

Background: ClpP1P2 is a novel protease complex essential for viability of Mycobacterium tuberculosis.
Results: Cleavage preferences of ClpP1P2 were defined, which allowed us to design potent substrate-based boronate inhibitors showing anti-mycobacterial activity.
Conclusion: Excellent new fluorogenic peptide substrates of ClpP1P2 were obtained, and novel enzyme properties were identified.
Significance: Selective inhibition of ClpP1P2 activity is a promising approach for drug development.
Keywords: Drug Development, Enzyme Inhibitor, Enzyme Mechanism, Infectious Disease, Peptide Chemical Synthesis, Protein Degradation
Abstract
The ClpP1P2 protease complex is essential for viability in Mycobacteria tuberculosis and is an attractive drug target. Using a fluorogenic tripeptide library (Ac-X3X2X1-aminomethylcoumarin) and by determining specificity constants (kcat/Km), we show that ClpP1P2 prefers Met ≫ Leu > Phe > Ala in the X1 position, basic residues or Trp in the X2 position, and Pro ≫ Ala > Trp in the X3 position. We identified peptide substrates that are hydrolyzed up to 1000 times faster than the standard ClpP substrate. These positional preferences were consistent with cleavage sites in the protein GFPssrA by ClpXP1P2. Studies of ClpP1P2 with inactive ClpP1 or ClpP2 indicated that ClpP1 was responsible for nearly all the peptidase activity, whereas both ClpP1 and ClpP2 contributed to protein degradation. Substrate-based peptide boronates were synthesized that inhibit ClpP1P2 peptidase activity in the submicromolar range. Some of them inhibited the growth of Mtb cells in the low micromolar range indicating that cleavage specificity of Mtb ClpP1P2 can be used to design novel anti-bacterial agents.
Introduction
Tuberculosis is a devastating disease that causes nearly 2 million deaths annually. About a third of all humans are infected with latent Mycobacterium tuberculosis (Mtb),2 which is becoming increasingly resistant to available antibiotics. Present treatments are not optimal; they require high doses of multiple agents for long periods and have numerous side effects. Consequently, it is important to identify and characterize new therapeutic targets and agents that selectively hit these targets. Recently, we characterized an enzyme that has the potential to be such a target: the protease complex ClpP1P2 (1, 2). ClpP1 and ClpP2 are both essential for viability and infectivity (3). Moreover, no enzyme homologous to ClpP1P2 is present in the cytosol of mammalian cells, where protein breakdown occurs by very different enzymes composing the ubiquitin proteasome pathway or autophagy-lysosome process. Also ClpP1P2 is quite different structurally from the ClpP complex in the mitochondrial matrix (4, 5).
The ClpP enzymes are a highly conserved family of multimeric serine proteases originally discovered and extensively characterized in Escherichia coli (6, 7). ClpP homologs exist in a wide range of bacteria as well as in chloroplasts and mitochondria in eukaryotes (4). The active enzyme is a tetradecamer composed of two heptameric rings that form a hollow cylinder with 14 proteolytic sites compartmentalized within its central chamber (8, 9). Most microorganisms possess a single clpP gene, whereas some, like Mtb, have two or more clpPs (4, 10, 11). By itself, E. coli ClpP is able to rapidly hydrolyze oligopeptides but not large globular protein without first forming a complex with an AAA ATPase, such as ClpA or ClpX in E. coli or ClpC in other species (12, 13). These hexameric ATPases associate with both ends of ClpP (12–14) and activate it but also selectively bind protein substrates, unfold them, and translocate them into the ClpP proteolytic chamber for degradation (14–18). Mtb contains two Clp ATPases, ClpC1 and ClpX, both of which are essential for viability (3). In fact, in collaborative studies, we recently showed that the cyclic peptide antibiotic lassomycin selectively kills mycobacteria by preventing ClpC1-dependent protein breakdown by ClpP1P2 protease (19).
We recently demonstrated that recombinant ClpP1 or ClpP2 by themselves form tetradecamers that lack proteolytic activity, but when mixed together, especially in the presence of low molecular weight peptide activators (N-terminal-blocked dipeptides or derivatives), they form a mixed tetradecameric complex that is very active in degrading peptides (1). The exact role of each ring in proteolysis is unclear and has been investigated here. The dramatic activation (up to >1000-fold) of the enzyme by the activator occurs in a unique fashion; they bind to the ClpP1 and CpP2 inactive homo-tetradecamers promoting their dissociation into heptameric rings and re-associate to form the mixed functional ClpP1P2 complex containing one ring of ClpP1 and one ring of ClpP2. This activation by synthetic dipeptides is reversible and presumably mimics the action of some novel activating factor (e.g. chemical chaperone) that functions in vivo.
In designing or identifying selective inhibitors for an enzyme, it is important to have some knowledge of its specificity. It has been known for 25 years that the activity of ClpP from E. coli could be readily assayed with the fluorescent peptide, Suc-Leu-Tyr-amc (6, 7), which has also been widely used to assay ClpPs from other bacteria and mitochondria. However, this compound is a rather poor substrate for Mtb ClpP1P2. Here, by using an N-acetyl tripeptide-aminomethylcoumarin library (Ac-X3X2X1-amc), we elucidated the cleavage preferences of Mtb ClpP1P2 and identified a number of novel substrates that are up to 1000-fold better than the standard substrate.
On this basis we investigated the specific roles of ClpP1 and ClpP2 in degrading peptides and proteins. In addition, we synthesized a series of substrate-based peptide boronate derivatives that inhibit ClpP1P2 peptidase activity and protein degradation in the presence of ClpC1 and ClpX ATPases. Some of these inhibitors prevented growth of Mtb selectively. These observations provide a platform for further development of potent inhibitors of Mtb ClpP1P2 with anti-tuberculosis activity.
EXPERIMENTAL PROCEDURES
Materials
Human 20 S proteasomes purified from red blood cells was purchased from Boston Biochem, Cambridge, MA. Individual substrates for kinetic analysis were custom-synthesized by AnaSpec and Biomol (Plymouth Meeting, PA).
Bacterial Strains, Plasmids, Expression, and Growth of Cells
H37Rv strain was used in all experiments involving Mtb. All Mtb proteins were expressed in an E. coli BL21 strain lacking endogenous ClpP and ClpX. For expression of mature forms of wild type and active site mutants of ClpP1 (lacking 6 N-terminal amino acids) and ClpP2 (lacking 11 N-terminal amino acids), pTetOR plasmid, which has an inducible tetracycline promoter, was used. Induction with anhydrotetracycline (100 ng/ml) was carried out overnight. ClpC1 was expressed using pET28a plasmid and induced for 3 h at 37 °C by 1 mm isopropyl 1-thio-β-d-galactopyranoside. A truncated form of ClpX (residues 60–426) was used throughout this study. It was expressed from pTrc99 in a 3-h induction at 16 °C with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside. All proteins had C-terminal His6 tags except ClpX, which contained an N-terminal His6 tag.
Characterization of Ac-X3X2X1-amc Library
The ChemRX Protease Profiler library of N-acetylated tripeptide-amc peptides was from Discovery Partners International (20). In addition to the 20 standard amino acids, l-citruline and l-ornithine were present in the X1 position. In the X2 and X3 positions, cysteine was replaced by l-ornithine in the 20-standard amino acid set. All peptides contained an N-terminal acetyl group. Of the possible 8800 tripeptides (22 X1 × 20 X2 × 20 X3) 5920 were useable based on yield and purity. The average purity was 85–90% for any individual substrate.
Purification of Mtb Enzymes
Purification of recombinant Mtb ClpP1, ClpP2, and their inactive mutants was carried out as described previously (1). Purification of Mtb ClpX and ClpC1 was carried out at 4 °C using buffer B (50 mm Tris-HCl, pH 7.6, 50 mm KCl, 0.1 mm DTT, 1 mm Mg-ATP, and 10% glycerol). Cells were resuspended in two volumes of buffer and lyzed with a French press at 1500 p.s.i. The extract was centrifuged at 100,000 × g and mixed with nickel-nitrilotriacetic acid-agarose. After a 4-h incubation, nickel-nitrilotriacetic acid-agarose resin was transferred to a column, and proteins were eluted using step gradient (25, 50, 100, and 200 mm) of imidazole in buffer B. The active fractions containing nearly homogeneous proteins were combined and concentrated to 1–3 mg/ml by Millipore MWCO 10-kDa cut filter and fractioned further by gel filtration on a column (2.5 × 22 cm) of Sephacryl S-300 equilibrated in the same buffer. The protein peak was collected, concentrated to ∼3 mg/ml, and stored at −80 °C. All enzymes purified migrated as a single band in the SDS-PAGE.
Peptidase Assay
All assays of peptidase activity were performed at 37 °C in black 384-well plates using a Plate Reader SpectraMax M5 (Molecular Devices). 2–3 min were allowed for the reaction mixtures to reach 37 °C. Each well contained 10 μm fluorogenic peptide, 20–50 nm ClpP1P2 in 80 μl of buffer A (20 mm phosphate buffer, pH 7.6, with 100 mm KCl, 5% glycerol, and 5 mm Z-Leu-Leu). DMSO concentration never exceeded 2%. The reaction was initiated by the addition of the enzyme, and peptidase activity was followed in the linear range by monitoring the rate of production of fluorescent 7-amino-4-methylcoumarin-amc from peptide-amc substrates at 460 nm (excitation at 380 nm). The deviation of fluorescence value in three independent measurements was not >5%.
Determination of Kinetic Constants for Peptide Substrates and Boronate Inhibitors
Specificity constant (kcat/Km) for peptide substrates was calculated from the linear plot of enzyme activity at 10 μm peptides concentrations, which is several hundred time less than the Km of peptide substrates. Ki for peptide boronates were calculated using the formula Ki = IC50/1 + [S]/Km (21). Under these conditions, Ki values equaled the IC50. Inhibitors were assayed at eight different concentrations, and the IC50 (Ki) was calculated from four points for nonlinear curves using SpectroMax 5 program.
Proteinase Assay
Mtb ClpP1P2 was also assayed continuously in 96-well plates using the fluorescent protein substrates, GFPssrA, and FITC-casein. To measure GFPssrA degradation by the ClpXP1P2 complex, each well contained 2 mm Mg-ATP, 500 nm GFPssrA, 75–100 nm ClpP1P2 tetradecamer, and 300–400 nm ClpX hexamer in 100 μl of buffer A, and GFPssrA fluorescence was measured at 510 nm (excitation at 395 nm). To measure FITC-casein degradation by ClpC1P1P2, each well contained 2 mm Mg-ATP, 150–200 nm ClpP1P2, 500–700 nm ClpC1 hexamer, and 1–1.2 μm FITC-casein in 100 μl of buffer A. FITC-casein fluorescence was monitored at 518 nm (excitation at 492 nm), and the deviation of fluorescence value in three independent measurements was not >10%.
Products of GFPssrA Digestion by ClpXP1P2
To determine the cleavage preferences of ClpP1P2 in proteins, 1 μm GFPssrA was incubated with 75–100 nm ClpP1P2, 400 nm ClpX hexamer, and 2 mm Mg-ATP in buffer A. After 50% degradation of protein, the products were extracted and analyzed by liquid chromatography and Tandem MS/MS according to Akopian et al. (1).
Determination of Peptidase and Proteinase Activity of ClpP1P2 Complexes in the Native Gel
WT or mutant forms of ClpP1 and ClpP2 (3 μg of each) were mixed together in buffer A in the presence of the peptide activator Z-LL to form WT ClpP1P2 or mutant ClpP1P2(S-A) or ClpP1(S-A)P2 complexes, which were then isolated using the native PAGE. The formation of mixed ClpP1P2 complexes was confirmed by mass spectroscopy. To determine the peptidase activity, the gel was incubated for 25 min in buffer A containing 10 μm Ac-PKM-amc and 5 mm Z-LL, and the appearance of amc fluorescence in the bands was detected in UV light using AlphaImager. To determine the proteinase activity, the gel was first incubated for 2 h with 0.1 mg/ml GFP-ssrA in buffer A to allow the GFP diffusion into the gel. Then the gel was incubated for 2 h with 0.02 mg/ml ClpX in buffer A with 5 mm Mg-ATP. The decrease of GFP fluorescence in proteolytically active ClpP1P2 bands was detected using AlphaImager.
ATPase Assay
ATP hydrolysis was measured with the enzyme-linked assay using pyruvate kinase and lactic dehydrogenase (PK/LDH). 2 μg of pure ClpC1 or ClpX were mixed with 100 μl of the assay buffer B containing 1 mm phosphoenolpyruvate, 1 mm NADH, 2 units of pyruvate kinase/lactic dehydrogenase, 4 mm MgCl2, and 1 mm ATP, and the ATPase activity was followed by measuring the oxidization of NADH to NAD spectrometrically at 340 nm. Measurements were performed in triplicate, which agreed within 5%.
Determination of MIC50 for Peptide Boronate Inhibitors against Mtb and Mycobacterium smegmatis
To determine the concentration of peptide boronates that inhibit cell growth, the colorimetric resazurin microtiter assay was used. 7H9 media (Difco) was dispensed in each well of a sterile flat-bottom 96-well plate. Peptide boronates were serially diluted 2-fold across the plate. Bacteria were diluted to A600 = 0.003, and equal volumes of culture were added to the serially diluted compounds. Plates were grown at 37 °C for 5 days for Mtb or overnight for M. smegmatis. At different times, 20 μl of resazurin (0.05% w:v in water) were added to each well, and plates were incubated at 37 °C for 1 day (for Mtb) or 6 h for M. smegmatis. Growth and metabolism of bacteria is reflected by the reduction of resazurin, a blue non-fluorescent dye, to resorufin, a pink highly fluorescent dye. MICs for each boronate were defined as the highest concentration that resulted in no color change in resazurin.
Synthesis of the Peptide Boronate Inhibitors
Except for N-(picolinoyl)-Lys-boroMet and N-(benzo[b]thiophene-7-carbonyl)-Lys-boroMet (see below), all compounds were well characterized and reported (22). The target compounds were purified by reverse-phase (RP)-HPLC using a Varian semi-preparative system with a Discovery C18 569226-U RP-HPLC column. The mobile phase was typically made by mixing water (0.1% TFA) with acetonitrile (0.08% TFA) in gradient concentration. Purities determined by HPLC analysis were >95%.
N-(Picolinoyl)-Lys-boroMet
1H NMR (D2O) δ 8.73 (d, J = 4.3 Hz, 1H), 8.27–8.19 (m, 2H), 7.90–7.80 (m, 1H), 2.99 (t, J = 7.5 Hz, 2H), 2.80–2.70 (m, 1H), 2.60–2.50 (m, 2H), 2.06–1.90 (m, 5H), 1.80–1.65 (m, 4H), 1.55–1.45 (m, 2H). MS (ESI+) m/z (relative intensity): 365.3 ([M − H2O + H]+, 100).
N-(Benzo[b]thiophene-7-carbonyl)-Lys-boroMet
1H NMR (D2O) δ 8.17 (d, J = 7.6 Hz, 1H), 7.94–7.85 (m, 1H), 7.75 (d, J = 5.6 Hz, 1H), 7.60–7.52 (m, 2H), 3.03 (t, J = 7.4 Hz, 2H), 2.85–2.75 (m, 1H), 2.54 (t, J = 7.4 Hz, 2H), 2.02–1.95 (m, 5H), 1.79–1.70 (m, 4H), 1.60–1.50 (m, 2H), 1.35 (d, J = 6.6 Hz, 1H). MS (ESI+) m/z (relative intensity): 420.1 ([M − H2O + H]+, 100).
RESULTS
Residues Preferred by ClpP1P2 in the X1 Position in the Tripeptide Library
During our initial characterization of ClpP1P2 protease using a small set of substrates (1), we observed that the complex had a preference for hydrophobic residues in the X1 position. To define more precisely the positional preferences of ClpP1P2, we used an N-acetylated tripeptide-amc fluorogenic substrate library consisting of individual peptides covering the Ac-X3X2X1-amc sequence space (20). First, the best substrate was identified for each of the 20 sublibraries with individual amino acid residues in the X1 position (Fig. 1A). Substrates with the hydrophobic residues, Leu, Phe, Ala, and especially Met in the X1 position were cleaved most efficiently by ClpP1P2 (Fig. 1A). Moreover, all of the 30 most rapidly hydrolyzed substrates (∼0.5% of the library) and 49 of the 60 top substrates contained a Met in the X1 position (data not shown). It is noteworthy that similar results were obtained when the average enzymatic activity for all substrates with specific residues in the X1 position was calculated (Fig. 1B, Table 1). By contrast, little or no enzymatic activity was detected with other amino acids in the X1 position, although some peptides with Lys, Arg, Glu, or Asp in this position could be cleaved much more slowly than ones with Met or other hydrophobic residues in X1 position (Table 2).
FIGURE 1.
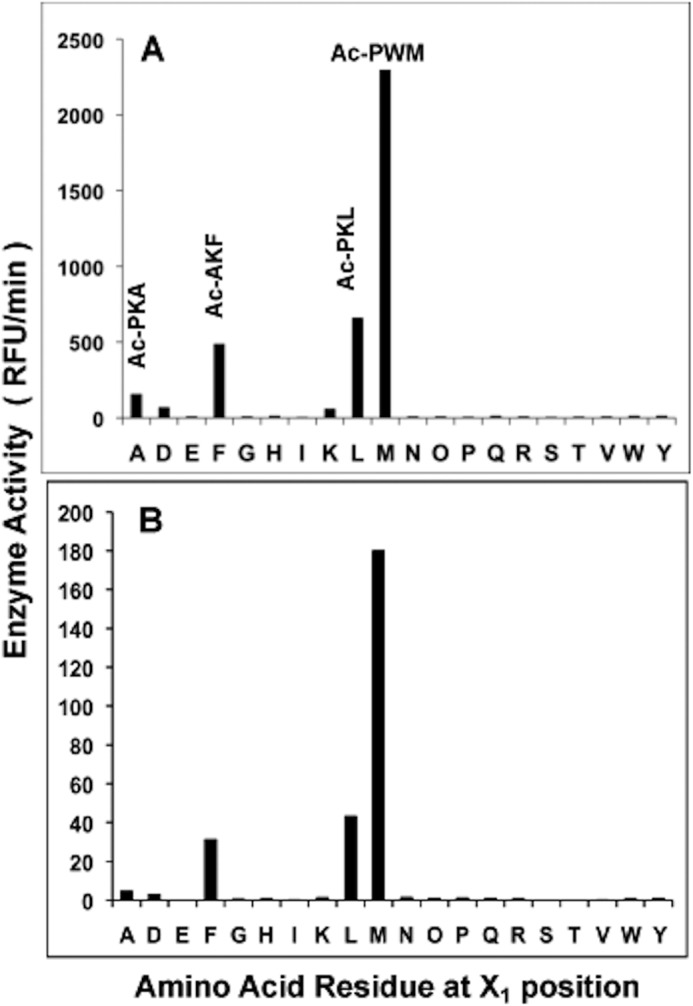
Mtb ClpP1P2 cleaves peptide-amc substrates mainly after Leu, Phe, Ala, and especially Met. A, to determine cleavage preferences for the X1 position, ClpP1P2 activity was measured continuously using Ac-X3X2X1-amc fluorogenic peptides library (at 10 μm) in buffer A containing 30 nm ClpP1P2. The ClpP1P2 activity against the best substrates with a given amino acid in X1 position is reflected in the graph. B, average rate of hydrolysis of substrates with a given amino acid in the X1 position. The activity was measured as in Fig. A. RFU, relative fluorescence units.
TABLE 1.
Cleavage preferences of Mtb ClpP1P2 for X2 and X3 positions with Met, Leu, Phe, or Ala at X1 position
RFU, relative fluorescence units; Or = ornithine.
| Ac-X3X2X1-amc | X3 | X2 | X1 | Peptidase activity (RFU) |
|---|---|---|---|---|
| PWM | Pro | Trp | Met | 2299 |
| HKM | His | Lys | Met | 1957 |
| PFM | Pro | Phe | Met | 1754 |
| PRM | Pro | Arg | Met | 1712 |
| PKM | Pro | Lys | Met | 1497 |
| POrM | Pro | OrR | Met | 1459 |
| ARM | Ala | Arg | Met | 1294 |
| AKM | Ala | Lys | Met | 1267 |
| AWM | Ala | Lys | Met | 1230 |
| GKM | Gly | Lys | Met | 1277 |
| PKL | Pro | Lys | Leu | 661 |
| WKL | Trp | Lys | Leu | 591 |
| WYL | Trp | Tyr | Leu | 478 |
| PLL | Pro | Leu | Leu | 385 |
| POrL | Pro | Or | Leu | 374 |
| PWL | Pro | Trp | Leu | 347 |
| WRL | Trp | Arg | Leu | 347 |
| PRL | Pro | Arg | Leu | 306 |
| YKL | Tyr | Lys | Leu | 276 |
| PSL | Pro | Ser | Leu | 272 |
| AKF | Ala | Lys | Phe | 489 |
| PKF | Pro | Lys | Phe | 454 |
| GWF | Gly | Trp | Phe | 440 |
| PRF | Pro | Arg | Phe | 423 |
| AWF | Ala | Trp | Phe | 320 |
| AYF | Ala | Tyr | Phe | 269 |
| PYF | Pro | Tyr | Phe | 265 |
| WKF | Trp | Lys | Phe | 238 |
| ARF | Ala | Arg | Phe | 235 |
| WYF | Trp | Tyr | Phe | 206 |
| PKA | Pro | Lys | Ala | 158 |
| PRA | Pro | Arg | Ala | 101 |
| VKA | Val | Lys | Ala | 87 |
| ARA | Ala | Arg | Ala | 52 |
| POrA | Pro | Or | Ala | 45 |
| AKA | Ala | Lys | Ala | 43 |
| FKA | Phe | Lys | Ala | 24 |
| AOrA | Ala | Or | Ala | 21 |
| PWA | Pro | Trp | Ala | 18 |
| IRA | Ile | Arg | Ala | 11 |
TABLE 2.
Specificity constant kcat/Km for Mtb ClpP1P2 and E. coli, B. subtilis, and S. aureus ClpPs against tripeptide-amc substrates
Nle, norleucine.
| Substrate X3X2X1-amc |
kcat/Km |
|||
|---|---|---|---|---|
| M. tuberculosis | S. aureus | B. subtilis | E. coli | |
| m−1 s−1 | ||||
| Chymotryptic | ||||
| Ac-PKM | 1327 | 331 | 176 | 2632 |
| Ac-PKNle | 411 | 69 | 239 | 2464 |
| Ac-KM | 144 | 56 | 36 | 630 |
| Ac-PWM | 1155 | 462 | 520 | 4745 |
| Ac-WM | 612 | 184 | 268 | 4095 |
| Ac-ARM | 410 | 236 | 526 | 1553 |
| Ac-HKM | 169 | 128 | 6 | 695 |
| Ac-PKL | 199 | 270 | 234 | 2021 |
| Ac-PQL | 63 | 183 | 247 | 2886 |
| Ac-PYL | 32 | 265 | 598 | 3302 |
| Ac-PAL | 42 | 127 | 282 | 1804 |
| Ac-PKF | 320 | 185 | 132 | 634 |
| Ac-PAF | 68 | 62 | 97 | 605 |
| Ac-PKA | 40 | 35 | 76 | 812 |
| Z-LLL | 0 | 14 | 0 | 46 |
| Z-LL | 0 | 8 | 2 | 58 |
| Suc-LY | 1 | 7 | 50 | 721 |
| Suc-LLVY | 2 | 2 | 1 | 26 |
| Tryptic | ||||
| Ac-WEK | 2 | 0 | 0 | 3 |
| Ac-FAK | 1 | 0 | 0 | 0 |
| YMK | 2 | 0 | 2 | 1 |
| Z-LLR | 5 | 0 | 1 | 1 |
| Z-FVR | 4 | 1 | 0 | 1 |
| Acidic | ||||
| Z-nLPnLD | 14 | 3 | 10 | 31 |
| Z-APnLD | 0 | 0 | 0 | 0 |
| Ac-PnLD | 0 | 4 | 0 | 0 |
| Ac-LWD | 2 | 1 | 0 | 14 |
| Ac-WID | 0 | 2 | 0 | 0 |
Residues Preferred by ClpP1P2 in the X2 and X3 Positions in the Tripeptide Library
The preferences at the X2 and X3 positions were calculated when the X1 residue was fixed with one of the residues identified above (Met, Leu, Phe, or Ala). There was a clear preference for basic residues, particularly Lys and aromatic residues, particularly Trp, in the X2 position, and Pro ≫ Ala > Trp residues in the X3 position (Fig. 2). Table 1 presents the 10 best tripeptide substrates with Met, Leu, Phe, or Ala in the X1 position, and most contained a basic residue in the X2 and Pro in X3 position. Of the 40 best substrates, 70% contain a positively charged amino acid in X2 position, half of which were Lys. For the X3 position, 58% contained Pro, and 20% contained Ala (Table 1).
FIGURE 2.

Mtb ClpP1P2 cleavage preferences for the X2 and X3 positions with fixed Met, Leu, Phe or Ala at the X1 position. ClpP1P2 activity was measured using Ac-X3X2X1-amc fluorogenic peptides library as in Fig. 1A. The size of the dots represent the ClpP1P2 activity against the given peptide and is calculated as percent of the maximal value for each panel. Maximal activity (relative fluorescence units) for the libraries with fixed Met in X1 position was 2300, with Leu 660, with Phe 490, or with Ala 160.
The Tripeptides Most Efficiently Cleaved by ClpP1P2
To confirm the data obtained using the peptide library and to determine the catalytic efficiency (kcat) for ClpP1P2 enzyme, a number of peptide substrates identified above were synthesized and retested. Due to limited solubility of many peptide-amc substrates, it was not possible to measure the Vmax, which requires high concentrations of the substrates (e.g. for Ac-Pro-Lys-Met-amc Km > 3 mm). To directly compare the enzyme against different substrates, we decided to determine the second order rate specificity constant kcat/Km (see “Experimental Procedures”). Two peptides, Ac-Pro-Lys-Met-amc and Ac-Pro-Trp-Met-amc, were determined to be the best substrates for ClpP1P2 (Table 2). Exchange of Met in the X1 position for its close analog Nle (norleucine) decreased substrate cleavage 3-fold. The C-terminal dipeptides derived from the best two substrates, Ac-Pro-Lys-Met-amc and Ac-Pro-Trp-Met-amc, were also good substrates for ClpP1P2, especially Ac-Trp-Met-amc. It is noteworthy that all these four substrates are 100–1000× more efficiently cleaved than the standard fluorogenic substrate, Suc-Leu-Tyr-amc (7, 23). Based on its kcat/Km value, Ac-Pro-Lys-Met-amc is the best substrate among the peptides with Met at the X1 position and was clearly better than Ac-Pro-Trp-Met-amc in contrast to our observations with the peptide library.
To be enzymatically active ClpP1P2 required the presence of the dipeptide activator Z-Leu-Leu, which induces dissociation of the inactive ClpP1 and ClpP2 complexes and reassociation into the active ClpP1P2 complex (1). Our prior observation that the amc derivatives of the peptide activators are not good substrates (1) suggested that the activators bind either to sites distinct from the active site or in a non-productive mode to the active site. Accordingly, we found here that kcat/Km values of the amc derivatives of peptide activators Z-Leu-Leu-amc and Z-Leu-Leu-Leu-amc are very low (Table 2), confirming our prior observations and conclusions.
These Tripeptide Substrates Are Rapidly Cleaved by Other ClpP Enzymes but Not by the Proteasome
We also tested whether the cleavage specificity observed here for ClpP1P2 is unique or is shared by other members of ClpP family. To compare the specificity constant of ClpP1P2 with those of other members of the ClpP family, it is important to know how efficiently ClpP1 and ClpP2 associate into the active ClpP1P2 complex. Analysis by native gel electrophoresis indicates that in the presence of the activator nearly all of ClpP1 and ClpP2 are engaged in the mixed complex with a distinct mobility (Fig. 3) (the presence of both proteins in the band was confirmed by mass spectroscopy).
FIGURE 3.
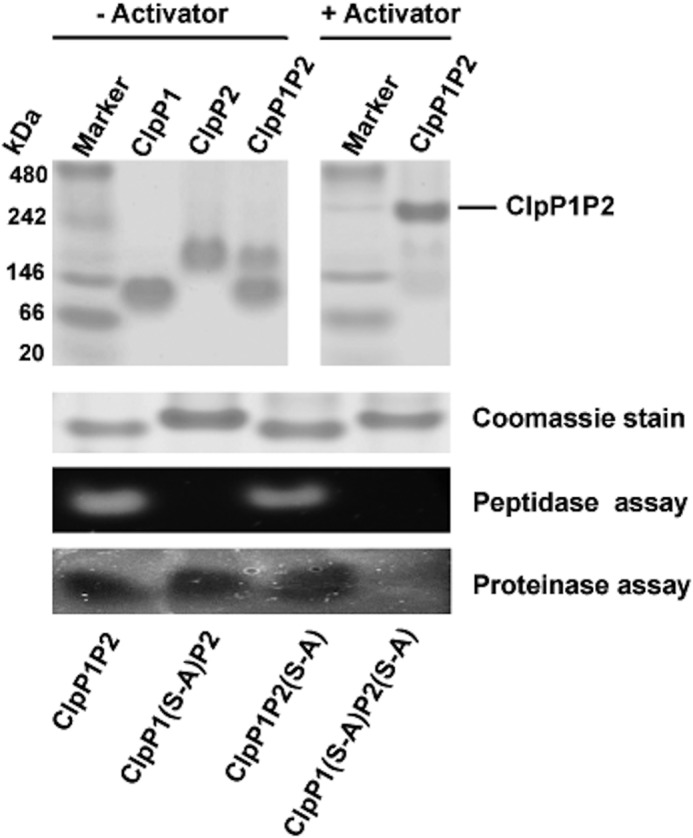
Isolation of WT or mutant forms of ClpP1P2 complexes in native gel and determining their enzymatic activity. ClpP1 and ClpP2 (3 μg of each) were mixed together in buffer A to form WT ClpP1P2 or mutant ClpP1P2(S-A) or ClpP1(S-A)P2 complexes and isolated using the native PAGE in the presence or absence of the peptide activator Z-LL. The peptidase (with Ac-PKM-amc) and proteinase (with GFPssrA) activities of the enzyme were then determined directly in the gel as described under “Experimental Procedures.”
We found that the best substrates for ClpP1P2 are also very good substrates for Bacillus subtilis, Staphylococcus aureus, and E. coli ClpPs (Table 2) and that they are much better than the standard substrate, Suc-Leu-Tyr-amc. However, some individual differences were evident. For instance, the best substrates for Mtb ClpP1P2 were Ac-Pro-Lys-Met-amc and Ac-Pro-Trp-Met-amc, whereas S. aureus and E. coli ClpPs preferred Ac-Pro-Trp-Met-amc, and B. subtilis preferred Ac-Pro-Tyr-Leu-amc. The standard ClpP substrate Suc-Leu-Tyr-amc is cleaved poorly by these enzymes except E. coli ClpP (Table 2).
Unlike most other bacteria, Mtb in addition to the ClpP1P2 expresses a 20 S proteasome that appears to be important for survival in the macrophage (24). Mtb proteasomes do not cleave tripeptides with Met in the X1 position (25), the preferred substrates of ClpP1P2. We further tested whether the tripeptide substrates preferred by the Mtb proteasome were hydrolyzed by ClpP1P2. The seven best proteasome substrates (e.g. Ac-Arg-Ala-Trp-amc or Ac-Tyr-Gly-Trp-amc (25)) were poorly cleaved by ClpP1P2 (with kcat/Km from 0 to 14) (data not shown).
If a ClpP1P2 inhibitor were to be used in human patients, it is critical that it should not inhibit intracellular human proteases, the most important of which are proteasomes. We, therefore, tested if these ClpP substrates are degraded by human proteasomes and whether commonly used proteasome substrates are cleaved by ClpP1P2. The results show that the cleavage specificity of ClpP1P2 is quite different from that of the human proteasome (Fig. 4). For example, the standard substrate used for the mammalian proteasome chymotrypsin-like site, Suc-Leu-Leu-Tyr-amc, is a very poor substrate for Mtb ClpP1P2. Therefore, it should be possible to selectively inhibit ClpP1P2 without interfering with the proteasome function.
FIGURE 4.

Cleavage preferences of Mtb ClpP1P2 are different from those of human proteasomes. Specificity constants kcat/Km were determined using 70 nm of ClpP1P2 or 8.5 nm concentrations of human proteasomes at peptide-amc substrate concentration of 10 μm.
Cleavage Preferences in Proteins Resemble Those in the Peptide Substrates
We next tested whether ClpP1P2 shows similar cleavage preferences for the X1 position in protein substrates as were observed with the tripeptide amc library. Rapid protein degradation requires protease association with a hexameric AAA ATPase complex, either ClpC1 or ClpX, which bind specific proteins and unfolds and translocates them into ClpP1P2 proteolytic chamber for processive degradation. We expressed His6-tagged Mtb ClpX and ClpC1 ATPases in E. coli, purified them using affinity and size exclusion chromatography (to near-homogeneity by SDS-PAGE), and identified a fluorescent model protein substrate for each. Together with ClpX, the Mtb ClpP1P2 degrades GFPssrA-like ClpXP from E. coli (18) in a process requiring ATP. With ClpC1, ClpP1P2 degrades FITC-casein in an ATP-dependent manner (although some very slow breakdown of FITC-casein by ClpP1P2 alone was also observed) (1).
To determine the cleavage site preference of ClpP1P2 in protein substrates, GFPssrA was digested by ClpXP1P2, and the resulting peptides were analyzed by liquid chromatography-tandem mass spectrometry (LS-MS/MS). Ninety-five unique peptides were identified covering >90% of the sequence of GFP. The sizes of the recovered peptides ranged from 4 to 26 residues in length, but most were 8–13 amino acids in length (Fig. 5A). Analysis of the C-terminal residues of these peptides indicate that ClpP1P2 cleaved GFPssrA most readily after Leu, Phe, and especially Met (Fig. 5B). Thus, in hydrolyzing proteins, ClpP1P2 showed similar preferences for the X1 position as in digesting the peptide library. The main difference was that in digestion of GFPssrA, cleavages often occurred after Asn residues, which was not observed with the tripeptide library. Analysis of N-terminal amino acid residues of peptides generated during GFPssrA degradation allowed us also to determine if ClpP1P2 showed any preference for the nature of the residue(s) after the cleavage site (X1′). As shown in Fig. 5C, ClpP1P2 seems to prefer positive amino acids (Arg, Lys, and His) and Ser in the X1′ position.
FIGURE 5.
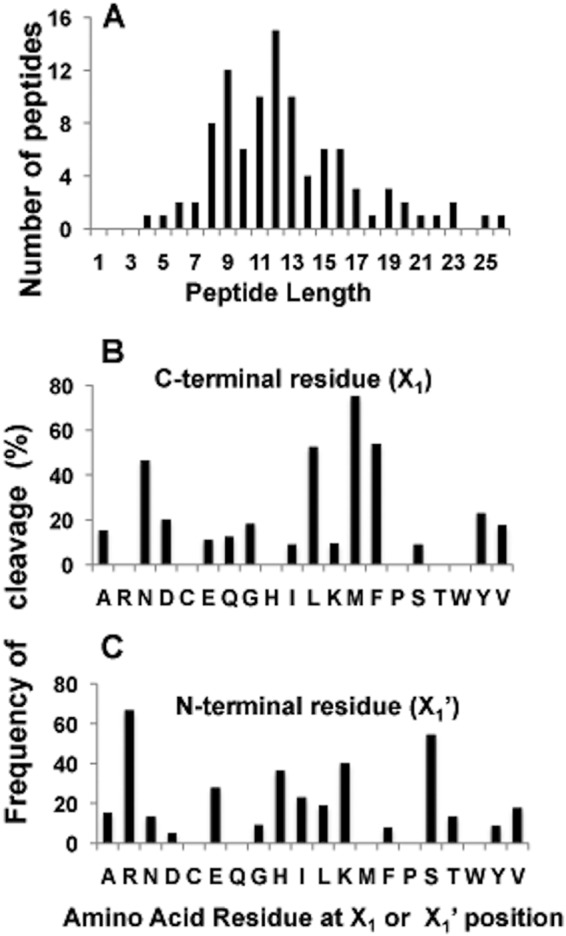
Size distribution of peptide products and cleavage preferences for ClpP1P2. A, size distribution of individual peptides generated after cleavage of GFPssrA. Protein substrate was digested by ClpP1P2 in buffer A containing 1 μm GFPssrA, 75–100 nm ClpP1P2, 300–400 nm ClpX, and 2 mm Mg-ATP. The produced peptides were separated by centrifugal filter (10 kDa) and analyzed using Liquid LS-MS/MS. B and C, cleavage preferences for X1 and X1′ positions during GFPssrA degradation. The number of cleavages after and before a given amino acid is presented as a percent of the total number of that amino acid in the protein sequence. Peptides were generated and analyzed as in A.
ClpP1 Accounts for Most of the Peptidase Activity Even though ClpP1 and ClpP2 Subunits Both Can Support Protein Breakdown
To understand the roles of ClpP1 and ClpP2 in the breakdown of proteins and peptides, we constructed ClpP1P2 complexes where only one of the subunits was enzymatically active. As we reported previously (1), both subunits must be present for enzymatic activity. Wild type and mutant ClpP1 and ClpP2 subunits were mixed together with the activator to form mixed complexes in which either of the subunits (or both) was inactive due to mutation of the active site serine to an alanine (S-A). When their abilities to degrade various tripeptide substrates were compared, surprisingly, the ClpP1P2(S-A) complex showed equal or even better activity than the WT enzyme (Fig. 6A). By contrast, ClpP1(S-A)P2 showed almost no activity against the best peptide substrates (even when tested at 1 mm concentration) and only limited activity against the caspase substrate, Z-Nle-Pro-Nle-Asp-amc (Nle is norleucine; Fig. 6A). As expected, the double ClpP1(S-A)P2(S-A) mutant was inactive against all the substrates tested. Thus, ClpP1 is responsible for nearly all the activity against the tripeptide-amc substrates, as was suggested previously using other peptide substrates (1).
FIGURE 6.
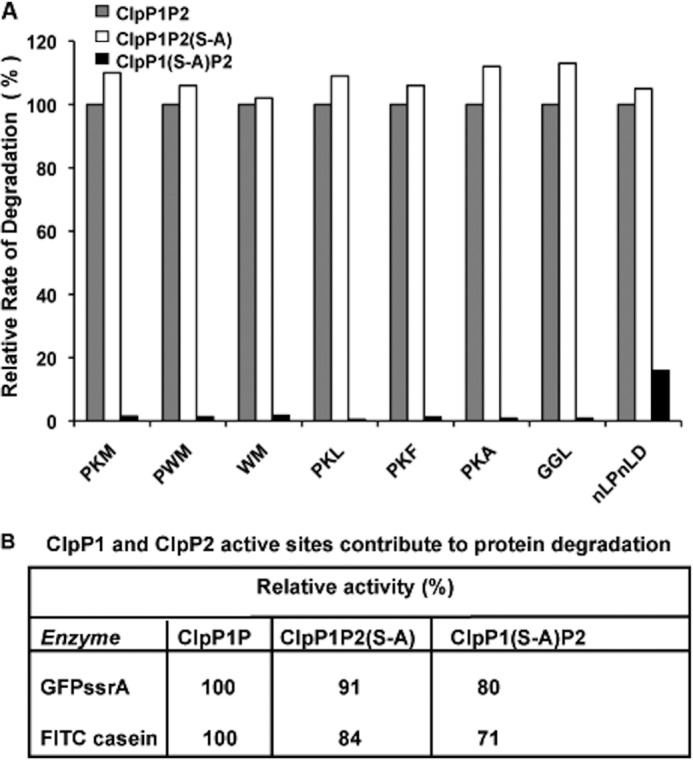
ClpP1 accounts for nearly all of the peptidase activity, although both ClpP1 and ClpP2 subunits contribute to protein breakdown. A, degradation of peptide-amc substrates by WT ClpP1P2 and mutant ClpP1P2(S-A) and ClpP1(S-A)P2 mutant complexes. To determine the contribution of each type of active site in peptide degradation, the peptidase activity of ClpP1P2 complexes was measured against the best peptide substrates as in Fig. 1A. The activity of the WT ClpP1P2 against each substrate was taken as 100%. B, degradation of protein substrates by WT ClpP1P2, ClpP1P2(S-A), and ClpP1(S-A)P2 mutant complexes. To determine the ClpP1 and ClpP2 contributions to protein degradation, the cleavage of fluorescent protein substrates, GFPssrA with ClpX present and FITC-casein with ClpC1 present, were measured, as described under “Experimental Procedures.” The rates of degradation by the WT ClpP1P2 are taken as 100%.
We then tested whether ClpP1 also played the predominant role in protein degradation. Surprisingly, in contrast to tripeptide degradation, ClpP1(S-A)P2 complexes degraded FITC-casein (with ClpC1) and GFPssrA (with ClpX) only slightly worse than ClpP1P2(S-A) or WT ClpP1P2 (Fig. 6B). Thus, even though ClpP1 and ClpP2 have very different activities against tripeptide substrates (Fig. 6A), the proteolytic activities of ClpP1 or ClpP2 do not determine the rates of degradation of protein substrates, which are determined by the ATPases, and in the presence of ATPases, either subunit has sufficient activity to digest these proteins almost as rapidly as the WT complex.
This conclusion was also confirmed by a different approach in which the wild type and two mutant ClpP1P2 complexes were isolated by native gel electrophoresis, and the peptidase activity against Ac-Pro-Lys-Met-amc and proteinase activity against GFPssrA in the presence of ClpX were determined directly in the gel (Fig. 3). As expected, the peptidase activity was detected only in the complexes containing active ClpP1, whereas the proteinase activity was detected in complexes containing either active ClpP1 or ClpP2.
It is noteworthy that both ATPases also accelerated tripeptide cleavage by ClpP1P2 and thus must be influencing ClpP1P2 structure/conformation and not just protein unfolding and translocation. ClpC1 increased the degradation of Ac-Pro-Lys-Met-amc about 1.5-fold and ClpX by 3.5-fold. With the longer peptide substrate, Mca-Gly-His-Gln-Gln-Tyr-Lys-Met-Lys-Dpa(2,4-dinitrophenol)-amide (where Mca is (7-methoxycoumarin-4yl)acetyl and Dpa is diaminopropionic acid), ClpC1 caused an approximately 2-fold increase, and ClpX caused an 8-fold increase in degradation rate. This activation was observed when ClpP1P2(S-A) was used in place of ClpP1P2 but not for ClpP1(S-A)P2. As was found for protein breakdown (1), the activation of tripeptide degradation by the ATPases was observed only in the presence of the activator peptides. Thus, although the ClpP2 active site cannot cleave tripeptide substrates but can digest proteins for reasons that are not clear, the association with the ATPase is, by itself, not sufficient to activate the ClpP2 active site against peptides. Presumably, the presence of the protein substrate somehow exposes activity of ClpP2.
Synthesis of Tripeptide Boronates Inhibitors of ClpP1P2
Our major goal in defining the specificity of ClpP1P2 was to lay the basis for the designing of new inhibitors that may selectively kill Mtb cells. Of the many peptide derivatives that inhibit serine proteases, a potent class is peptides with a boronic-based electrophile in their C termini (26–28). These highly specific transition state inhibitors can bind to the active sites of both serine and threonine proteases. For example, the peptide boronate proteasome inhibitor, bortezomib (Velcade), is now used as the preferred treatment for multiple myeloma (27). Using our best substrates identified in Table 1, we synthesized a number of corresponding boronate derivatives. Some of these peptide boronates (e.g. Ac-Pro-Lys-boroMet (Table 3, #1), Ac-His-Lys-boroMet (Table 3, #2), and Ac-Ala-Lys-boroMet (Table 3, #3)) inhibited the peptidase activity of ClpP1P2 with a Ki of <1 μm (Table 3). To test how the nature of the N-terminal blocking group might affect inhibitor potency, we synthesized several tripeptide boronates with an N-terminal picolinoyl group in place of the acetyl one. The inhibitory activity of picolinoyl-Ala-Lys-boroMet (Table 3, #6) and picolinoyl-Pro-Lys-boroMet (Table 3, #7) had similar or somewhat greater activities to those of the corresponding acetyl compounds. Interestingly, picolinoyl-Trp-Lys-boroMet (Table 3, #5) was a very potent inhibitor (Ki = 0.18 μm). Because the dipeptide substrates Ac-Lys-Met-amc and Ac-Trp-Met-amc are rapidly cleaved by ClpP1P2 (Table 2), we also synthesized dipeptide boronate Lys-boroMet with several types of N-terminal blocking groups. As shown in Table 3, several dipeptide boronates were the most effective inhibitors of ClpP1P2 activity, especially N-(2-(3,5 difluorophenyl)acetyl)-Lys-boroMet ((Table 3, #11), Ki = 65 nm).
TABLE 3.
Selected potent boronates inhibit ClpP1P2 stronger than human mitochondrial ClpP and prevent the growth of Mtb
| Inhibitor |
Ki |
MIC50a | |
|---|---|---|---|
| Human | Mtb | ||
| μm | μm | ||
| #1, Ac-Pro-Lys-boroMet | 2.4 | 0.46 | 12 |
| #2, Ac-His-Lys-boroMet | 6.8 | 0.78 | 6 |
| #3, Ac-Ala-Lys-boroMet | 2.1 | 0.80 | 3 |
| #4, Ala(1-naphtyl)-Lys-boroLeu | 1.46 | 3.2 | 12 |
| #5, N-(Picolinoyl)-Trp-Lys-boroMet | 0.96 | 0.18 | 3 |
| #6, N-(Picolinoyl)-Ala-Lys-boroMet | 1.7 | 0.24 | 3 |
| #7, N-(Picolinoyl)-Pro-Lys-boroMet | 4.2 | 0.82 | 24 |
| #8, N-(Picolinoyl)-Lys-boroMet | 5.8 | 0.34 | 6 |
| #9, N-(3-Phenyl)propanoyl)-Lys-boroMet | 0.9 | 0.23 | 3 |
| #10, N-(Benzyl)-Lys-boroMet | 2.4 | 0.18 | 6 |
| #11, N-(2-(3,5-Difluorophenyl)acetyl)-Lys-boroMet | 0.75 | 0.065 | 6 |
| #12, N-(2-(3,5-Difluorophenyl)acetyl-Trp-boroMet | 0.34 | 0.29 | 12 |
| #13, N-(1H-benzo(b)thiophene-7-carbonyl-Lys-boroMet | 0.4 | 0.12 | 12 |
| #14, N-(Phenylmetanesulfonyl)-Lys-boroMet | 11.6 | 13 | 200 |
a MIC of all listed inhibitors against E. coli and S. aureus were >200 μm.
Although peptide boronic acid inhibitor may often dissociate extremely slowly from their target enzymes (e.g. bortezomib from proteasomes), inhibition is nevertheless usually fully reversible. Thus, when ClpP1P2 was incubated with the inhibitors for 2 h at room temperature, enzyme activity could be restored to >90% of the original activity by either centrifugation using filters with 10,000 Mr cut off or simply by dilution.
Effects of Peptide Boronate Inhibitors on ClpP1 and ClpP2 Activity
We then tested how the peptide boronate inhibitors affect peptide and protein degradation by ClpP1P2 complexes and by each of its two types of active sites. In hydrolyzing the peptide substrates, ClpP1P2(S-A) was even more sensitive to the peptide boronate inhibitor than WT ClpP1P2 (Fig. 7A). This result is in agreement with our earlier finding (Ref. 1, Fig. 6A) that ClpP1 active sites catalyzed most of hydrolysis of the tripeptide substrates. ClpP1(S-A)P2, whose peptidase activity was only 1% that of WT ClpP1P2 (Fig. 6A), was much less sensitive to boronate inhibitors. Thus its IC50 against Ac-PKM-amc was >10 times higher than that of the WT enzyme (Fig. 7A). Together, these findings indicate that the peptide boronates inhibit preferentially ClpP1 and apparently have much lower affinity to ClpP2 for reasons that are not clear.
FIGURE 7.
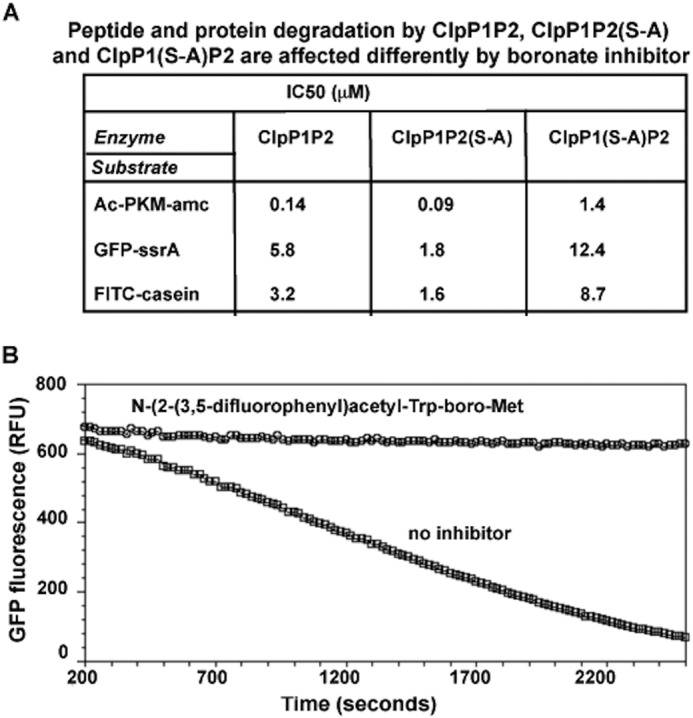
A, peptide boronates inhibit the ClpP1 activity more strongly than the ClpP2 activity. Effects of peptide boronate inhibitor (N-(1H-benzo(b)thiophene-7-carbonyl)-Lys-boro-Met) on degradation of peptide (Ac-Pro-Lys-Met-amc) and protein (GFPssrA and FITC-casein) substrates by WT ClpP1P2, ClpP1P2(S-A), and ClpP1(S-A)P2 complexes are presented as IC50 values. Enzymatic activity of ClpP1P2 complexes was measured as described under “Experimental Procedures.” B, substrate-based boronate derivatives inhibit protein degradation by ClpXP1P2. N-(2-(3,5-difluorophenyl)acetyl-Trp-boroMet (20 μm) prevents the degradation of GFPssrA by Mtb ClpXP1P2 (the degradation assay was carried out as in Fig. 5A). RFU, relative fluorescence units.
Because both ClpP1 and ClpP2 active sites contribute to protein degradation (Fig. 6B) and the ClpP2 active site has low affinity for the tripeptide substrates (Fig. 6A), we initially anticipated that the peptide boronate inhibitors would have little effect on protein degradation. Surprisingly, FITC-casein degradation by ClpC1P1P2 and GFPssrA degradation by ClpXP1P2 were both inhibited by peptide boronates (Fig. 7, A and B), although these agents showed 10 times lower potency against proteins than against Ac-PKM-amc, presumably reflecting the lower ability to inhibit ClpP2. Accordingly, protein degradation by the complex with only ClpP1 active was inhibited most strongly (Fig. 7A). With only ClpP2 active, the enzyme was inhibited by the boronates ∼2 times worse than the WT ClpP1P2, further indicating that ClpP2 is also important in protein degradation by the complex (Fig. 7A). These observations predict that some peptide boronates, if taken up by Mtb, should block ClpP1P2-dependent protein degradation and be toxic to these cells.
Peptide Boronates Are Toxic to Mtb but Not to Other Bacteria or Mammalian Cells
To test the effects of these peptide boronates on the growth of Mtb cells, we used the resazurin cytotoxicity assay. Many of these potent inhibitors of ClpP1P2 were very effective against Mtb. We did not see a direct correlation between the potency of the inhibitors in vitro (Ki value) and cytotoxicity. For instance, the boronate derivative of the best substrate, Ac-Pro-Lys-boroMet (Table 3, #1; Ki = 0.46 μm), had an MIC50 = 12 μm, whereas a relatively less potent inhibitor, Ac-Ala-Lys-boroMet (Table 3, #3; Ki = 0.8 μm), showed higher cytotoxicity with MIC50 = 3 μm (Table 3). Also, the very potent inhibitor, picolinoyl-Trp-Lys-boroMet (Table 3, #5; Ki = 0.18 μm) had an MIC50 = 3 μm against Mtb (Table 3). This lack of a direct correlation is not surprising as cytotoxicity must depend also on drug uptake and metabolic stability in bacteria and on the inhibitor's capacity to block the degradation of certain cell proteins (not peptide substrates) and thus on the degree of inhibition of ClpP1P2, which these peptidase assays do not assess.
As noted above, some dipeptide boronates are also very potent inhibitors of ClpP1P2. Interestingly, the dipeptide boronate, picolinoyl-Lys-boroMet (Table 3, #8), was as effective as its tripeptide derivative against ClpP1P2 peptidase activity (Ki = 0.34 μm) but appeared somewhat less toxic to cells (MIC50 = 6 μm) (Table 3). The dipeptide (Table 3, #9) with N-(3-phenyl)propanoyl) instead of piconoyl was more potent with Ki = 0.23 μm and MIC50 = 3 μm. Thus, dipeptide derivatives can also be used as anti-Mtb agents. Several other types of N-terminal blocking groups were also tested that appeared to increase inhibitory potency of the dipeptide Lys-boroMet but reduced its toxicity against Mtb. In the case of N-(phenylmethanesulfonyl)-Lys-boroMet (Table 3, #14), both inhibitory efficiency and toxicity dramatically decreased. Thus, the blocking group is an important determinant of cytotoxicity, presumably by affecting inhibitor uptake or metabolism.
All these boronate inhibitors tested in the viability assay were only toxic to Mtb. At the same time, even at 200 μm concentration, they did not affect the growth of E. coli or S. aureus, whose ClpP are not essential for viability (Table 3). It is noteworthy that only some of these boronates (e.g. Ala(1-naphtyl)-Lys-boro-Leu) that were toxic to Mtb had an effect on growth of M. smegmatis (probably due to the differences in permeability between these two related species).
We also tested whether some of these potent boronate inhibitors affected mammalian cells. When myeloma cells (MM1.S) were grown overnight in the presence of 10 μm N-(picolinoyl)-Ala-Lys-boroMet, N-(picolinoyl)-Pro-Lys-boroMet, and Ala(1-naphtyl)-Lys-boroLeu, no inhibition of growth was observed, whereas N-(picolinoyl)-Trp-Lys-boroMet caused only slight inhibition (25%). By contrast, the proteasome inhibitor bortezomib in parallel experiments caused 50% inhibition of growth at 5 nm. The weak effect of peptide boronate inhibitors on mammalian cells could be in part explained by the relatively lower potency of these inhibitors against human mitochondrial ClpP (Table 3).
Peptide Boronates Inhibit ClpP1P2 Function in Vivo
To confirm that these peptide boronate inhibitors actually can enter in mycobacteria and inhibit ClpP1P2, we tested whether these inhibitors could slow the degradation of GFP-ssrA by ClpP1P2 in Mtb. A strain constitutively expressing GFP-ssrA was incubated with N-(3-phenyl)propanoyl)Lys-boroMet, a potent ClpP1P2 inhibitor with 3× MIC (Table 3, #9). The accumulation of the substrate could be observed much earlier (within 10 h) than when the toxic effects of inhibitor become evident (5 days). The fluorescence of GFP was followed from 3 to 42 h during logarithmic growth at 37 °C and normalized to cell number. Although hardly any GFP fluorescence was detected in the absence of the inhibitors (the same baseline was observed for the strain without GFPssrA), GFP fluorescence markedly increased after 10 h of incubation with N-(3-phenyl)propanoyl)Lys-boroMet (Table 3, #9) and stayed at the elevated levels for at least 40 h (Fig. 8, A and B). These data indicate that the ClpXP1P2 is indeed a target of this inhibitor in vivo.
FIGURE 8.
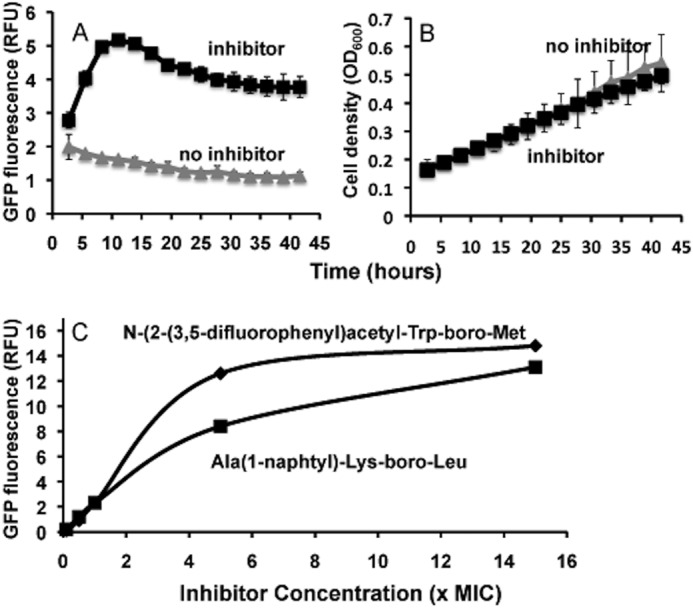
ClpP1P2 is the target for boronate inhibitors in mycobacteria. A, GFPssrA substrate accumulates in Mtb in the presence of ClpP1P2 inhibitor. H37Rv strain expressing PMV261::GFP-ssrA was incubated with and without 10 μm concentrations of inhibitor (N-(3-phenyl)propanoyl)-Lys-boroMet). Fluorescence measurements were normalized by cell density. Fluorescence measurements were taken every 2.75 h for 42 h. Error bars represent 95% confidence intervals of triplicate experiment. B, growth curves for Mtb in the presence and absence of the inhibitor. C, ClpP1P2 is the target for boronate inhibitors in M. smegmatis. M. smegmatis constitutively expressing GFP-ssrA was grown to stationary phase then diluted to an A600 = 0.003 and dispensed into a sterile flat-bottom 96-well plate. Peptide boronates (Ala(1-naphtyl)-Lys-boroLeu (Table 3, #4) or N-(2-(3,5-difluorophenyl)acetyl-Trp-boroMet) (Table 3, #12) at different concentrations were added to wells. The fluorescence of GFP was measured after 6 h at 37 °C and normalized to cell density. RFU, relative fluorescence units.
Similar experiments were also carried out with non-pathogenic fast-growing-related species M. smegmatis to determine the concentration dependence of the inhibition. A strain constitutively expressing GFP-ssrA (2) was incubated with different concentrations of either Ala(1-naphtyl)-Lys-boroLeu (Table 3, #4), which is the most potent agent against M. smegmatis (with an MIC50 = 1.5 μm), and an MIC50 against Mtb of 25 μm or N-(2-(3,5-difluorophenyl)acetyl-Trp-boroMet (Table 3, #12), which had an MIC50 = 12 μm against both Mtb and M. smegmatis. After 6 h in the presence of both inhibitors at concentrations above the MIC50, the fluorescence of GFP markedly increased (Fig. 8C). These findings indicate that in both species of mycobacteria ClpP1P2 represents a target for boronate inhibitors.
DISCUSSION
Distinct Specificity of Peptide Bond Hydrolysis by ClpP1P2
Although the ClpP family of proteases has been known for almost 30 years and its biological roles, ATP dependence, and molecular architecture extensively studied, its cleavage site specificity had not been investigated previously. To develop specific inhibitors of ClpP1P2 that may serve as lead compounds in drug development, we first defined the protease's sequence preference using a tripeptide diversity library. ClpP1P2 strongly preferred Met over other hydrophobic amino acids (Leu > Phe > Ala) in the X1 position (Fig. 1). This preference for cleavages after Met was also observed in an analysis of the peptides generated during protein degradation by the ClpXP1P2 complex (Fig. 5). Interestingly, during maturation of ClpP from E. coli (29) and Mtb (1), their N-terminal sequence are autolytically cleaved after Met to produce the mature proteins. This specificity appears quite unusual; in fact, we know of no endoprotease with a similar preference for cleavages after Met.
The substrates for Mtb ClpP1P2 identified during the screening of a tripeptide library have greatly improved the assay of this family of enzymes. For example, Ac-Pro-Lys-Met-amc and Ac-Pro-Trp-Met-amc have specificity constant kcat/Km values 1000 times higher than those of Suc-Leu-Tyr-amc (the most widely used substrate for 25 years) and >40 times higher than those of the Z-Gly-Leu-Leu-amc, which we used initially to characterize Mtb ClpP1P2 (1). Most of these new ClpP1P2 substrates are also very rapidly hydrolyzed by ClpPs from E. coli, B. subtilis, and S. aureus (Table 2). Although the substrate library screened most possible tripeptides of the form X1-X2-X3-amc, cleavage specificity of some serine proteases is determined in part by residues both preceding and following the scissile bond (30) (e.g. the hepatitis C virus NS3/4 protease). Interestingly, the protein GFP was most frequently cleaved before a positively charged residue or a Ser (Fig. 5C). Thus, the nature of the X′ position may influence cleavage rates.
The two Clp ATPases, ClpX and ClpC, clearly promote degradation of different types of cell proteins. In the presence of ClpX, ClpP1P2, like ClpPs from other species, degraded GFPssrA but not GFP lacking the ssrA tag. Although the ClpC1P1P2 complex does not support ATP-dependent degradation of GFPssrA, under similar conditions it did catalyze degradation of casein, which was not a substrate for ClpXP1P2. The proteolytic sites of most if not all ATP-dependent proteases like ClpPs or proteasomes are compartmentalized within a distinct proteolytic chamber. Therefore, protein substrates transported into the chamber cannot exit readily and are hydrolyzed in a processive manner to produce a spectrum of peptide products of different lengths (1, 31, 32). The peptides released during GFPssrA degradation by ClpXP1P2 varied in length from 4 to 26 residues (Fig. 5A), with most ranging between 8 and 16. Thus the mean size of the products is significantly larger than that generated by the archaeal or mammalian proteasome, where most peptide products are <8 residues long (33). Because product size seems to depend on a kinetic competition between further cleavages and diffusion of peptides out of the proteolytic chamber, the larger size of products probably reflects the easier diffusion of larger peptides out of the 2-ring ClpP than of the 4-ring proteasomes. Interestingly, homologous homooligomeric ClpPs exist in either an extended state with open active sites and closed equatorial pores or in a compressed state with closed active sites and open pores for product release (34).
Roles of ClpP1 and P2 in Proteolysis
Although the presence of both ClpP1 and ClpP2 is essential for enzymatic activity, their active sites contribute to the degradation process in very different manners. Although the ClpP1P2(S-A) complex was as active as the wild type enzyme against tripeptide substrates, ClpP1(S-A)P2 showed only 0.2–1% of its activity (Fig. 6A). Thus, ClpP2 lacks activity against these types of tripeptides, although Z-nLPnLD-amc was hydrolyzed by ClpP1(S-A)P2 slightly faster than other substrates. Interestingly, in another bacterium containing ClpP1 and ClpP2 subunits, Listeria monocytogenes, the relative roles of ClpP1 and ClpP2 were quite different, with ClpP2 complexes proteolytically active by themselves, whereas ClpP1 subunits were only activated in the mixed ClpP1P2 complex (35). Despite their very different activities in tripeptide degradation, surprisingly, we found that both ClpP1 and ClpP2 subunits made major contributions to protein degradation. Thus, ClpP2 clearly plays a catalytic and not just a structural role, although its active sites for some reason appear latent except when Clp ATPases and a protein substrate were present. Perhaps the low peptidase activity of ClpP2 is associated with the requirement for additional contacts with the substrate on both ends of the scissile bond. Alternatively, the ClpC1 or ClpX ATPases and/or the protein substrate may induce the proteolytically active conformation in ClpP2. In human mitochondrial ClpP, the association with ClpX alters the conformation of ClpP to promote the formation of the active tetradecamer (5), and substrate binding is known to alter the conformations of the Clp ATPases (23). In Mtb, unlike most bacteria, ClpX and ClpC1 are both essential for viability and infectivity, which makes them attractive targets as we noted previously (36). In fact, two novel natural product antibiotics have been identified recently that act on ClpC1 and cause selective killing of mycobacteria (19, 37). For example, the cyclic peptide lassomycin uncouples ATPase activity from ATP-dependent proteolysis by ClpC1P1P2 (19).
While we were preparing this manuscript for publication, Schmitz and Sauer (38) also reported studies of the degradation of GFPssrA by Mtb ClpXClpP1P2 complex. They confirmed our findings (1) that the active form of the protease is the mixed ClpP1P2 complex and also observed that inactivation of ClpP1 or ClpP2 still allows rapid GFPssrA breakdown. However, they reported that GFPssrA could also be degraded slowly by ClpC1P1P2, perhaps because they used 10-fold more protease in their degradation assay than were used here. Another clear inconsistency is that those authors observed peptide degradation only in the presence of both the ATPase and the activator (which they designated by the pharmacological or physiological term, “agonist”), whereas in our studies peptides were efficiently degraded in the presence of the dipeptide activator alone.
Peptide Boronate Inhibitors and Therapeutic Potential
This characterization of cleavage specificity of Mtb ClpP1P2 indicated a novel active site preference and strongly suggested that synthesis of substrate-based inhibitors would be a valuable approach for development of selective anti-Mtb agents. We have focused on peptide boronates because many potent and highly specific peptide boronic acid inhibitors have been synthesized for serine and threonine proteases (26). For example, bortezomib is a potent, highly selective inhibitor of human proteasomes that is now the preferred treatment worldwide for multiple myeloma, and two other boro-peptides are presently in clinical trials. Peptide boronates were shown to inhibit the Mtb proteasome and Lon from Salmonella enterica and mitochondria (28, 39). Unlike ClpP or the proteasome, Lon is inhibited by these agents and other covalent inhibitors only in the ATP-bound state, which is necessary for its active site formation.
Although many peptide boronates have been synthesized previously, these agents with a methionine in the X1 position are a novel type of protease inhibitors and appear relatively selective for ClpP family members (40). The ability of these substrate-based inhibitors to block protein hydrolysis as well as to stop Mtb growth strongly suggests that cleavage specificity of ClpP1P2 is an accurate reflection of enzyme function in vivo. However, a clear limitation of this approach was apparently the surprising lack of activity of ClpP2 on the tripeptide substrates, and because our tripeptide boronates were synthesized based upon substrate preference, it is not surprising that they are less potent in blocking ClpP2-catalyzed protein breakdown than peptide hydrolysis. Presumably, if these peptide boronates were more potent against ClpP2, they would be even more effective against protein substrates and in killing Mtb.
Our most potent substrate-based boronate derivatives and their analogs inhibited the peptidase activity of ClpP1P2 with Ki of <1 μm, with some as low as 65 nm. Most importantly, they blocked Mtb growth with MIC50 value from 3 to 12 μm (Table 3), apparently through their abilities to inhibit ClpP1P2. Accordingly, these inhibitors did not affect the growth of bacteria where ClpP is not essential, E. coli and S. aureus. Moreover, in Mtb and M. smegmatis, these agents blocked ClpP1P2 activity as they caused GFPssrA to accumulate (Fig. 8) in a similar way as was observed upon ClpP1P2 depletion genetically (2). Not surprisingly, the efficacy of these peptide boronates in preventing mycobacterial growth correlated only roughly with their potency against peptide substrates, presumably because cytotoxicity requires blocking the degradation of endogenous proteins, whose accumulation is toxic.
Although we utilized here a blocked tripeptide-amc library, the dipeptide derivatives (Ac-Trp-Met-amc and Ac-Lys-Met-amc) were also very good substrates for ClpP1P2. Consequently several blocked dipeptide boronates were synthesized. They were found to be as effective as their tripeptide analogs against ClpP1P2 and showed similar or slightly lower toxicity against Mtb (Table 3). The N-terminal blocking group also had significant effects on inhibitor efficacy. Curiously, use of a picolinoyl group in place of the standard acetyl group resulted in a higher affinity for ClpP1P2 in the case of Ala-Lys-boro-Met with similar cytotoxicity, but this substitution reduced both inhibitory activity and cytotoxicity in the case of Pro-Lys-boro-Met for reasons that are unclear. Many further modifications are clearly possible to further enhance potency in vitro and efficacy in vivo, which presumably is limited by rates of drug uptake and metabolism as well as their limited ability to inhibit the ClpP2 and ClpP1 active sites.
Unlike other eubacteria, Mtb and other actinomycetes in addition to ClpP1P2 express a 20 S proteasome (11) that is much simpler in organization than proteasomes of eukaryotes and has quite different cleavage preferences (41–43). Unlike ClpP1P2, the Mtb proteasome is not essential but appears to be important for survival in the macrophage (24) and protection against free radicals (44). Peptide boronates that inhibit with high affinity ClpP1P2 and proteasomes could, therefore, be even more potent anti-Mtb agents, although this possibility does not seem feasible, as our data show that these two enzymes have quite different cleavage preferences (Fig. 4). A successful agent against Mtb would have to have little or no effect on important mammalian enzymes. It is noteworthy that the cleavage preferences of ClpP1P2 are quite different from those of the human proteasome, the main protein degradation system in human cytosol. The best ClpP1P2 substrates (Ac-Pro-Lys-Met-amc and Ac-Pro-Trp-Met-amc) are quite poor proteasomal substrates, and the best proteasomal substrates are cleaved very slowly by ClpP1P2 (Fig. 4). Therefore, these inhibitors are unlikely to affect protein degradation in human cells. Indeed, peptide boronates did not affect the growth of mammalian cells tested here.
These peptide boronates are also potent inhibitors of other ClpP family members, and inhibitors of S. aureus ClpP are potentially of therapeutic interest. Even though ClpP is not essential in S. aureus, Lewis and co-workers recently showed that ClpP-deficient S. aureus were more sensitive to standard antibiotics and less able to enter the persister state than wild type strains (45). Because S. aureus infections are common in hospitals and difficult to treat with conventional antibiotics, inhibitors of ClpP may prove valuable in fighting S. aureus infections. These findings on enzyme specificity provide a rational basis for synthesis of more potent substrate-based inhibitors of ClpP1P2 from Mtb and perhaps other bacteria. Clearly, the success achieved thus far in generating anti-Mtb agents validates this target choice, biochemical rationale, and synthetic strategy for development of new types of agents cytotoxic for Mtb.
Acknowledgment
We are grateful to Dr. Zhe Sha for help in experiments with myeloma cell lines.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM051923-17A1. This work was also supported by a Harvard Catalyst Grant, a grant from Roche Applied Science (to A. L. G.), and by Bill and Melinda Gates Foundation Grant OPP1024065 (to E. J. R.).
- Mtb
- M. tuberculosis
- amc
- aminomethylcoumarin
- MIC
- minimal inhibitory concentration
- BoroMet
- methionine boronic acid
- Z
- benzyloxy-carbonyl.
REFERENCES
- 1. Akopian T., Kandror O., Raju R. M., Unnikrishnan M., Rubin E. J., Goldberg A. L. (2012) The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 31, 1529–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raju R. M., Unnikrishnan M., Rubin D. H., Krishnamoorthy V., Kandror O., Akopian T. N., Goldberg A. L., Rubin E. J. (2012) Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 8, e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sassetti C. M., Boyd D. H., Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 [DOI] [PubMed] [Google Scholar]
- 4. Porankiewicz J., Wang J., Clarke A. K. (1999) New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Mol. Microbiol. 32, 449–458 [DOI] [PubMed] [Google Scholar]
- 5. Kang S. G., Dimitrova M. N., Ortega J., Ginsburg A., Maurizi M. R. (2005) Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J. Biol. Chem. 280, 35424–35432 [DOI] [PubMed] [Google Scholar]
- 6. Hwang B. J., Park W. J., Chung C. H., Goldberg A. L. (1987) Escherichia coli contains a soluble ATP-dependent protease (Ti) distinct from protease La. Proc. Natl. Acad. Sci. U.S.A. 84, 5550–5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Katayama-Fujimura Y., Gottesman S., Maurizi M. R. (1987) A multiple-component, ATP-dependent protease from Escherichia coli. J. Biol. Chem. 262, 4477–4485 [PubMed] [Google Scholar]
- 8. Flanagan J. M., Wall J. S., Capel M. S., Schneider D. K., Shanklin J. (1995) Scanning transmission electron microscopy and small-angle scattering provide evidence that native Escherichia coli ClpP is a tetradecamer with an axial pore. Biochemistry 34, 10910–10917 [DOI] [PubMed] [Google Scholar]
- 9. Wang J., Hartling J. A., Flanagan J. M. (1997) The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell 91, 447–456 [DOI] [PubMed] [Google Scholar]
- 10. Andersson F. I., Tryggvesson A., Sharon M., Diemand A. V., Classen M., Best C., Schmidt R., Schelin J., Stanne T. M., Bukau B., Robinson C. V., Witt S., Mogk A., Clarke A. K. (2009) Structure and function of a novel type of ATP-dependent Clp protease. J. Biol. Chem. 284, 13519–13532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Butler S. M., Festa R. A., Pearce M. J., Darwin K. H. (2006) Self-compartmentalized bacterial proteases and pathogenesis. Mol. Microbiol. 60, 553–562 [DOI] [PubMed] [Google Scholar]
- 12. Kress W., Maglica Z., Weber-Ban E. (2009) Clp chaperone-proteases: structure and function. Res. Microbiol. 160, 618–628 [DOI] [PubMed] [Google Scholar]
- 13. Maurizi M. R. (1991) ATP-promoted interaction between Clp A and Clp P in activation of Clp protease from Escherichia coli. Biochem. Soc. Trans. 19, 719–723 [DOI] [PubMed] [Google Scholar]
- 14. Hoskins J. R., Pak M., Maurizi M. R., Wickner S. (1998) The role of the ClpA chaperone in proteolysis by ClpAP. Proc. Natl. Acad. Sci. U.S.A. 95, 12135–12140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ortega J., Singh S. K., Ishikawa T., Maurizi M. R., Steven A. C. (2000) Visualization of substrate binding and translocation by the ATP-dependent protease, ClpXP. Mol. Cell 6, 1515–1521 [DOI] [PubMed] [Google Scholar]
- 16. Ishikawa T., Beuron F., Kessel M., Wickner S., Maurizi M. R., Steven A. C. (2001) Translocation pathway of protein substrates in ClpAP protease. Proc. Natl. Acad. Sci. U.S.A. 98, 4328–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reid B. G., Fenton W. A., Horwich A. L., Weber-Ban E. U. (2001) ClpA mediates directional translocation of substrate proteins into the ClpP protease. Proc. Natl. Acad. Sci. U.S.A. 98, 3768–3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baker T. A., Sauer R. T. (2012) ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 1823, 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gavrish E., Sit C. S., Cao S., Kandror O., Spoering A., Peoples A., Ling L., Fetterman A., Hughes D., Bissell A., Torrey H., Akopian T., Mueller A., Epstein S., Goldberg A., Clardy J., Lewis K. (2014) Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 21, 509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sheppeck J. E., 2nd, Kar H., Gosink L., Wheatley J. B., Gjerstad E., Loftus S. M., Zubiria A. R., Janc J. W. (2000) Synthesis of a statistically exhaustive fluorescent peptide substrate library for profiling protease specificity. Bioorg. Med. Chem. Lett. 10, 2639–2642 [DOI] [PubMed] [Google Scholar]
- 21. Cer R. Z., Mudunuri U., Stephens R., Lebeda F. J. (2009) IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 37, W441–W445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coutts S. J., Kelly T. A., Snow R. J., Kennedy C. A., Barton R. W., Adams J., Krolikowski D. A., Freeman D. M., Campbell S. J., Ksiazek J. F., Bachovchin W. W. (1996) Structure-activity relationships of boronic acid inhibitors of dipeptidyl peptidase IV. 1. Variation of the P2 position of Xaa-boroPro dipeptides. J. Med. Chem. 39, 2087–2094 [DOI] [PubMed] [Google Scholar]
- 23. Hwang B. J., Woo K. M., Goldberg A. L., Chung C. H. (1988) Protease Ti, a new ATP-dependent protease in Escherichia coli, contains protein-activated ATPase and proteolytic functions in distinct subunits. J. Biol. Chem. 263, 8727–8734 [PubMed] [Google Scholar]
- 24. MacMicking J., Xie Q. W., Nathan C. (1997) Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350 [DOI] [PubMed] [Google Scholar]
- 25. Lin G., Tsu C., Dick L., Zhou X. K., Nathan C. (2008) Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J. Biol. Chem. 283, 34423–34431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smoum R., Rubinstein A., Dembitsky V. M., Srebnik M. (2012) Boron containing compounds as protease inhibitors. Chem. Rev. 112, 4156–4220 [DOI] [PubMed] [Google Scholar]
- 27. Goldberg A. L. (2012) Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 199, 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu G., Lin G., Wang M., Dick L., Xu R. M., Nathan C., Li H. (2006) Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol. Microbiol. 59, 1417–1428 [DOI] [PubMed] [Google Scholar]
- 29. Maurizi M. R., Clark W. P., Katayama Y., Rudikoff S., Pumphrey J., Bowers B., Gottesman S. (1990) Sequence and structure of Clp P, the proteolytic component of the ATP-dependent Clp protease of Escherichia coli. J. Biol. Chem. 265, 12536–12545 [PubMed] [Google Scholar]
- 30. Ingallinella P., Bianchi E., Ingenito R., Koch U., Steinkühler C., Altamura S., Pessi A. (2000) Optimization of the P′-region of peptide inhibitors of hepatitis C virus NS3/4A protease. Biochemistry 39, 12898–12906 [DOI] [PubMed] [Google Scholar]
- 31. Akopian T. N., Kisselev A. F., Goldberg A. L. (1997) Processive degradation of proteins and other catalytic properties of the proteasome from Thermoplasma acidophilum. J. Biol. Chem. 272, 1791–1798 [DOI] [PubMed] [Google Scholar]
- 32. Thompson M. W., Singh S. K., Maurizi M. R. (1994) Processive degradation of proteins by the ATP-dependent Clp protease from Escherichia coli. Requirement for the multiple array of active sites in ClpP but not ATP hydrolysis. J. Biol. Chem. 269, 18209–18215 [PubMed] [Google Scholar]
- 33. Cascio P., Hilton C., Kisselev A. F., Rock K. L., Goldberg A. L. (2001) 26 S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. EMBO J. 20, 2357–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Geiger S. R., Böttcher T., Sieber S. A., Cramer P. (2011) A conformational switch underlies ClpP protease function. Angew. Chem. Int. Ed. Engl. 50, 5749–5752 [DOI] [PubMed] [Google Scholar]
- 35. Zeiler E., List A., Alte F., Gersch M., Wachtel R., Poreba M., Drag M., Groll M., Sieber S. A. (2013) Structural and functional insights into caseinolytic proteases reveal an unprecedented regulation principle of their catalytic triad. Proc. Natl. Acad. Sci. U.S.A. 110, 11302–11307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Raju R. M., Goldberg A. L., Rubin E. J. (2012) Bacterial proteolytic complexes as therapeutic targets. Nat. Rev. Drug Discov. 11, 777–789 [DOI] [PubMed] [Google Scholar]
- 37. Schmitt E. K., Riwanto M., Sambandamurthy V., Roggo S., Miault C., Zwingelstein C., Krastel P., Noble C., Beer D., Rao S. P., Au M., Niyomrattanakit P., Lim V., Zheng J., Jeffery D., Pethe K., Camacho L. R. (2011) The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed. Engl. 50, 5889–5891 [DOI] [PubMed] [Google Scholar]
- 38. Schmitz K. R., Sauer R. T. (2014) Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol. Microbiol. 93, 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frase H., Lee I. (2007) Peptidyl boronates inhibit Salmonella enterica serovar Typhimurium Lon protease by a competitive ATP-dependent mechanism. Biochemistry 46, 6647–6657 [DOI] [PubMed] [Google Scholar]
- 40. Bachovchin D. A., Koblan L. W., Wu W., Liu Y., Li Y., Zhao P., Woznica I., Shu Y., Lai J. H., Poplawski S. E., Kiritsy C. P., Healey S. E., DiMare M., Sanford D. G., Munford R. S., Bachovchin W. W., Golub T. R. (2014) A high-throughput, multiplexed assay for superfamily-wide profiling of enzyme activity. Nat. Chem. Biol. 10, 656–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Voges D., Zwickl P., Baumeister W. (1999) The 26 S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068 [DOI] [PubMed] [Google Scholar]
- 42. Coux O., Tanaka K., Goldberg A. L. (1996) Structure and functions of the 20 S and 26 S proteasomes. Annu. Rev. Biochem. 65, 801–847 [DOI] [PubMed] [Google Scholar]
- 43. Goldberg A. L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899 [DOI] [PubMed] [Google Scholar]
- 44. Darwin K. H., Ehrt S., Gutierrez-Ramos J. C., Weich N., Nathan C. F. (2003) The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302, 1963–1966 [DOI] [PubMed] [Google Scholar]
- 45. Conlon B. P., Nakayasu E. S., Fleck L. E., LaFleur M. D., Isabella V. M., Coleman K., Leonard S. N., Smith R. D., Adkins J. N., Lewis K. (2013) Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503, 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]