

Background: β-Cell apoptosis, a critical contributor to T1D, involves iPLA2β activation and is suppressed by Bcl-x(L).
Results: iPLA2β-derived lipids activate an alternative 5′-splice site, reducing protective Bcl-x(L) protein.
Conclusion: Modulation of Bcl-x splicing is another key mechanism by which iPLA2β-derived lipids promote β-cell apoptosis.
Significance: Delineation of molecular mechanisms underlying iPLA2β-regulated splicing will elucidate novel strategies to counter β-cell death in T1D.
Keywords: Alternative Splicing, Apoptosis, Diabetes, Phospholipase A, Signal Transduction, Bcl-x, β-Cell
Abstract
Diabetes is a consequence of reduced β-cell function and mass, due to β-cell apoptosis. Endoplasmic reticulum (ER) stress is induced during β-cell apoptosis due to various stimuli, and our work indicates that group VIA phospholipase A2β (iPLA2β) participates in this process. Delineation of underlying mechanism(s) reveals that ER stress reduces the anti-apoptotic Bcl-x(L) protein in INS-1 cells. The Bcl-x pre-mRNA undergoes alternative pre-mRNA splicing to generate Bcl-x(L) or Bcl-x(S) mature mRNA. We show that both thapsigargin-induced and spontaneous ER stress are associated with reductions in the ratio of Bcl-x(L)/Bcl-x(S) mRNA in INS-1 and islet β-cells. However, chemical inactivation or knockdown of iPLA2β augments the Bcl-x(L)/Bcl-x(S) ratio. Furthermore, the ratio is lower in islets from islet-specific RIP-iPLA2β transgenic mice, whereas islets from global iPLA2β−/− mice exhibit the opposite phenotype. In view of our earlier reports that iPLA2β induces ceramide accumulation through neutral sphingomyelinase 2 and that ceramides shift the Bcl-x 5′-splice site (5′SS) selection in favor of Bcl-x(S), we investigated the potential link between Bcl-x splicing and the iPLA2β/ceramide axis. Exogenous C6-ceramide did not alter Bcl-x 5′SS selection in INS-1 cells, and neutral sphingomyelinase 2 inactivation only partially prevented the ER stress-induced shift in Bcl-x splicing. In contrast, 5(S)-hydroxytetraenoic acid augmented the ratio of Bcl-x(L)/Bcl-x(S) by 15.5-fold. Taken together, these data indicate that β-cell apoptosis is, in part, attributable to the modulation of 5′SS selection in the Bcl-x pre-mRNA by bioactive lipids modulated by iPLA2β.
Introduction
Accumulating evidence suggests that β-cell apoptosis underlies the pathogenesis of both type 1 (T1D)3 and type 2 diabetes (1–6). Reduced β-cell mass has been observed both in animal models of diabetes and in autopsies of type 2 diabetes subjects, and this has been linked to increased apoptosis rather than reduced proliferation (7–9). For example, β-cells of the diabetic Akita and NOD mouse strains are hypersensitive to pro-apoptotic stimuli (10–12), and pro-inflammatory cytokines induce β-cell apoptosis, a critical event contributing to the development of autoimmune T1D (13, 14). In addition, accumulation of misfolded insulin in the ER is reported to promote ER stress and lead to β-cell apoptosis and diabetes (2, 15). Despite the mounting evidence connecting β-cell apoptosis to diabetes mellitus, the underlying biochemical and molecular mechanisms contributing to β-cell apoptosis have yet to be completely elucidated.
Our ongoing work reveals prominent roles for β-cell-derived lipid signals in processes that eventually lead to apoptosis of the β-cells. In particular, we demonstrated that the group VIA phospholipase A2 (iPLA2β) plays a key role in this event. The iPLA2β is a member of the phospholipase A2 family of enzymes, which hydrolyze the sn-2 fatty acid from membrane phospholipids to release a free fatty acid and a lysolipid (16). In pancreatic islets, iPLA2β is predominantly localized in β-cells (17–19), and our studies reveal that expression and activity of iPLA2β are increased when β-cells undergo ER stress-induced apoptosis (12, 17, 18, 20, 21). Activation of iPLA2β also induces neutral sphingomyelinase 2 (NSMase2), resulting in accumulation of pro-apoptotic ceramides (12, 17, 18, 22). Various strategies (selective inhibitors, siRNA, and genetic-modulation) indicate a role for iPLA2β and subsequent NSMase2-derived ceramides in ER stress-induced β-cell apoptosis (12, 17, 18, 20–23). Furthermore, we recently demonstrated that iPLA2β inhibition reduces T1D incidence (24).
ER stress-induced β-cell apoptosis is mediated through the intrinsic pathway, which is dependent on mitochondrial dysfunction and activation of caspase-9 (20, 25). Our studies indicate that activation of iPLA2β in β-cells undergoing ER stress promotes loss of mitochondrial membrane potential and resultant apoptosis of the β-cells (12, 17, 18, 23). The intrinsic apoptosis pathway is regulated by members of the Bcl-2 family of proteins that can be pro- or anti-apoptotic, depending on the spectrum of Bcl-2 homology (BH) domains that they contain. Among the anti-apoptotic Bcl-2 family members is Bcl-x(L), which associates with mitochondrial membranes and prevents their permeabilization, an early step in the intrinsic apoptosis pathway (26, 27). Overexpression of Bcl-x(L) has been correlated with increased survival of a variety of cells and tissues (28), including islet β-cells (29–31). Bcl-x(L)-null β-cells are hypersensitive to pro-apoptotic stimuli, and reduced expression of Bcl-x(L) protein correlates with β-cell apoptosis in response to immunosuppressive drugs or high glucose (29–32). Conversely, overexpression of exogenous Bcl-x(L) protects β-cells from pro-inflammatory cytokine- and thapsigargin-induced apoptosis (32, 33). These observations suggest that stabilization of the Bcl-x(L) protein mass could be a key to preserving β-cell viability. However, very little is known of the processes that regulate endogenous Bcl-x(L) protein in β-cells.
Modulation of Bcl-x(L) expression is a complex mechanism consisting of both transcriptional and post-transcriptional processes. Among the well studied regulatory mechanisms in non-β-cell systems is alternative splicing of Bcl-x pre-mRNA. This is a common process among the regulators of apoptosis and often leads to generation of both pro- and anti-apoptotic proteins from a single pre-mRNA (28, 34). Bcl-x(L) is the most abundant variant of the Bcl-x pre-mRNA, but other species can be generated at the expense of the mature mRNA encoding this anti-apoptotic protein (28). For instance, a well documented Bcl-x splicing event is the one that determines whether Bcl-x(L) or Bcl-x(S) is generated. Bcl-x(S) is produced by activation of an upstream 5′-splice site (5′SS) within the Bcl-x exon 2 and blockage of the downstream (Bcl-x(L)-specific) 5′SS in Bcl-x exon 2. RNA oligonucleotides targeted to the downstream 5′SS induce Bcl-x(S) expression, down-regulate Bcl-x(L) protein, and sensitize tumor cells to chemotherapy (35, 36). Thus, regulation of the 5′SS selection within the Bcl-x exon 2 is a critical factor in determining whether a cell is susceptible or resistant to apoptosis. Although molecular mechanisms controlling Bcl-x splicing have been studied, the mechanisms differ substantially depending on the cell system. For example, de novo ceramide generation in response to chemotherapeutics and apoptotic agonists (e.g. Fas ligand) has been implicated in the activation of the Bcl-x(S) 5′SS in transformed cells (37). In contrast, Chabot and co-workers (38) have implicated a classical protein kinase C mechanism for regulating Bcl-x RNA splicing in nontransformed cells. Hence, the signaling mechanism in a particular cell system must be considered, and to date, Bcl-x RNA splicing has not been investigated in the β-cell, especially in the context of β-cell apoptosis and diabetes mellitus.
The experiments described herein were designed to test our hypothesis that iPLA2β regulates Bcl-x(L) splicing and promotes usage of the alternative 5′SS. We demonstrate that both chemical inactivation and genetic ablation or knockdown of iPLA2β shift Bcl-x splicing in favor of anti-apoptotic Bcl-x(L) and that iPLA2β inactivation largely prevents the shift in Bcl-x splicing that occurs upon ER stress-induced apoptosis. Unexpectedly, the effects of iPLA2β are found to be largely independent of ceramide but are modulated by bioactive metabolites of arachidonic acid. These observations reveal a novel role for iPLA2β in survival of β-cells.
EXPERIMENTAL PROCEDURES
Materials
The following were obtained: 1° antibody against Bcl-x (BD Biosciences); (S)-BEL, C6-ceramide, EPA, GW4869, 5(S)-HETE, and thapsigargin (Cayman Chemical Co.); 1° antibodies against actin and activated caspase 3 (Cell Signaling Technology); oligonucleotides (Integrated DNA Technologies and Life Technologies, Inc.); 2° antibody coupled to Cy3 to detect insulin (Jackson ImmunoResearch); Accuprime Taq Polymerase System, 2° antibody Alexa Fluor 594 to detect iPLA2β, Lipofectamine 2000, Opti-MEM, RPMI 1640 medium, Superscript III One-Step RT-PCR System, SYBR Gold, Thermoscript RT-PCR System, and TRIzol LS (Life Technologies, Inc.); HRP-coupled secondary antibodies and SuperSignal West Femto substrate (Pierce); T-14 anti-iPLA2β (Santa Cruz Biotechnology); CellLytic M buffer (Sigma); and control and rat iPLA2β-targeted siRNA (Thermo Scientific Dharmacon).
INS-1 Cell Culture
Empty vector and iPLA2β-overexpressing INS-1 cells were generated and maintained, as described (39). The cells (4 × 105/well) were seeded in 12-well plates and cultured overnight before treatment. Cell viability was quantified by trypan blue exclusion assay.
Akita Cell Culture and Treatment
The Akita and wild-type (WT) β-cells were gifts from Dr. Akio Koizuma (Dept. of Health and Environmental Sciences, Kyoto University Graduate School of Medicine, Kyoto, Japan). The cells were cultured in DMEM with 10 μl of β-mercaptoethanol/200 ml, at 37 °C in 95% air, 5% CO2 as described (40). Cells were grown to 80% confluency in cell culture dishes before treatment.
Transfection
INS-1 cells (4 × 105/well) were seeded in 12-well plates and transfected with 20 nm siRNA 24 h after plating. Lipofectamine 2000-siRNA complexes were prepared in Opti-MEM according to the manufacturer's instructions, using 4 μl of Lipofectamine/transfection. Cells were incubated with Lipofectamine 2000-siRNA complexes overnight and were then treated before analysis of endogenous rat Bcl-x splice variants. For co-transfection protocols, 0.5 ng of human Bcl-x minigene was included in the complexes. The minigenes were prepared and characterized, as described (41). For minigene experiments, cells were transfected for 7 h; Lipofectamine 2000-nucleic acid complexes were removed, and cells were transferred to fresh media for additional treatments.
Islet Isolation and Culture
iPLA2β-deficient (KO) and RIP-iPLA2β-Tg mice breeders generously provided by Dr. John Turk (Washington University School of Medicine (WUSM), St. Louis, MO) were used to generate wild-type (WT), KO, and Tg mouse colonies at the University of Alabama at Birmingham (UAB). RIP-iPLA2β-Tg is a tissue-specific transgenic mouse line that selectively overexpresses iPLA2β in β-cells (42). The generation and characterization of this line and the global iPLA2β-KO line have been described previously (43). Islets were also isolated from Akita mice, which spontaneously develop ER stress in β-cells, leading to β-cell apoptosis and consequential diabetes (10, 11). Murine islets were isolated and cultured, as described (18). All mouse studies were performed according to protocols approved by the IACUC at WUMS and UAB.
Immunoblot Analyses
Protein extracts were prepared in CellLytic M buffer, resolved by 10% SDS-PAGE, and transferred to nitrocellulose membranes. The blots were blocked with 5% nonfat dry milk in TBS and then incubated overnight with 1° antibody directed against Bcl-x (1:1000), iPLA2β (1:200), or loading control, actin (1:5000). The 1° antibody-protein complexes were detected with HRP-coupled secondary antibodies at 1:5000. Bcl-x was detected with anti-rabbit, actin with anti-mouse IgM, and iPLA2β with anti-goat. HRP signals detected with the SuperSignal West Femto substrate were captured on x-ray film and quantified with a ChemiDoc XRS+ imager from Bio-Rad. Target protein signals were normalized to loading control.
Immunocytochemistry Analyses
Paraffin sections (10 μm) of pancreata were processed for immunostaining, as described (18). The sections were incubated overnight with 1° antibody (1:25), washed with PBS (four times for 30 min), incubated for 3 h with 2° antibodies (1:100 of Cy3 for insulin and Alexa Fluor 594 for iPLA2β), and washed with PBS (three times for 10 min each). Nuclear DAPI stain (25 μl) was then added, and the sections sealed with a coverslip using nail polish. Fluorescence was recorded using a Nikon Eclipse TE300 microscope, and images were captured (×40 magnification).
RT-PCR and qPCR Analyses of iPLA2β and Bcl-x Splice Variants
TRIzol LS was used to extract RNA from INS-1 cells and isolated islets. For RT-PCR assessment of endogenous Bcl-x splice variants, 0.35 μg of RNA was converted to cDNA, and RT-PCR was performed with the SuperScript III One Step RT-PCR system. For RT-PCR assessment of minigene splice variants, 1 μg of RNA was converted to cDNA with the Thermoscript RT-PCR First Strand cDNA System, and then splice variants were amplified with the Accuprime Taq polymerase system. PCR products were separated on 6.2% acrylamide gels and detected with SYBR® Gold. A Bio-Rad ChemiDoc XRS+ imager was used to quantify the signals, and data were analyzed using ImageQuant software. In all cases, conditions were adjusted to ensure that chemiluminescent signals were within the linear range of the response. The data are reported as the ratio of the Bcl-x(L)/Bcl-x(S) signal. For qPCR analyses, RNA was converted to cDNA with the Thermoscript kit. Bcl-x splice variants were quantified with the TaqMan Universal PCR Master Mix from Life Technologies, Inc. iPLA2β mRNA was quantified with Power SYBR Green PCR Master Mix from ABI. PCR primers sequences are shown in Table 1.
TABLE 1.
Primer sequences used for the various targets examined
F is forward, R is reverse, and P is probe.
| Target | Primer sequence |
|---|---|
| Rat Bcl-x (RT-PCR) | F, 5′ GGA GAG CAT TCA GTG ATC 3′ |
| R, 5′ CAA TGG TGG CTG AAG AGA 3′ | |
| Mouse Bcl-x (RT-PCR) | F, 5′ CCA GCT TCA CAT AAC CCC AG 3′ |
| R, 5′ CCG TAG AGA TCC ACA AAA GTG TC 3′ | |
| Human Bcl-x minigene (RT-PCR) | F, 5′GGA GCT GGT GGT TGA CTT TCT 3′ |
| R, 5′TAG AAG GCA CAG TCG AGG 3′ | |
| Rat Bcl-x(L) (qPCR) | F, 5′ GCG TAG ACA AGG AGA TGC AG 3′ |
| R, 5′ TGT TCC CGT AGA GAT CCA CA 3′ | |
| P, 5′ AAG TGT CCC AGC CGC CGT TC 3′ | |
| Rat Bcl-x(s) (qPCR) | F, 5′CAG CAG TGA AGC AAG CGC TGA 3′ |
| R, 5′ AAC CAG CGG TTG AAA CGC TC 3′ | |
| P, 5′ TGA ACA GGA CAC TTT TGT GGA TCT CTA CGG G 3′ | |
| Rat iPLA2β (PCR) | F, 5′ GCC CTG GCC ATT CTA CAC A 3′ |
| R, 5′ CAC CTC ATC CTT CAT ACG GA 3′ |
Mass Spectrometry Analysis of Lipids
Eicosanoids were analyzed from culture medium as described previously (44, 45). Briefly, 10% methanol and glacial acetic acid were added to 4 ml of medium. An internal standard ((d4)-6-keto-prostaglandin F1α) was added to each sample; (d4) prostaglandin E2 (PGE2), (d4) prostaglandin D2 (PGD2), (d8) 5-HETE, (d8) 15-HETE, (d8) 14,15-epoxyeicosatrienoic acid, and (d8) arachidonic acid were added. Strata-X SPE columns (Phenomenex) were washed with methanol and distilled water before samples were applied to the columns. Eicosanoids were eluted with isopropyl alcohol; the eluent was dried under vacuum, and then the samples were reconstituted in 50:50 ethanol-distilled water for LC/MS/MS analyses. The reconstituted eicosanoids were analyzed via HPLC ESI-MS/MS. Eicosanoids were separated via reversed-phase LC method utilizing a Kinetex C18 column (100 × 2.1 mm, 2.6 μm; flow rate of 200 μl/min at 50 °C). The column was equilibrated with 100% solvent A (acetonitrile/water/formic acid (40:60:0.02, v/v/v)) before the sample was injected, and then 100% solvent A was used for the 1st min of elution. Solvent B (acetonitrile/isopropyl alcohol (50:50, v/v)) was increased in a linear gradient of 25% solvent B for 3 min, 45% for 11 min, 60% for 13 min, 75% for 18 min, and 100% for 20 min. 100% solvent B was held for 25 min, decreased to 0% in a linear gradient for 26 min, and then held for 30 min. Eicosanoids were then analyzed using a tandem quadrupole mass spectrometer (AB Sciex 4000 QTRAP®, Applied Biosystems) via multiple-reaction monitoring in the negative-ion mode. Eicosanoids were monitored using the analyte-specific precursor → product multiple reaction monitoring pairs that we have reported previously (44). The mass spectrometer parameters used were as follows: curtain gas, 30; collisionally activated dissociation (CAD), high; ion spray voltage, −3,500 V; temperature, 500 °C; gas 1, 40; gas 2, 60; declustering potential, collision energy, and cell exit potential vary per transition.
Statistical Analyses
Data from independent experiments were converted to means ± S.E. and compared using analysis of variance or Student's t test. Significant differences were reflected by p values ≤ 0.05.
RESULTS
Chemically induced ER Stress Correlates with Reduced Expression of Anti-apoptotic Bcl-x(L) in INS-1 Cells
To assess the impact of ER stress on Bcl-x(L) expression, INS-1 cells were treated with the ER stressor thapsigargin. In previous reports, we demonstrated that these conditions promote ER stress-induced apoptosis in INS-1 cells, as assessed by accumulation of various ER stress factors, cleaved caspase-3, and loss of mitochondrial membrane potential (12, 18, 21–23). Thapsigargin-induced ER stress resulted in induction of iPLA2β (Fig. 1A) as we have demonstrated previously (17, 18). To investigate the impact of ER stress on anti-apoptotic Bcl-x(L), protein and RNA were harvested from cells treated with thapsigargin for 13 h, and Bcl-x protein levels and RNA splicing, respectively, were assessed. We found that following exposure to thapsigargin, Bcl-x(L) protein was significantly reduced, relative to vehicle-treated cells (Fig. 1B).
FIGURE 1.
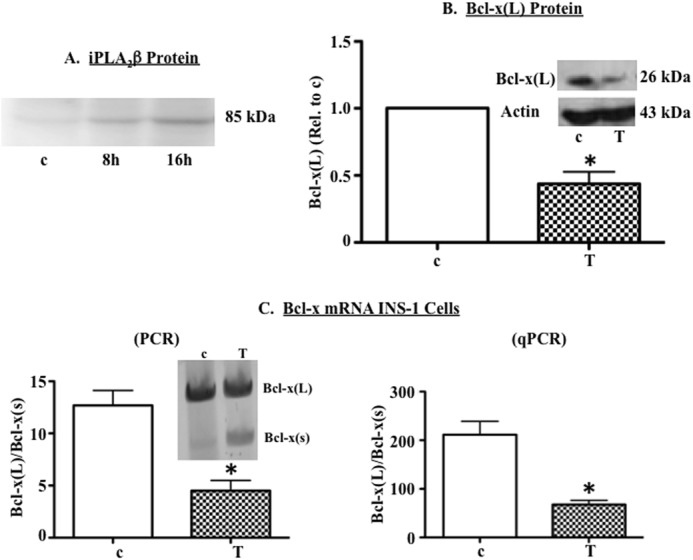
Chemically induced ER stress correlates with reduced expression of anti-apoptotic Bcl-x(L) in β-cells. A, INS-1 cells were treated with 1 μm thapsigargin (T) or DMSO (c), and protein was extracted and used for immunoblot analysis of iPLA2β protein. A representative experiment is shown. B and C, INS-1 cells were cultured for 13 h in the presence of DMSO (c) or thapsigargin (T, 1 μm) and then RNA and protein were extracted. B, representative immunoblot analysis of Bcl-x(L) protein in c- and Tg-treated cells and quantification of three independent immunoblots. Each replicate was derived from an independent experiment that started with freshly plated cells. C, analysis of Bcl-x splice variants in a representative RT-PCR experiment (left panel inset), quantification of Bcl-x(L)/Bcl-x(S) ratio in four independent RT-PCR experiments, and quantification of Bcl-x(L)/Bcl-x(S) ratio in three independent qPCR experiments (right panel). (*, Tg group is significantly different from the c group, p < 0.05.).
We next examined whether the loss of Bcl-x(L) protein correlated with a shift in pre-mRNA splicing away from Bcl-x(L) and in favor of Bcl-x(S), which lacks the 3′ end of exon 2 and does not encode an anti-apoptotic protein (28, 34). RT-PCR and qPCR analyses (Fig. 1C) revealed that vehicle-treated INS-1 cells expressed high levels of Bcl-x(L) mRNA and relatively little Bcl-x(S). However, exposure to thapsigargin resulted in a dramatic shift in Bcl-x RNA splicing as reflected by an ∼75% reduction in the ratio of Bcl-x(L)/Bcl-x(S). The RT-PCR (Fig. 1C, left panel) and qPCR (right panel) analyses were performed on different samples. Although the absolute value of the Bcl-x(L)/Bcl-x(S) ratio differed with the two analyses, thapsigargin-induced fold-changes, relative to vehicle treatment, were comparable (RT-PCR, 3.8 ± 1.5; qRT-PCR, 3.3 ± 1.4).
Spontaneous ER Stress Correlates with Reduced Expression of Anti-apoptotic Bcl-x(L)
To preclude the effects of chemically induced ER stress on Bcl-x 5′-splice site (5′SS) selection, we quantified Bcl-x splice variants in a β-cell line derived from pancreatic islets of Akita mice. β-Cells in Akita islets undergo spontaneous ER stress and subsequent apoptosis, due to a mutation in the INS2 gene and accumulation of pre-proinsulin in the ER (12, 46). As we reported previously (12), spontaneous ER stress in Akita β-cells was associated with increased iPLA2β (Fig. 2A). Analyses of Bcl-x splice variants revealed abundant expression of Bcl-x(L) in both WT and Akita β-cell lines (Fig. 2B) and pancreatic islet β-cells (Fig. 2C). However, Bcl-x(S) expression was barely detectable in the WT preparations but was nearly 3-fold higher in the Akita preparations. Taken together, these data indicate that ER stress is associated with a loss of anti-apoptotic Bcl-x(L) protein, which is secondary to a shift in Bcl-x 5′SS selection.
FIGURE 2.
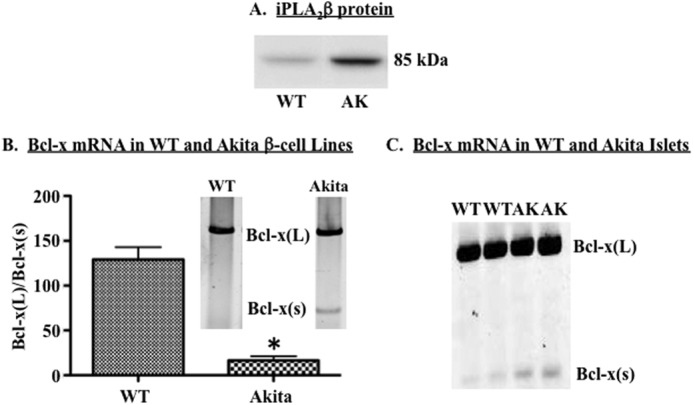
Spontaneous ER stress correlates with reduced expression of anti-apoptotic Bcl-x(L) in β-cells. A, representative immunoblot comparing iPLA2β in wild-type and Akita (AK) β-cells. B, WT and Akita β-cell lines were cultured for 8 h, and RNA was then extracted and RT-PCR used to amplify Bcl-x splice variants. Shown are a representative experiment (inset) and quantification of Bcl-x(L)/Bcl-x(S) ratio in three independent experiments. C, islets were harvested from wild-type (WT) and Akita (AK) mice. RNA was extracted and RT-PCR used to amplify murine Bcl-x splice variants. Two independent experiments are shown. Each quantification is presented as mean ± S.E. (*, Akita (AK) group is significantly different form the WT group, p < 0.05.)
Effects of Chemical Ablation and Overexpression of iPLA2β on 5′SS Selection in Human Bcl-x Minigene
In view of the increases in iPLA2β associated with ER stress, we hypothesized that iPLA2β-derived bioactive lipids might promote the ER stress-induced shift in 5′SS selection in Bcl-x exon 2, leading to reduced Bcl-x(L) mRNA. To further test this, INS-1 cells containing empty vector or a plasmid encoding rat iPLA2β (OE) were transfected with a functional human Bcl-x minigene that we used previously to investigate the effects of ceramides on Bcl-x splicing (41). As expected (39), OE INS-1 cells express nearly 5-fold higher iPLA2β than do empty-vector transfected INS-1 cells (Fig. 3A). We find that in the presence of the iPLA2β-selective inhibitor, (S)-BEL, the ratio of human minigene Bcl-x(L)/Bcl-x(S) mRNA was shifted in favor of Bcl-x(L) (Fig. 3B). In contrast, iPLA2β OE INS-1 cells exhibited a significant decrease in the ratio of Bcl-x(L)/Bcl-x(S) mRNA, relative to vector-transfected cells. Treatment of OE cells with (S)-BEL augmented Bcl-x(L) and restored the ratio of Bcl-x(L)/Bcl-x(S) mRNA to levels observed in vector cells.
FIGURE 3.
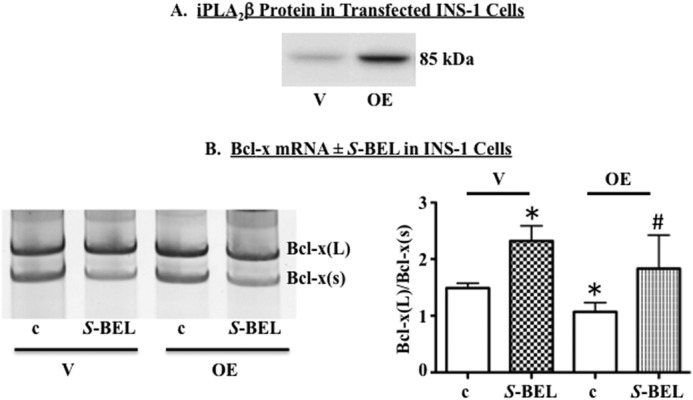
Chemical ablation of iPLA2β promotes and iPLA2β overexpression suppresses selection of downstream 5′SS in human Bcl-x minigene. A, representative immunoblot showing iPLA2β protein levels in INS-1 cells transfected with empty vector (V) or iPLA2β cDNA (OE). B, empty vector and OE INS-1 cells were transfected with plasmid expressing a functional human Bcl-x minigene. Cells were cultured for 13–16 h in the presence of DMSO (c) or (S)-BEL (10 μm). RNA was harvested, and RT-PCR performed to amplify minigene splice variants. Shown are a representative experiment (left panel) and the quantification of four independent experiments (right panel). (*, significantly different from c-treated INS-1 vector cells, p < 0.05; #, significantly different from c-treated INS-1 cells, p < 0.05.) Each quantification is presented as mean ± S.E.
Effects of Genetic Ablation of iPLA2β on 5′SS Selection in Human Bcl-x Minigene
To preclude nonspecific effects of (S)-BEL on Bcl-x splicing, we used iPLA2β-targeted siRNA to specifically reduce iPLA2β expression (Fig. 4A). As we reported previously, iPLA2β siRNA protected INS-1 cells against thapsigargin-induced cell death (Fig. 4B). Co-transfection of INS-1 cells with the siRNA and the functional human Bcl-x minigene promoted an increase in the ratio of Bcl-x(L)/Bcl-x(S), as compared with cells transfected with control siRNA (Fig. 4C). Consistent with this, iPLA2β siRNA largely prevented the thapsigargin-induced accumulation of Bcl-x(S) mRNA in the spontaneous ER stress model (Fig. 4D). Collectively, these data support our hypothesis that iPLA2β modulates Bcl-x 5′SS selection and biases splicing in favor of Bcl-x(S).
FIGURE 4.

Genetic ablation of iPLA2β promotes selection of the downstream Bcl-x 5′ SS in human Bcl-x minigene. INS-1 cells were transfected with control (c) or iPLA2β (iPLA2β) siRNA. A, representative immunoblot analysis of iPLA2β protein in transfected cells. B, INS-1 cells were transfected with control or iPLA2β siRNA and then treated with DMSO (c) or 1 μm thapsigargin (T). Cell death was quantified through trypan blue exclusion assays. Shown are mean ± S.E. from four independent experiments. (*, T group significantly different from control-c or iPLA2β-c, p < 0.0001; #, iPLA2β-T group significantly different from control T group, p < 0.0001.) C, INS-1 cells were co-transfected with Bcl-x minigene and c- or iPLA2β-siRNA. Cells were cultured for 13 h, and then RNA was harvested and RT-PCR performed to amplify minigene splice variants. Shown are a representative experiment (left panel) and quantification (right panel) of four independent experiments (mean ± S.E.). (*, significantly different from control siRNA treatment group, p < 0.05.) D, wild-type (WT) and Akita β-cells were transfected with control (left)- or iPLA2β (right)-siRNA and then treated with 1 μm thapsigargin for 4–16 h. RNA was extracted and RT-PCR performed to amplify Bcl-x splice variants. A representative experiment is shown. Each representative experiment was performed at least twice.
iPLA2β Modulates Use of 5′SS of Endogenous Bcl-x in Islets
The availability of genetically modified mice offered the means to confirm iPLA2β regulation of 5′SS selection in endogenous Bcl-x in primary β-cells. We have demonstrated that iPLA2β is predominantly expressed in insulin-producing islet β-cells and that ER stress-induced apoptosis of those cells is exquisitely sensitive to iPLA2β levels (18). Here, islets isolated from age-matched wild-type (WT), iPLA2β-KO, and RIP-iPLA2β-Tg mice were treated with DMSO (vehicle) or thapsigargin and subsequently screened for endogenous murine Bcl-x splice variants. Consistent with our previous report, iPLA2β expression was verified as being primarily associated with insulin-producing β-cells, expressed at higher levels in RIP-iPLA2β-Tg islets, and absent from iPLA2β-KO islets (Fig. 5A). Consistent with expression in vehicle-treated INS-1 cells, WT islets contained very little Bcl-x(S) RNA under resting conditions (Fig. 5B). However, the ratio of Bcl-x(L)/Bcl-x(S) decreased (∼75%) in response to the ER stressor. In comparison, resting RIP-iPLA2β-Tg islets had higher levels of Bcl-x(S) RNA than WT islets, and the basal ratios of Bcl-x(L)/Bcl-x(S) in RIP-iPLA2β-Tg islets were comparable with those detected in thapsigargin-treated WT islets. Exposure to thapsigargin, however, did not further decrease the Bcl-x(L)/Bcl-x(S) ratio in RIP-iPLA2β-Tg islets. In contrast, under basal conditions KO islets exhibited almost undetectable levels of Bcl-x(S) RNA, as reflected by Bcl-x(L)/Bcl-x(S) ratios of >100, and thapsigargin failed to augment Bcl-x(S). A spurious PCR product was observed in some but not in all amplifications of mouse Bcl-x splice variants. Although this product does not co-migrate with Bcl-x(S), we cannot rule out the possibility that it is another previously unidentified splice variant of Bcl-x. These data provide additional support for our hypothesis that iPLA2β regulates Bcl-x 5′SS selection and promotes the use of the upstream 5′SS that generates Bcl-x(S).
FIGURE 5.
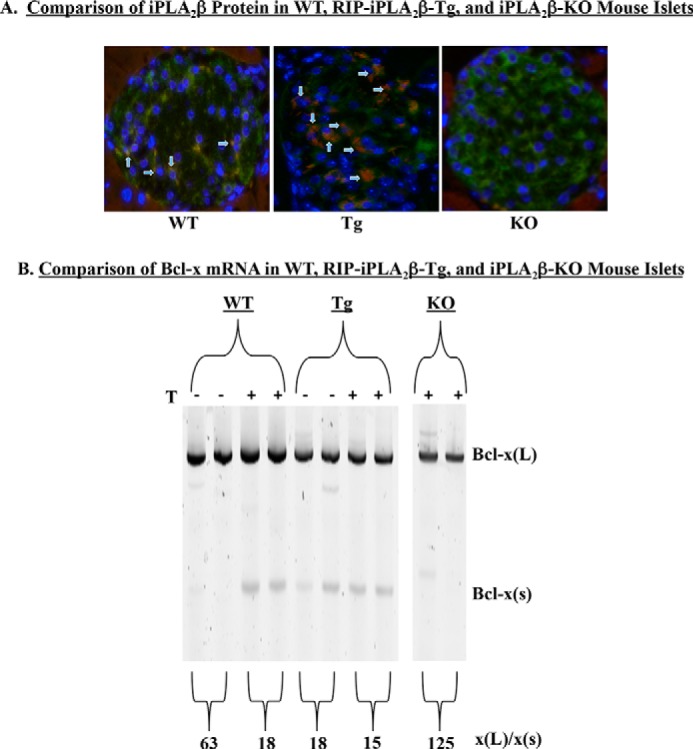
iPLA2β promotes selection of the upstream alternative 5′SS in endogenous islet Bcl-x. Islets were harvested from wild-type (WT), RIP-iPLA2β-Tg (Tg), and iPLA2β−/− (KO) mice. A, immunohistochemistry analysis of iPLA2β (red) and insulin (green). Islets were counterstained with DAPI (blue) to mark the nuclei of individual cells. Merged images are presented, where arrowheads indicate co-expression of iPLA2β and insulin. B, islets were isolated and then cultured in the presence of DMSO (−) or 2 μm thapsigargin (+). RNA was harvested and used for RT-PCR to amplify Bcl-x splice variants. Two mice were studied in each group, and results from both mice are shown. The average ratio of Bcl-x(L)/Bcl-x(S) is shown for each treatment group.
iPLA2β Regulates Bcl-x 5′-Splice Site Selection through Both Ceramide-dependent and Ceramide-independent Mechanisms
In previous studies, we demonstrated the following. (a) ER stress leads to accumulation of ceramides in β-cells. (b) This accumulation is inhibited by (S)-BEL and siRNA targeted to iPLA2β. (c) ER stress-induced ceramide accumulation did not occur via the de novo or salvage pathways but is blocked by chemical inhibition or knockdown of neutral NSMase2. (d) iPLA2β inactivation suppresses NSMase2 induction and ceramide accumulation. (e) Ceramide promotes activation of the alternative 5′SS that generates Bcl-x(S) in A549 lung carcinoma cells (21, 29–31, 48, 52, 54). These observations prompted us to test the possibility that iPLA2β might regulate Bcl-x splicing through ceramides. INS-1 cells were treated with DMSO (c) or 50 μm C6-ceramide (Cer) for 13 h; RNA was harvested; cDNA was prepared and RT-PCR performed to amplify the endogenous β-cell Bcl-x splice variants. In contrast to our previous report in lung carcinoma cells, exogenous ceramide had no significant effect on Bcl-x 5′SS selection in INS-1 cells (Fig. 6A). We next examined whether the exogenous ceramide could overcome effects of iPLA2β inactivation on Bcl-x 5′SS selection. As with the human minigene (Fig. 3B), iPLA2β inactivation largely prevented the shift in 5′SS selection of the endogenous rat Bcl-x in INS-1 cells undergoing ER stress. However, exogenous C6-ceramide did not overcome the effects of (S)-BEL (Fig. 6B).
FIGURE 6.
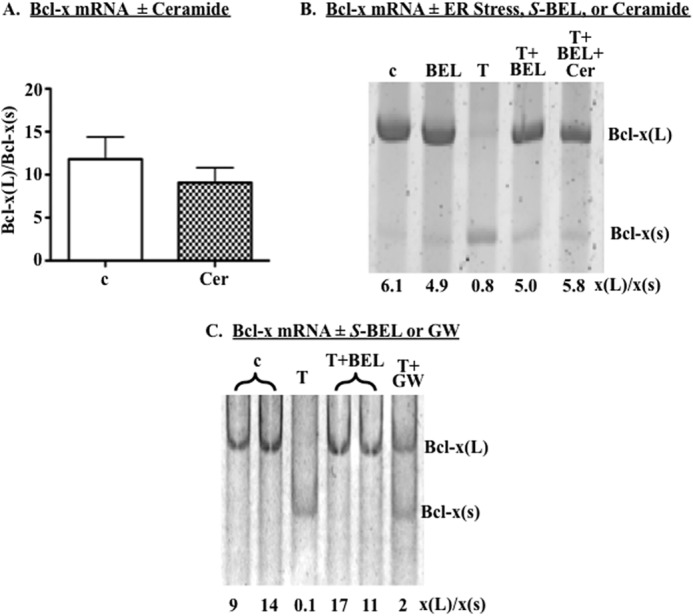
iPLA2β regulates Bcl-x 5′SS selection through both ceramide-dependent and -independent mechanisms. A, INS-1 cells were treated with DMSO (c) or 50 μm C6-ceramide (Cer) for 24 h. RNA was harvested and RT-PCR performed to amplify Bcl-x splice variants. Shown is the ratio of Bcl-x(L)/Bcl-x(S) in four independent experiments. B, INS-1 cells were treated with DMSO (c), 10 μm (S)-BEL (BEL), or 1 μm thapsigargin (T) alone or pretreated with (S)-BEL prior to treatment with thapsigargin ± 50 μm C6-ceramide (Cer) for 13 h. RNA was isolated and RT-PCR performed to amplify Bcl-x splice variants. A representative experiment is shown. C, INS-1 cells were treated with DMSO (c) or thapsigargin alone or pretreated with (S)-BEL (BEL) or 10 μm GW4869 (GW) prior to treatment with thapsigargin for 13 h, and Bcl-x RNAs were amplified. A representative experiment is shown. Each representative experiment was performed at least twice. Each quantification is presented as mean ± S.E.
To further assess ceramide involvement, we tested the possibility that the Bcl-x(L)/Bcl-x(S) ratio could be restored in ER-stressed INS-1 cells by preventing endogenous ceramide accumulation via NSMase2-catalyzed hydrolysis of sphingomyelins. INS-1 cells were therefore treated with a selective inhibitor of NSMase2 (GW4869), which we previously demonstrated as being able to inhibit NSMase2 and completely block ceramide accumulation in β-cells (29–31). Subsequent RT-PCR analyses revealed that GW4869 only partially restored the Bcl-x(L)/x(s) ratio in ER-stressed INS-1 cells (Fig. 6C). This is in contrast to near complete restoration of the Bcl-x(L)/Bcl-x(S) ratio in cells treated with (S)-BEL (Fig. 6, B and C). These observations suggest that iPLA2β likely modulates Bcl-x 5′SS selection through both ceramide-dependent and ceramide-independent mechanisms.
Role of iPLA2β-regulated Lipids in Bcl-x 5′ SS Selection
iPLA2β generates a variety of bioactive lipids that might modulate 5′SS selection in Bcl-x exon 2. We considered the possibility that Bcl-x 5′SS selection was regulated by lysophosphatidylcholine (LPC), a lysolipid product of the reaction catalyzed by iPLA2β. However, exogenous LPC had no effect on Bcl-x splicing in INS-1 cells (data not shown). As we and others have found, ER stress, glucose, and pro-inflammatory cytokines promote arachidonic acid hydrolysis and eicosanoid production in pancreatic islets (47–49), and these responses are mediated by iPLA2β (19, 50, 51). Arachidonic acid has been shown to modulate pre-mRNA splicing in primary rat hepatocytes (52), and β-cell glycerophospholipids are enriched in arachidonic acid (53–55). These observations suggested that arachidonic acid or its metabolites might modulate Bcl-x splicing in β-cells. To address this possibility, we used ESI-MS/MS to quantify polyunsaturated fatty acids (PUFAs) and eicosanoids in culture supernatants of INS-1 cells treated with vehicle, thapsigargin (Tg), or (S)-BEL. INS-1 cells produced a variety of polyunsaturated fatty acids, including prostanoids and other derivatives. Although none of the lipids exhibited statistically significant differences among the three treatment groups, EPA exhibited a trend to accumulate in culture supernatants of Tg-treated INS-1 cells (Fig. 7A, p = 0.075, one-way analysis of variance). We also observed a trend for accumulation of 5-HETE in both thapsigargin and (S)-BEL-treated cells (Fig. 7B). Notably, the ratio of Bcl-x splice variants (Bcl-x(L)/Bcl-x(S)) directly correlated with the ratio of 5-HETE/EPA (Fig. 7C, p = 0.003).
FIGURE 7.
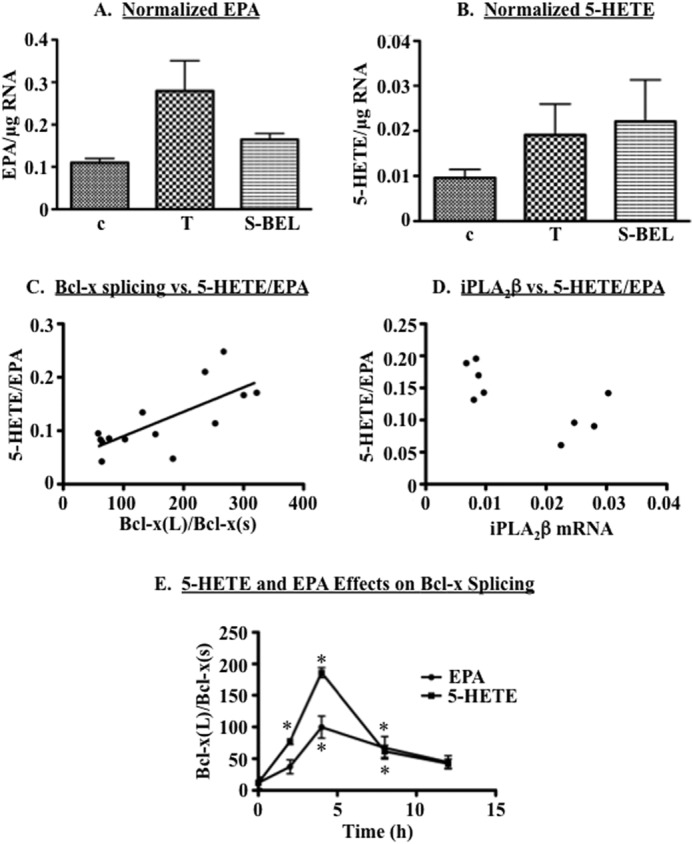
iPLA2β modulates lipid mediators involved in Bcl-x 5′SS selection. A and B, INS-1 cells were treated with DMSO (c), 1 μm thapsigargin (T), or 10 μm (S)-BEL for 13 h. Culture supernatants were harvested, and ESI-MS/MS was performed to quantify polyunsaturated fatty acids and their derivatives. The lipids were normalized to RNA retrieved from cells that conditioned the media. Shown are means ± S.E. of EPA (A) or 5-HETE (B) in three replicates. C, Bcl-x(L) and Bcl-x(S) mRNAs were quantified in samples from A and B, and the Bcl-x(L)/Bcl-x(S) ratio is plotted against the ratio of 5-HETE/EPA. Linear regression analysis indicated a significant correlation (p = 0.003). D, iPLA2β mRNA was quantified in samples from A and B and is plotted against the ratio of 5-HETE/EPA. The 5-HETE/EPA ratio was significantly higher in cells with low iPLA2β expression (p < 0.005, Wilcoxon rank sum test). E, INS-1 cells were treated with vehicle (DMSO), 10 μm 5-HETE, or 10 μm EPA for up to 13 h. RNA was isolated and qPCR performed to quantify Bcl-x splice variants. The data are derived from three independent treatments. (*, significantly different from vehicle control treatment group, p < 0.05.).
Given the inverse correlation between iPLA2β and the Bcl-x(L)/Bcl-x(S) ratio, we postulated that the 5-HETE/EPA ratio would be elevated in cells with low levels of iPLA2β. To test this hypothesis, we measured iPLA2β expression in RNAs isolated from the cells in Fig. 7, A–C. Consistent with our hypothesis, the 5-HETE/EPA ratio was indeed highest in cells with the lowest levels of iPLA2β (Fig. 7D). When taken together, these observations suggest that high iPLA2β activity is associated with a lipid profile that promotes the use of the upstream 5′SS that generates Bcl-x(S) mRNA. They also suggest 5-HETE and EPA as candidate lipids that activate the downstream and upstream 5′SS in Bcl-x exon 2, respectively. To test these possibilities, we treated INS-1 cells with exogenous 5-HETE or EPA and then used qPCR to quantify Bcl-x splice variant mRNAs. We found that 5-HETE robustly increased the Bcl-x(L)/Bcl-x(S) ratio (Fig. 7E). Unexpectedly, EPA also shifted Bcl-x splicing in favor of Bcl-x(L), although to a more modest degree than did 5-HETE. These findings suggest a critical role for 5-HETE in promoting the use of the downstream 5′SS that generates Bcl-x(L) mRNA.
DISCUSSION
Bcl-x(L) protein is an anti-apoptotic and negative regulator of the intrinsic apoptotic pathway (26, 27). Alternative splicing of the Bcl-x pre-mRNA generates Bcl-x(S), which does not encode an anti-apoptotic protein (28). In view of previous studies linking ceramide to alternative splicing of Bcl-x pre-mRNA and iPLA2β to ceramide accumulation and ER stress-induced apoptosis of β-cells (12, 17, 18, 22, 37, 41), we hypothesized that iPLA2β might regulate Bcl-x splicing in β-cells. To test this, we amplified Bcl-x mRNA splice variants in β-cells. Our experiments indicate that high levels of iPLA2β expression/activity promote the use of the alternative 5′SS, as reflected by decreases in the ratio of Bcl-x(L)/Bcl-x(S). Conversely, the conventional 5′SS is favored in cells treated with iPLA2β inhibitor or iPLA2β-targeted siRNA. Both thapsigargin-induced and spontaneous ER stress are associated with a reduced Bcl-x(L)/Bcl-x(S) ratio and lower expression of anti-apoptotic Bcl-x(L) protein. Together, these data are evidence that iPLA2β participation in β-cell apoptosis occurs, in part, through modulation of Bcl-x splicing.
Bcl-x(L), a member of the Bcl-2 family of proteins, suppresses apoptosis when it associates with mitochondrial membranes and prevents their permeabilization, release of cytochrome c, and induction of the intrinsic apoptosis pathway (26, 27). For many years, it has been known that Bcl-x(L) overexpression protects tumor cells from apoptosis induced by chemotherapeutic agents. More recent studies have correlated Bcl-x(L) with increased viability of pancreatic islets. Bcl-x(L)−/− islets are hypersensitive to a variety of pro-apoptotic stimuli, including thapsigargin (29). Immunosuppressive drugs used in transplant therapy reduce islet viability, and this has been correlated with reduced expression of Bcl-x(L) and other Bcl-2 family members (30). Similarly, both cytokine- and high glucose-induced β-cell death are associated with reduced Bcl-x(L) protein (31, 56). Conversely, transduction of full-length Bcl-x(L) or its BH4 domain protects human islets from apoptosis induced by cytokines, staurosporine, or serum deprivation (32, 56). Islets from transgenic mice overexpressing Bcl-x(L) are protected from thapsigargin-induced apoptosis, although the mice exhibit reduced glucose tolerance due to a defect in insulin secretion (33).
Our studies add to this body of literature by demonstrating that β-cell apoptosis in the presence of ER stress is also associated with reduced Bcl-x(L) protein mass. To our knowledge, ours is the first study to correlate reduced Bcl-x(L) protein mass with spontaneous ER stress. Although chemical inactivation, knockdown, and genetic ablation of iPLA2β were all associated with increases in the ratio of Bcl-x(L)/Bcl-x(S) RNA, they did not restore Bcl-x(L) protein in thapsigargin-treated cells (data not shown). These observations suggest that the impact of iPLA2β on Bcl-x 5′SS selection is subtle and cannot overcome the overwhelming effects of thapsigargin. It is also possible that other iPLA2β/splicing-independent mechanisms contribute to the loss of Bcl-x(L) protein in β-cells undergoing ER stress. We also recognize the possibility that SERCA-1 activity could modulate Bcl-x splicing through mechanisms that are independent of ER stress in the thapsigargin-driven model.
Our study is also among the first to delineate molecular mechanisms regulating endogenous Bcl-x(L) protein mass in β-cells, and to our knowledge we are the first to investigate Bcl-x splicing in β-cells. We demonstrate that both thapsigargin-induced and spontaneous ER stress correlate with reduced ratios of both endogenous Bcl-x(L)/Bcl-x(S) RNA in rat insulinoma and murine β-cells and of RNA derived from a human Bcl-x minigene expressed in INS-1 cells. The Bcl-x(S) protein contains BH3 and BH4 domains and is suggested to be pro-apoptotic due it its ability to heterodimerize and neutralize the anti-apoptotic actions of Bcl-x(L) (57, 58). Although endogenous Bcl-x(S) protein has been detected in some cell types (37, 59, 60), evidence for the pro-apoptotic actions of Bcl-x(S) comes primarily from overexpression studies (57, 58, 61–63). Despite the increased abundance of Bcl-x(S) mRNA in INS-1 cells undergoing ER stress, we were unable to detect Bcl-x(S) protein in thapsigargin-treated INS-1 cells or primary islets from mice. It is possible that Bcl-x(S) mRNA is not efficiently translated into protein in β-cells. Alternatively, the Bcl-x(S) protein may be unstable and therefore not accumulate to levels detectable by immunoblot analyses. We therefore suggest that ER stress-induced apoptosis of β-cells is mediated through reduced levels of Bcl-x(L) protein rather than accumulation of Bcl-x(S) protein.
We have not yet fully delineated the biochemical and molecular mechanisms underlying the iPLA2β-regulated splicing of Bcl-x or whether iPLA2β-derived lipids activate the upstream alternative 5′SS, block the downstream conventional 5′SS, or both. Our investigation of regulation of Bcl-x splicing by iPLA2β was prompted by reports from us and others that ceramide promotes selection of the alternative 5′SS that generates Bcl-x(S) (37, 41, 64–66). We demonstrated that ER stress-induced apoptosis is associated with iPLA2β-dependent accumulation of ceramide in INS-1 cells and murine and human islets and that this accumulation results from an iPLA2β-dependent induction of NSMase2 and not increased de novo ceramide synthesis (12, 17, 18, 22). Given this, it seemed likely that the ER stress-induced shift in Bcl-x splicing was mediated by the pool of ceramides that accumulated downstream of iPLA2β. Although we cannot rule out the possibility that endogenous ceramides regulate Bcl-x splicing in β-cells, our data argue for additional ceramide-independent mechanisms on several levels, based on the following. (a) Exogenous C6-ceramide has no effect on the ratio of Bcl-x(L)/Bcl-x(S) in INS-1 cells. (b) The ER stress-induced shift in Bcl-x splicing is only partially reversed by chemical inhibition of NSMase2. (c) Exogenous C6-ceramide does not overcome the effects of (S)-BEL on Bcl-x splicing in INS-1 cells. (d) In sharp contrast to our recent report that ceramide mass is comparable in wild-type and iPLA2β-KO islets (18), the ratio of Bcl-x(L)/Bcl-x(S) is 2–7 times larger in the knock-out islets. These observations indicate that the regulation of Bcl-x 5′SS selection is tissue-specific and may be controlled differently in β-cells than in other cells, which is likely due to tissue-specific expression of RNA-binding proteins that regulate 5′SS selection. Although ceramides may have a limited role in Bcl-x 5′SS selection in β-cells, they are clearly involved in ER stress-induced apoptosis in this cell type (18, 22, 23). In addition to their roles in 5′SS selection, ceramides likely promote β-cell apoptosis through splicing-independent mechanisms that may or may not involve Bcl-x(L).
Our present studies suggest that Bcl-x splice 5′SS selection is regulated by another bioactive lipid downstream of iPLA2β. As arachidonic acid has been linked to alternative splicing of the glucose-6-phosphatase dehydrogenase pre-mRNA (52) and β-cell glycerophospholipids are enriched in arachidonic acid (53–55), we considered the possibility that arachidonic acid, another PUFA, or metabolite might modulate Bcl-x 5′SS selection in INS-1 cells. 12-Lipoxygenase is expressed in human and murine islets (67, 68), and a variety of studies have linked this enzyme and its product, 12-S-HETE, to β-cell apoptosis and dysfunction (69–74). Thapsigargin-induced apoptosis of MIN6 mouse insulinoma cells is suppressed by inhibition of lipoxygenase but not cyclooxygenase activity (74). Given this, we considered 12-HETE and 15-HETE to be attractive candidates for iPLA2β-derived lipids that regulated Bcl-x 5′SS selection. However, neither 12-HETE nor 15-HETE correlated with iPLA2β activity or Bcl-x splicing in INS-1 cells (data not shown). In contrast, we observed a trend toward increased 5-HETE accumulation in (S)-BEL-treated INS-1 cells, and exogenous 5-HETE induced a significant increase (15.5-fold) in the ratio of Bcl-x(L)/Bcl-x(S). These findings suggest the following: (a) iPLA2β modulates the production of bioactive lipids, promoting a profile that is deficient in 5-HETE and (b) 5-HETE promotes use of the downstream 5′SS, resulting in generation of mRNA encoding anti-apoptotic Bcl-x(L). We cannot rule out the possibility that iPLA2β modulates additional bioactive lipids that control Bcl-x 5′SS selection as well. At present, we are focusing on identifying iPLA2β-modulated lipids that activate the alternative upstream 5′SS that generates Bcl-x(S) mRNA. Unexpectedly, our initial candidate (EPA) modestly augmented the Bcl-x(L)/Bcl-x(S) ratio when added exogenously to INS-1 cells.
Likely, iPLA2β-derived lipids modulate expression, post-translational modification, or localization of one or more RNA-binding proteins that regulate Bcl-x 5′SS selection. A variety of splicing factors have been implicated in the regulation of Bcl-x alternative splicing (38, 59, 60, 64, 75, 76). We are currently performing studies to determine whether these proteins are modulated in response to 5-HETE and other bioactive lipids in β-cells.
Our observations contribute to a growing body of evidence linking iPLA2β with apoptosis of β-cells. The link between iPLA2β and apoptosis was first recognized in the 1990s, when Atsumi et al. (77) demonstrated activation of the enzyme upon caspase 3-mediated proteolysis. iPLA2β is highly expressed in insulin producing β-cells and is induced in response to both thapsigargin-stimulated and spontaneous ER stress (12, 17–19, 21). Chemical inhibition and genetic knockdown of iPLA2β in β-cells reduces both ER stress and apoptosis (12, 17, 18, 21–23). Although autophagy can be cytoprotective, like ER stress, this process can contribute to apoptosis when poorly controlled (78). In a recent paper, we demonstrate that thapsigargin-stimulated ER stress induces autophagy in murine islets and that iPLA2β amplifies this process (18). These observations are consistent with increased recognition of cross-talk between autophagy and apoptosis (78). Importantly, Bcl-x(L) suppresses both processes (26, 27, 78, 79). In addition to its ability to associate with and stabilize mitochondrial membranes, Bcl-x(L) also binds the BH3 domain of beclin and prevents it from associating with Vps-34 to induce autophagy (78, 79). We speculate that iPLA2β augments both apoptosis and autophagy by modulating Bcl-x splicing and limiting the availability of Bcl-x(L) protein. Additional experiments are required to test this hypothesis and determine the molecular events underlying iPLA2β regulation of Bcl-x splicing in β-cells. There is accumulating evidence that pro-inflammatory cytokines (critical promoters of auto-immune destruction of β-cells and development of T1D) not only induce ER stress in islet β-cells of diabetes-prone mice (80) and human islet β-cells (81) but also up-regulate the expression/activity of iPLA2β (81). Given this, further elucidation of the mechanism(s) by which iPLA2β activation contributes to β-cell death during the onset and progression of T1D is clearly warranted.
Acknowledgments
We gratefully acknowledge the technical contributions of Sheng Zhang and Ying Gai.
This work was supported, in whole or in part, by National Institutes of Health Grant DK-69455 from NIDDK (to S. R.), Grant HL91388 from NHLBI (to S. E. B.), Grants HL072925 (to C. E. C.), CA154314 (to C. E. C.), and NH1C06-RR17393 (to Virginia Commonwealth University), and P30 CA016059 from NCI Cancer Center. This work was also supported by the American Diabetes Association (to S. R.), National Science Foundation Grant MCB 0544068 (to S. E. B.), Virginia Commonwealth University Presidential Research Incentive Program (to S. E. B.), Veterans Affairs Merit Award BX001792 (to C. E. C.), a Research Career Scientist Award (to C. E. C.), and Grant BSF#2011380 from the United States-Israel Bi-National Science Foundation (to C. E. C.).
- T1D
- type I diabetes
- ER
- endoplasmic reticulum
- (S)-BEL
- bromoenol lactone selective inhibitor of iPLA2β
- c
- control (DMSO)
- iPLA2β
- group VIA phospholipase A2β
- iPLA2β-KO
- global iPLA2β knock out
- OE
- iPLA2β overexpressing INS-1 cells
- RIP-iPLA2β-Tg
- islet-specific iPLA2β transgenic
- 5′SS
- 5′-splice site
- 5-HETE
- 5-hydroxytetraenoic acid
- EPA
- eicosapentaenoic acid
- qPCR
- quantitative PCR
- NSMase2
- neutral sphingomyelinase 2
- Tg
- thapsigargin
- BH
- Bcl-2 homology
- EPA
- eicosapentaenoic acid.
REFERENCES
- 1. Cohen G. M. (1997) Caspases: the executioners of apoptosis. Biochem. J. 326, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oyadomari S., Koizumi A., Takeda K., Gotoh T., Akira S., Araki E., Mori M. (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 109, 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Socha L., Silva D., Lesage S., Goodnow C., Petrovsky N. (2003) The role of endoplasmic reticulum stress in nonimmune diabetes: NOD.k iHEL, a novel model of β-cell death. Ann. N.Y. Acad. Sci. 1005, 178–183 [DOI] [PubMed] [Google Scholar]
- 4. Delépine M., Nicolino M., Barrett T., Golamaully M., Lathrop G. M., Julier C. (2000) EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 25, 406–409 [DOI] [PubMed] [Google Scholar]
- 5. Yamada T., Ishihara H., Tamura A., Takahashi R., Yamaguchi S., Takei D., Tokita A., Satake C., Tashiro F., Katagiri H., Aburatani H., Miyazaki J., Oka Y. (2006) WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic β-cells. Hum. Mol. Genet. 15, 1600–1609 [DOI] [PubMed] [Google Scholar]
- 6. Iwawaki T., Oikawa D. (2013) The role of the unfolded protein response in diabetes mellitus. Semin. Immunopathol. 35, 333–350 [DOI] [PubMed] [Google Scholar]
- 7. Butler A. E., Janson J., Bonner-Weir S., Ritzel R., Rizza R. A., Butler P. C. (2003) β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 [DOI] [PubMed] [Google Scholar]
- 8. Butler A. E., Janson J., Soeller W. C., Butler P. C. (2003) Increased β-cell apoptosis prevents adaptive increase in β-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52, 2304–2314 [DOI] [PubMed] [Google Scholar]
- 9. Sesti G. (2002) Apoptosis in the beta cells: Cause or consequence of insulin secretion defect in diabetes? Ann. Med. 34, 444–450 [DOI] [PubMed] [Google Scholar]
- 10. Kayo T., Koizumi A. (1998) Mapping of murine diabetogenic gene mody on chromosome 7 at D7Mit258 and its involvement in pancreatic islet and beta-cell development during the perinatal period. J. Clin. Invest. 101, 2112–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoshioka M., Kayo T., Ikeda T., Koizumi A. (1997) A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 46, 887–894 [DOI] [PubMed] [Google Scholar]
- 12. Lei X., Zhang S., Barbour S. E., Bohrer A., Ford E. L., Koizumi A., Papa F. R., Ramanadham S. (2010) Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. J. Biol. Chem. 285, 6693–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mandrup-Poulsen T. (2001) Beta-cell apoptosis: stimuli and signaling. Diabetes 50, S58–S63 [DOI] [PubMed] [Google Scholar]
- 14. Rabinovitch A., Suarez-Pinzon W. L. (1998) Cytokines and their roles in pancreatic islet β-cell destruction and insulin-dependent diabetes mellitus. Biochem. Pharmacol. 55, 1139–1149 [DOI] [PubMed] [Google Scholar]
- 15. Araki E., Oyadomari S., Mori M. (2003) Impact of endoplasmic reticulum stress pathway on pancreatic β-cells and diabetes mellitus. Exp. Biol. Med. 228, 1213–1217 [DOI] [PubMed] [Google Scholar]
- 16. Gijón M. A., Leslie C. C. (1997) Phospholipases A2. Semin. Cell Dev. Biol. 8, 297–303 [DOI] [PubMed] [Google Scholar]
- 17. Lei X., Zhang S., Bohrer A., Barbour S. E., Ramanadham S. (2012) Role of calcium-independent phospholipase A2β in human pancreatic islet β-cell apoptosis. Am. J. Physiol. Endocrinol. Metab. 303, E1386–E1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lei X., Bone R. N., Ali T., Wohltmann M., Gai Y., Goodwin K. J., Bohrer A. E., Turk J., Ramanadham S. (2013) Genetic modulation of islet beta-cell iPLA2β expression provides evidence for its impact on beta-cell apoptosis and autophagy. Islets 5, 29–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gross R. W., Ramanadham S., Kruszka K. K., Han X., Turk J. (1993) Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry 32, 327–336 [DOI] [PubMed] [Google Scholar]
- 20. Lei X., Barbour S. E., Ramanadham S. (2010) Group VIA Ca2+-independent phospholipase A2 (iPLA2β) and its role in β-cell programmed cell death. Biochimie 92, 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramanadham S., Hsu F. F., Zhang S., Jin C., Bohrer A., Song H., Bao S., Ma Z., Turk J. (2004) Apoptosis of insulin-secreting cells induced by endoplasmic reticulum stress is amplified by overexpression of group VIA calcium-independent phospholipase A2 (iPLA2β) and suppressed by inhibition of iPLA2β. Biochemistry 43, 918–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lei X., Zhang S., Bohrer A., Bao S., Song H., Ramanadham S. (2007) The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry 46, 10170–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lei X., Zhang S., Bohrer A., Ramanadham S. (2008) Calcium-independent phospholipase A2 (iPLA2β)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J. Biol. Chem. 283, 34819–34832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bone R. N., Gai Y., Magrioti V., Kokotou M. G., Ali T., Lei X., Tse H. M., Kokotos G., Ramanadham S. (2015) Inhibition of Ca2+-independent phospholipase A2 (iPLA2β) ameliorates islet infiltration and incidence of diabetes in NOD mice. Diabetes 64, 541–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vannuvel K., Renard P., Raes M., Arnould T. (2013) Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 228, 1802–1818 [DOI] [PubMed] [Google Scholar]
- 26. Michels J., Kepp O., Senovilla L., Lissa D., Castedo M., Kroemer G., Galluzzi L. Functions of Bcl-x(L) at the interface between cell death and metabolism. Int. J. Cell Biol. 2013. 705294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams J. M., Cory S. (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yip K. W., Reed J. C. (2008) Bcl-2 family proteins and cancer. Oncogene 27, 6398–6406 [DOI] [PubMed] [Google Scholar]
- 29. Carrington E. M., McKenzie M. D., Jansen E., Myers M., Fynch S., Kos C., Strasser A., Kay T. W., Scott C. L., Allison J. (2009) Islet beta-cells deficient in Bcl-xL develop but are abnormally sensitive to apoptotic stimuli. Diabetes 58, 2316–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hui H., Khoury N., Zhao X., Balkir L., D'Amico E., Bullotta A., Nguyen E. D., Gambotto A., Perfetti R. (2005) Adenovirus-mediated XIAP gene transfer reverses the negative effects of immunosuppressive drugs on insulin secretion and cell viability of isolated human islets. Diabetes 54, 424–433 [DOI] [PubMed] [Google Scholar]
- 31. Federici M., Hribal M., Perego L., Ranalli M., Caradonna Z., Perego C., Usellini L., Nano R., Bonini P., Bertuzzi F., Marlier L. N., Davalli A. M., Carandente O., Pontiroli A. E., Melino G., et al. (2001) High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: a potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes 50, 1290–1301 [DOI] [PubMed] [Google Scholar]
- 32. Klein D., Ribeiro M. M., Mendoza V., Jayaraman S., Kenyon N. S., Pileggi A., Molano R. D., Inverardi L., Ricordi C., Pastori R. L. (2004) Delivery of Bcl-XL or its BH4 domain by protein transduction inhibits apoptosis in human islets. Biochem. Biophys. Res. Commun. 323, 473–478 [DOI] [PubMed] [Google Scholar]
- 33. Zhou Y.-P., Pena J. C., Roe M. W., Mittal A., Levisetti M., Baldwin A. C., Pugh W., Ostrega D., Ahmed N., Bindokas V. P., Philipson L. H., Hanahan D., Thompson C. B., Polonsky K. S. (2000) Overexpression of Bcl-x(L) in beta-cells prevents cell death but impairs mitochondrial signal for insulin secretion. Am. J. Physiol. Endocrinol. Metab. 278, E340–E351 [DOI] [PubMed] [Google Scholar]
- 34. Schwerk C., Schulze-Osthoff K. (2005) Regulation of apoptosis by alternative pre-mRNA splicing. Mol. Cell 19, 1–13 [DOI] [PubMed] [Google Scholar]
- 35. Mercatante D. R., Bortner C. D., Cidlowski J. A., Kole R. (2001) Modification of alternative splicing of Bcl-x pre-mRNA in prostate and breast cancer cells. analysis of apoptosis and cell death. J. Biol. Chem. 276, 16411–16417 [DOI] [PubMed] [Google Scholar]
- 36. Bauman J. A., Li S.-D., Yang A., Huang L., Kole R. (2010) Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 38, 8348–8356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chalfant C. E., Rathman K., Pinkerman R. L., Wood R. E., Obeid L. M., Ogretmen B., Hannun Y. A. (2002) De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells–dependence on protein phosphatase-1. J. Biol. Chem. 277, 12587–12595 [DOI] [PubMed] [Google Scholar]
- 38. Revil T., Pelletier J., Toutant J., Cloutier A., Chabot B. (2009) Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-x(S) splice isoform. J. Biol. Chem. 284, 21458–21467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma Z., Ramanadham S., Wohltmann M., Bohrer A., Hsu F. F., Turk J. (2001) Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2β) indicate a signaling rather than a housekeeping role for iPLA2β. J. Biol. Chem. 276, 13198–13208 [DOI] [PubMed] [Google Scholar]
- 40. Nozaki Ji., Kubota H., Yoshida H., Naitoh M., Goji J., Yoshinaga T., Mori K., Koizumi A., Nagata K. (2004) The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/Akita pancreatic β-cells. Genes Cells 9, 261–270 [DOI] [PubMed] [Google Scholar]
- 41. Massiello A., Salas A., Pinkerman R. L., Roddy P., Roesser J. R., Chalfant C. E. (2004) Identification of two RNA cis-elements that function to regulate the 5′-splice site selection of Bcl-x pre-mRNA in response to ceramide. J. Biol. Chem. 279, 15799–15804 [DOI] [PubMed] [Google Scholar]
- 42. Bao S., Jacobson D. A., Wohltmann M., Bohrer A., Jin W., Philipson L. H., Turk J. (2008) Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2β in pancreatic β-cells and in iPLA2β-null mice. Am. J. Physiol. Endocrinol. Metab. 294, E217–E229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bao S., Miller D. J., Ma Z., Wohltmann M., Eng G., Ramanadham S., Moley K., Turk J. (2004) Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J. Biol. Chem. 279, 38194–38200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wijesinghe D. S., Brentnall M., Mietla J. A., Hoeferlin L. A., Diegelmann R. F., Boise L. H., Chalfant C. E. (2014) Ceramide kinase is required for a normal eicosanoid response and the subsequent orderly migration of fibroblasts. J. Lipid Res. 55, 1298–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simanshu D. K., Kamlekar R. K., Wijesinghe D. S., Zou X., Zhai X., Mishra S. K., Molotkovsky J. G., Malinina L., Hinchcliffe E. H., Chalfant C. E., Brown R. E., Patel D. J. (2013) Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 500, 463–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., Takeuchi T., Tanaka S., Kubo S. K., Kayo T., Lu D., Takata K., Koizumi A., Izumi T. (1999) A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Invest. 103, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Turk J., Mueller M., Bohrer A., Ramanadham S. (1992) Arachidonic acid metabolism in isolated pancreatic islets. VI. Carbohydrate insulin secretagogues must be metabolized to induce eicosanoid release. Biochim. Biophys. Acta 1125, 280–291 [DOI] [PubMed] [Google Scholar]
- 48. Turk J., Hughes J. H., Easom R. A., Wolf B. A., Scharp D. W., Lacy P. E., McDaniel M. L. (1988) Arachidonic acid metabolism and insulin secretion by isolated human pancreatic islets. Diabetes 37, 992–996 [DOI] [PubMed] [Google Scholar]
- 49. Hughes J. H., Easom R. A., Wolf B. A., Turk J., McDaniel M. L. (1989) Interleukin 1-induced prostaglandin E2 accumulation by isolated pancreatic islets. Diabetes 38, 1251–1257 [DOI] [PubMed] [Google Scholar]
- 50. Ramanadham S., Wolf M. J., Jett P. A., Gross R. W., Turk J. (1994) Characterization of an ATP-stimulatable Ca2+-independent phospholipase A2 from clonal insulin-secreting HIT cells and rat pancreatic islets: a possible molecular component of the beta-cell fuel sensor. Biochemistry 33, 7442–7452 [DOI] [PubMed] [Google Scholar]
- 51. Ramanadham S., Gross R. W., Han X., Turk J. (1993) Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in beta-cell cytosolic calcium ion concentration. Biochemistry 32, 337–346 [DOI] [PubMed] [Google Scholar]
- 52. Tao H., Szeszel-Fedorowicz W., Amir-Ahmady B., Gibson M. A., Stabile L. P., Salati L. M. (2002) Inhibition of the splicing of glucose-6-phosphate dehydrogenase precursor mRNA by polyunsaturated fatty acids. J. Biol. Chem. 277, 31270–31278 [DOI] [PubMed] [Google Scholar]
- 53. Ramanadham S., Bohrer A., Mueller M., Jett P., Gross R. W., Turk J. (1993) Mass spectrometric identification and quantitation of arachidonate-containing phospholipids in pancreatic islets: prominence of plasmenylethanolamine molecular species. Biochemistry 32, 5339–5351 [DOI] [PubMed] [Google Scholar]
- 54. Ramanadham S., Bohrer A., Gross R. W., Turk J. (1993) Mass spectrometric characterization of arachidonate-containing plasmalogens in human pancreatic islets and in rat islet beta-cells and subcellular membranes. Biochemistry 32, 13499–13509 [DOI] [PubMed] [Google Scholar]
- 55. Nowatzke W., Ramanadham S., Ma Z., Hsu F. F., Bohrer A., Turk J. (1998) Mass spectrometric evidence that agents that cause loss of Ca2+ from intracellular compartments induce hydrolysis of arachidonic acid from pancreatic islet membrane phospholipids by a mechanism that does not require a rise in cytosolic Ca2+ concentration. Endocrinology 139, 4073–4085 [DOI] [PubMed] [Google Scholar]
- 56. Holohan C., Szegezdi E., Ritter T., O'Brien T., Samali A. (2008) Cytokine-induced beta-cell apoptosis is NO-dependent, mitochondria-mediated and inhibited by Bcl-x(L). J. Cell. Mol. Med. 12, 591–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Minn A. J., Boise L. H., Thompson C. B. (1996) Bcl-x(S) antagonizes the protective effects of Bcl-x(L). J. Biol. Chem. 271, 6306–6312 [DOI] [PubMed] [Google Scholar]
- 58. Braun T., Dar S., Vorobiov D., Lindenboim L., Dascal N., Stein R. (2003) Expression of Bcl-x(S) in Xenopus oocytes induces BH3-dependent and caspase-dependent cytochrome c release and apoptosis. Mol. Cancer Res. 1, 186–194 [PubMed] [Google Scholar]
- 59. Paronetto M. P., Achsel T., Massiello A., Chalfant C. E., Sette C. (2007) The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 176, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Merdzhanova G., Edmond V., De Seranno S., Van den Broeck A., Corcos L., Brambilla C., Brambilla E., Gazzeri S., Eymin B. (2008) E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 15, 1815–1823 [DOI] [PubMed] [Google Scholar]
- 61. Ealovega M. W., McGinnis P. K., Sumantran V. N., Clarke M. F., Wicha M. S. (1996) Bcl-xs gene therapy induces apoptosis of human mammary tumors in nude mice. Cancer Res. 56, 1965–1969 [PubMed] [Google Scholar]
- 62. Han J. S., Núñez G., Wicha M. S., Clarke M. F. (1998) Targeting cancer cell death with a Bcl-x(S) adenovirus. Springer Semin. Immunopathol. 19, 279–288 [DOI] [PubMed] [Google Scholar]
- 63. Mitra R. S., Benedict M. A., Qian D., Foreman K. E., Ekhterae D., Nickoloff B. J., Nuñez G. (2001) Killing of sarcoma cells by pro-apoptotic Bcl-X(S): role of the BH3 domain and regulation by Bcl-x(L). Neoplasia 3, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Massiello A., Roesser J. R., Chalfant C. E. (2006) SAP155 Binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5′-splice site selection of Bcl-x pre-mRNA. FASEB J. 20, 1680–1682 [DOI] [PubMed] [Google Scholar]
- 65. Yang H., Sadda M. R., Li M., Zeng Y., Chen L., Bae W., Ou X., Runnegar M. T., Mato J. M., Lu S. C. (2004) S-Adenosylmethionine and its metabolite induce apoptosis in HepG2 cells: role of protein phosphatase 1 and Bcl-x (s). Hepatology 40, 221–231 [DOI] [PubMed] [Google Scholar]
- 66. Xiao Q., Ford A. L., Xu J., Yan P., Lee K. Y., Gonzales E., West T., Holtzman D. M., Lee J. M. (2012) Bcl-x pre-mRNA splicing regulates brain injury after neonatal hypoxia-ischemia. J. Neurosci. 32, 13587–13596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Green-Mitchell S. M., Tersey S. A., Cole B. K., Ma K., Kuhn N. S., Cunningham T. D., Maybee N. A., Chakrabarti S. K., McDuffie M., Taylor-Fishwick D. A., Mirmira R. G., Nadler J. L., Morris M. A. (2013) Deletion of 12/15-lipoxygenase alters macrophage and islet function in NOD-Alox15(null) mice, leading to protection against type 1 diabetes development. PLoS One 8, e56763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Persaud S. J., Muller D., Belin V. D., Kitsou-Mylona I., Asare-Anane H., Papadimitriou A., Burns C. J., Huang G. C., Amiel S. A., Jones P. M. (2007) The role of arachidonic acid and its metabolites in insulin secretion from human islets of Langerhans. Diabetes 56, 197–203 [DOI] [PubMed] [Google Scholar]
- 69. Dobrian A. D., Lieb D. C., Cole B. K., Taylor-Fishwick D. A., Chakrabarti S. K., Nadler J. L. (2011) Functional and pathological roles of the 12- and 15-lipoxygenases. Prog. Lipid Res. 50, 115–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ma K., Nunemaker C. S., Wu R., Chakrabarti S. K., Taylor-Fishwick D. A., Nadler J. L. (2010) 12-Lipoxygenase products reduce insulin secretion and β-cell viability in human islets. J. Clin. Endocrinol. Metab. 95, 887–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Prasad K. M., Thimmalapura P. R., Woode E. A., Nadler J. L. (2003) Evidence that increased 12-lipoxygenase expression impairs pancreatic beta-cell function and viability. Biochem. Biophys. Res. Commun. 308, 427–432 [DOI] [PubMed] [Google Scholar]
- 72. Weaver J. R., Holman T. R., Imai Y., Jadhav A., Kenyon V., Maloney D. J., Nadler J. L., Rai G., Simeonov A., Taylor-Fishwick D. A. (2012) Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol. Cell. Endocrinol. 358, 88–95 [DOI] [PubMed] [Google Scholar]
- 73. Bleich D., Chen S., Zipser B., Sun D., Funk C. D., Nadler J. L. (1999) Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J. Clin. Invest. 103, 1431–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou Y. P., Teng D., Dralyuk F., Ostrega D., Roe M. W., Philipson L., Polonsky K. S. (1998) Apoptosis in insulin-secreting cells. Evidence for the role of intracellular Ca2+ stores and arachidonic acid metabolism. J. Clin. Invest. 101, 1623–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cloutier P., Toutant J., Shkreta L., Goekjian S., Revil T., Chabot B. (2008) Antagonistic effects of the SRp30c protein and cryptic 5′-splice sites on the alternative splicing of the apoptotic regulator Bcl-x. J. Biol. Chem. 283, 21315–21324 [DOI] [PubMed] [Google Scholar]
- 76. Leu S., Lin Y. M., Wu C. H., Ouyang P. (2012) Loss of Pnn expression results in mouse early embryonic lethality and cellular apoptosis through SRSF1-mediated alternative expression of Bcl-x(S) and ICAD. J. Cell Sci. 125, 3164–3172 [DOI] [PubMed] [Google Scholar]
- 77. Atsumi G., Tajima M., Hadano A., Nakatani Y., Murakami M., Kudo I. (1998) Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2 which undergoes proteolytic inactivation. J. Biol. Chem. 273, 13870–13877 [DOI] [PubMed] [Google Scholar]
- 78. Zhou F., Yang Y., Xing D. (2011) Bcl-2 and Bcl-x(L) play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 278, 403–413 [DOI] [PubMed] [Google Scholar]
- 79. Kang R., Zeh H. J., Lotze M. T., Tang D. (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tersey S. A., Nishiki Y., Templin A. T., Cabrera S. M., Stull N. D., Colvin S. C., Evans-Molina C., Rickus J. L., Maier B., Mirmira R. G. (2012) Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61, 818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lei X., Bone R. N., Ali T., Zhang S., Bohrer A., Tse H. M., Bidasee K. R., Ramanadham S. (2014) Evidence of contribution of iPLA2β-mediated events during islet beta-cell apoptosis due to pro-inflammatory cytokines suggests a role for iPLA2β in T1D development. Endocrinology 155, 3352–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]