

Background: Phospholipase D toxins from brown spider venoms can cause disease in humans.
Results: Different toxin family members show specificity for lipid substrates with choline or ethanolamine headgroups or can be ambiguous.
Conclusion: Spider phospholipase D toxins have evolved diverse substrate preferences.
Significance: The diverse substrate preference may be significant for predation and the mammalian toxicity of venom.
Keywords: Crystal Structure, Enzyme Mechanism, Phospholipase D, Sphingomyelinase, Substrate Specificity, Toxin, Loxosceles, Sicarius, Loxoscelism
Abstract
Venoms of the sicariid spiders contain phospholipase D enzyme toxins that can cause severe dermonecrosis and even death in humans. These enzymes convert sphingolipid and lysolipid substrates to cyclic phosphates by activating a hydroxyl nucleophile present in both classes of lipid. The most medically relevant substrates are thought to be sphingomyelin and/or lysophosphatidylcholine. To better understand the substrate preference of these toxins, we used 31P NMR to compare the activity of three related but phylogenetically diverse sicariid toxins against a diverse panel of sphingolipid and lysolipid substrates. Two of the three showed significantly faster turnover of sphingolipids over lysolipids, and all three showed a strong preference for positively charged (choline and/or ethanolamine) over neutral (glycerol and serine) headgroups. Strikingly, however, the enzymes vary widely in their preference for choline, the headgroup of both sphingomyelin and lysophosphatidylcholine, versus ethanolamine. An enzyme from Sicarius terrosus showed a strong preference for ethanolamine over choline, whereas two paralogous enzymes from Loxosceles arizonica either preferred choline or showed no significant preference. Intrigued by the novel substrate preference of the Sicarius enzyme, we solved its crystal structure at 2.1 Å resolution. The evolution of variable substrate specificity may help explain the reduced dermonecrotic potential of some natural toxin variants, because mammalian sphingolipids use primarily choline as a positively charged headgroup; it may also be relevant for sicariid predatory behavior, because ethanolamine-containing sphingolipids are common in insect prey.
Introduction
Envenomation by brown spiders in the genus Loxosceles can induce necrotic breakdown of mammalian tissue and can occasionally lead to renal failure, circulatory shock, and even death. These cutaneous and systemic disease states are known as loxoscelism (1–5).
Loxoscelism is caused primarily by phospholipase D (PLD)2 enzyme toxins in the spider venom. Purified PLD toxins from both venom and recombinant sources induce a disease state similar to loxoscelism in animal models (6–14). The PLD toxins cleave the phosphodiester linkage between the phosphate and headgroup of certain phospholipids, forming an alcohol (often choline) and a cyclic phosphate (Scheme 1) (15). Formally, the cyclic phosphate is formed by intramolecular nucleophilic attack of a hydroxyl nucleophile at the phosphorus center, with displacement of the headgroup. This chemistry restricts the toxins to phospholipid substrates that contain a hydroxyl moiety, including ceramide-based lipids like sphingomyelin (SM) and lysophospholipids such as lysophosphatidylcholine (LPC) (Scheme 1) (16–20).
SCHEME 1.
Relevant substrates and products for our study of SicTox enzymes. The reaction at left shows conversion of sphingolipid substrates to cyclic ceramide phosphate; the reaction at right shows conversion of lysophospholipid substrates to cyclic phosphatidic acid. R1, R2, and R3 represent alkyl chains of varying length. The four different headgroup alcohols (XOH) represented here are choline (SM and LPC), ethanolamine (CPE and LPE), glycerol (LPG), and serine (LPS).
Loxosceles spiders belong to the spider family Sicariidae, which also contains the genus Sicarius. Sicarius species are native to Southern Africa and South/Central America. The venom of African Sicarius can also induce loxoscelism in animal models (21–23). Although most research attention focuses on Loxosceles, Sicarius individuals also carry PLD toxins in their venoms (24). The gene family comprising the sicariid PLD toxins has been named “SicTox” to reflect this distribution and to distinguish them from nonhomologous PLDs that typically serve more general housekeeping functions.
SicTox family members show variable levels of sphingomyelinase D (SMase D) activity (Fig. 1). Phylogenetic analysis of the SicTox family resolves two clades, α and β, of the venom-expressed members (24). The most thoroughly studied toxins are members of the α clade, which are abundant in venoms of New World Loxosceles. These show high SMase D activity and can induce loxoscelism in animal models. By contrast, some β clade members, generally belonging to the βI subclade, show little to no SMase D activity and have diminished toxic effects in animal models (7–10, 25, 26). SicTox proteins with confirmed low SMase D activity have also been termed “class IIb” PLDs (27, 28); all these are members of the SicTox β clade.
FIGURE 1.

Simplified cladogram of SicTox gene products that have been biochemically characterized. The tree, which is based on a previously reported phylogeny (24), includes all SicTox gene products that have been tested for PLD activity following isolation from whole venom or purification from recombinant sources. The names of toxins investigated in this study are highlighted in purple. The ingroup includes two major clades (α and β). IsSMase from Ixodes scapularis (74) is included as an outgroup. Homologs with >95% amino acid sequence identity are considered isoforms and grouped under the same name. Names follow the SicTox nomenclature (24), but alternative names used in the literature are also given, and PDB codes are given for known protein structures. Branch colors correspond to species groups as previously presented (24): red, L. reclusa; green, Loxosceles spadicea; purple, Loxosceles gaucho; blue, L. laeta, and pink, South America Sicarius species. Branches lacking terminal names represent clades for which no member has been characterized biochemically. Except for toxins characterized in this study, high or low SMase D activity is based on comparisons with the activity of whole venom (at the same protein amount) from the toxin source species. In the case of β1A1 (SMase II) from L. laeta, SMase D activity has been observed but with a 3-fold higher Km value and a reported 2-fold lower reaction rate at saturating substrate concentrations compared with αIII1 (SMase I) from the same species (26). References for structure and activity data are as follows: IsSMase (74); LiRecDT6 (11); LiRecDT7 (14); SMase I (8, 18, 27, 35, 59, 64, 75); Ll2 (8); LiRecDT4 (9); LgRec1 (76); Lr1/Lb1/Lb2 (8, 25); L. arizonica αIB2bi (15, 30); Lr2 (19, 25); P1/P2 (77–79); LiRecDT1 (6, 7, 12, 13, 20, 60); 3RLG (61); 3RLH (28); LiRecDT2 (7); LiRecDT5 (9); Lb3 (8, 25); LiRecDT3 (7, 10); and SMase II (26).
Sicarius SicTox genes are particularly interesting with respect to functional diversity within this gene family, because only β clade paralogs have been found in Sicarius, and whole venoms of some species of New World Sicarius exhibit diminished SMase D activity compared with Loxosceles and Old World Sicarius venom. Even though there is variability in SMase D activity, all Sicarius species express diverse β clade paralogs in their venom (24, 29). βI clade (class IIb) PLDs could be generally less active or inactive SMase D enzymes, but it seems more likely that they are specific for non-SM substrates that have yet to be identified. In support of this hypothesis, they conserve all key residues thought to be important for enzyme activity (Fig. 2) and are present in diverse sicariid venoms. In addition, βI clade enzymes are present in multiple New World species of Loxosceles (see Figs. 1 and 2), although in lower abundance than α clade enzymes. We propose that the βI clade (class IIb) proteins perform some important but undiscovered function in the venom mixture.
FIGURE 2.

Multiple amino acid sequence alignment of selected SicTox enzymes. Residues 1–285 are shown. Amino acid residues deemed essential for catalytic activity as previously inferred from the structures of Ll_αIII1 and Li_αIA1a (PDB 1XX1 and 3RLH, respectively) are indicate by red boxes. Despite variation in SMase D activity, all 11 of these residues are perfectly conserved in the family. Conserved cysteine residues involved in disulfide bonds are indicated by yellow boxes. Amino acid residues mutated in this study are indicated by blue boxes.
Identifying molecular target(s) for these toxins is important for understanding the evolution of functional specificity within the SicTox gene family, the biology of the sicariid spiders, and the molecular mechanism underlying the diminished toxicity of the βI clade toxins in mammalian models. Here, we utilize 31P NMR-based substrate screening to identify substrates and substrate preferences for two diverse βI clade members as follows: St_βIB1i from Sicarius terrosus, a New World species that lacks venom SMase D activity in vitro and has no known α clade toxins (24); and La_βID1 from Loxosceles arizonica, a species that is known to cause loxoscelism and does produce α clade toxins (30–32). We include parallel characterization of La_αIB2bi, a previously characterized α clade toxin from L. arizonica (15, 30), both as a positive control for activity against SM and LPC (Fig. 2) and to provide a direct comparison between substrate preferences of α clade and βI clade members. Inclusion of La_αIB2bi also provides an intraspecies comparison with La_βID1.
We report that St_βIB1i, like other βI/class IIb toxins, has low activity against SM and LPC; however, it selectively turns over both ceramide and lysolipid substrates containing ethanolamine headgroups. La_βID1, by contrast, shows little preference between choline and ethanolamine, and La_αIB2bi shows a preference for choline. Our results show for the first time that 1) SicTox enzymes vary widely in substrate selectivity and ambiguity and 2) that some βI/class IIb enzymes with little to no SMase D activity are actually active enzymes that prefer other substrates. To further investigate substrate preference by St_βIB1i, we determined its crystal structure, the first of a class IIb PLD as well as the first structure of a SicTox PLD from a Sicarius species. Substrate docking studies using this structure did not reveal a clear basis for its different activity but did shed light on the enzyme mechanism.
EXPERIMENTAL PROCEDURES
Materials
The following phospholipid substrates were purchased from Avanti Polar Lipids (Alabaster, AL): 6:0 SM (6:0 SM, N-hexanoyl-d-erythrosphingosylphosphorylcholine); 8:0 LPC (1-octanoyl-2-hydroxy-sn-glycero-3-phosphocholine); 14:0 LPC (1-myristoyl-2-hydroxy-sn-glycero-3-phosphocholine); 14:0 LPE (1-myristoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine); 12:0 SM (N-lauroyl-d-erythrosphingosylphosphorylcholine); CPE (N-lauroyl-d-erythrosphingosylphosphoethanolamine); 14:0 LPG (1-myristoyl-2-hydroxy-sn-glycero-3-phospho-(1′-rac-glycerol)), and LPS (l-α-lysophosphatidylserine from porcine brain). QuikChange site-directed mutagenesis kit was purchased from Stratagene (La Jolla, CA). BugBuster and Benzonase nuclease were purchased from Novagen (Madison, WI). Nickel-nitrilotriacetic acid resin was purchased from Qiagen (Hilden, Germany). All other chemicals were obtained from standard sources.
Cloning of Coding Sequence of SicTox Isolates
Generation of cDNA libraries of SicTox homologs has been described previously (24). To compare the levels of enzyme ambiguity in the SicTox PLD family, and to investigate substrate specificity in the β clade, we chose a set of three SicTox PLD from two different sicariid species. The three toxins in this study are as follows: La_αIB2bi (gb:AY699703); La_βID1 from L. arizonica (gb:KM884812); St_βIB1i from S. terrosus (gb:KM884813). La_αIB2bi has been identified and studied previously (15, 30) and is used here as a representative of the SicTox α clade. St_βIB1i from S. terrosus was chosen as a representative of the β clade. St_βIB1i is a close homolog to a previously reported (24) S. terrosus sequence, βIB1 (gb: FJ171474.1). The full-length clone was isolated from a previously obtained cDNA pool (24) using a 5′-rapid amplification of cDNA ends kit (Invitrogen). The GSP2 primer was designed to specifically amplify the missing 5′ end of St_βIB1 (5′-CAAATGTGCTCACGAATGC-3′), and St_βIB1i was amplified using the using the manufacturer's protocol. Amplicons were TOPO-cloned (Invitrogen) and sequenced. The amino acid sequence of St_βIB1i differs from St_βIB1 at five positions (N9I, I10M, A11G, E116D, and I142T). St_βIB1i shares 69% sequence identity with LiRecDT3 (LiSicTox-βIA1i) from Loxosceles intermedia (7), a SicTox PLD reported to have diminished SMase D activity. St_βIB1i is the first SicTox PLD toxin from a Sicarius species to be described and characterized, and St_βIB1i shares >60% identity with all previously characterized βI/class IIb enzymes (Fig. 1).
To obtain a divergent β clade paralog from L. arizonica, allowing for comparison of two intraspecific paralogs, an Illumina cDNA library of L. arizonica cephalothorax (including venom glands) was searched for the motifs DNPW, HMVN, and HXXPCDCXRXC. Transcript 1239 was isolated and phylogenetically resolved in the β clade. This transcript is confirmed to be venom expressed based on hits to venom proteins using proteomic analysis (MudPIT, data not shown). The full-length clone was then amplified from the venom gland cDNA pool using two primers designed from the initial transcript sequence, TOPO cloned (Invitrogen) and sequenced. The predicted amino acid sequence of locus 1239 shares 85% sequence identity with LiRecDT5 (LiSicTox-βID1) from L. intermedia (9), another SicTox PLD reported to have diminished SMase D activity compared with α clade isoforms from the same species. Because of this relatedness (Fig. 1), we named the 1239 clone “La_βID1” as outlined in the SicTox nomenclature (24).
Signal sequences and mature protein sequences were predicted by analysis of the primary sequence with SignalP (33). Cloning of DNA sequences, encoding the mature PLD toxins St_βIB1i and La_βID1 obtained from cDNA libraries, into pHis8 expression vectors (34) was performed as described previously for La_αIB2bi (15, 30). Sequence variants of St_ βIB1i were generated using QuikChange site-directed mutagenesis according to the manufacturer's protocol.
Heterologous Expression and Purification of SicTox PLD Toxins
N-terminally His8-tagged recombinant proteins were expressed from pHis8-toxin constructs in Escherichia coli strain BL21(λDE3), closely following published methods (15, 35). Freshly transformed cells were grown with shaking at 250 rpm at 37 °C in 1 liter of 2× YT media (20 mg/ml tryptone, 10 mg/ml Bacto yeast extract, 5 mg/ml NaCl (pH 7)) containing 30 μg/ml kanamycin. When the cultures reached an A600 of ∼0.6, overexpression was induced by addition of isopropyl β-d-1-thiogalactopyranoside to a concentration of 0.1 mg/ml. After 2–3 h, cells were harvested by centrifugation at 5,000 × g for 10 min. To extract soluble proteins, pelleted cells were lysed with BugBuster Protein Extraction Reagent (Novagen. Madison, WI) according to the manufacturer's protocol. Cleared lysate was brought to ∼20 mm imidazole by addition of NE250 buffer (0.1 m Tris-HCl (pH 7.4), 0.2 m NaCl, and 0.25 m imidazole) and loaded onto a gravity flow column containing 3 ml of nickel-nitrilotriacetic acid-agarose resin (Qiagen) equilibrated in NW20 buffer (0.1 m Tris-HCl (pH 7.4), 0.2 m NaCl, and 20 mm imidazole). Resin was washed twice with 30 ml of NW20 buffer following collection of flow through. His8-tagged PLD toxin was eluted with 2× 10-ml aliquots of NE250 buffer. Eluate fractions containing >80% pure protein, as judged by inspection of Coomassie-stained SDS-polyacrylamide gels run under nonreducing conditions, were pooled, centrifugally concentrated to a volume of 2 ml, and loaded at a flow rate of 0.5 ml/min onto a HiPrep 16/60 Sephacryl S-100 HR column (GE Healthcare) equilibrated in TBS buffer (0.1 m Tris-HCl (pH 7.4), 0.2 m NaCl). Fractions containing monomeric, purified protein were identified by SDS-PAGE analysis, pooled, and centrifugally concentrated (molecular mass cutoff ∼10 kDa) to an appropriate volume and exchanged into a low ionic strength buffer (25 mm Tris-HCl (pH 8), 50 mm NaCl). Protein concentration was determined by ultraviolet absorption at 280 nm using appropriate estimated extinction coefficients (36). For 31P NMR assays, concentrated protein was stored in 50% glycerol at −20 °C until use. For crystallization trials, concentrated protein was either used immediately, or flash-frozen and stored at −80 °C. All enzymes could be heterologously expressed in Escherichia coli systems and >95% enzyme purity was achieved by nickel affinity purification and size exclusion chromatography.
31P NMR Enzymatic Assays
All one-dimensional 31P NMR spectra were recorded at 310 K on a Bruker DRX-500 spectrometer equipped with a BBO-500 MHz S2 5-mm probe with a Z gradient. Spectra were acquired at a spectrometer frequency of 202.11 MHz with 1H decoupling. Data points (32 K) were acquired per spectrum at a spectral width of 80 ppm, with signal averaging over 99 scans. Spectra were obtained with a 60° pulse width and a 7-s relaxation delay for quantitative signal integration (15). Samples for initial assays on short chain lipid substrates contained either 2 mm 6:0 SM solubilized with 2 mm Triton X-100 or 80 mm 8:0 LPC, in 100 mm Tris-HCl (pH 7.4) containing 10 mm MgCl2, 1 mm trimethyl phosphate as an internal chemical shift and concentration standard, and 10% D2O (15). Samples for assays on long-chain lipids were similar except that they contained 2 mm phospholipid substrate solubilized in 50 mg/ml CHAPS detergent. Each sample contained a volume of 0.6 ml in a 5-mm thin wall NMR sample tube. Following an initial spectrum obtained in the absence of enzyme, 10 μg of pure recombinant enzyme was added to the sample. After a delay of 5 min to allow for magnet shimming, five consecutive spectra were obtained at 15-min intervals. Samples were then stored at 37 °C for 24 h, and an NMR spectrum was obtained at that time. Data were processed and analyzed with MestReNova 7.1.1 (Mestrelab Research, Santiago de Compostela, Spain). A baseline correction was applied to the spectra, and quantitative peak integration was performed with the autodetect function in MestReNova. All integrated peaks were normalized to the 1 mm trimethyl phosphate standard. For hexanoyl SM (6:0 SM) and octanoyl LPC (8:0 LPC), percent loss of substrate at each time point was calculated by dividing the normalized integrated peak area of substrate resonance by the initial substrate signal. Where no product signal was observed, a zero was assigned to indicate that substrate to product turnover was not detected in our assay. For the CHAPS assay, product yield for each time point was calculated by dividing the normalized integrated peak area of product resonance by the initial substrate signal. Substrate and product resonances were identified based on analogous results in a previous study (15).
Crystallization Trials of St_βIB1i
Initial crystallization screens were carried out with a Phoenix robotic liquid dispenser (Art Robbins Instruments, Sunnyvale, CA) in a 96-well Intelli-Plate using the Index Crystallization Screen (Hampton Research, Aliso Viejo, CA). Each sitting drop in the screen contained a mixture of purified St_βIB1i and precipitant solution at a ratio of 1:2 or 2:1 in a total drop volume of 200 nl. Tetragonal rods appeared within 24–48 h in 0.2 m magnesium chloride hexahydrate, 0.1 m BisTris-HCl (pH 5.5), 25% w/v PEG-3350. Crystal growth was optimized in 24-well plates using the hanging drop method with 2 μl of 3 mg/ml purified St_βIB1i to 2 μl of precipitant containing between 10 and 25% PEG 3350 buffered with 0.1 m BisTris (pH 5.5) and 0.2 m magnesium chloride hexahydrate. Thin rectangular crystals grew in 6–8 days from precipitant solution containing 14% PEG 3350. Crystals were crushed and added to decreasing concentrations of PEG 3350 for seeding trials. Diffraction quality crystals of St_βIB1i grew in a precipitant solution containing 12% PEG 3350, 0.1 m BisTris (pH 5.5), 0.2 m magnesium chloride hexahydrate. Large single crystals (0.2 × 0.3 × 0.4 mm) were flash frozen using 40% PEG 3350 as a cryoprotectant.
Diffraction and Model Building of St_βIB1i
Diffraction data for crystals of St_βIB1i were collected remotely on beam line 7-1 at the Standard Synchrotron Radiation Laboratory using Blu-Ice software (37). The phase problem was solved with molecular replacement using the programs Chainsaw and MR phaser of the CCP4 suite (38). The crystal structure of a homologous toxin from L. intermedia (PDB code 3RLH, chain A) was used as a starting model. Manual model building and modification were performed using COOT (39). Bulk solvent correction and standard refinement with Refmac5 (40) yielded an Rcrys of 18% and an Rfree of 24%. Optimum TLS groups were obtained from the TLS motion determination server (41, 42). Refinement including TLS groups yielded a new Rcrys of 16% and a new Rfree of 20%. Coordinates have been deposited in the RCSB Protein Data Bank under PDB code 4Q6X.
In Silico Docking Studies
In silico docking of substrates to StSicTox-βIB1i was performed using Autodock Vina (43). PDBQT files for StSicTox-βIB1i receptor and substrate ligands were prepared using UCSF Chimera 1.8.1 (44) and/or AutoDockTools-1.5.6 in the MGLTools package. St_βIB1i contains two active site histidine residues, His-12 and His-47. In one set of runs, His-12 was deprotonated, whereas His-47 was protonated; this is the starting configuration in the mechanism proposed by Shi et al. (45) for GDPD enzymes. In a second set of runs, the opposite configuration was used. For docking of LPE and LPC substrates, the dihedral angles for the phosphoglycerol moiety of the ligand were restricted to a conformation in which the 2-hydroxyl group was poised to attack the phosphorus atom, leading to CPA formation. This conformation was based on the observed geometry of sn-glycerol-3-phosphate found in the active site of a glycerol phosphodiester phosphodiesterase from Oleispira antarctica (PDB code 3QVQ). Substrate molecules were docked into a 30 × 30 × 30 Å box centered on the magnesium atom in the active site of St_βIB1i. The charge on magnesium was manually set at +2. The binding mode search was conducted at maximal exhaustiveness setting. For each ligand in each protonation state of the enzyme, 30–60 binding modes were calculated, depending upon the strength of structural consensus observed among the lowest five modes in a smaller initial test calculation (strong for LPE; less strong for LPC). In the docking of LPE, the lowest overall energy binding modes calculated for the two histidine protonation states were practically identical. For LPC, however, docking to the His-47-protonated version of St_βIB1i yielded a highly improbable lowest energy mode in which the phosphate group was ∼10 Å from the magnesium center. Results were visualized using UCSF Chimera.
RESULTS
Enzymatic Activity and Substrate Preference of Three Diverse SicTox Proteins
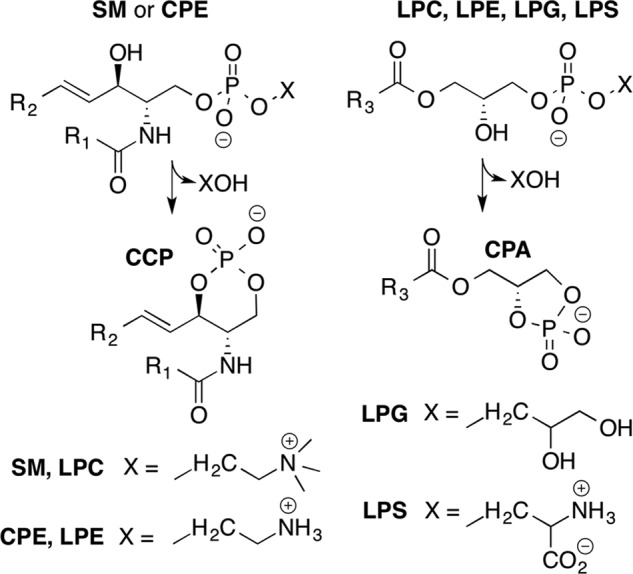
We initially tested the three selected SicTox homologs for PLD activity on hexanoyl SM/Triton X-100 and octanoyl-LPC micelles using previously described 31P NMR assays (15). These short chain substrates give small micelles and thus minimize resonance linewidths (46). Both enzymes from L. arizonica (La_αIB2bi and La_βID1) showed high activity against both SM and LPC, whereas St_βIB1i showed much lower activity (Fig. 3A). The L. arizonica enzymes consumed all the SM within the dead time of the experiment (∼10 min), whereas more than 50% of this substrate remained after ∼80 min of incubation with the S. terrosus enzyme. Similar differential activity was also observed with 8:0 LPC as substrate (Fig. 3A). Both L. arizonica enzymes also consumed substantial amounts of 8:0 LPC after 80 min (initial rate of ∼0.24 mm/min for La_αIB2bi and 0.8 mm/min for La_βID1), whereas St_βIB1i showed no activity against 8:0 LPC even after 24 h. These initial experiments demonstrate that the homologs have variable PLD activity against SM and LPC and are consistent with other studies showing reduced SMase D activity among some β clade members (Fig. 1).
FIGURE 3.
31P NMR assay results of three different SicTox PLD toxins with different phospholipid substrates. A, preliminary assessment of activity against SM and LPC using 10 μg of pure enzyme with either 2 mm hexanoyl sphingomyelin (6:0 SM) and 2 mm Triton X-100, or 80 mm octanoyl lysophosphatidylcholine (8:0 LPC). St_βIB1i shows much lower activity toward the two substrates than La_αIB2bi and La_βID1, (B–D). Panel assays using 10 μg of pure enzyme with ∼2 mm each of six different phospholipid substrates solubilized in CHAPS detergent. Fitted slopes of product accumulation during the linear portion of the reaction were used to extract initial reaction rates. Error bars have been added to points used for slope determination; these are based on statistical analysis of instrument precision and represent standard deviations. In some cases, no product was observed after 75 min but did appear after 24 h. Substrate conversion and product yield were derived from integration of NMR signals relative to a trimethyl phosphate internal standard, as described under “Experimental Procedures.”
We then tested the three enzymes against a wider panel of commercially available long-chain lysolipid and phosphosphingolipid substrates with various headgroups. In addition to LPC, the panel included the lysolipids LPE, LPG, and LPS, which are analogous to LPC but contain ethanolamine, glycerol, and serine headgroups, respectively, in place of choline (see Scheme 1). In addition to SM, the panel included the phosphosphingolipid ceramide phosphoethanolamine (CPE), which differs from SM only in replacement of the choline headgroup with ethanolamine (see Scheme 1). The ethanolamine-containing substrates LPE and CPE were particularly interesting to us as possible alternative substrates to LPC and SM, because ethanolamine is a common headgroup in insect lipids (47–50). We included LPG and LPS as examples of lipids with neutral/zwitterionic headgroups. These lipids have been previously reported as substrates for Loxosceles reclusa protein LrSicTox-αIB1 (19).
For our panel comparison (Fig. 3, B–D), we utilized 31P NMR in mixed micelles with the nondenaturing detergent CHAPS (51). CHAPS effectively solubilizes the long-chain lipids and has precedent in phospholipase activity studies (52). The CHAPS NMR assay allows comparison of diverse long-chain lipid substrates at a common concentration of ∼2 mm and allows direct detection of sharp, well resolved substrate and product resonances in all cases. It is less well suited to extraction of detailed primary kinetic parameters, partly because of the low sensitivity of NMR (53) and because such measurements are frequently problematic for interfacial enzymes in substrate/detergent mixtures (54). We regard these strengths and limitations as an acceptable compromise in an initial panel study of substrate preference across multiple SicTox family members. We thus characterize apparent substrate preferences (Table 1) by comparing initial reaction rates at a single initial concentration of substrate, both in single-substrate assays (Fig. 3) and in competition assays of selected substrate pairs (Fig. 4).
TABLE 1.
Initial reaction rates (μm/min) of SicTox PLD toxins with various phospholipid substrates
Reported rates are slopes of product accumulation in single substrate assays (see Fig. 3). Rates in parentheses derive from direct competition assays (SM/CPE or LPC/LPE) based on disappearance of the substrate signal (see Fig. 4). The two bottom rows represent rate ratios for SM and CPE or LPC and LPE, based either on comparison of the rates in single substrate assays or relative rates in a competition assay (values in parentheses). Note that values derived from single substrate and competition assays are similar. All rates were directly measured at 10 μg of enzyme and ∼2 mm initial substrate concentration, with the exception of St_βIB1i with CPE. In this case, the rate was too fast to be measured, so a rate was measured at 1 μg of enzyme and multiplied by 10. In cases where the reported rate is <1 μm/min, no substrate decay was evident after ∼75 min (and in the case of the single substrate assays, no product formation was evident either). For these cases, the rates were estimated from the 24-h time points, unless no product was seen even at 24 h, in which case the rate was reported as <0.1 μm/min (see Fig. 3).
| Phospholipid | La_αIB2bi | La_βID1 | St_βIB1i |
|---|---|---|---|
| SM | 11 (9) | 31 (20) | 0.2 (0.8) |
| CPE | 0.3 (0.5) | 12 (10) | 410 (>90) |
| LPC | 0.4 (0.7) | 13 (14) | <0.1 |
| LPE | 0.1 (0.1) | 5.2 (5.3) | 21 (16) |
| SM/CPE | 36.7 (18) | 2.6 (2) | 0.0005 (<0.009) |
| LPC/LPE | 5 (7.8) | 2.5 (2.6) | <0.005 (<0.006) |
FIGURE 4.

Stacked 31P NMR spectra from competition assays of three SicTox PLD enzymes with choline- and ethanolamine-containing substrates. Left panels show SM/CPE competition experiments, and right panels show LPC/LPE competition experiments. The spectra illustrate the preference, or lack thereof, of the different enzymes for choline (SM and LPC) or ethanolamine (CPE and LPE) headgroups. Assays contained 10 μg of each SicTox PLD and ∼2 mm of each substrate. In the SM/CPE assay, cleavage of either substrate yields cyclic ceramide phosphate (CCP), a six-membered ring phosphate with an upfield 31P resonance. In the LPC/LPE assay, cleavage of either substrate yields cyclic phosphatidic acid (CPA), a five-membered ring phosphate with a far downfield 31P resonance.
The substrate preferences of the three enzymes measured from single-substrate assays have two common features (Fig. 3 and Table 1). First, all three enzymes show activity against lysophospholipids with positively charged headgroups (LPC and/or LPE) but show no detectable activity against neutral/zwitterionic headgroups (LPG and LPS). Second, the enzymes show more rapid turnover of ceramide-based substrates (SM or CPE) versus the corresponding lysophospholipids (LPC or LPE, respectively). This latter preference is marginal in the case of La_βID1 (∼2-fold difference in measured rate) but increases to an order of magnitude or more for St_βIB1i and La_αIB2bi.
The enzymes have strikingly different substrate preferences for choline and ethanolamine, the two positively charged headgroups. La_βID1 shows very little discrimination between choline and ethanolamine. Meanwhile, La_αIB2bi is specific for choline and St_βIB1i is specific for ethanolamine. La_αIB2bi turns over SM faster than CPE by an estimated 1 to 2 orders of magnitude at ∼2 mm initial substrate concentration (Table 1). La_αIB2bi also prefers LPC over LPE, although neither substrate is turned over rapidly in this assay. St_βIB1i, however, turns over CPE and LPE at least 3 and 2 orders of magnitude faster than SM and LPC, respectively. The CPE/SM comparison is particularly striking; all CPE substrate is gone within the first 10 min, while no turnover of SM is evident up to 80 min (Fig. 3D).
Direct competition experiments between substrates with choline and ethanolamine headgroups show similar substrate preferences to those seen in single-substrate assays (Fig. 4 and Table 1). For each enzyme, we set up two competition experiments using either phosphosphingolipids (CPE versus SM) or lysolipids (LPC versus LPE). The most notable result is that we continue to observe rapid turnover of CPE, and extremely slow turnover of SM, by St_βIB1i in direct competition experiments. In sum, our experiments show for the first time that these venom toxins have variable substrate specificity and reveal that toxins with low sphingomyelinase activity, such as St_βIB1i, may be highly specific for ethanolamine-containing phospholipids.
Crystal Structure of St_βIB1i
To begin exploring the basis of differences in substrate specificity, we solved the structure of SicTox protein βIB1i from S. terrosus using crystallographic methods (Table 2). Refinement of the St_βIB1i structure converged to a crystallographic residual of 17% (Rfree = 22%) for all data between 38.5 and 2.14 Å. The refined model of St_βIB1i contains 278 amino acid residues, 122 solvent water molecules, and 1 Mg2+ ion in the active site. Model quality was assessed with SFCHECK (55), and all stereochemical parameters are within acceptable ranges. A Ramachandran diagram of the model generated with RAMPAGE (56) indicates that all main-chain dihedral angles are within the most favored and additionally allowed regions.
TABLE 2.
Data collection and refinement statistics for St_βIB1i (PDB code 4Q6X)
| Space group | Trigonal, P32 |
|---|---|
| Unit cell parameters (Å, °) | a = b = 49.22, c = 90.11; α = β = 90, γ = 120 |
| Z (molecules/au) | 1 |
| Data collection | |
| X-ray source | SSRL BL7-1 |
| Wavelength (Å) | 1.127 |
| Detector | CCD-ADSQ Quantum 315 |
| Data refinement | |
| Resolution range (Å)a | 38.56–2.14 (2.25–2.14) |
| Measured reflectionsa | 49,403 (5,484) |
| Unique reflectionsa | 13,352 (1,828) |
| Multiplicitya | 3.7 (3.0) |
| Completeness (%)a | 99.2 |
| Rmergea,b | 0.08 (0.44) |
| I/σ(I)a | 13.9 (1.79) |
| Structure refinement | |
| Rcrysa,c | 0.17 (0.21) |
| Rfreea,c | 0.22 (0.32) |
| r.m.s.d.d bonds (Å) | 0.010 |
| r.m.s.d. angles (°) | 1.39 |
| 〈B〉 (Å2) | 39.0 |
| Wilson B-factor (Å2) | 27.3 |
| Ramachandran plot | |
| Most favored regions | 97.8% |
| Additional allowed regions | 2.2% |
| Generously allowed regions | 0% |
| In disallowed regions | 0% |
a Overall/outermost shell is indicated.
b Rmerge = Σhkl Σi|Ii(hkl) − 〈I(hkl)〉|Σhkl Σi I(hkl), where 〈I(hkl)〉 is the mean intensity of all symmetry-related reflections Ii(hkl).
c Rcryst = (Σ|Fobs − Fcalc|)/ΣFobs. Rfree as for Rcryst, using a random subset of the data (5%) not included in the refinement.
d r.m.s.d., is root mean square deviation.
Structural Comparison of St_βIB1i with Other SicTox Proteins
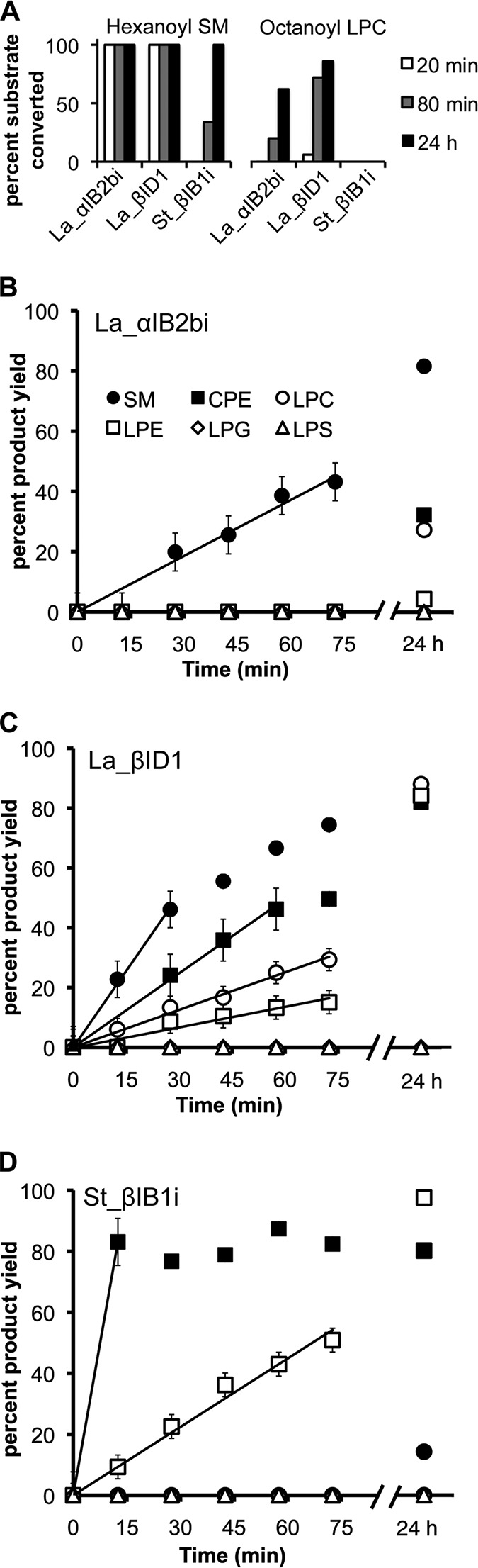
St_βIB1i has a canonical TIM (α/β)8 barrel fold with close structural homology to LiRecDT1 (Fig. 5A) (PDB code 3RLH (28)). St_βIB1i and LiRecDT1 superimpose with a global root mean square deviation of 1.37 Å for 275 Cα atoms (57), a sequence similarity of 63.3%, and a DALI Z-score of 44.3 (58). St_βIB1i is slightly less similar to SMase I (PDB code 1XX1 (59)), with a DALI Z-score of 40.6, but both comparisons indicate close structural homology.
FIGURE 5.
Structural and amino acid sequence comparison of the SicTox PLD toxins. A, superposition of St_β1B1i from S. terrosus (purple; PDB code 4Q6X) on Li_αIA1a/LiRecDT1 from L. intermedia (gold; PDB code 3RLH), and Ll_αIII1/SMase I from L. laeta (green; PDB code 1XX1). B, active sites of the same three enzymes, showing putatively important amino acid residues for catalysis and/or substrate binding (see also Fig. 2). The essential magnesium ion cofactor from the structure of St_β1B1i is shown as a small red sphere, along with a sulfate ion from the L. laeta SMase I structure. With the exception of Asp-52, in the flexible/variable catalytic loop, all the amino acid residues have a similar location and conformation in St_β1B1i, a β clade member, as in the two α clade representatives. Residue numbering is referenced to Ll_αIII1/SMase I.
St_βIB1i contains two disulfide bonds as follows: one between Cys-51 and Cys-57, part of a hairpin turn in the catalytic loop, and one between Cys-53 and Cys-197, linking the catalytic loop to a second flexible loop region (27). This disulfide bonding pattern, along with low SMase D activity, places St_βIB1i in the class IIb group (27, 28). The flexible, catalytic, and variable loop regions of St_βIB1i all exhibit large temperature factors, as also seen in both LiRecDT1 and SMase I. These apparently dynamic regions are likely to be involved in substrate recognition and formation of the membrane interface (2, 27, 28, 61). Modeling of these highly flexible regions in St_βIB1i required TLS refinement (40, 41, 62).
The active site of St_βIB1i contains all the residues previously identified as essential for catalytic activity (Figs. 2 and 5B) (2, 27, 28, 59). With the exception of Asp-52, which is in the highly flexible loop region of the molecule (Fig. 5B), the catalytic side chains of St_βIB1i also superimpose closely with those of LiRecDT1 and SMase I. The Mg2+ cofactor is coordinated by six groups: the OD1 atom of Asp-33 (Asp-34 in SMase I numbering); the OE2 atom of Glu-31 (Glu-32 in SMase I numbering); the OD2 atom of Asp-91, and three water molecules. Thus, the inner hydration shell of the divalent magnesium is observed in the St_βIB1i structure (63). The active site of St_βIB1i does not show clear electron density for any ligand other than Mg2+; all residual electron density in the active site was attributed to water molecules.
Docking and Mutational Studies
To examine possible substrate binding modes, we docked LPE and LPC ligands into the S. terrosus βIB1i active site (Fig. 6) using Autodock Vina (see under “Experimental Procedures”). We focused on lysophospholipid substrates rather than sphingolipids because of their simpler structure, and because other structural data yielded clues to the binding mode for lysophospholipids. Specifically, the structure of a bacterial GDPD from O. antarctica (PDB code 3QVQ) contains a glycerol 3-phosphate bound in a mode where the 2-hydroxyl is poised to attack the phosphorus center and make a five-membered ring (see Fig. 8, below). GDPDs are distantly related to the SicTox proteins (27, 64, 65), and their mechanism has been proposed to proceed through a five-membered cyclic phosphate intermediate (45) analogous to the product of the toxin action on lysophospholipids (15). We felt that the conformation of the glycerol phosphate in the GDPD active site was a reasonable model for the phosphoglycerol portion of LPC or LPE bound to the toxin in a catalytically competent mode. Consequently, we simplified our docking calculations by restricting the dihedral angles of the phosphoglycerol moiety of LPE and LPC to mimic the glycerol phosphate structure. The lowest energy binding modes for LPE and LPC are highly similar (Fig. 6A) and suggest that the headgroup is accommodated within a pocket bordered by Val-89, Asp-91, Asn-95, Ser-132, Glu-134, Asp-165, Ser-167, Gly-195, Tyr-228, Trp-230, and Met-251 (Fig. 6B). Part of this region (residues 89, 132, 134, 165, 167 and 195) has previously been named the “distal binding pocket” and is suggested to be important for recognition of different substrates (24). This pocket is quite well conserved between St_βIB1i and the α SMase D toxins of known structure. Only residues 134 (Glu versus Pro) and 195 (Gly versus Ser) differ (Figs. 6B and 2). In the lowest energy binding modes for LPE and LPC, neither position directly contacts the headgroup. Thus, the docking studies do not generate a strong sense that the active sites of these proteins should preferentially bind more strongly to choline than to ethanolamine headgroups or vice versa.
FIGURE 6.

In silico docking of LPE and LPC into the active site of St_βIB1i. A, surface rendering of St_βIB1i (tan), with lysophosphatidylcholine (blue) and lysophosphatidylethanolamine (lavender) docked into the active site. The magnesium cofactor is shown as a small green sphere interacting with the phosphate group of each substrate. For both substrates, the headgroup is situated in a binding pocket that has been previously identified as potentially important for substrate specificity (24). B, side chains in the putative headgroup binding pocket of St_βIB1i (tan; PDB code 4Q6X) compared with those of LiRecDT1, an α clade protein from L. intermedia (green; PDB code 3RLH). The two pockets are quite similar but contain amino acid sequence differences at positions 95, 134, and 195 (see also amino acid residues in blue boxes in Fig. 2). Residue numbering is referenced to Ll_αIII1/SMase I.
FIGURE 8.

Two plausible mechanisms of LPC cyclization by SicTox enzymes, based on two possible substrate binding modes. A, active site pocket of St_βIB1i with glycerol 3-phosphate bound as seen in a GDPD enzyme from O. antarctica (PDB code 3QVQ). B, this binding mode places the nucleophilic 2′-OH of the glycerol moiety adjacent to His-12. The putative leaving group position is not evident, as glycerol 3-phosphate is the product of removal of the leaving group by a GDPD enzyme. However, the phosphate oxygen opposite the nucleophile (see arrow) is likely to represent the leaving group oxygen of substrate. C, mechanism for LPC/LPE cyclization derived from the glycerol 3-phosphate binding mode. His-12 deprotonates the 2′-hydroxyl generating the nucleophile for attack on the phosphate. His-47 acts as a general acid to protonate the leaving group. D, active site pocket of St_βIB1i with LPE docked as in Fig. 6. E, this binding mode inverts the orientation of the glycerol phosphate moiety relative to that shown for glycerol 3-phosphate in A–C, and places the 2′-OH of glycerol adjacent to His-47. F, mechanism for LPC/LPE cyclization based on the LPC/LPE docking. His-47 activates the 2′-OH for in-line attack on the phosphodiester. His-12 protonates the leaving group.
Nonetheless, we tested a variant of St_βIB1i containing the substitutions E134P and G195S. In a competition experiment between SM and CPE, the variant does have measurable activity against SM during the 1st h, unlike the wild type, but it still turns over CPE much faster (Fig. 7). We also see no evidence that the variant favors LPC over LPE (data not shown). Interestingly, the rate of SM consumption in the SM/CPE competition slows markedly after the 1st h (44% SM consumed after 24 h) and is also very slow in an experiment with pure SM (data not shown). This suggests not only that activity of the variant against SM is limited, but also that it may depend on the presence of CPE.
FIGURE 7.
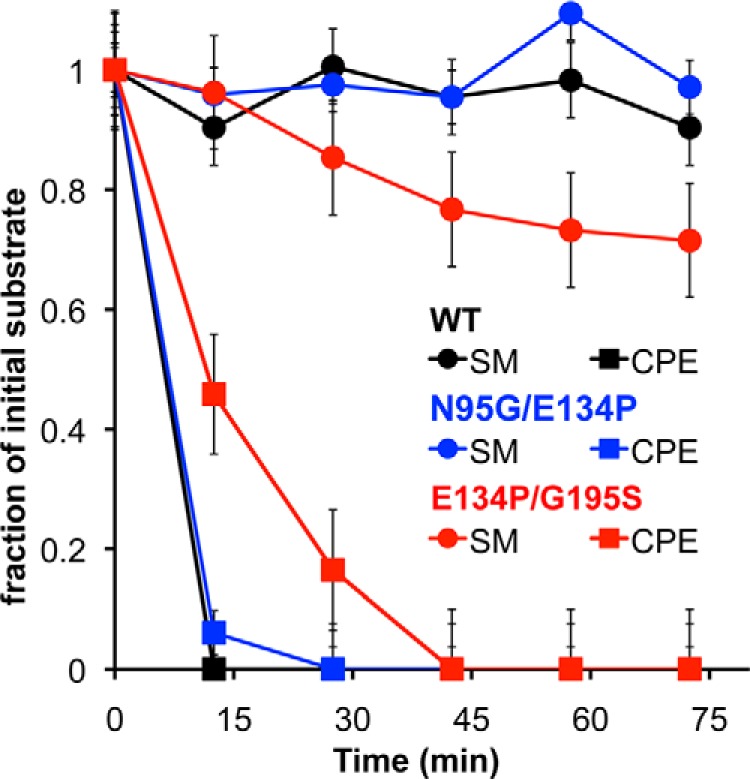
31P NMR competition assays of St_βIB1i variants against SM and CPE. The data for wild-type St_βIB1i (black) derive from the spectra shown in Fig. 4 and the N95G/E134P (blue) and E134P/G195S (red) variants that were assayed in the same manner. All three variants turn over CPE considerably faster than SM, although E134P/G195S may exhibit a decreased preference for CPE. See “Experimental Procedures” for details of assay conditions. Error bars are based on statistical analysis of instrument precision and represent standard deviations.
Asn-95, a residue adjacent to Glu-134 (Fig. 6B), has also been proposed (along with Glu 134) to help exclude choline-containing substrates from the active site of class IIb enzymes (27). This residue is conserved as Gly in the α clade toxins (see Fig. 2). To test the importance of these residues, we engineered a E134P/N95G variant of St_βIB1i. The variant showed rapid consumption of CPE and no measurable activity against SM in a competition experiment (Fig. 7). Because the rates are outside the dynamic range of the experiment for both the variant and the wild type, we cannot exclude some mutational effect on the relative rates of turnover. We conservatively concluded that site-directed mutations in or near the putative headgroup binding region may have a limited impact on substrate specificity, but they do not alter it qualitatively.
Mechanism of SicTox PLD Activity
Although the above analysis of St_βIB1i does not explain its substrate specificity, it does provide an opportunity to reconsider the general mechanism of the SicTox enzymes. The currently accepted mechanism (2, 27, 59) is based on the mechanisms of two other phosphodiesterases, a bacterial SMase C (66) and hydrolytic PLD enzymes with a conserved HKD motif (67). The first step is proposed to involve nucleophilic attack by a conserved histidine at the scissile phosphodiester bond, followed by displacement of the headgroup, leading to formation of a penta-coordinated covalent intermediate. In the second step, deprotonation of water forms a nucleophile, which attacks the covalent adduct to release a monoester product. This mechanism must be reexamined in light of our recent discovery that SicTox enzymes catalyze cyclic phosphate formation rather than hydrolysis (15), combined with the lack of an evolutionary relationship between SicTox and HKD PLD enzymes (67).
One possibility is that the SicTox mechanism is homologous to the first step of the mechanism of GDPD enzymes, which are distantly related (27, 64, 65). GDPDs hydrolyze phosphoglycerol substrates and have been proposed to form a cyclic intermediate analogous to the lipid product of the action of SicTox enzymes on lysophospholipids (45). In the proposed cyclization step, one histidine (His-17) acts as a general base, accepting a proton from the 2′-hydroxyl of the glycerol moiety, generating a nucleophile; a second histidine (His-59) acts as a general acid, donating a proton to the leaving group. This mechanism is supported by both the conformation and positioning of glycerol 3-phosphate product bound to O. antarctica GDPD (PDB code 3QVQ).
A reasonable GDPD-like mechanism for cyclization of LPC by the SicTox proteins is shown in Fig. 8C. To visualize the probable GDPD cyclization step in the context of a SicTox protein, we superimposed the O. antarctica and S. terrosus structures to position the phosphoglycerol moiety in the active site of St_βIB1i (Fig. 8, A and B). The superposition yields a reasonable binding mode for the phosphoglycerol moiety of LPC or LPE, with the two active site histidines (His-12 and His-47) nicely positioned for general base and acid catalysis, respectively.
Our in silico docking of full-length lipid substrates suggests a related but different mechanism in which the orientation of the phosphoglycerol moiety is reversed (Fig. 8, D–F). In the GDPD-like mechanism, the orientation of the phosphoglycerol moiety implies that the lipid chain of LPC or LPE would face deep into the binding pocket, although the headgroup would protrude from the active site (Fig. 8, A–C). The docking studies imply essentially the opposite positioning of lipid chain and headgroup. One advantage of this configuration is that the lipid chain may not easily fit into the deep part of the binding pocket in the GDPD-like mechanism. The reversed orientation also leads to a swapping of the roles of the two histidines as follows: His-47 is now in a position to activate the nucleophile, whereas His-12 can protonate the leaving group. His-47 could also be assisted by the adjacent Asp-52 residue (Fig. 8E). A final difference is that in the GDPD-like mechanism, the nucleophilic hydroxyl occupies one coordination site of magnesium; in the docking-based mechanism, the oxygen of the leaving group occupies this position. In either case, the magnesium ion could accelerate the reaction by stabilizing a developing negative charge.
DISCUSSION
We have biochemically characterized three diverse SicTox PLD toxins to compare substrate preference and ambiguity among venom-expressed enzymes in the SicTox gene family. The results demonstrate for the first time that a wide range of preference exists for choline versus ethanolamine headgroups. Most strikingly, we found that St_βIB1i, a class IIb toxin with low PLD activity against SM and LPC, is actually a highly active enzyme with strong specificity for ethanolamine-containing substrates. We determined the structure of St_βIB1i, the first of a class IIb enzyme as well as the first structure of a SicTox PLD from a Sicarius species. The structure led to new insights into substrate binding mode and mechanism, but it did not immediately reveal the basis for differences in specificity. Nevertheless, our findings show that SicTox proteins are functionally diverse, which may in turn help to explain their variable mammalian toxicity (7–9, 25, 26).
The specificity of St_βIB1i for ethanolamine headgroups may explain why other class IIb enzymes tend to show diminished sphingomyelinase activity compared with whole venoms. St_βIB1i shares over 60% sequence identity with all previously reported class IIb enzymes (Figs. 1 and 2), and phylogenetic analysis groups these toxins within the βI clade of the SicTox gene family (24). It is therefore plausible that many βI SicTox PLD toxins have evolved specificity for ethanolamine-containing targets such as CPE.
Our test set also included two paralogs, La_αIB2bi (15, 30) and La_βID1, from the venom of L. arizonica. La_αIB2bi, an α clade member, strongly preferred choline-containing over ethanolamine-containing substrates. The preference of La_αIB2bi for LPC over LPE resembles the specificity previously observed for another α clade toxin, αIA1 from L. reclusa (19). Surprisingly, La_βID1 also slightly preferred choline-containing substrates, despite belonging to the βI clade like St_βIB1i. The observation of robust SMase D activity in La_βID1 is intriguing given that it shares 84% sequence identity with LiRecDT5 (βID1) from L. intermedia, an enzyme with very limited SMase D activity compared with α clade toxins from the same species (9). Direct experimental comparison of La_βID1 and LiRecDT5 could further illuminate whether these two enzymes differ in SMase D activity and/or substrate specificity. Functional differences between these two proteins would suggest that the βID clade is functionally diverse and represents a transition toward ethanolamine specificity in the βI-ABC clades, beginning from either low specificity or a preference for SM in the ancestral sequences (see Fig. 1). They would also potentially lead to identification of limited sequence changes that modulate function.
At present, the determinants of substrate specificity remain a mystery. The structure of St_βIB1i, including the active site, is quite similar to the previously determined structures of a class I enzyme (PDB code 1XX1) and a class IIa enzyme (PDB code 3RLH). As a class IIb enzyme, St_βIB1i is most similar to the latter, as all class II enzymes contain two disulfide bonds. The largest structural variations were found in the flexible loop regions, as observed previously in comparisons of class I and class IIa structures (2, 28). Docking experiments with LPC and LPE substrates and St_βIB1i suggested a location for the headgroup recognition pocket deep in the active site, away from variable loops. The sequence and structure of this pocket are largely conserved between the different enzymes, and both substrates docked in a similar mode. Mutation of variable residues in this region of St_βIB1 led to at most minor alterations in its specificity. If these enzymes use active site interactions to discriminate between substrates, it is not obvious how they do so.
Because PLDs are interfacial catalysts (67), an alternative source of specificity might involve interactions between the i-face of the enzyme (68) and a lipid interface. The i-face of the PLD toxins has not been identified experimentally but has been proposed to consist of the catalytic loop (visible in the lower left corner of Fig. 5B) and the loops following the fifth and sixth strands of the barrel (27). These regions could be targets for future mutational studies. Ethanolamine and choline could present very different interaction surfaces to the i-face of an interfacial catalyst. Although both are positively charged headgroups, they differ in hydrogen bonding capability, hydrophobicity, and size. For example, unlike their choline counterparts, ethanolamine-containing phospholipids can form a hydrogen-bonded ring structure between the positively charged amine group and a negatively charged phosphate oxygen (69).
Substrate preferences could result from changes in kinetics and thermodynamics of the enzyme-lipid interactions at the i-face, including tighter binding and/or longer residence times for processive catalysis. They may also result from allosteric coupling between the i-face and the active site, which can affect substrate binding as well as catalytic steps (70). Because of these complexities, apparent substrate preferences may themselves depend somewhat on the form of the lipid interface, e.g. micelles versus vesicles, present in the assay. This is a caveat in our study, which uses detergent-mixed micelles; we note, however, that the trends we observed were similar in most single substrate and mixed substrate assays and that the enzymes showed similar trends in SMase/LPCase activity in different micelle mixtures (Figs. 3 and 4 and Table 1).
This study suggests generally stronger substrate specificity for the SicTox enzymes in comparison with previous studies. Specifically, the three PLD toxins showed moderate to strong preference for sphingolipid over lysolipid substrates, as well as a very strong preference for positively charged over neutral headgroups. By contrast, for an α clade enzyme from L. reclusa, Lee and Lynch (19) reported very similar kinetics for sphingolipid (SM) and lysolipid (LPC) substrates, and observed strong activity against LPG and LPS. van Meeteren et al. (18) also reported similar levels of turnover of SM and LPC for an α clade enzyme from Loxosceles. laeta. These different findings may reflect variable substrate preference within the SicTox family. Indeed, one of our three proteins, La_βID1, shows considerably lower preference for sphingolipids, and less discrimination between choline and ethanolamine, than the other two. The general preference for sphingolipids found in our study, however, may have biological significance; sphingolipid substrates exist at higher concentrations than lysolipids in the lipidome of insect prey (49, 50).
Discrimination between choline and ethanolamine headgroups may also be biologically significant for both mammalian toxicity and predation. CPE is a major phosphosphingolipid in insects (47), the main prey item of the sicariid spiders (30); SM, by contrast, dominates in mammals (71, 72). As noted above, some βI clade members show diminished toxicity in mammalian models (7–10, 25, 26). Our work suggests that this reduced toxicity may relate to specificity for ethanolamine-containing lipids like CPE. Given the specificity of St_βIB1i and the paucity of CPE in humans, we also propose that St_βIB1i, or similar toxins, have potential as safe insecticides if utilized in a transgenic system (73).
As for predation, lipidome and genetic analyses suggest that most arthropods can produce both CPE and SM, but some insects (e.g. the Brachycera) may produce only CPE (47). The diversification of headgroup specificity in the SicTox gene family may allow some spider species to tailor venom activity specifically toward prey species in which either SM or CPE is the predominant phosphosphingolipid. A question for future research is whether variation in whole venom specificity for SM or CPE correlates with the levels of each lipid in typical prey.
Purified La_αIB2bi acts as a paralytic neurotoxin in crickets (30), and this may be one general function of the SicTox proteins, whether they are specific for SM or CPE or act on both. CPE is concentrated in neural tissue of certain insects, comprising about 4% of the total lipid content of brain tissue (49) in Drosophila and about 6–7% of total lipid content of the nervous system in Musca domestica (50). In Drosophila, CPE is required by glial cells to properly insulate axons, and lack of CPE leads to neuropathy (48). Another future research question is whether the neurotoxic effects of isolated SicTox proteins on different prey will depend on a match between enzyme specificity and the content of SM and/or CPE in neurological tissue.
Acknowledgments
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health Grant P41GM103393 from NIGMS.
This work was supported, in whole or in part, by National Institutes of Health Grants R15-GM-097676-01 (to G. J. B. and P. Z. T.) and R01 GM72623 (to V. B.). This work was also supported by a pilot project grant from the Bio5 Institute (to M. H. J. C., G. J. B., and V. B.) and by National Science Foundation CAREER Award IOB 0546858 (to G. J. B.).
The atomic coordinates and structure factors (code 4Q6X) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PLD
- phospholipase D
- LPC
- lysophosphatidylcholine
- SM
- sphingomyelin
- SMase
- sphingomyelinase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- GDPD
- glycerophosphoryl phosphodiesterase
- PDB
- Protein Data Bank
- LPE
- 1-myristoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine
- LPG
- 1-myristoyl-2-hydroxy-sn-glycero-3-phospho-(1′-rac-glycerol)
- LPS
- l-α-lysophosphatidylserine.
REFERENCES
- 1. Tambourgi D. V., Gonçalves-de-Andrade R. M., van den Berg C. W. (2010) Loxoscelism: From basic research to the proposal of new therapies. Toxicon 56, 1113–1119 [DOI] [PubMed] [Google Scholar]
- 2. Gremski L. H., Trevisan-Silva D., Ferrer V. P., Matsubara F. H., Meissner G. O., Wille A. C., Vuitika L., Dias-Lopes C., Ullah A., de Moraes F. R., Chávez-Olortegui C., Barbaro K. C., Murakami M. T., Arni R. K., Senff-Ribeiro A., et al. (2014) Recent advances in the understanding of brown spider venoms: from the biology of spiders to the molecular mechanisms of toxins. Toxicon 83, 91–120 [DOI] [PubMed] [Google Scholar]
- 3. Hogan C. J., Barbaro K. C., Winkel K. (2004) Loxoscelism: old obstacles, new directions. Ann. Emerg. Med. 44, 608–624 [DOI] [PubMed] [Google Scholar]
- 4. da Silva P. H., da Silveira R. B., Appel M. H., Mangili O. C., Gremski W., Veiga S. S. (2004) Brown spiders and loxoscelism. Toxicon 44, 693–709 [DOI] [PubMed] [Google Scholar]
- 5. Swanson D. L., Vetter R. S. (2006) Loxoscelism. Clin. Dermatol. 24, 213–221 [DOI] [PubMed] [Google Scholar]
- 6. Chaim O. M., Sade Y. B., da Silveira R. B., Toma L., Kalapothakis E., Chávez-Olórtegui C., Mangili O. C., Gremski W., von Dietrich C. P., Nader H. B., Sanches Veiga S. (2006) Brown spider dermonecrotic toxin directly induces nephrotoxicity. Toxicol. Appl. Pharmacol. 211, 64–77 [DOI] [PubMed] [Google Scholar]
- 7. da Silveira R. B., Pigozzo R. B., Chaim O. M., Appel M. H., Dreyfuss J. L., Toma L., Mangili O. C., Gremski W., Dietrich C. P., Nader H. B., Veiga S. S. (2006) Molecular cloning and functional characterization of two isoforms of dermonecrotic toxin from Loxosceles intermedia (brown spider) venom gland. Biochimie 88, 1241–1253 [DOI] [PubMed] [Google Scholar]
- 8. Olvera A., Ramos-Cerrillo B., Estévez J., Clement H., de Roodt A., Paniagua-Solís J., Vázquez H., Zavaleta A., Arruz M. S., Stock R. P., Alagón A. (2006) North and South American Loxosceles spiders: development of a polyvalent antivenom with recombinant sphingomyelinases D as antigens. Toxicon. 48, 64–74 [DOI] [PubMed] [Google Scholar]
- 9. da Silveira R. B., Pigozzo R. B., Chaim O. M., Appel M. H., Silva D. T., Dreyfuss J. L., Toma L., Dietrich C. P., Nader H. B., Veiga S. S., Gremski W. (2007) Two novel dermonecrotic toxins LiRecDT4 and LiRecDT5 from brown spider (Loxosceles intermedia) venom: from cloning to functional characterization. Biochimie 89, 289–300 [DOI] [PubMed] [Google Scholar]
- 10. Ribeiro R. O., Chaim O. M., da Silveira R. B., Gremski L. H., Sade Y. B., Paludo K. S., Senff-Ribeiro A., de Moura J., Chávez-Olórtegui C., Gremski W., Nader H. B., Veiga S. S. (2007) Biological and structural comparison of recombinant phospholipase D toxins from Loxosceles intermedia (brown spider) venom. Toxicon 50, 1162–1174 [DOI] [PubMed] [Google Scholar]
- 11. Appel M. H., da Silveira R. B., Chaim O. M., Paludo K. S., Silva D. T., Chaves D. M., da Silva P. H., Mangili O. C., Senff-Ribeiro A., Gremski W., Nader H. B., Veiga S. S. (2008) Identification, cloning and functional characterization of a novel dermonecrotic toxin (phospholipase D) from brown spider (Loxosceles intermedia) venom. Biochim. Biophys. Acta 1780, 167–178 [DOI] [PubMed] [Google Scholar]
- 12. Kusma J., Chaim O. M., Wille A. C., Ferrer V. P., Sade Y. B., Donatti L., Gremski W., Mangili O. C., Veiga S. S. (2008) Nephrotoxicity caused by brown spider venom phospholipase-D (dermonecrotic toxin) depends on catalytic activity. Biochimie 90, 1722–1736 [DOI] [PubMed] [Google Scholar]
- 13. Chaim O. M., da Silveira R. B., Trevisan-Silva D., Ferrer V. P., Sade Y. B., Bóia-Ferreira M., Gremski L. H., Gremski W., Senff-Ribeiro A., Takahashi H. K., Toledo M. S., Nader H. B., Veiga S. S. (2011) Phospholipase-D activity and inflammatory response induced by brown spider dermonecrotic toxin: endothelial cell membrane phospholipids as targets for toxicity. Biochim. Biophys. Acta 1811, 84–96 [DOI] [PubMed] [Google Scholar]
- 14. Vuitika L., Gremski L. H., Belisário-Ferrari M. R., Chaves-Moreira D., Ferrer V. P., Senff-Ribeiro A., Chaim O. M., Veiga S. S. (2013) Brown spider phospholipase-D containing a conservative mutation (D233E) in the catalytic site: identification and functional characterization. J. Cell Biochem. 114, 2479–2492 [DOI] [PubMed] [Google Scholar]
- 15. Lajoie D. M., Zobel-Thropp P. A., Kumirov V. K., Bandarian V., Binford G. J., Cordes M. H. (2013) Phospholipase D toxins of brown spider venom convert lysophosphatidylcholine and sphingomyelin to cyclic phosphates. PLoS One 8, e72372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kurpiewski G., Campbell B. J., Forrester L. J., Barrett J. T. (1981) Alternate complement pathway activation by recluse spider venom. Int. J. Tissue React. 3, 39–45 [PubMed] [Google Scholar]
- 17. Forrester L. J., Barrett J. T., Campbell B. J. (1978) Red blood cell lysis induced by the venom of the brown recluse spider: the role of sphingomyelinase D. Arch. Biochem. Biophys. 187, 355–365 [DOI] [PubMed] [Google Scholar]
- 18. van Meeteren L. A., Frederiks F., Giepmans B. N., Pedrosa M. F., Billington S. J., Jost B. H., Tambourgi D. V., Moolenaar W. H. (2004) Spider and bacterial sphingomyelinases D target cellular lysophosphatidic acid receptors by hydrolyzing lysophosphatidylcholine. J. Biol. Chem. 279, 10833–10836 [DOI] [PubMed] [Google Scholar]
- 19. Lee S., Lynch K. R. (2005) Brown recluse spider (Loxosceles reclusa) venom phospholipase D (PLD) generates lysophosphatidic acid (LPA). Biochem. J. 391, 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wille A. C., Chaves-Moreira D., Trevisan-Silva D., Magnoni M. G., Boia-Ferreira M., Gremski L. H., Gremski W., Chaim O. M., Senff-Ribeiro A., Veiga S. S. (2013) Modulation of membrane phospholipids, the cytosolic calcium influx and cell proliferation following treatment of B16-F10 cells with recombinant phospholipase-D from Loxosceles intermedia (brown spider) venom. Toxicon 67, 17–30 [DOI] [PubMed] [Google Scholar]
- 21. Newlands G., Atkinson P. (1988) Review of southern African spiders of medical importance, with notes on the signs and symptoms of envenomation. S. Afr. Med. J. 73, 235–239 [PubMed] [Google Scholar]
- 22. Richardson-Boedler C. (1999) Sicarius (six-eyed crab spider): a homeopathic treatment for Ebola haemorrhagic fever and disseminated intravascular coagulation? Br. Homeopath. J. 88, 24–27 [DOI] [PubMed] [Google Scholar]
- 23. Van Aswegen G., Van Rooyen J. M., Van der Nest D. G., Veldman F. J., De Villiers T. H., Oberholzer G. (1997) Venom of a six-eyed crab spider, Sicarius testaceus (Purcell, 1908), causes necrotic and haemorrhagic lesions in the rabbit. Toxicon 35, 1149–1152 [DOI] [PubMed] [Google Scholar]
- 24. Binford G. J., Bodner M. R., Cordes M. H., Baldwin K. L., Rynerson M. R., Burns S. N., Zobel-Thropp P. A. (2009) Molecular evolution, functional variation, and proposed nomenclature of the gene family that includes sphingomyelinase D in sicariid spider venoms. Mol. Biol. Evol. 26, 547–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramos-Cerrillo B., Olvera A., Odell G. V., Zamudio F., Paniagua-Solís J., Alagón A., Stock R. P. (2004) Genetic and enzymatic characterization of sphingomyelinase D isoforms from the North American fiddleback spiders Loxosceles boneti and Loxosceles reclusa. Toxicon 44, 507–514 [DOI] [PubMed] [Google Scholar]
- 26. de Santi Ferrara G. I., Fernandes-Pedrosa Mde F., Junqueira-de-Azevedo Ide L., Gonçalves-de-Andrade R. M., Portaro F. C., Manzoni-de-Almeida D., Murakami M. T., Arni R. K., van den Berg C. W., Ho P. L., Tambourgi D. V. (2009) SMase II, a new sphingomyelinase D from Loxosceles laeta venom gland: molecular cloning, expression, function and structural analysis. Toxicon 53, 743–753 [DOI] [PubMed] [Google Scholar]
- 27. Murakami M. T., Fernandes-Pedrosa M. F., de Andrade S. A., Gabdoulkhakov A., Betzel C., Tambourgi D. V., Arni R. K. (2006) Structural insights into the catalytic mechanism of sphingomyelinases D and evolutionary relationship to glycerophosphodiester phosphodiesterases. Biochem. Biophys. Res. Commun. 342, 323–329 [DOI] [PubMed] [Google Scholar]
- 28. de Giuseppe P. O., Ullah A., Silva D. T., Gremski L. H., Wille A. C., Chaves Moreira D., Ribeiro A. S., Chaim O. M., Murakami M. T., Veiga S. S., Arni R. K. (2011) Structure of a novel class II phospholipase D: catalytic cleft is modified by a disulphide bridge. Biochem. Biophys. Res. Commun. 409, 622–627 [DOI] [PubMed] [Google Scholar]
- 29. Lopes P. H., Bertani R., Gonçalves-de-Andrade R. M., Nagahama R. H., van den Berg C. W., Tambourgi D. V. (2013) Venom of the Brazilian spider Sicarius ornatus (Araneae, Sicariidae) contains active sphingomyelinase D: potential for toxicity after envenomation. PLoS Negl. Trop. Dis. 7, e2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zobel-Thropp P. A., Kerins A. E., Binford G. J. (2012) Sphingomyelinase D in sicariid spider venom is a potent insecticidal toxin. Toxicon 60, 265–271 [DOI] [PubMed] [Google Scholar]
- 31. Bey T. A., Walter F. G., Lober W., Schmidt J., Spark R., Schlievert P. M. (1997) Loxosceles arizonica bite associated with shock. Ann. Emerg. Med. 30, 701–703 [DOI] [PubMed] [Google Scholar]
- 32. Russell F. E., Waldron W. G., Madon M. B. (1969) Bites by the brown spiders Loxosceles unicolor and Loxosceles arizonica in California and Arizona. Toxicon 7, 109–117 [DOI] [PubMed] [Google Scholar]
- 33. Petersen T. N., Brunak S., von Heijne G., Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 34. Jez J. M., Ferrer J. L., Bowman M. E., Dixon R. A., Noel J. P. (2000) Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction mechanism of a plant polyketide synthase. Biochemistry 39, 890–902 [DOI] [PubMed] [Google Scholar]
- 35. Fernandes Pedrosa Mde F., Junqueira de Azevedo Ide L., Gonçalves-de-Andrade R. M., van den Berg C. W., Ramos C. R., Ho P. L., Tambourgi D. V. (2002) Molecular cloning and expression of a functional dermonecrotic and haemolytic factor from Loxosceles laeta venom. Biochem. Biophys. Res. Commun. 298, 638–645 [DOI] [PubMed] [Google Scholar]
- 36. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 37. McPhillips T. M., McPhillips S. E., Chiu H. J., Cohen A. E., Deacon A. M., Ellis P. J., Garman E., Gonzalez A., Sauter N. K., Phizackerley R. P., Soltis S. M., Kuhn P. (2002) Blu-ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406 [DOI] [PubMed] [Google Scholar]
- 38. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Painter J., Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 42. Painter J., Merritt E. A. (2005) A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Crystallogr. D Biol. Crystallogr. 61, 465–471 [DOI] [PubMed] [Google Scholar]
- 43. Trott O., Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 45. Shi L., Liu J. F., An X. M., Liang D. C. (2008) Crystal structure of glycerophosphodiester phosphodiesterase (GDPD) from Thermoanaerobacter tengcongensis, a metal ion-dependent enzyme: insight into the catalytic mechanism. Proteins 72, 280–288 [DOI] [PubMed] [Google Scholar]
- 46. Stafford R. E., Fanni T., Dennis E. A. (1989) Interfacial properties and critical micelle concentration of lysophospholipids. Biochemistry 28, 5113–5120 [DOI] [PubMed] [Google Scholar]
- 47. Vacaru A. M., van den Dikkenberg J., Ternes P., Holthuis J. C. (2013) Ceramide phosphoethanolamine biosynthesis in Drosophila is mediated by a unique ethanolamine phosphotransferase in the Golgi lumen. J. Biol. Chem. 288, 11520–11530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghosh A., Kling T., Snaidero N., Sampaio J. L., Shevchenko A., Gras H., Geurten B., Göpfert M. C., Schulz J. B., Voigt A., Simons M. (2013) A global in vivo Drosophila RNAi screen identifies a key role of ceramide phosphoethanolamine for glial ensheathment of axons. PLoS Genet. 9, e1003980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carvalho M., Sampaio J. L., Palm W., Brankatschk M., Eaton S., Shevchenko A. (2012) Effects of diet and development on the Drosophila lipidome. Mol. Syst. Biol. 8, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bridges R. G. (1973) Lipid composition of larval nervous-system of Musca domestica–comparison between insects susceptible and resistant to cyclodiene insecticides. Comp. Biochem. Physiol. B 44, 191–203 [DOI] [PubMed] [Google Scholar]
- 51. Banerjee P., Joo J. B., Buse J. T., Dawson G. (1995) Differential solubilization of lipids along with membrane proteins by different classes of detergents. Chem. Phys. Lipids 77, 65–78 [DOI] [PubMed] [Google Scholar]
- 52. el-Sayed M. Y., Roberts M. F. (1985) Charged detergents enhance the activity of phospholipase C (Bacillus cereus) toward micellar short-chain phosphatidylcholine. Biochim. Biophys. Acta 831, 133–141 [DOI] [PubMed] [Google Scholar]
- 53. Roberts M. F. (1991) Nuclear-magnetic-resonance spectroscopy to follow phospholipase kinetics and products. Methods Enzymol. 197, 31–48 [DOI] [PubMed] [Google Scholar]
- 54. Berg O. G., Gelb M. H., Tsai M. D., Jain M. K. (2001) Interfacial enzymology: the secreted phospholipase A(2)-paradigm. Chem. Rev. 101, 2613–2654 [DOI] [PubMed] [Google Scholar]
- 55. Vaguine A. A., Richelle J., Wodak S. J. (1999) SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 56. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Structure validation by Calpha geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 57. Maiti R., Van Domselaar G. H., Zhang H., Wishart D. S. (2004) SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 32, W590–W594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Murakami M. T., Fernandes-Pedrosa M. F., Tambourgi D. V., Arni R. K. (2005) Structural basis for metal ion coordination and the catalytic mechanism of sphingomyelinases D. J. Biol. Chem. 280, 13658–13664 [DOI] [PubMed] [Google Scholar]
- 60. Chaves-Moreira D., Souza F. N., Fogaça R. T., Mangili O. C., Gremski W., Senff-Ribeiro A., Chaim O. M., Veiga S. S. (2011) The relationship between calcium and the metabolism of plasma membrane phospholipids in hemolysis induced by brown spider venom phospholipase-D toxin. J. Cell Biochem. 112, 2529–2540 [DOI] [PubMed] [Google Scholar]
- 61. Ullah A., de Giuseppe P. O., Murakami M. T., Trevisan-Silva D., Wille A. C., Chaves-Moreira D., Gremski L. H., da Silveira R. B., Sennf-Ribeiro A., Chaim O. M., Veiga S. S., Arni R. K. (2011) Crystallization and preliminary x-ray diffraction analysis of a class II phospholipase D from Loxosceles intermedia venom. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 234–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rivero E. R., Mesquita R. A., de Sousa S. C., Nunes F. D. (2006) Detection of TLS/FUS-CHOP fusion transcripts in a case of oral liposarcoma. Ann. Diagn. Pathol. 10, 36–38 [DOI] [PubMed] [Google Scholar]
- 63. Maguire M. E., Cowan J. A. (2002) Magnesium chemistry and biochemistry. Biometals 15, 203–210 [DOI] [PubMed] [Google Scholar]
- 64. Catalán A., Cortés W., Muñoz C., Araya J. E. (2014) Tryptophan and aspartic acid residues present in the glycerophosphoryl diester phosphodiesterase (GDPD) domain of the Loxosceles laeta phospholipase D are essential for substrate recognition. Toxicon 81, 43–47 [DOI] [PubMed] [Google Scholar]
- 65. Cordes M. H., Binford G. J. (2006) Lateral gene transfer of a dermonecrotic toxin between spiders and bacteria. Bioinformatics 22, 264–268 [DOI] [PubMed] [Google Scholar]
- 66. Matsuo Y., Yamada A., Tsukamoto K., Tamura H., Ikezawa H., Nakamura H., Nishikawa K. (1996) A distant evolutionary relationship between bacterial sphingomyelinase and mammalian DNase I. Protein Sci. 5, 2459–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Selvy P. E., Lavieri R. R., Lindsley C. W., Brown H. A. (2011) Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 111, 6064–6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Verheij H. M., Slotboom A. J., de Haas G. H. (1981) Structure and function of phospholipase A2. Rev. Physiol. Biochem. Pharmacol. 91, 91–203 [DOI] [PubMed] [Google Scholar]
- 69. Henderson T. O, Glonek T., Myers T. C. (1974) Phosphorus-31 nuclear magnetic-resonance spectroscopy of phospholipids. Biochemistry 13, 623–628 [DOI] [PubMed] [Google Scholar]
- 70. Jain M. K., Berg O. G. (2006) Coupling of the i-face and the active site of phospholipase A2 for interfacial activation. Curr. Opin. Chem. Biol. 10, 473–479 [DOI] [PubMed] [Google Scholar]
- 71. Fyrst H., Saba J. D. (2010) An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat. Chem. Biol. 6, 489–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vacaru A. M., Tafesse F. G., Ternes P., Kondylis V., Hermansson M., Brouwers J. F., Somerharju P., Rabouille C., Holthuis J. C. (2009) Sphingomyelin synthase-related protein SMSr controls ceramide homeostasis in the ER. J. Cell Biol. 185, 1013–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. King G. F., Hardy M. C. (2013) Spider-venom peptides: structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 58, 475–496 [DOI] [PubMed] [Google Scholar]
- 74. Alarcon-Chaidez F. J., Boppana V. D., Hagymasi A. T., Adler A. J., Wikel S. K. (2009) A novel sphingomyelinase-like enzyme in Ixodes scapularis tick saliva drives host CD4 T cells to express IL-4. Parasite Immunol. 31, 210–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Catalán A., Cortes W., Sagua H., González J., Araya J. E. (2011) Two new phospholipase D isoforms of Loxosceles laeta: cloning, heterologous expression, functional characterization, and potential biotechnological application. J. Biochem. Mol. Toxicol. 25, 393–403 [DOI] [PubMed] [Google Scholar]
- 76. Magalhães G. S., Caporrino M. C., Della-Casa M. S., Kimura L. F., Prezotto-Neto J. P., Fukuda D. A., Portes-Junior J. A., Neves-Ferreira A. G., Santoro M. L., Barbaro K. C. (2013) Cloning, expression and characterization of a phospholipase D from Loxosceles gaucho venom gland. Biochimie 95, 1773–1783 [DOI] [PubMed] [Google Scholar]
- 77. de Andrade S. A., Murakami M. T., Cavalcante D. P., Arni R. K., Tambourgi D. V. (2006) Kinetic and mechanistic characterization of the sphingomyelinases D from Loxosceles intermedia spider venom. Toxicon 47, 380–386 [DOI] [PubMed] [Google Scholar]
- 78. Tambourgi D. V., de F Fernandes Pedrosa M., van den Berg C. W., Gonçalves-de-Andrade R. M., Ferracini M., Paixão-Cavalcante D., Morgan B. P., Rushmere N. K. (2004) Molecular cloning, expression, function and immunoreactivities of members of a gene family of sphingomyelinases from Loxosceles venom glands. Mol. Immunol. 41, 831–840 [DOI] [PubMed] [Google Scholar]
- 79. Tambourgi D. V., Magnoli F. C., van den Berg C. W., Morgan B. P., de Araujo P. S., Alves E. W., Da Silva W. D. (1998) Sphingomyelinases in the venom of the spider Loxosceles intermedia are responsible for both dermonecrosis and complement-dependent hemolysis. Biochem. Biophys. Res. Commun. 251, 366–373 [DOI] [PubMed] [Google Scholar]