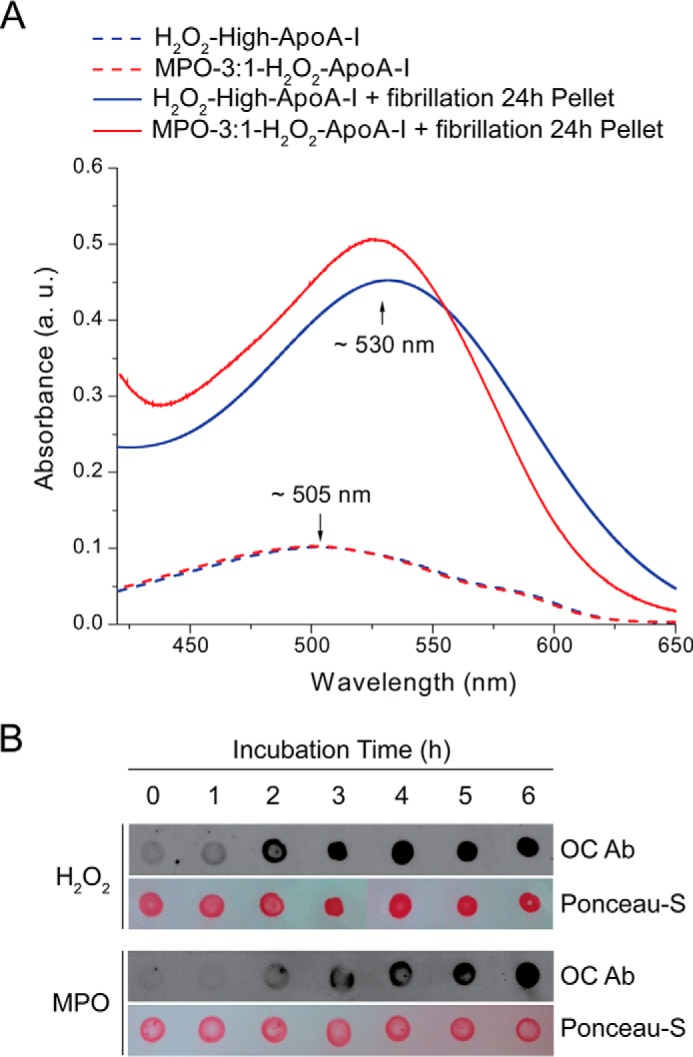
Congo red binding assay and dot-blot analysis of H2O2-high-apoA-I and MPO-3:1-H2O2-apoA-I. Panel A, absorbance spectra of CR added to oxidized apoA-I samples before exposure to fibrillation conditions (dash lines) and to aggregated material isolated from samples incubated under fibrillation conditions for 24 h (solid lines). a.u., arbitrary units, Panel B, dot-blot analysis of H2O2-high-apoA-I (H2O2) and MPO-3:1-H2O2-apoA-I (MPO) using the anti-amyloid fibril specific OC antibody. Samples incubated under fibrillation conditions were deposited on a nitrocellulose membrane at the indicated time points and stained with OC as described under “Experimental Procedures.
Oxidized apoA-I samples did not react with the anti-amyloid fibril specific OC antibody (Fig. 7B, 0 h). However, upon incubation under fibrillation conditions, reactivity developed rapidly and in a time-dependent manner (Fig. 7B). The difference in the time course of antibody reactivity between H2O2-high-apoA-I and MPO-3:1-H2O2-apoA-I (faster and slower, respectively) was also consistent with the kinetic results obtained by ThT binding (Fig. 2). CR binding and OC dot-blot results further support the conclusion that both H2O2-oxidized and MPO-oxidized apoA-I produce amyloid fibrils upon incubation under fibrillation conditions.
Oxidation of Specific Met Residues and Amyloid Fibril Formation
The kinetics of amyloid formation (Fig. 2) by differently oxidized apoA-I (Table 1) suggest that the three Met(O)s do not equally contribute to the amyloid formation mechanism. To dissect how oxidation of each of the Met residues promotes amyloid formation, we generated three apoA-I variants in which one Met was selectively substituted with Leu. Wild-type recombinant apoA-I and the three single Met-to-Leu protein variants were oxidized with a 750:1 molar excess of H2O2 as described for plasma apoA-I and incubated under fibrillation conditions for up to 5 days. Kinetics of amyloid fibril formation were constructed by measuring the time-dependent increase in ThT fluorescence (Fig. 8A), and t½ was calculated by data fitting (Fig. 8B). Strikingly, substitution of Met-148 with a non-oxidable Leu severely increased the half-time of amyloid formation (t½ = 3.7 and 70.1 h for H2O2-WT-rapoA-I and H2O2-M148L-rapoA-I, respectively). The longer t½ was primarily due to a dramatic increase in the lag phase (<1 min and ∼60 h for H2O2-WT-rapoA-I and H2O2-M148L-rapoA-I, respectively). Oxidation of Met-86 also contributed to the amyloid formation process, as substitution of Met-86 with Leu significantly impacted the half-time (t½ = 38.2 h) through a large increase in the lag phase (∼30 h for H2O2-M86L-rapoA-I). Notably, the half-time of amyloid formation by H2O2-M112L-rapoA-I was not significantly different from that of H2O2-WT-rapoA-I (Fig. 8B), indicating that Met(O)-112 does not participate in the process of structural destabilization and amyloid fibril formation.
FIGURE 8.
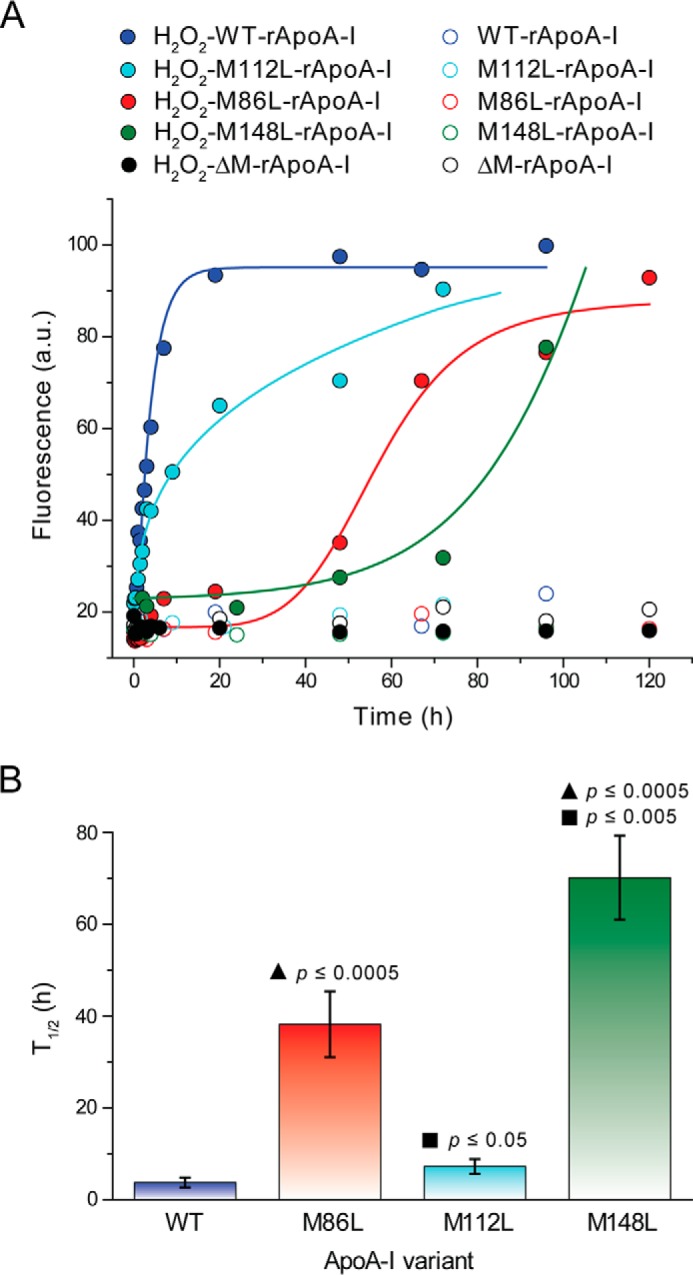
Kinetics of amyloid fibril formation by Met-to-Leu apoA-I variants after oxidation with a 750:1 molar excess of H2O2. ThT fluorescence was measured as described in Fig. 2 and under “Experimental Procedures.” Panel A, representative kinetics of amyloid fibril formation by H2O2-WT-rapoA-I, H2O2-M86L-rapoA-I, H2O2-M112L-rapoA-I, H2O2-M148L-rapoA-I, H2O2-ΔM-rapoA-I (solid symbols), and the corresponding non-oxidized controls (open symbols). Solid lines are the fit of the experimental values by exponential growth curves for H2O2-WT-rapoA-I and H2O2-M112L-rapoA-I and sigmoidal curves for H2O2-M86L-rapoA-I and H2O2-M148L-rapoA-I (logistic model and dose-response model, respectively). a.u., arbitrary units. Panel B, t½ were calculated from the data fitting and defined as the time at which the ThT fluorescence reached 50% of the maximal value measured for H2O2-WT-rapoA-I. Reported values are the means and S.E. for 3–11 independent experiments, as defined under “Experimental Procedures.” ▴, probability that the reported mean value is not different from t½ of H2O2-WT-rapoA-I. ■, probability that the reported mean value is not different from t½ of H2O2-M86L-rapoA-I.
No significant oxidative modifications other than Met oxidation (e.g. Tyr chlorination and Trp oxidation) were reported in apoA-I when the protein was exposed to chemical or enzymatic oxidation conditions similar to those used for this study (10, 30, 36). In agreement with the literature results, mass spectrometry survey of the tryptic peptides containing Trp and Tyr residues did not reveal significant modifications upon chemical or enzymatic oxidation. However, to conclusively demonstrate that no other protein modifications were responsible for the amyloidogenic outcome of oxidized apoA-I, all three Met were substituted with Leu to obtain a Met-null variant (ΔM-rapoA-I). When incubated under fibrillation conditions, the H2O2-ΔM-rapoA-I sample remained clear, and no significant increase in ThT fluorescence was detected for up to a 5-day incubation (Fig. 8A). Similar results were obtained for MPO-3:1-H2O2-ΔM-rapoA-I (data not shown). Thus, in the absence of oxidable Met residues, the oxidative conditions used in this study did not produce amyloidogenic modifications in apoA-I. Furthermore, an apoA-I variant in which all Trp were substituted with Phe (ΔW-rapoA-I) (35) also formed amyloid fibrils upon oxidation by H2O2 and incubation under fibrillation conditions (Fig. 9), indicating that Trp oxidation is not necessary for amyloid fibril formation. In summary, these results illustrate that oxidation of Met-148 and Met-86 is exclusively responsible for the amyloidogenic outcome of apoA-I with a prominent contribution from oxidation of Met-148.
FIGURE 9.
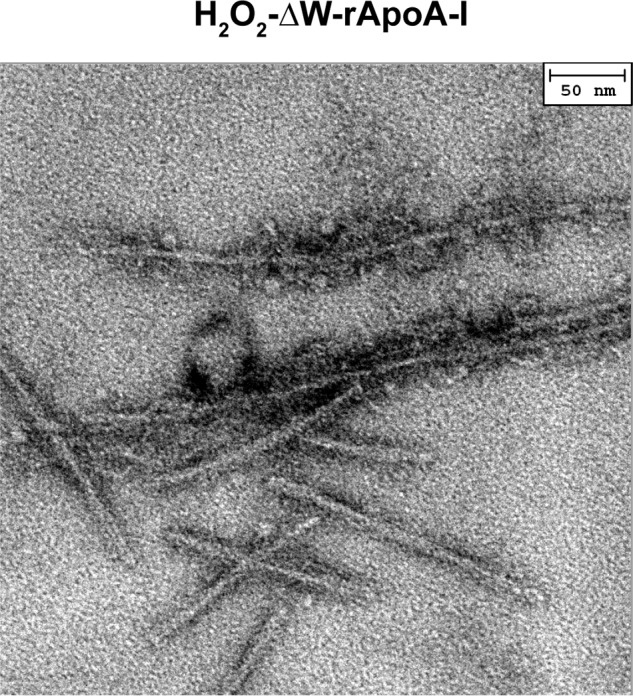
Fibrillar morphologies in H2O2-oxidized ΔW-rapoA-I, a protein variant in which all Trp were substituted with Phe. Negative staining EM micrographs were obtained after 24 h of incubation under fibrillation conditions.
Seeding Experiments and Physiological Significance
In vitro, amyloid fibril formation by oxidized apoA-I occurred exclusively at pH 6.0. When H2O2-oxidized apoA-I was incubated under fibrillation conditions at physiological pH (7.0–7.4), no structural changes or increase in ThT binding was observed for up to 10 days of incubation. However, when apoA-I fibrils preformed at pH 6.0 were incubated under fibrillation conditions (37 °C, with continuous vortexing) at pH 7.0 in the presence of a molar excess of H2O2-rapoA-I (10:1), amyloid features developed rapidly in H2O2-rapoA-I, as illustrated by the large increase in ThT fluorescence starting at 3 day incubation (Fig. 10). A similar seeding effect also occurred when H2O2-rapoA-I and preformed apoA-I fibrils where incubated at higher molar excess (100:1), although the increase in ThT fluorescence occurred with longer lag time (4 days) (Fig. 10). The ability of 1% of amyloid fibrils to transfer amyloid characteristics to the rest of the oxidized protein in physiological conditions is very relevant to the in vivo mechanism of amyloid fibril formation.
FIGURE 10.
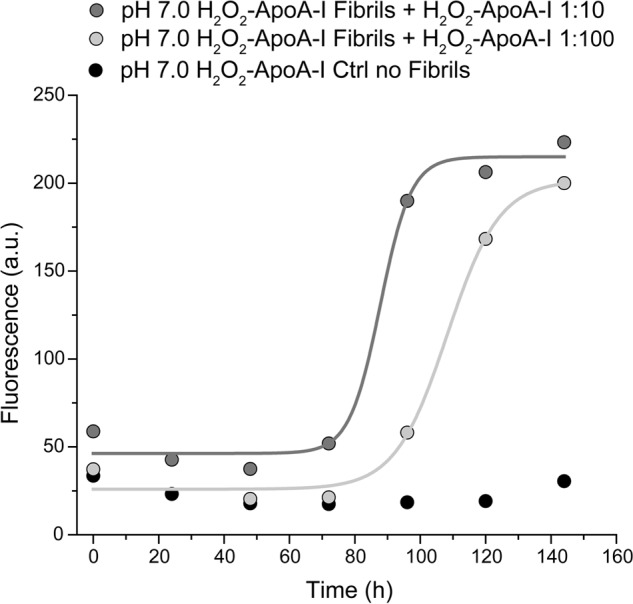
Seeding at pH 7. 0 of H2O2-rapoA-I with (molar) 10% (dark gray) and 1% (light gray) of preformed apoA-I amyloid fibrils. In a control sample (black), H2O2-rapoA-I was incubated in the absence of pre-formed apoA-I amyloid fibrils. Total protein concentration in all samples was 0.8 mg/ml. The samples were incubated at pH 7.0 and 37 °C with continuous vortexing. ThT fluorescence was measured at the indicated time points as described in Fig. 2 and “Experimental Procedures.” Solid lines are the fit of the experimental values by sigmoidal curves (dose-response model). a.u., arbitrary units.
DISCUSSION
The current study demonstrates that apoA-I Met oxidation promoted by MPO levels similar to those occurring in atherosclerotic lesions induces amyloid fibril formation. Although the level of apoA-I Met oxidation in human atherosclerotic lesions is unknown, compelling evidence suggests that in atherosclerotic plaques, apoA-I Met oxidation could reach levels similar to those produced by MPO oxidation in this study. Recently, the levels of apoA-I harboring a 3-nitrotyrosine (NO2-Tyr) in position 166 or a 2-hydroxy-l-tryptophan (2-OH-Trp) in position 72 were quantified in human plasma and in atherosclerotic plaques (10, 11). The levels of NO2-Tyr-apoA-I and 2-OH-Trp-apoA-I in atherosclerotic lesions were respectively ∼50- and 3000-fold higher than in apoA-I from plasma and normal aortic tissue (10, 11). Results from Heinecke and co-workers (8) also showed that the percentages of 3-chloro-Tyr and NO2-Tyr detected in HDL-associated apoA-I from atherosclerotic carotid lesions were 4 and 22% compared with 1% and <1% in HDL-associated apoA-I of plasma origin. Although our knowledge of in vivo apoA-I Tyr and Trp modifications has been improving, information relative to apoA-I Met oxidation remains scarce. A recent study has revealed that although the levels of Tyr chlorination in HDL-associated apoA-I from plasma of CVD patients are too low to produce a global reduction of HDL functionality, Met(O) levels (∼20% Met-86, ∼9% Met-112, ∼12% Met-148) are high enough to impact the efficiency of HDL-mediated cellular cholesterol metabolism systemically (12). No direct information about the levels of Met oxidation in atherosclerosis plaques is available to date, but if we assume a similar enrichment of Met(O), compared with plasma, as observed for OH-Trp-apoA-I and NO2-Tyr-apoA-I, the levels of Met(O) in the atherosclerotic lesions could easily amount to those generated by MPO oxidation and promoting amyloid fibrils formation in the current study.
A survey of the reported cases of non-hereditary (acquired) and hereditary apoA-I amyloidoses indicates that N-terminal rather than full-length apoA-I is the main component of the amyloid fibrils (typically < 1–80 N-terminal fragments in acquired amyloidosis). Whether apoA-I amyloid fibril formation requires fragmentation of the wild-type protein or N-terminal fragmentation is a secondary step after amyloid fibril formation remains unknown. However, in some cases of acquired amyloidosis, full-length wild-type apoA-I has been detected either as the sole component of amyloid fibrils or in association with N-terminal fragments of the protein (23, 26). A reverse argument can be formulated by analyzing the case of a patient homozygote for a non-sense mutation at position 84 (50). The absence of amyloid-related organ failure in this case suggests that the presence of an N-terminal truncated form of apoA-I in plasma does not necessarily promote the amyloid sequelae. Our current results and Howlett and co-workers previous study (30) support the hypothesis that N-terminal fragmentation of apoA-I is not necessary to promote amyloid formation, as no signs of protein degradation were observed in fibrils produced by oxidized apoA-I up to 6 months after fibril formation. These results suggest that oxidation of full-length apoA-I suffices to initiate amyloid fibril formation even in the absence of lysis at the protein N terminus. It is to be noted that in vivo, atherosclerosis-associated amyloids may build up and reside in the atherosclerotic plaques for decades. Therefore, in vivo, apoA-I N-terminal fragments could accumulate due to proteolysis or other lytic events occurring with aging of the amyloid material.
It is also important to notice that the curvilinear fibrillar structures reported in Fig. 6, A, B, and D, may not represent the final form of apoA-I amyloid deposits in vivo but rather a morphological state enhanced in our in vitro experimental conditions. A recent paper from Miti et al. (51) clearly illustrates how different amyloid morphologies can be formed by egg white lysozyme under the control of a variety of factors, such as protein and salt concentration, pH, and temperature. In their experimental model for instance, the thermodynamically favored linear fibrils, similar to the one reported for apoA-I in Fig. 6C, are predominant at lower protein concentrations.
The ability of 1 mol of apoA-I fibrils to seed amyloid formation in 100 mol of oxidized-apoA-I indicates how the oxidized protein contains metastable intermediates that can easily undergo structural rearrangements upon interaction with fully formed amyloid β-structures. FTIR analysis of the clarified supernatant from H2O2-high-apoA-I incubated under fibrillation conditions detected only α-helical/random-coil structures (Fig. 4). We can conclude that no β-structure-bearing intermediate species are present in the soluble fraction of the aggregating solution. Therefore, it can be hypothesized that conversion from α-helical/random-coil to β-structure occurs at the fibril growth interface. For the first time, our results demonstrate that a stable seed is more critical for fibril formation by oxidized apoA-I at neutral pH than the stability of the monomeric protein. In vivo, the presence of serum amyloid P, glycosaminoglycans, proteoglycans, collagen, and other amyloid relevant factors may stabilize early seeds, providing a surface onto which assembly competent molecules can bind.
Met residues are the principal players in amyloid formation by apoA-I. However, results from our Met substitution study indicate that Met-112, despite being the most prone to oxidation of the three Met residues, contributes to a lesser extent to the amyloid formation process, which depends on the presence of a sulfoxide moiety on Met-86 and -148. In particular, when either Met-86 or Met-148 were substituted with Leu, the much longer nucleation phase of the protein variants (lag phase of ∼30 and ∼60 h compared with < 1 min for WT-rapoA-I) illustrates how oxidation of these two residues is critical for seed formation. In later stages of fibrillation however, the process normalizes, as the final morphologies of amyloid fibrils from all the protein variants and WT-apoA-I were similar.
In conclusion, oxidation of Met-148 and -86 by physiological levels of MPO promotes rapid nucleation of the oxidized full-length apoA-I and formation of amyloid fibrils. The ability of a small concentration of apoA-I fibrils to catalyze aggregation of a larger amount of protein has in vivo pathophysiological implications. A minimal amount of fibrils could be produced in specific microenvironments of the atheroma and transfer to the surrounding tissues to promote more extended amyloid deposition by the large pool of oxidized apoA-I. Our results identify the mechanistic link between local inflammation and deposition of amyloid in the cardiovascular system. The impact on cardiovascular disease of such a conspicuous deposition of amyloids in the arterial intima deserves further investigation.
Acknowledgments
We thank the biophysical facility of the Department of Physiology and Biophysics at Boston University School of Medicine for granting access to some of their instruments. We are grateful to Virginia Gildengorin for expert assistance in statistical analysis.
This work was supported, in whole or in part, by National Institutes of Health Grants HL113059 (to G. C.) and DK79895 (to M. T. Z.).
- MPO
- myeloperoxidase
- Met(O)
- methylsulfoxide
- ThT
- Thioflavin T
- FB
- fibrillation buffer
- OB
- oxidation buffer
- CR
- Congo red
- rapoA-I
- recombinant apoA-I.
REFERENCES
- 1. Smith J. D. (2010) Myeloperoxidase, inflammation, and dysfunctional high-density lipoprotein. J. Clin. Lipidol. 4, 382–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daugherty A., Dunn J. L., Rateri D. L., Heinecke J. W. (1994) Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Invest. 94, 437–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nicholls S. J., Zheng L., Hazen S. L. (2005) Formation of dysfunctional high-density lipoprotein by myeloperoxidase. Trends Cardiovasc. Med. 15, 212–219 [DOI] [PubMed] [Google Scholar]
- 4. Shao B., Oda M. N., Bergt C., Fu X., Green P. S., Brot N., Oram J. F., Heinecke J. W. (2006) Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J. Biol. Chem. 281, 9001–9004 [DOI] [PubMed] [Google Scholar]
- 5. Shao B., Tang C., Heinecke J. W., Oram J. F. (2010) Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J. Lipid Res. 51, 1849–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng L., Settle M., Brubaker G., Schmitt D., Hazen S. L., Smith J. D., Kinter M. (2005) Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J. Biol. Chem. 280, 38–47 [DOI] [PubMed] [Google Scholar]
- 7. Shao B., Cavigiolio G., Brot N., Oda M. N., Heinecke J. W. (2008) Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc. Natl. Acad. Sci. U.S.A. 105, 12224–12229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shao B., Pennathur S., Heinecke J. W. (2012) Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J. Biol. Chem. 287, 6375–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DiDonato J. A., Huang Y., Aulak K. S., Even-Or O., Gerstenecker G., Gogonea V., Wu Y., Fox P. L., Tang W. H., Plow E. F., Smith J. D., Fisher E. A., Hazen S. L. (2013) Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 128, 1644–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang Y., DiDonato J. A., Levison B. S., Schmitt D., Li L., Wu Y., Buffa J., Kim T., Gerstenecker G. S., Gu X., Kadiyala C. S., Wang Z., Culley M. K., Hazen J. E., Didonato A. J., Fu X., Berisha S. Z., Peng D., Nguyen T. T., Liang S., Chuang C. C., Cho L., Plow E. F., Fox P. L., Gogonea V., Tang W. H., Parks J. S., Fisher E. A., Smith J. D., Hazen S. L. (2014) An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 20, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DiDonato J. A., Aulak K., Huang Y., Wagner M., Gerstenecker G., Topbas C., Gogonea V., DiDonato A. J., Tang W. H., Mehl R. A., Fox P. L., Plow E. F., Smith J. D., Fisher E. A., Hazen S. L. (2014) Site-specific nitration of apolipoprotein A-I at tyrosine 166 is both abundant within human atherosclerotic plaque and dysfunctional. J. Biol. Chem. 289, 10276–10292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shao B., Tang C., Sinha A., Mayer P. S., Davenport G. D., Brot N., Oda M. N., Zhao X. Q., Heinecke J. W. (2014) Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ. Res. 114, 1733–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eriksson M., Schönland S., Yumlu S., Hegenbart U., von Hutten H., Gioeva Z., Lohse P., Büttner J., Schmidt H., Röcken C. (2009) Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: identification of three novel mutations in the APOA1 gene. J. Mol. Diagn. 11, 257–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raimondi S., Guglielmi F., Giorgetti S., Di Gaetano S., Arciello A., Monti D. M., Relini A., Nichino D., Doglia S. M., Natalello A., Pucci P., Mangione P., Obici L., Merlini G., Stoppini M., Robustelli P., Tartaglia G. G., Vendruscolo M., Dobson C. M., Piccoli R., Bellotti V. (2011) Effects of the known pathogenic mutations on the aggregation pathway of the amyloidogenic peptide of apolipoprotein A-I. J. Mol. Biol. 407, 465–476 [DOI] [PubMed] [Google Scholar]
- 15. Rowczenio D., Dogan A., Theis J. D., Vrana J. A., Lachmann H. J., Wechalekar A. D., Gilbertson J. A., Hunt T., Gibbs S. D., Sattianayagam P. T., Pinney J. H., Hawkins P. N., Gillmore J. D. (2011) Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am. J. Pathol. 179, 1978–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Obici L., Franceschini G., Calabresi L., Giorgetti S., Stoppini M., Merlini G., Bellotti V. (2006) Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid 13, 191–205 [DOI] [PubMed] [Google Scholar]
- 17. Benson M. D. (2005) Ostertag revisited: the inherited systemic amyloidoses without neuropathy. Amyloid 12, 75–87 [DOI] [PubMed] [Google Scholar]
- 18. Obici L., Bellotti V., Mangione P., Stoppini M., Arbustini E., Verga L., Zorzoli I., Anesi E., Zanotti G., Campana C., Viganò M., Merlini G. (1999) The new apolipoprotein A-I variant Leu-174 → Ser causes hereditary cardiac amyloidosis, and the amyloid fibrils are constituted by the 93-residue N-terminal polypeptide. Am. J. Pathol. 155, 695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amarzguioui M., Mucchiano G., Häggqvist B., Westermark P., Kavlie A., Sletten K., Prydz H. (1998) Extensive intimal apolipoprotein A1-derived amyloid deposits in a patient with an apolipoprotein A1 mutation. Biochem. Biophys. Res. Commun. 242, 534–539 [DOI] [PubMed] [Google Scholar]
- 20. Mangione P., Sunde M., Giorgetti S., Stoppini M., Esposito G., Gianelli L., Obici L., Asti L., Andreola A., Viglino P., Merlini G., Bellotti V. (2001) Amyloid fibrils derived from the apolipoprotein A1 Leu174Ser variant contain elements of ordered helical structure. Protein Sci. 10, 187–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murphy C., Kestler D., Weiss D., Solomon A. (2011) Non-hereditary apolipoprotein AI-associated pulmonary amyloid. Amyloid 18, 219–220 [DOI] [PubMed] [Google Scholar]
- 22. Solomon A., Murphy C. L., Kestler D., Coriu D., Weiss D. T., Makovitzky J., Westermark P. (2006) Amyloid contained in the knee joint meniscus is formed from apolipoprotein A-I. Arthritis Rheum. 54, 3545–3550 [DOI] [PubMed] [Google Scholar]
- 23. Loavenbruck A. J., Chaudhry V., Zeldenrust S. R., Spinner R. J., Theis J. D., Klein C. J. (2012) Mass spectrometry analysis reveals non-mutated apolipoprotein A1 lumbosacral radiculoplexus amyloidoma. Muscle Nerve 46, 817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Audet A., Côté N., Couture C., Bossé Y., Després J. P., Pibarot P., Mathieu P. (2012) Amyloid substance within stenotic aortic valves promotes mineralization. Histopathology 61, 610–619 [DOI] [PubMed] [Google Scholar]
- 25. Westermark P., Mucchiano G., Marthin T., Johnson K. H., Sletten K. (1995) Apolipoprotein A1-derived amyloid in human aortic atherosclerotic plaques. Am. J. Pathol. 147, 1186–1192 [PMC free article] [PubMed] [Google Scholar]
- 26. Mucchiano G. I., Häggqvist B., Sletten K., Westermark P. (2001) Apolipoprotein A-1-derived amyloid in atherosclerotic plaques of the human aorta. J. Pathol. 193, 270–275 [DOI] [PubMed] [Google Scholar]
- 27. Mucchiano G. I., Jonasson L., Häggqvist B., Einarsson E., Westermark P. (2001) Apolipoprotein A-I-derived amyloid in atherosclerosis. Its association with plasma levels of apolipoprotein A-I and cholesterol. Am. J. Clin. Pathol. 115, 298–303 [DOI] [PubMed] [Google Scholar]
- 28. Cornwell G. G., 3rd, Murdoch W. L., Kyle R. A., Westermark P., Pitkänen P. (1983) Frequency and distribution of senile cardiovascular amyloid: a clinicopathologic correlation. Am. J. Med. 75, 618–623 [DOI] [PubMed] [Google Scholar]
- 29. Mucchiano G., Cornwell G. G., 3rd, Westermark P. (1992) Senile aortic amyloid: evidence for two distinct forms of localized deposits. Am. J. Pathol. 140, 871–877 [PMC free article] [PubMed] [Google Scholar]
- 30. Wong Y. Q., Binger K. J., Howlett G. J., Griffin M. D. (2010) Methionine oxidation induces amyloid fibril formation by full-length apolipoprotein A-I. Proc. Natl. Acad. Sci. U.S.A. 107, 1977–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tubb M. R., Smith L. E., Davidson W. S. (2009) Purification of recombinant apolipoproteins A-I and A-IV and efficient affinity tag cleavage by tobacco etch virus protease. J. Lipid Res. 50, 1497–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schumaker V. N., Puppione D. L. (1986) Sequential flotation ultracentrifugation. Methods Enzymol. 128, 155–170 [DOI] [PubMed] [Google Scholar]
- 33. Wetterau J. R., Jonas A. (1982) Effect of dipalmitoylphosphatidylcholine vesicle curvature on the reaction with human apolipoprotein A-I. J. Biol. Chem. 257, 10961–10966 [PubMed] [Google Scholar]
- 34. Rogers D. P., Roberts L. M., Lebowitz J., Datta G., Anantharamaiah G. M., Engler J. A., Brouillette C. G. (1998) The lipid-free structure of apolipoprotein A-I: effects of amino-terminal deletions. Biochemistry 37, 11714–11725 [DOI] [PubMed] [Google Scholar]
- 35. Jayaraman S., Abe-Dohmae S., Yokoyama S., Cavigiolio G. (2011) Impact of self-association on function of apolipoprotein A-I. J. Biol. Chem. 286, 35610–35623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shao B., Heinecke J. W. (2008) Using tandem mass spectrometry to quantify site-specific chlorination and nitration of proteins: model system studies with high-density lipoprotein oxidized by myeloperoxidase. Methods Enzymol. 440, 33–63 [DOI] [PubMed] [Google Scholar]
- 37. Coelho-Cerqueira E., Pinheiro A. S., Follmer C. (2014) Pitfalls associated with the use of thioflavin-T to monitor anti-fibrillogenic activity. Bioorg. Med. Chem. Lett. 24, 3194–3198 [DOI] [PubMed] [Google Scholar]
- 38. Mehta R., Gantz D. L., Gursky O. (2003) Human plasma high-density lipoproteins are stabilized by kinetic factors. J. Mol. Biol. 328, 183–192 [DOI] [PubMed] [Google Scholar]
- 39. Klunk W. E., Jacob R. F., Mason R. P. (1999) Quantifying amyloid β-peptide (Aαβ) aggregation using the Congo red-Aβ (CR-aβ) spectrophotometric assay. Anal. Biochem. 266, 66–76 [DOI] [PubMed] [Google Scholar]
- 40. Srinivasan S., Patke S., Wang Y., Ye Z., Litt J., Srivastava S. K., Lopez M. M., Kurouski D., Lednev I. K., Kane R. S., Colón W. (2013) Pathogenic serum amyloid A 1.1 shows a long oligomer-rich fibrillation lag phase contrary to the highly amyloidogenic non-pathogenic SAA2.2. J. Biol. Chem. 288, 2744–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Andreola A., Bellotti V., Giorgetti S., Mangione P., Obici L., Stoppini M., Torres J., Monzani E., Merlini G., Sunde M. (2003) Conformational switching and fibrillogenesis in the amyloidogenic fragment of apolipoprotein a-I. J. Biol. Chem. 278, 2444–2451 [DOI] [PubMed] [Google Scholar]
- 43. Brahms S., Brahms J., Spach G., Brack A. (1977) Identification of β,β-turns and unordered conformations in polypeptide chains by vacuum ultraviolet circular dichroism. Proc. Natl. Acad. Sci. U.S.A. 74, 3208–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zandomeneghi G., Krebs M. R., McCammon M. G., Fändrich M. (2004) FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 13, 3314–3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Buchanan L. E., Dunkelberger E. B., Tran H. Q., Cheng P. N., Chiu C. C., Cao P., Raleigh D. P., de Pablo J. J., Nowick J. S., Zanni M. T. (2013) Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient β-sheet. Proc. Natl. Acad. Sci. U.S.A. 110, 19285–19290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Middleton C. T., Marek P., Cao P., Chiu C. C., Singh S., Woys A. M., de Pablo J. J., Raleigh D. P., Zanni M. T. (2012) Two-dimensional infrared spectroscopy reveals the complex behaviour of an amyloid fibril inhibitor. Nat. Chem. 4, 355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim Y. S., Liu L., Axelsen P. H., Hochstrasser R. M. (2009) 2D IR provides evidence for mobile water molecules in β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 106, 17751–17756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moran S. D., Woys A. M., Buchanan L. E., Bixby E., Decatur S. M., Zanni M. T. (2012) Two-dimensional IR spectroscopy and segmental 13C labeling reveals the domain structure of human γD-crystallin amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 3329–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Petrlova J., Duong T., Cochran M. C., Axelsson A., Mörgelin M., Roberts L. M., Lagerstedt J. O. (2012) The fibrillogenic L178H variant of apolipoprotein A-I forms helical fibrils. J. Lipid Res. 53, 390–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsunaga T., Hiasa Y., Yanagi H., Maeda T., Hattori N., Yamakawa K., Yamanouchi Y., Tanaka I., Obara T., Hamaguchi H. (1991) Apolipoprotein A-I deficiency due to a codon 84 nonsense mutation of the apolipoprotein A-I gene. Proc. Natl. Acad. Sci. U.S.A. 88, 2793–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miti T., Mulaj M., Schmit J. D., Muschol M. (2015) Stable, metastable, and kinetically trapped amyloid aggregate phases. Biomacromolecules 16, 326–335 [DOI] [PMC free article] [PubMed] [Google Scholar]