

Background: Mmp13 is vital to bone homeostasis and controlled by a plethora of stimuli.
Results: Mmp13 is modulated by distinct distal enhancers for basal (−30 kb) and vitamin D regulation (−10 kb).
Conclusion: Enhancer deletions lead to altered transcription factor occupancy and expression for Mmp13.
Significance: Specific CRISPR deletions reveal the repressive secondary effect of VDR, coordinated multi-enhancer gene control, and distal basal regulation of Mmp13.
Keywords: CCAAT Enhancer-binding Protein (C/EBP), ChIP-Sequencing (ChIP-seq), CRISPR/Cas, Histone Modification, Matrix Metalloproteinase (MMP), Osteoblast, Mmp13, RUNX2, VDR, Collagenase-3
Abstract
Matrix metalloproteinase 13 (Mmp13, collagenase-3) plays an essential role in bone metabolism and mineral homeostasis. It is regulated by numerous factors, including BMP-2, parathyroid hormone, and 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3), through transcription factors such as Runt-related transcription factor 2 (RUNX2), CCAAT/enhancer-binding protein β (C/EBPβ), OSX, and vitamin D receptor (VDR). During osteoblast maturation, the basal expression of Mmp13 and its sensitivity to 1,25(OH)2D3 are strikingly increased. In this report, ChIP-sequencing analysis in mouse preosteoblasts revealed that the Mmp13 gene was probably regulated by three major enhancers located −10, −20, and −30 kb upstream of the gene promoter, occupied by activated VDR and prebound C/EBPβ and RUNX2, respectively. Initially, bacterial artificial chromosome clone recombineering and traditional mutagenesis defined binding sites for VDR and RUNX2. We then employed a CRISPR/Cas9 gene editing approach to delete the −10 and −30 kb Mmp13 enhancers, a region proximal to the promoter, and VDR or RUNX2. VDR-mediated up-regulation of Mmp13 transcription was completely abrogated upon removal of the −10 kb enhancer, resulting in a 1,25(OH)2D3-directed repression of Mmp13. Deletion of either the −30 kb enhancer or RUNX2 resulted in a complete loss of basal transcript activity and a ChIP-identified destabilization of the chromatin enhancer environment and factor binding. Whereas enhancer deletions only affected Mmp13 expression, the RUNX2 deletion led to changes in gene expression, a reduction in cellular proliferation, and an inability to differentiate. We conclude that the Mmp13 gene is regulated via at least three specific distal enhancers that display independent activities yet are able to integrate response from multiple signaling pathways in a model of activation and suppression.
Introduction
The primary function of Mmp13 (collagenase-3, Mmp-13) in osteoblasts and chondrocytes is to facilitate the breakdown of extracellular proteins, such as collagen during bone remodeling (1). However, Mmp13 also plays non-skeletal roles and has been implicated in the invasiveness and progression of different types of cancers, including breast, lung, esophageal, multiple myeloma, and chondrosarcomas (2–4). The Mmp13 gene was first cloned from a human breast tumor (5) and was found to be part of a larger family of matrix metalloproteases. The Mmp13 knock-out mouse displays profound defects in growth plate cartilage and endochondral ossification and exhibits increased interstitial collagen accumulation (6). Studies also link the loss of Mmp13 in an ApoE null background to an increase in atherosclerosis due to this same collagen accumulation (7). Importantly, Mmp13 can be regulated by a litany of different regulatory factors and cellular stimuli. Early studies found that Mmp13 was regulated by BMP2 (8), PTH2 (9), estrogens (10), and heparin (11). It was also shown to be responsive in human chondrosarcoma cells to FGF-2 (fibroblast growth factor 2) (2). Mmp13 expression is similarly increased by cytokines, such as IL-1β and TNFα, activated by cFOS through an AP-1 site located near the gene promoter (12), yet inhibited in osteoblast cultures by IGF-1 (13). In studies of PTH action, Mmp13 was found to be regulated by Runt-related transcription factor-2 (RUNX2) through a Runt binding domain DNA sequence located near an AP-1 site adjacent to the Mmp13 promoter (9). Additional studies revealed that both PKC and PKA signaling pathways are active at the AP-1 and RUNX2 binding sites, respectively (14, 15). This ongoing work has focused much of the molecular investigation of Mmp13 regulation by PTH and other hormones on these specific promoter proximal sites in bone cells.
RUNX2 is an essential transcription factor for the generation of osteoblasts and for the formation of chondrocytes because mice lacking this transcription factor are unable to complete these required tasks for normal bone formation (1, 16, 17). RUNX2 and its family members, RUNX1 and RUNX3, bind to DNA using a common heterodimer partner CBFβ to coordinate the expression of numerous genes (18). RUNX2 is known to drive the expression of genes responsible for the osteoblast phenotype, such as osteopontin (Spp1), osteocalcin (Bglap), vitamin D receptor (Vdr), osterix (Sp7), and Runx2 itself (19). Similar to Mmp13, aberrant expression of RUNX2 manifests in numerous cancerous cells (20, 21). The Mmp13 gene was also found to be regulated by the VDR-activating ligand 1,25(OH)2D3 in rats as well as in mouse MC3T3-E1 cells (22, 23). This latter study suggested that the induction of Mmp13 by 1,25(OH)2D3 was due to an interaction at an AP-1 site located near the transcriptional start site (TSS) but not at an adjacent RUNX2 site. However, no VDR DNA binding response element (VDRE) was identified near Mmp13 in that investigation.
Recent ChIP-seq studies suggest that many if not most genes are regulated through multiple elements located distal to promoters, often within introns and in surrounding intergenic regions many kilobases from the promoters of the genes that they regulate (24, 25). We have recently cataloged genome-wide binding patterns for RUNX2 and C/EBPβ as a function of osteoblast differentiation and have shown that the binding sites for these transcription factors are predominantly located tens if not hundreds of kilobases from their target genes and that the cistromes (collection of all cis-acting transcriptional elements) for both of these master regulators change in a dynamic fashion during the differentiation process (19, 24). Interestingly, the binding of RUNX2 and C/EBPβ can occur either to a single enhancer of a target gene or to separate regulatory enhancers that contribute to the modulation of the same gene but in both cases integrate the transcriptional output. We also discovered that these two factors were often bound to genes that were in turn regulated by 1,25(OH)2D3 and that the VDR was also frequently present at these distal enhancer sites as well (19, 24), suggesting the presence of a general “osteoblast enhancer complex.” These observations, together with the absence of an unbiased analysis of the Mmp13 landscape, raise the possibility that in addition to the promoter proximal elements investigated over the past decade, this gene may also be controlled by more distal regulatory regions.
In this report, we identified three specific distal enhancers located −10, −20, and −30 kb upstream of the Mmp13 TSS using ChIP-seq data obtained from previous studies (26). Subsequent analyses revealed that the VDR bound almost exclusively to the −10 kb enhancer, and that this enhancer mediated the actions of 1,25(OH)2D3 at the Mmp13 gene, strongly supported by evidence from traditional mutagenesis, bacterial artificial chromosome (BAC) clone recombineered reporters, and CRISPR/Cas9 genomic deletion. The basal level of expression of the Mmp13 gene was highly regulated by RUNX2; this control, however, was exerted via transcription factor binding at the enhancer located −30 kb from the promoter proximal region. Both deletion of this enhancer and genetic knockout of RUNX2 in the cell line abrogated virtually all basal transcriptional activity of Mmp13. Finally, ChIP analysis in daughter cells containing the specific enhancer deletions revealed that the loss of the −30 kb region or deletion of RUNX2 expression not only eliminated RUNX2 binding but disrupted transcription factor interaction at both the −10 kb enhancer and the promoter proximal region. Here we describe a complex mechanism wherein the interplay between RUNX2, C/EBPβ, and the VDR is evident at individual enhancers that control Mmp13 expression. This interplay is capable of mediating both induction and repression of the gene, mechanisms that may have broad implications for 1,25(OH)2D3-mediated regulation in additional target cells.
EXPERIMENTAL PROCEDURES
Reagents
1,25(OH)2D3 was obtained from SAFC Global (Madison, WI). All ChIP antibodies were purchased from Santa Cruz Biotechnology, Inc., Millipore (Billerica, MA), or Abcam (Cambridge, MA), as reported previously (24). All quantitative real-time PCR (qPCR) reagents (Fast Start SYBR Green Master Mix (with rox)) were obtained from Roche Applied Science, and TaqMan gene expression assays were obtained from Life Technologies, Inc. (Applied Biosystems, Foster City, CA). All qPCR was conducted on a StepOnePlus from Applied Biosystems. Primers were obtained from IDT (Coralville, IA), and the sequences are available upon request and on our laboratory's website.
Cell Culture and Differentiation
MC3T3-E1 cells (preosteoblast; POB) (early passage line from Sudo et al. (27)) were cultured in minimum Eagle's medium α (MEMα) modification supplemented with 10% heat-inactivated fetal bovine serum from Hyclone (Logan, UT) and 1% penicillin-streptomycin from Invitrogen. UAMS-32 (28) and UAMS-32PB (UAMS-PB, blasticidin-resistant PTH receptor expressing UAMS) (29) (gift from Charles O'Brien, University of Arkansas Medical School) were cultured in MEMα with 10% fetal bovine serum and 1% penicillin-streptomycin. For differentiation, cells were grown to confluence and then transferred to differentiation medium (10 mm β-glycerophosphate and 50 μg/ml ascorbic acid) for the indicated lengths, replenishing the medium every 2–3 days until the assay. Cells were stained for calcium content with Alizarin Red as reported previously (19). Calcium content was quantitatively measured by decalcifying the cells with 0.6 n HCl at 4 ºC for 24 h, followed by a triplicate analysis using the o-cresolphthalein complexone method (Sigma-Aldrich) normalized to total protein content of the calcium-stripped cells.
Gene Expression Analysis
Cells were treated for 24 h with the indicated factors, and 1 μg of isolated total RNA was reverse transcribed using the High Capacity cDNA Kit (Applied Biosystems) and then diluted to 100 μl with RNase/DNase I-free water. qPCR was performed using primers specific to a select set of genes by TaqMan analyses.
Molecular Cloning
The pCH110-β galactosidase reporter plasmid was described previously (30). All gene-specific pTK plasmids were constructed by cloning the appropriate mouse DNA fragments obtained through DNA amplification of genomic MC3T3-E1 DNA into the pTK-luc vector using BamHI, SalI, and/or HindIII restriction sites. Mutations were introduced using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) as per the manufacturer's protocol and recommendations. BAC luciferase reporter constructs were created via recombineering using the BAC clone RP23-355F15 and methods established previously (31, 32). All CRISPR plasmids were constructed by cloning into the BbsI site as described previously (33). RUNX2 lentiviral overexpression plasmid (pLeGO-RUNX2–2A-GFP) was created through Gibson isothermal assembly (34) of PCR cDNA for mouse RUNX2 derived from pCMV-RUNX2 (35) and pLeGO-G (36) (Addgene 27347). The RUNX2 cDNA was inserted together with a P2A sequence (GGATCCGGAGCCACGAACTTCTCTCTGTTAAAGCAAGCAGGAGACGTGGAAGAAAACCCCGGTCCT) prior to the GFP of the LeGO-G plasmid. RUNX2 was PCR-amplified using the primers available on our laboratory's Web site, gel-purified, and combined through Gibson assembly with an alkaline phosphatase-pretreated, BamHI-digested LeGO-G plasmid. The resulting clones were sequenced for verification of the appropriate RUNX2–2A insertion and confirmed by Western blot for size (slight shift due to additional P2A sequence 3′) and appropriate expression.
Analysis of Enhancer Activity Using Reporter Genes
MC3T3-E1 cells were seeded into 24-well plates in MEMα containing 10% FBS at a concentration of 5.0 × 104 cells/well and transfected 24 h later with Lipofectamine PLUS (Invitrogen) in serum- and antibiotic-free medium. Individual wells were co-transfected with 250 ng of a luciferase reporter vector and 50 ng of pCH110-βgal. After transfection, the cells were cultured in medium supplemented with 20% FBS and 100 nm 1,25(OH)2D3 or ethanol vehicle. Cells were harvested 18 h after treatment, and the lysates were assayed for luciferase and β-galactosidase activities in triplicate ± S.E. as described previously (37). Luciferase activity was normalized to β-galactosidase activity in all cases.
CRISPR/Cas9 Genome Editing
CRISPR/Cas9 reagents pX330-U6-Chimeric_BB-CBh-hSpCas9 (pX330, Addgene 42230) and its derivative pSpCas9(BB)-2A-GFP (pX458, Addgene 48138), were obtained from the Zhang laboratory via Addgene (33), as were the CRISPR design tools based on the mm9 genome assembly for a 20-nucleotide sequence followed by a protospacer adjacent motif (PAM) from Streptococcus pyogenes of NGG. All guides were selected for the least number of potential off-target sites and, where possible, the fewest potential off-target sites within coding exons. Guide sequences (sequence-PAM) were as follows (a “g” was added preceding the sequence as necessary for the U6 promoter): Mmp13-Pro (338-bp deletion), G1-gTTCTGCCACAAACCACACTT-AGG and G2-GCCTTCAAGGAAATACAGCA-AGG; Mmp13 −10k (427-bp deletion), G1-GGTCCTGGCCTTAGGTGAGC-GGG and G2-gCTGAGGCCAACTGGTTCAA-AGG; Mmp13 −30k (582-bp deletion), G1-gAGAAGCAACCTACCTACTCA-TGG and G2-GCTGTAGCCCTCGTGAGTCC-AGG; VDRKO (exon 3, 55-bp deletion): G1-gAGTGTGTGGAGACCGAGCCA-CGG and G2-gCGGTCAAAGTCACCAGGGTC-AGG; RUNX2KO (exon 3, 33-bp deletion), G1- gCTGTGGTTACCGTCATGGCC-GGG and G2-gCCCATCTGGTACCTCTCCGA-GGG. Guide sequences (without PAM) were cloned into pX330 or pX458 and co-transfected into UAMS-PB cells using FuGENE HD (Promega, Madison, WI) as per the manufacturer's recommendations scaled to a 10-cm plate. After 48 h, transfected cells were subjected to fluorescence-activated cell sorting (FACS) to acquire GFP-positive cells (130-μm tip and 15 p.s.i.) that were individually placed into single wells of a 96-well plate by the University of Wisconsin Madison Flow Cytometry Core. Isolated cell clones were grown to confluence and tested using genotyping primers (available upon request) that spanned the deleted genomic region, both junctions, and an internal set of primers. The VDR and RUNX2 KO clones were further tested by Western blot analysis. Clones were also PCR-amplified and sequenced to verify the deleted genomic material.
Lentiviral Expression and Cell Proliferation
The pLeGO-RUNX2–2A-GFP or parental pLeGO-GFP plasmids were co-transfected with packaging plasmids pMD2.6 (Addgene 12259) and psPAX (Addgene 12260) into 293FT cells (Invitrogen) using FuGene HD (Promega) in MEMα. After 24 h, medium containing virus was removed from transfected 293FT cells, filtered (Millex 0.33-μm filter, Millipore), and placed on the UAMS-PB or target cells using 4 μg/ml Polybrene (Millipore) for 24 h. Infection of UAMS-PB or target cells was carried out for 72 h, replenishing with 293FT viral medium every 24 h. Cells were passaged a minimum of 7 times prior to FACS sorting for GFP-positive cells in standard non-viral medium. GFP-positive cells were collected and propagated for Western blot, gene expression, or cell proliferation assay. All cells were plated at 5,000 cells/well in a 24-well plate for the cell proliferation assay. Triplicate plated cells were trypsinized and counted with a standard hemocytometer over 5 days.
Western Blot Analysis
Cells were treated as indicated, and Western blot analysis was performed as described recently for VDR and RUNX2 (19, 24). Primary antibodies were as indicated for VDR (9A7; 1:2,000), RUNX2 (D130-3, MBL International; 1:1,000), MMP13 (ab39012, Abcam; 1:5,000), or β-tubulin (H-235, Santa Cruz Biotechnology; 1:5,000). Secondary antibodies used were goat α-rat IgG-HRP (Santa Cruz Biotechnology; 1:2,500), goat α-mouse IgG2b-HRP (Santa Cruz Biotechnology; 1:10,000), and goat α-rabbit IgG-HRP (Santa Cruz Biotechnology; 1:5,000) for MMP13 and β-tubulin. PVDF membrane was stripped three times at room temperature with 50 mm glycine, pH 2.5, 1% SDS) and then washed three times with PBST prior to reexamination for β-tubulin. Detection was completed with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) on an ImageQuant LAS-4000 (GE Healthcare).
ChIP-seq
Chromatin immunoprecipitation was performed as described previously (38). Briefly, UAMS-PB or CRISPR-deleted clonal cells were treated for 3 h with vehicle or 100 nm 1,25(OH)2D3 as reported previously (39). Kidney and intestine samples were collected after a 1-h 10 μg/kg dose of 1,25(OH)2D3 into C57bl/6 mice as described previously (40). Samples were subjected to immunoprecipitation using either a control IgG antibody or the indicated experimental antibody (19, 24). All ChIP and ChIP-seq methodologies, including statistical information and data processing, were performed as reported recently (19).
RESULTS
Mmp13 Is Regulated by Three Distinct Distal Enhancers Located Upstream of the TSS
Mmp13 transcription is up-regulated during early precursor cell differentiation into mature, mineralizing osteoblasts (14) and modulated by a myriad of regulatory factors that include 1,25(OH)2D3 (23). These conclusions are confirmed by the observations in Fig. 1, which demonstrate that basal expression of Mmp13 in preosteoblastic MC3T3-E1 cells (POBs) was indeed increased substantially in differentiated osteoblasts (OBs) following a 15-day exposure to osteogenic medium and that Mmp13 expression was up-regulated by 1,25(OH)2D3 in both states of differentiation. Interestingly, however, the absolute level of Mmp13 up-regulation was much more robust in mature cells, as reported previously for a cohort of additional osteoblastic genes (24). To determine the molecular basis for this complex regulation, we searched for active binding sites for RUNX2, C/EBPβ, and VDR at the Mmp13 gene locus as documented in Fig. 2 using ChIP-seq data sets that we had obtained previously (19, 24). These data sets also included analyses of the levels of key histone marks across the genome that represent signatures of both enhancer structure and activity (histone H3 Lys-4 monomethylation and histone H4 Lys-5 acetylation) and transcriptional output (histone H3 Lys-36 trimethylation). The ChIP-seq data tracks for these features at the Mmp13 locus in either POBs or OBs are depicted in Fig. 2A, where each track is composed of a normalized tag density data stream in the presence of either vehicle (yellow) or 1,25(OH)2D3 (blue) with overlapping occupancy appearing green. As can be seen, three major upstream regions of activity in POBs were apparent for VDR, C/EBPβ, and RUNX2 centered at −10, −20, and −30 kb, respectively. C/EBPβ was at the −30 kb region as well, and a small amount of RUNX2 binding was similarly apparent at the −10 kb region and particularly near the promoter but largely restricted to the differentiated OBs. Whereas both C/EBPβ and RUNX2 were prebound to these sites (green), DNA binding of the VDR required the presence of 1,25(OH)2D3 (blue). Of major significance, while histone H3 Lys-36 trimethylation strongly decorated the Mmp13 gene body as was expected of this mark, each of the three factor binding regions was reproducibly marked by elevated levels of histone H3 Lys-4 monomethylation and histone H4 Lys-5 acetylation, modifications that often specify the location of regulatory enhancers. VDR binding decreased at the −10 kb region in OBs, due to changes in the epigenetic landscape but also to a decrease in VDR expression in OBs (24). This finding raises the possibility that, like other gene targets in this cell type, the more robust response of Mmp13 to 1,25(OH)2D3 was probably facilitated through increased cooperative interactions among the three enhancers. Finally, the results documented in Fig. 2B revealed that although VDR binding across the Mmp13 locus was similar in other osteoblast lineage cells (mouse mesenchymal stem cells and osteoblastic UAMS-PB cells), receptor binding at the −10 kb region was specific for bone cells and absent in murine intestine and kidney tissues, where Mmp13 is expressed at extremely low levels. These analyses suggest that at least three distal enhancers are likely to play a significant role in both the basal and 1,25(OH)2D3-regulated expression of the Mmp13 gene.
FIGURE 1.
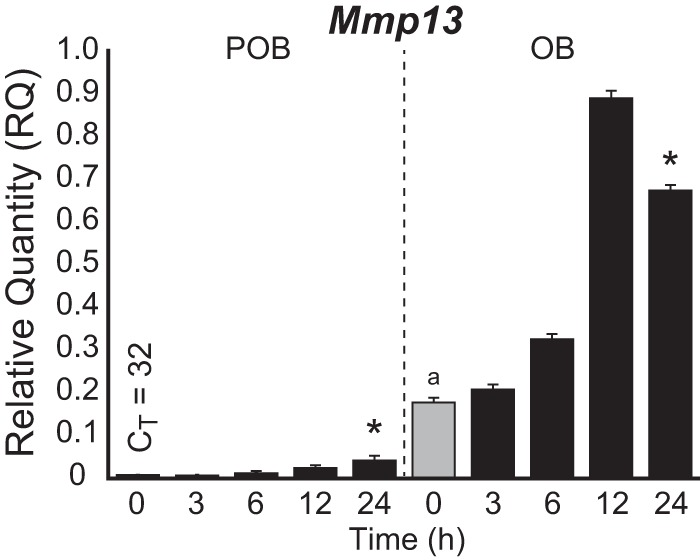
Mmp13 expression is increased during differentiation and as a function of 1,25(OH)2D3. Time-dependent induction of Mmp13 gene expression in response to 1,25(OH)2D3 treatment (100 nm) in POB and OB cells as assessed by TaqMan RT-qPCR. Data are displayed as relative quantitation (RQ) compared with β-actin ± S.E. (error bars). *, p < 0.05, 24 h versus 0 h within cell type by one-way ANOVA. a, p < 0.05, POB 0 h versus OB 0 h by two-way ANOVA with Dunnett's post-test.
FIGURE 2.

Mmp13 contains three putative transcription factor enhancer binding regions. A, overlaid ChIP-seq tag density tracks at the Mmp13 gene locus in MC3T3-E1 cells for VDR, RUNX2, C/EBPβ, acetylated histone H4 Lys-5 (H4K5Ac), monomethylated histone H3 Lys-4 (H3K4me1), and trimethylated histone H3 Lys-36 (H3K36me3) binding in the absence (yellow) and presence of 1,25(OH)2D3 (blue), where the overlap is green in POB and OB cells. B, overlaid ChIP-seq tag density tracks in the absence or presence of 1,25(OH)2D3 for the VDR in mesenchymal stem cells (MSC), UAMS-PB, mouse intestine, and kidney. Genomic location and scale are indicated, and maximum height of tag sequence density for each data track is indicated on the y axis (normalized to input and 107 tags). Gene transcriptional direction is indicated by an arrow, and exons are indicated by boxes. Putative enhancer regions and locations are indicated below the tracks (Pro, −10k, −20k, and −30k) in A.
VDR-mediated Up-regulation of Mmp13 Expression Is Controlled through the −10 kb Region
Although the VDR was found predominantly bound to the −10 kb region, modest amounts of the VDR were also localized to the more distal regions of the gene (−20 and −30 kb) as well as to the promoter proximal region. As a consequence, we cloned small DNA segments marked by each of these peaks of activity (500–1,000 bp) into a luciferase reporter vector, transfected the plasmids into POBs, and assessed their relative contribution to the up-regulation of Mmp13 expression in response to 1,25(OH)2D3. In Fig. 3A, although the −10 kb region conferred a striking dose-dependent transcriptional up-regulation of the reporter, none of the other constructs exhibited such independent regulatory capability. These data suggest that this enhancer probably plays a dominant role in 1,25(OH)2D3 activation of Mmp13 and that VDR binding to the regions outside the −10 kb location are incapable of mediating such regulation and could be due to chromatin cross-linking artifacts. A position weight matrix-based analysis via MatInspector (Genomatix) was used to identify repeated DR3-type VDREs in the −10 kb region (41, 42). Two candidates of the sequence, AGGTGAgcgGGTTCA (VDRE1) and GGTTCAaacGGAATT (VDRE2), that were separated by ∼400 bp were found located directly under the ChIP-seq peak maxima (Fig. 3B). To confirm the functionality of these potential binding sites, we introduced separate and combinatorial mutations (AGG to TTT in VDRE1 and GGT to AAA in VDRE2) into the cloned −10 kb segment and contrasted the ability of these mutant segments with that of the unmodified parental vector to mediate response to 1,25(OH)2D3. In Fig. 3C, mutations in either VDRE1 or VDRE2 strongly reduced response to the hormone, and the combination of mutations in both VDREs completely abrogated the ability of 1,25(OH)2D3 to induce an up-regulation of the TK reporter construct. These results suggest that two VDREs located in the −10 kb region are the exclusive mediators of 1,25(OH)2D3 action in our constructs.
FIGURE 3.
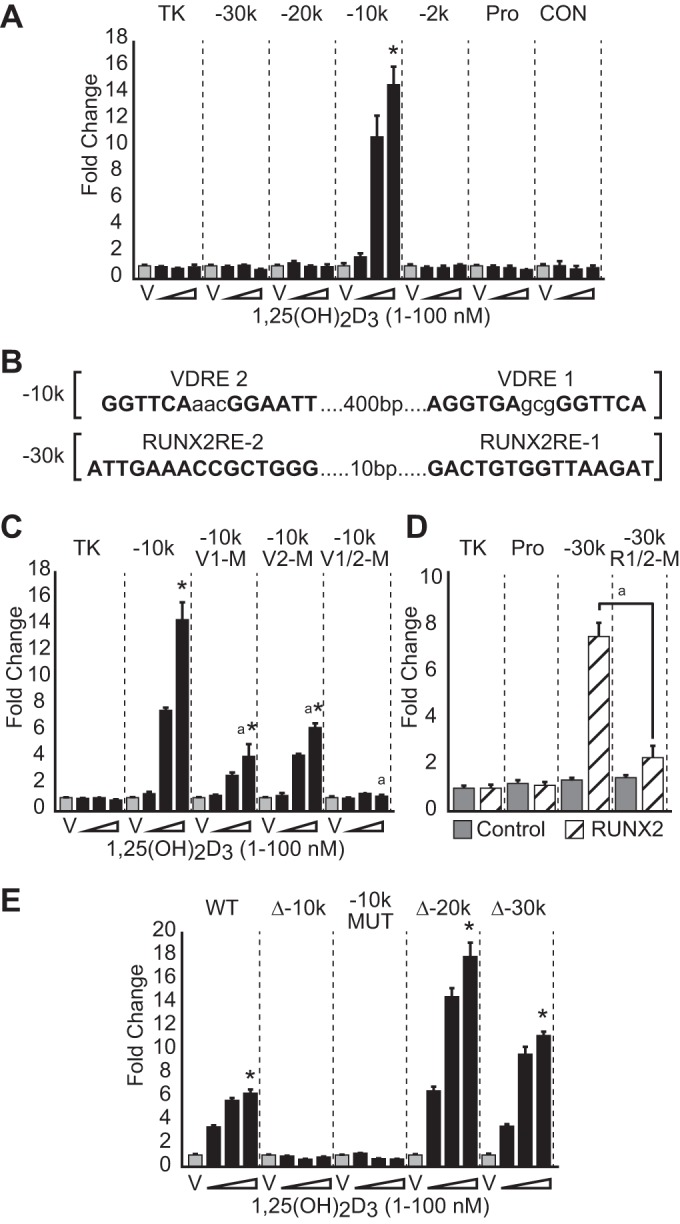
Traditional reporter assays implicate the −10 kb region as transcriptionally responsive to 1,25(OH)2D3. A, cloned pTK-luciferase enhancer reporter constructs (TK-empty, −30k, −20k, −10k, −2k, Pro, and +5k control (CON)) were transfected into MC3T3-E1 cells, treated with vehicle (gray) or increasing concentrations of 1,25(OH)2D3 (black) as indicated, and evaluated for luciferase activity 24 h later. Results are displayed as relative light units (RLU) normalized to β-galactosidase in a triplicate set of assays ± S.E. (error bars) as -fold change versus vehicle for each construct. *, p < 0.05 1,25(OH)2D3 compared with vehicle within each construct by one-way ANOVA. B, VDREs and RUNX2-REs were discovered using a position weight matrix search. Response elements were mutated AGG to TTT in VDRE1 and GGT to AAA in VDRE2, GTGG to TTTT in RUNX2-RE-1, and CCGC to CTTT in RUNX2-RE-2. C, TK reporter constructs containing wild type and mutated VDREs were transfected as in A and treated with increasing concentrations of 1,25(OH)2D3 (1–100 nm) for 24 h, and the data were treated as in A. *, p < 0.05 1,25(OH)2D3 compared with vehicle within each construct by one-way ANOVA. a, p < 0.05, 100 nm 1,25(OH)2D3 between constructs by two-way ANOVA. D, TK reporter constructs containing wild type and mutated for RUNX2-REs were transfected as in A in the absence or presence of pCMV-RUNX2 or control pCMV-empty plasmid (50 ng/well). a, p < 0.05 RUNX2 overexpression between constructs by Student's t test. E, BAC clone luciferase reporters were created for the Mmp13 locus (BAC RP23-355F15) with deletion (Δ) of −10k, −20k, and −30k along with mutations as in C. BAC clones were stably transfected, and the cell lines were selected using neomycin. Individual cell lines were treated with increasing concentrations of 1,25(OH)2D3 (1–100 nm) for 24 h. Luciferase was measured as relative light units (RLU) normalized to total protein and displayed as -fold change versus vehicle for each stable cell line. *, p < 0.05, 1,25(OH)2D3 compared with vehicle within each construct by one-way ANOVA.
RUNX2 Activity Is Mediated through the −30 kb Region of the Mmp13 Gene
A similar approach was used to identify response elements for the RUNX2 transcription factor (RUNX2-REs). Accordingly, a pair of highly ranked RUNX2-REs identified by position weight matrix analysis was discovered 10 bp apart in the −30 kb enhancer directly under the peak maxima, as illustrated in Fig. 3B. We tested the activity of the parental construct as well as a version in which the two RUNX2-REs located at the center of the −30 kb region had been mutated (GTGG to TTTT in RUNX2-RE-1 and CCGC to CTTT in RUNX2-RE-2) by co-transfecting these reporter plasmids together with a pCMV-RUNX2 expression construct (or pCMV-empty control) into MC3T3-E1 cells. In Fig. 3D, although increased reporter activity was evident in the parental construct when exogenous RUNX2 was present, this activity was significantly reduced following the introduction of mutations at the RUNX2-REs. These experiments localize functional regulatory elements for RUNX2 to the −30 kb region and indicate that at this site, RUNX2 is fully capable of activity independent of its actions at other sites. The absence of RUNX2-inducible activity at the promoter proximal region of Mmp13 was surprising in light of the minor appearance of RUNX2 at this region and of previously reported studies as well (9). The majority of RUNX2 binding at this site and at the −10 kb region is largely observed only in mature OBs. Thus, it is possible that RUNX2 regulatory activity manifests itself at these sites only after differentiation because RUNX2 binding is indeed dynamic.
The −10 kb Enhancer Mediates 1,25(OH)2D3 Action in the Context of the Mmp13 Gene Locus
The above approach assesses enhancer activity that was manifested independently of both the Mmp13 gene transcription unit and its natural chromatin environment. To address this final issue, we obtained a 182-kb BAC clone spanning the entire Mmp13 transcription unit and its associated upstream regulatory segments (chr9:7,171,215–7,353,797), inserted a luciferase reporter cassette (P2A-luciferase-TK-neomycin) into the 3′-untranslated region of the gene, and stably introduced this construct into MC3T3-E1 cells. Mutant versions of this construct containing either deletions of the −10, −20, and −30 kb enhancers or a specific mutation altering the two VDREs were also stably integrated and examined for 1,25(OH)2D3 response as well. As can be seen in Fig. 3E, although the recombineered parental BAC together with versions containing deletions at −20 and −30 kb were fully capable of mediating response to 1,25(OH)2D3, neither the version containing the −10 kb deletion nor that containing mutations of the VDREs was active. These results strongly suggest that the enhancer identified by ChIP-seq analysis −10 kb upstream of the Mmp13 gene and the VDREs localized within mediate the actions of 1,25(OH)2D3 and that the sequences within the Mmp13 promoter proximal region do not appear to be involved in this regulation.
CRISPR Genomic Editing Confirms the Mechanism for Mmp13 Regulation by 1,25(OH)2D3
The above experiments involving the VDR were made possible because of the ligand-inducible nature of the receptor's action at the gene, where -fold change in activity can be analyzed independent of the construct's basal activity. The activity of transcription factors like RUNX2 contributes to the basal expression of genes such as Mmp13, however, and are not ligand-activated per se. Thus, their contribution to the basal activity of stable BAC clones is difficult to interpret given the random integration of individual clones into the genome, the uncertain influence of the environment at these sites of integration, and the potential for the presence of multiple copies. To overcome these pitfalls, the direct genome editing technique of the CRISPR/Cas9 system was employed (33) to target and excise specific enhancer regions of the Mmp13 gene using separate CRISPR guide RNAs or to delete the expression of factors such as the VDR or RUNX2, schematically outlined in Fig. 4A. Interestingly, CRISPR-targeted RNAs that bound directly at the two separate VDREs in the −10 kb region could be used because the NGG PAM sequence requirement for guide RNAs was fulfilled by the sequence of the canonical VDRE half-site, AGGTCA. We turned to UAMS-PB cells for this purpose because this line was similar to MC3T3-E1 cells with respect to differentiation (24) and because it displayed similar VDR ChIP-seq patterns (Fig. 2B). UAMS-PB cells offer several additional distinct advantages; they represent a clonally isolated cell line, stably express the PTH receptor, and, perhaps most importantly, appear to be more efficient at CRISPR non-homologous end joining recombination, thereby facilitating recovery of homozygous daughter cell lines. CRISPR −10 kb guide plasmids 1 and 2 were transfected into the UAMS-PB cells, GFP-positive cells were isolated through FACS sorting into 96-well plates, and the genotypes of the isolated clones were screened via PCR and confirmed via sequencing as described under “Experimental Procedures.” The frequency of desired homozygous non-homologous end joining enhancer deletions was ∼1.5–2%, and 2–4 genetically modified clones, as well as at least one intact experimental wild type clone (E/WT), were recovered and tested for expression of Mmp13 and other genes. In Fig. 4B, both the UAMS-PB wild type (WT) and an E/WT clone retained temporal responsiveness to 1,25(OH)2D3 (100 nm) and response to treatment with FGF2 (50 ng/ml) but not BMP2 (50 ng/ml), compounds known to modulate Mmp13 activity (2, 8). Mmp13 was strongly inhibited by FGF2 and does not appear on the scale. The −10 kb enhancer-deleted daughter cell line, on the other hand, was no longer inducible by 1,25(OH)2D3, as was anticipated, although it retained repressive response to FGF2. These effects were confirmed in a second clone containing a larger −10 kb enhancer deletion as well as in replicate clones (data not shown). In contrast, however, virtually all additional target genes examined were unaffected by the deletion, including Sp7 and Runx2 as well as others. Surprisingly (and to be explored below), we also found that in the absence of the −10 kb enhancer, Mmp13 expression was now potently down-regulated in a time-dependent manner by 1,25(OH)2D3. These data at the endogenous Mmp13 gene locus confirm previous observations that VDR-mediated activity is contained within the −10 kb enhancer while raising the possibility of a secondary independent activity of 1,25(OH)2D3 to suppress Mmp13 expression.
FIGURE 4.

Deletion of the −10 kb region using CRISPR/Cas9 gene editing completely eliminates VDR-mediated up-regulation of Mmp13. A, top, schematic depiction of CRISPR-targeted −10 kb enhancer sequence. VDREs are underlined in black, CRISPR guide sequences (g1 and g2) are highlighted in blue, and the PAMs are indicated in red. A, bottom, the targeted region (red bar) was eliminated in non-homologous end joining DNA repair. B, CRISPR-deleted and clonally isolated cell lines for WT, E/WT, and −10 kb region deletion (−10k KO) were treated with 1,25(OH)2D3 (100 nm) from 0 (gray bar) to 24 h (black bars) or with BMP2 (50 ng/ml for 24 h; green) or FGF2 (50 ng/ml for 24 h; orange), and the RNA was then isolated and assayed for Mmp13, Vdr, Spp1, Sp7, Runx2, and RankL transcripts using TaqMan qPCR analysis. Data are displayed as relative quantitation (RQ) normalized to Gapdh in a triplicate set of assays ± S.E. (error bars). *, p < 0.05, 24 h versus 0 h within cell type by one-way ANOVA. a, p < 0.05, 100 nm 1,25(OH)2D3 between cell types by two-way ANOVA. #, p < 0.05, FGF2 or BMP2 treatment compared with vehicle by Student's t test.
Elimination of the −30 kb Region or RUNX2 Expression Disrupts Basal Mmp13 Transcription
Based upon the initial success with the CRISPR approach, we created an additional series of UAMS-PB daughter cell lines, as outlined in Fig. 5A. First, a region located proximal to yet distinct from the Mmp13 promoter (−98 to −454 bp, Pro) was deleted in the UAMS-PB cell line. This region contained several highly ranked RUNX2-REs identified through in silico analysis that were also highly conserved in the rat Mmp13 promoter (43, 44). Second, a daughter cell line was created that contained two deletions, one at the −10 kb enhancer (−10k KO) and the other proximal to the Mmp13 promoter (Pro, −98 to −454 bp). Third, we deleted the region comprising the −30 kb enhancer (−30k KO). Finally, we also created several additional deletions to eliminate VDR expression (VDR KO), either in the parent cell line or in combination with the −10 kb deleted daughter cell line (−10k/VDR KO) and to eliminate RUNX2 expression (RUNX2 KO). These latter gene knockouts were obtained by targeting exon 3 of both the VDR (Fig. 5B) and the RUNX2 genes (Fig. 5C), which resulted in frameshift mutations within the genes due to small ∼50-bp deletions that ensured the loss of not only VDR and RUNX2 protein expression but of all putative splice variants as well.
FIGURE 5.

Basal Mmp13 activities are controlled by RUNX2 and the −30 kb enhancer region. A, schematic depiction of CRISPR-targeted sequences in the Mmp13 locus. Deleted regions (red) are expanded showing relative protein binding activities that correlated with ChIP-seq peak occupancy for RUNX2 (yellow), C/EBPβ (green), and VDR (blue). Guide RNAs (g1 and g2) are shown in blue with the genomic sequence deleted on chromosome 9 indicated below each region. B and C, CRISPR-targeted guide RNA design and location for exon 3 of the VDR (B) and RUNX2 (C) with corresponding Western blot analysis of whole cell extracts for VDR, RUNX2, and a β-tubulin control. Replicate samples are displayed for each WT and KO sample. CRISPR-deleted and clonally isolated cell lines for WT, E/WT, −10 kb region deletion (−10k KO), combination −10 kb and VDR deletion (−10 k/VDRKO), promoter proximal region deletion (Pro KO), combination −10 kb and promoter proximal region deletion (−10k/Pro KO), −30 kb region (−30k KO), RUNX2 deletion (RUNX2 KO), and VDR deletion (VDR KO) were treated with vehicle (gray) or 1,25(OH)2D3 (100 nm) for 24 h (black) prior to RNA isolation, reverse transcription, and TaqMan qPCR analysis for Mmp13 (expanded region shown to the right) (D) or Vdr, Spp1, Sp7, and Runx2 (E). Data are displayed as relative quantitation (RQ) normalized to Gapdh in a triplicate set of assays ± S.E. (error bars). *, p < 0.05 24 h versus 0 h within cell type by Student's t test. a, p < 0.05 basal vehicle of each line versus WT by Student's t test. F, gene regulatory model for 1,25(OH)2D3 and RUNX2 positive (+) and negative (−) actions on Mmp13 and Sp7 genes toward osteoblast maturation gene programs. VDR is shown in blue, and RUNX2 is shown in yellow.
With these cell lines established, we then examined the consequence of each deletion on both basal and 1,25(OH)2D3-regulated expression of Mmp13. Importantly, as was noted in Fig. 4 and seen explicitly in Fig. 5D, deletion of the −10 kb enhancer led not only to a reduction in the basal expression of Mmp13 but to a surprising 1,25(OH)2D3-mediated suppression of Mmp13 transcripts relative to the induction seen in the wild type and experimental control cell lines. This suppression was dose-dependent (data not shown). No changes, as seen in Fig. 5E, were again observed in the expression of control RNAs for Vdr, Runx2, Sp7, or Spp1. In contrast, however, genetic deletion of the VDR either from the wild type UAMS-PB parental cell line or from the −10 kb enhancer daughter cell line (−10k/VDR KO), resulted in a complete abrogation of the regulatory effects of 1,25(OH)2D3 at all target genes, including the ability of the hormone to induce Mmp13 in the parental cell line and to suppress the gene in the absence of the −10 kb enhancer. Surprisingly, the loss of VDR in the wild type UAMS-PB cells resulted in a more pronounced suppression of the basal level of Mmp13 transcripts than that seen in the absence of the −10 kb enhancer, although it was clear in each case that both the VDR and the enhancer itself contribute either directly or indirectly to the basal expression of Mmp13. It is also worth noting that despite the CRISPR deletion and frameshift mutation of the exon 3 region, small amounts of Vdr mRNA were still detectable in the VDR KO line via RT-PCR analysis, although VDR protein was not produced. The same was true of the Runx2 mRNA in the RUNX2 KO line.
Interestingly, deletion of the promoter proximal segment also caused a minor reduction in basal expression of Mmp13, although the gene's inducibility by 1,25(OH)2D3 still remained evident. Curiously, however, although elimination of both the promoter proximal and the −10 kb region led to a striking loss of basal Mmp13 expression, repression by 1,25(OH)2D3 was still apparent. This result lends credence to the idea that the promoter proximal region contains an important basal regulatory component for the Mmp13 gene. Additional evidence, however, suggests that this region may not be essential for controlling the expression of Mmp13: 1) elimination of the −30 kb enhancer caused a much more profound reduction in basal Mmp13 activity, as seen in Fig. 5D, and 2) loss of RUNX2 expression was equally debilitating to baseline Mmp13 transcription. Importantly, however, despite a loss in basal activity, the daughter cell lines containing either a deletion of the −30 kb enhancer or elimination of RUNX2 expression retained Mmp13 inducibility by 1,25(OH)2D3, demonstrating that the −10 kb region remained functional despite the change in basal expression of Mmp13 due either to loss of the −30 kb enhancer or loss of its RUNX2-dependent activity. As expected, the loss of RUNX2 expression also influenced to variable degrees the basal expression of other osteoblastic genes, including Runx2, Vdr, Sp7, and Spp1. It was also notable that the loss of RUNX2 expression has a dramatic impact on Sp7 expression, a dependence that has been described previously (45). These CRISPR data revealed that Mmp13 regulation by RUNX2 and VDR is mediated through the −30 and −10 kb regions, respectively. They also show that the effects of 1,25(OH)2D3 on Mmp13 expression are complex. Thus, whereas 1,25(OH)2D3 can induce Mmp13 expression through direct effects mediated by VDR DNA binding, the hormone also appears able to exert secondary effects on Mmp13 gene activity by down-regulating Runx2 and Sp7 expression and, in so doing, reduces basal Mmp13 expression, as summarized in the speculative model documented in Fig. 5F. It is also possible that the VDR may interact directly with RUNX2 itself, as has been shown (46). In either case, CRISPR-mediated enhancer deletion analysis has unveiled what appears to be a secondary effect of 1,25(OH)2D3 on Mmp13.
Loss of RUNX2, Not Mmp13, Adversely Affects Osteoblast Differentiation
To assess the impact that elimination of either RUNX2, VDR, or MMP13 has on the osteoblast differentiation, parental UAMS-PB cells (WT, E/WT), or the −10k KO, −30k KO, VDR KO, and RUNX2 KO cell lines were examined for their expression of these factors by Western blot and assessed for their ability to differentiate after 15 days in osteogenic medium. As can be seen in Fig. 6A, the basal level of expression of the Mmp13 protein was generally consistent with the transcript levels that were seen in the cell lines examined in Fig. 5D, with lower MMP13 levels evident in the −30k KO and RUNX2 KO lines. As seen in Fig. 6B, however, although the parental line exposed to osteogenic medium differentiated fully into mineralized osteoblasts, the presence or absence of MMP13 protein in several of the mutant daughter cell lines, in which RUNX2 expression was unaffected, had no impact on the differentiation process. In contrast, deletion of RUNX2 but not deletion of the −30 kb enhancer completely prevented osteoblast differentiation and mineralization, as documented in Fig. 6C. As seen in Fig. 6D, loss of RUNX2 resulted in a dramatic reduction in cellular proliferation, which was fully rescued when RUNX2 expression was restored. The results of this experiment also show that re-expression of RUNX2 via lentivirus expression is capable of rescuing the differentiation process and mineralization as well, although this effect was not fully restored despite a rather dramatic overexpression of the RUNX2 protein (Fig. 6E). The expressions of genes, including Sp7, were partially rescued as well (Fig. 6F). We conclude that the dramatic down-regulation of Mmp13 as a result of enhancer deletion had no effect on cellular growth or differentiation, although detailed features of cell function were not examined. RUNX2 deletion, on the other hand, uncovered a dramatic consequence of the protein on osteoblast proliferation, differentiation, and mineralization, effects that are consistent with previous studies of the loss of RUNX2 expression both in vitro and in vivo (16).
FIGURE 6.

RUNX2, not MMP13, controls osteoblast differentiation and maturation. A, Western blot analysis for MMP13, RUNX2, and VDR expression in the UAMS-PB cell lines designated WT, −10k KO, −30k KO, RUNX2 KO, and VDR KO. All samples are displayed in duplicate and normalized to β-tubulin levels. B, all of the indicated derivative UAMS-PB cell lines were differentiated for 15 days in osteogenic medium and then stained with Alizarin Red. Quantitative measurement for calcium content (μg of Ca2+/μg of total protein) is indicated in triplicate ± S.E. (error bars). C, CRISPR RUNX2-deleted cells (RUNX2 KO) and wild type cells were infected with lentivirus control vector (pLeGO-G) or RUNX2 overexpression vector (pLeGO-RUNX2-P2A-GFP), cells were selected for GFP expression, and then cells were cultured for 15 days in osteogenic medium. Alizarin Red staining and calcium content quantitation was performed as in B. D, cell proliferation assay for CRISPR clones. Cells were plated at low density and monitored by cell count over 5 days. *, p < 0.05 between samples. E, Western blot analysis for RUNX2 in WT and RUNX2 KO cells transduced with either lentivirus LeGO-G control (−) or RUNX2 overexpression vector LeGO-RUNX2-P2A-GFP (+). All samples are displayed in duplicate and normalized to β-tubulin levels. F, Sp7 expression is recovered with RUNX2 overexpression. CRISPR-deleted and clonally isolated cell lines for WT and RUNX2 deletion (RUNX2 KO) with LeGO-G control (−) or RUNX2 overexpression vector LeGO-RUNX2-P2A-GFP (+) were treated with vehicle (gray) or 1,25(OH)2D3 (100 nm) for 24 h (black) prior to RNA isolation, reverse transcription, and TaqMan qPCR analysis for Sp7. Data are displayed as relative quantitation (RQ) normalized to Gapdh in a triplicate set of assays ± S.E. *, p < 0.05, 1,25(OH)2D3 versus vehicle within cell type by Student's t test. a, p < 0.05, +RUNX2 sample of each line versus WT by Student's t test.
Genomic Organization of the Mmp13 Gene Relies upon −30 kb Enhancer and the Presence of RUNX2
Loss of either the −30 or −10 kb enhancer had a profound effect on basal Mmp13 transcription (Fig. 5D), which was probably the consequence of direct genome binding alterations. To examine this possibility, a ChIP-qPCR analysis of the Mmp13 locus at −30 kb, −20 kb, −10 kb, −2 kb, and the promoter proximal region was performed for the UAMS-PB parental WT and E/WT cells and the CRISPR daughter cell lines −10k KO, −30k KO, and RUNX2 KO. Cells were treated with either vehicle or 1,25(OH)2D3 for 3 h and then subjected to ChIP analysis using antibodies to the VDR (blue), RUNX2 (red), C/EBPβ (green), and IgG (black) (Fig. 7A). qPCR was performed using primers located within the individual peak regions, as defined by ChIP-seq analysis; therefore, regions deleted by CRISPR were not detected (N.D. in Fig. 7). As documented in Fig. 7A, ChIP analysis of RUNX2, VDR, and C/EBPβ in both the parental line (WT) and the experimental clone (E/WT) confirmed observations made by ChIP-seq analysis, which demonstrated that the highest level of RUNX2 binding was present at the −30 kb region, C/EBPβ was predominantly bound to both the −20 and −30 kb regions, and 1,25(OH)2D3-inducible VDR binding was localized to the −10 kb region. Very modest but measureable levels of VDR, RUNX2, and C/EBPβ binding were also observed near the promoter, consistent with data obtained by ChIP-seq analysis. Importantly, although the recruitment of VDR and C/EBPβ at their respective sites across the gene (−30 kb, −20 kb, and promoter proximal) was generally unaffected by deletion of the −10 kb enhancer as compared with the control cell lines, the removal of this enhancer had a modest inhibitory effect on RUNX2 binding at both the −30 kb and the promoter proximal regions. This effect on RUNX2 binding appears to be exaggerated when the overall expression of the VDR was eliminated. This suggests that genomic organization and transcription factor binding activity across the Mmp13 locus are affected as a result of loss of the −10 kb segment, but only nominally. In contrast, however, loss of the −30 kb enhancer had a significant effect on transcription factor occupancy at other sites across the Mmp13 gene locus. Accordingly, C/EBPβ, VDR, and RUNX2 binding at the −20 kb, the −10 kb, and the promoter proximal regions, respectively, were all reduced from their residual or inducible levels in the parent cell line. Almost identical effects at these enhancer regions were obtained when RUNX2 expression was eliminated, thus lowering recruitment of VDR and C/EBPβ to the −10 kb and −20 kb enhancer regions. The specificity of these data were confirmed through analysis of the control gene MyoD family inhibitor (Mdfi, I-mfa), which is known to be regulated by 1,25(OH)2D3 in osteoblasts (24, 47). At this gene, strong VDR and RUNX2 binding as well as weaker C/EBPβ binding was observed ∼8 kb upstream of the Mdfi gene TSS in MC3T3-E1 cells by ChIP-seq analysis (Fig. 7B) and confirmed by ChIP-qPCR in the parental UAMS-PB cells (Fig. 7C). RUNX2 binding was compromised only in the RUNX2 KO line, whereas the VDR and C/EBPβ binding remained unchanged. These data suggest that RUNX2 is essential for the overall expression of Mmp13 and that its binding to the −30 kb region is required for the interaction of VDR, C/EBPβ, and RUNX2 at the −10, −20, −30 kb, and promoter proximal regions, which are key for appropriate transcriptional regulation at the Mmp13 gene locus. Similar effects of VDR binding at the −10 kb region appear to selectively impact RUNX2 but not C/EBPβ binding at the −30 kb enhancer and the promoter proximal regions. These potential interactions are illustrated in the model shown in Fig. 7D.
FIGURE 7.

Elimination of the −30 kb region affects VDR, RUNX2, and C/EBPβ binding at other Mmp13 enhancers. A, CRISPR cell lines for WT, E/WT, −10 kb region deletion (−10k KO), −30 kb region (−30k KO), and RUNX2 deletion (RUNX2 KO) were treated with vehicle or 1,25(OH)2D3 (100 nm) for 3 h prior to ChIP-qPCR analysis for VDR (blue), RUNX2 (red), C/EBPβ (green), and IgG (black) using primers located at −30 kb, −20 kb, −10 kb, −2 kb, and the promoter proximal region (Pro). Data are displayed as quantity normalized to input in triplicate ± S.E. (error bars). *, p < 0.05 1,25(OH)2D3 versus vehicle within cell type and antibody by Student's t test. a, p < 0.05, 1,25(OH)2D3 of each line versus WT cell line within antibody by Student's t test. B, overlaid ChIP-seq tag density tracks upstream of the Mdfi gene locus for VDR, RUNX2, and C/EBPβ (vehicle (yellow), 1,25(OH)2D3 (blue), and overlap (green)) in MC3T3-E1 cells. Genomic location and scale are indicated, and maximum height of tag sequence density for the data track is indicated on the y axis (normalized to input and 107 tags). Gene transcriptional direction is indicated by the arrow. C, ChIP-qPCR at the −8 kb region of the Mdfi gene with specific details as in A above. D, enhancer interaction model for Mmp13 activation. Schematic depiction of enhancers shown clustered (−30k (blue), −20k (yellow), and −10k (green)) and interacting near the Mmp13 with the promoter proximal region (Pro, gray). The general transcription assembly (GTA, brown) is shown positioned at the Mmp13 TSS. Exons are indicated by black boxes, and the direction of gene transcription is indicated by the arrow.
DISCUSSION
Collagenase-3, the product of the Mmp13 gene, is required for proper formation of cartilage and bone but also serves a multitude of additional physiological roles in non-skeletal tissues and pathological roles in metastatic cancer. Accordingly, its expression is regulated by a diverse set of local as well as endocrine stimuli, although the molecular mechanisms that underlie these regulatory events are not well understood and have been limited to traditional studies focused on elements located near the Mmp13 promoter (9–15). In the present studies using unbiased ChIP-seq analyses, we challenged the idea that regulation of Mmp13 gene expression in bone cells is mediated through promoter proximal elements and show that this gene is regulated primarily through the actions of three distal enhancers located −10, −20, and −30 kb upstream of the Mmp13 promoter that enable the activity of the VDR, C/EBPβ, and RUNX2. Analysis of Mmp13 transcripts revealed promoter-independent roles for the distal −10 and −30 kb enhancers in mediating the activities of VDR and RUNX2, respectively, while supporting a minor involvement of the regulatory region proximal to the promoter in basal Mmp13 expression. Nevertheless, both the loss of the −30 kb enhancer and the elimination of RUNX2 expression had similar devastating effects on basal Mmp13 transcript production, seemingly diminishing the importance of RUNX2 binding at the promoter relative to its interaction at the distal −30 kb enhancer site.
The studies described made use of CRISPR/Cas9 gene editing to prepare daughter UAMS-PB cell lines in which one or more enhancers were removed from the Mmp13 gene as well as removal of either RUNX2 or VDR. Preparation of these daughter cell lines required the transfection of multiple plasmids, target cell enrichment, and extensive genotype screening using PCR to identify homozygous clones. As indicated, ∼1.5–2% of the cell lines examined contained homozygous enhancer deletions, although an extremely high percentage of cells contained either a single cut or the expected deletion at a single allele. Based upon this and reported literature, we determined that the low efficiency of obtaining homozygous cell lines was due to the need for two sets of guide RNAs to obtain deletions of the desired size (300–500 bp), whereas the gene deletions (VDR/RUNX2 KO at 50 bp) occurred at a higher rate (∼10%). Targeting of the deletions was not observed to be perfect either, probably due to fill back by the DNA repair machinery. Indeed, many of the deletions extended several nucleotides beyond the cut site in either direction; however, because the enhancer deletions were arbitrary, based upon the general span covered by the peak obtained through ChIP-seq analysis, this minor imprecision was not problematic. Surprisingly, initial attempts using the MC3T3-E1 cell line were unsuccessful. We speculate that this may to be due to an aberrantly polyploid genome shared by many cultured cell lines, an overactive DNA repair machinery, or simply an increased efficiency of CRISPR-mediated non-homologous end joining recombination in UAMS-PB cells. Finally, we routinely obtained and examined multiple daughter cell lines for each deletion to ensure that the general basal properties of each line were similar to those of the parental UAMS-PB line and that control genes were unaffected by the Mmp13 enhancer deletions. Despite the clonal isolation of the UAMS-PB line, selection and regrowth of single cells frequently altered the properties of some of the daughter cell lines, making it imperative that each line be thoroughly examined for proliferation and differentiation capabilities as well as gene expression profiles.
The results of the enhancer deletion analyses suggest that the three enhancers and the promoter proximal regulatory region are highly interactive and do not function in isolation. For example, deletion of the −30 kb enhancer and thus loss of RUNX2 regulation exclusively at this site exerted a major negative impact on VDR binding and regulatory activity at the −10 kb enhancer, loss of C/EBPβ binding at the −20 and −30 kb enhancers, and loss of a small amount of RUNX2 binding near the promoter. However, loss of cellular RUNX2 expression also strongly affected both C/EBPβ and VDR binding at these same sites. These observations lead to the conclusion that it is not the loss of the −30 kb enhancer per se but rather the presence of RUNX2 at the site that is key to the three-dimensional interaction. Similarly, deletion of the −10 kb enhancer and loss of VDR regulation at the site also reduced RUNX2 binding at the −30 kb enhancer and at the promoter proximal region. Although this effect on RUNX2 binding was modest, it was exaggerated in cells in which VDR expression had been eliminated. In contrast, C/EBPβ binding at its cognate sites was unaffected by the either the loss of the −10 kb enhancer or the loss of VDR expression. Nevertheless, similar conclusions can be drawn as well regarding the role of VDR in facilitating enhancer interaction. It is possible that small amounts of factor binding seen at −30 kb, −20 kb, and the promoter proximal region by ChIP-seq analysis, as exemplified by the VDR, may arise as a result of the cross-linking of proteins from complex formation rather than direct DNA binding to each enhancer. This idea, in the case of the VDR, appears to be reinforced by the fact that aside from the −10 kb segment, none of the remaining regions were capable of mediating 1,25(OH)2D3 activity when examined in isolation, although alternative explanations are also possible. Overall, however, these studies support the concept of a striking looping model as seen in Fig. 7D for the Mmp13 gene, wherein protein complexes at each of the distal enhancers are brought into proximity of the Mmp13 promoter region via protein-protein interaction.
Observations at the Mmp13 gene also provide additional support for the idea that changes in the binding and activity of transcription factors, such as RUNX2 and C/EBPβ, and the modifications to the epigenome that occur during differentiation leading to an altered transcriptome may cause dynamic transitions in both transcription factor binding and response to secondary factors, such as 1,25(OH)2D3. This change is seen at many genes but is exemplified by the enhanced response of the Mmp13 gene to 1,25(OH)2D3 following differentiation as seen in Fig. 1. The observation that loss of a dominant regulatory enhancer (−10 kb) for the VDR unveils a secondary effect of 1,25(OH)2D3 to suppress Mmp13 expression is also informative because it highlights an underlying mechanism through which 1,25(OH)2D3 can induce expression of a gene under one condition and suppress the same gene under another, as was seen for Bmp2 regulation (24). We speculate that this effect may be due to the ability of the VDR to suppress RUNX2 expression (Figs. 4 and 5), although a direct protein-protein interaction between RUNX2 and the VDR is also possible (46). In fact, the results in our control gene Mdfi suggest that VDR is required for RUNX2 action at that gene but perhaps not vice versa. This provides one mechanism for gene suppression by 1,25(OH)2D3 for which little is known. Interestingly, this mechanism in association with RUNX2 activity is not limited to secondary suppression because RUNX2 can be a suppressor of the expression of many genes (48). Accordingly, the down-regulation of RUNX2 expression or activity at a particular gene could result in increased responsiveness to 1,25(OH)2D3. Further studies of these interactions at genes regulated by RUNX2 will be illuminating with regard to this mechanism.
Our previous studies using ChIP-seq analysis defined an osteoblast enhancer complex composed of a single enhancer to which RUNX2, C/EBPβ, and the VDR bind in an organized fashion to control the expression of genes integral to the osteoblast phenotype (24). The present studies support a more complex version of the osteoblast enhancer complex in which linearly separated enhancers for a single target gene can bind these same three transcription factors independently yet interact to form a complex at the Mmp13 promoter that is capable of integrating the activities of the signaling pathways that regulate these factors. This finding has considerable implications for the mechanisms through which a gene can be regulated but also points to the complexity of identifying activity that is inherent to a particular enhancer when deletion of that enhancer affects indirectly the activity of another. Unfortunately, the contribution of C/EBPβ to the osteoblast enhancer complex or Mmp13 expression is even less well understood. However, C/EBPβ is known to play a role in osteoblast function in vivo, has been shown to interact directly with RUNX2, and may serve as a structural scaffold for basal activity of Mmp13 expression (49–51). Because the elimination of RUNX2 and the deletion of the −30 kb enhancer yielded the same results despite a large amount of C/EBPβ binding, it is difficult to determine what contribution C/EBPβ has, much less the contribution by the −20 kb enhancer, which may be completely structural for enhancer looping.
Previous studies have suggested that Mmp13 expression is regulated by PTH via the binding of RUNX2 to a site(s) near the Mmp13 promoter (9). Further studies have indicated that 1,25(OH)2D3 also regulates Mmp13 expression via elements similarly located (23) as well as by the estrogen receptor (10) at the promoter proximal region. The focus of virtually all of these studies was limited to the Mmp13 promoter, however, utilizing traditional approaches that included transient transfection analysis and mutagenesis to explore the cis-acting components within this regulatory region and more recently direct PCR ChIP analysis to detect the presence of specific proteins. Our studies using unbiased alternative approaches indicate that more upstream components are certainly involved in the regulation of Mmp13 expression in osteoblastic cells. These approaches involved the identification of transcription factor binding sites, the dissection of fundamental regulatory regions located within the endogenous gene via CRISPR deletion, and the analysis of transcriptional activity through direct measurement of the output of authentic Mmp13 transcripts from the endogenous gene. They also involved correlative deletion of both RUNX2 and VDR expression. These studies suggest that some activity of RUNX2 may be present at the promoter proximal region, although it is clear that this activity (either with or without our deletion studies) is neither exclusive to the regulation nor dominant when compared with the activity of RUNX2 at sites distal to the Mmp13 promoter. Similarly, we speculate that our understanding of human MMP13 gene regulation is probably incomplete as well. There have been several studies focused on the gene's promoter proximal region for induction, repression, or inhibition (52, 53). Furthermore, upstream segments of both mouse and human MMP13 share segments of homologous sequence (data not shown). However, the roles of the conserved regions are likely to remain uncertain until we are able to examine the human MMP13 gene in detail via ChIP-seq and CRISPR in relevant cell lines. These findings highlight the need for re-examination of genes as exemplified through Mmp13 to ensure that the understanding of their mechanisms of regulation is contemporary.
Taken together, the regulation of Mmp13 expression appears to be mediated via a complex arrangement of enhancers that interact with VDR, RUNX2, and C/EBPβ. Most strikingly, the 1,25(OH)2D3-mediated regulation is present at the −10 kb enhancer, whereas most basal activity is attributed to the −30 kb enhancer. This mechanism of regulation appears to be present in mouse cells of mesenchymal origin, and we speculate that subsets of these regulatory regions may participate in the control of Mmp13 expression in other tissues, including metastatic cancer cells, where both RUNX2 and other isoforms of RUNX are expressed. The mechanisms of distal regulation studied here open new avenues for investigation toward a more complete understanding of Mmp13 regulation.
Acknowledgments
We thank the members of the Pike laboratory for helpful discussion and contributions to the manuscript. We also thank Charles A. O'Brien for providing the UAMS-32PB osteoblastic cells.
This work was supported, in whole or in part, by National Institutes of Health, NIDDK, Grant DK-072281 (to J. W. P.).
- PTH
- parathyroid hormone
- qPCR
- quantitative real-time PCR
- TSS
- transcriptional start site
- C/EBP
- CCAAT/enhancer-binding protein
- RUNX2
- Runt-related transcription factor 2
- VDR
- vitamin D receptor
- VDRE
- VDR DNA binding response element
- 1,25(OH)2D3
- 1α,25-dihydroxyvitamin D3
- CRISPR
- clustered regularly interspaced short palindromic repeats
- PAM
- protospacer adjacent motif
- Cas9
- CRISPR-associated protein 9
- ChIP-seq
- ChIP followed by sequencing
- BAC
- bacterial artificial chromosome
- POB
- preosteoblast
- OB
- osteoblast
- MEMα
- minimum Eagle's medium α
- RUNX2-RE
- RUNX2 response element
- E/WT
- experimental WT
- ANOVA
- analysis of variance
- TK
- thymidine kinase.
REFERENCES
- 1. Inada M., Yasui T., Nomura S., Miyake S., Deguchi K., Himeno M., Sato M., Yamagiwa H., Kimura T., Yasui N., Ochi T., Endo N., Kitamura Y., Kishimoto T., Komori T. (1999) Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn. 214, 279–290 [DOI] [PubMed] [Google Scholar]
- 2. Uría J. A., Balbín M., López J. M., Alvarez J., Vizoso F., Takigawa M., López-Otín C. (1998) Collagenase-3 (MMP-13) expression in chondrosarcoma cells and its regulation by basic fibroblast growth factor. Am. J. Pathol. 153, 91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao P., Yang J. L., Zhao H., You J. H., Hu Y. (2014) Common polymorphism in the MMP-13 gene may contribute to the risk of human cancers: a meta-analysis. Tumour Biol. 35, 10137–10148 [DOI] [PubMed] [Google Scholar]
- 4. Etoh T., Inoue H., Yoshikawa Y., Barnard G. F., Kitano S., Mori M. (2000) Increased expression of collagenase-3 (MMP-13) and MT1-MMP in oesophageal cancer is related to cancer aggressiveness. Gut 47, 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freije J. M., Díez-Itza I., Balbín M., Sánchez L. M., Blasco R., Tolivia J., López-Otín C. (1994) Molecular cloning and expression of collagenase-3, a novel human matrix metalloproteinase produced by breast carcinomas. J. Biol. Chem. 269, 16766–16773 [PubMed] [Google Scholar]
- 6. Inada M., Wang Y., Byrne M. H., Rahman M. U., Miyaura C., López-Otín C., Krane S. M. (2004) Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc. Natl. Acad. Sci. U.S.A. 101, 17192–17197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deguchi J. O., Aikawa E., Libby P., Vachon J. R., Inada M., Krane S. M., Whittaker P., Aikawa M. (2005) Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation 112, 2708–2715 [DOI] [PubMed] [Google Scholar]
- 8. Varghese S., Canalis E. (1997) Regulation of collagenase-3 by bone morphogenetic protein-2 in bone cell cultures. Endocrinology 138, 1035–1040 [DOI] [PubMed] [Google Scholar]
- 9. Selvamurugan N., Chou W. Y., Pearman A. T., Pulumati M. R., Partridge N. C. (1998) Parathyroid hormone regulates the rat collagenase-3 promoter in osteoblastic cells through the cooperative interaction of the activator protein-1 site and the runt domain binding sequence. J. Biol. Chem. 273, 10647–10657 [DOI] [PubMed] [Google Scholar]
- 10. Achari Y., Lu T., Katzenellenbogen B. S., Hart D. A. (2009) Distinct roles for AF-1 and -2 of ER-α in regulation of MMP-13 promoter activity. Biochim. Biophys. Acta 1792, 211–220 [DOI] [PubMed] [Google Scholar]
- 11. Peeters-Joris C., Hammani K., Singer C. F. (1998) Differential regulation of MMP-13 (collagenase-3) and MMP-3 (stromelysin-1) in mouse calvariae. Biochim. Biophys. Acta 1405, 14–28 [DOI] [PubMed] [Google Scholar]
- 12. Borden P., Solymar D., Sucharczuk A., Lindman B., Cannon P., Heller R. A. (1996) Cytokine control of interstitial collagenase and collagenase-3 gene expression in human chondrocytes. J. Biol. Chem. 271, 23577–23581 [DOI] [PubMed] [Google Scholar]
- 13. Rydziel S., Delany A. M., Canalis E. (1997) Insulin-like growth factor I inhibits the transcription of collagenase 3 in osteoblast cultures. J. Cell. Biochem. 67, 176–183 [PubMed] [Google Scholar]
- 14. Winchester S. K., Bloch S. R., Fiacco G. J., Partridge N. C. (1999) Regulation of expression of collagenase-3 in normal, differentiating rat osteoblasts. J. Cell. Physiol. 181, 479–488 [DOI] [PubMed] [Google Scholar]
- 15. Winchester S. K., Selvamurugan N., D'Alonzo R. C., Partridge N. C. (2000) Developmental regulation of collagenase-3 mRNA in normal, differentiating osteoblasts through the activator protein-1 and the runt domain binding sites. J. Biol. Chem. 275, 23310–23318 [DOI] [PubMed] [Google Scholar]
- 16. Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R. T., Gao Y. H., Inada M., Sato M., Okamoto R., Kitamura Y., Yoshiki S., Kishimoto T. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 [DOI] [PubMed] [Google Scholar]
- 17. Lian J. B., Gordon J. A., Stein G. S. (2013) Redefining the activity of a bone-specific transcription factor: novel insights for understanding bone formation. J. Bone Miner. Res. 28, 2060–2063 [DOI] [PubMed] [Google Scholar]
- 18. Cohen M. M., Jr. (2009) Perspectives on RUNX genes: an update. Am. J. Med. Genet. A 149A, 2629–2646, 10.1002/ajmg.a.33021 [DOI] [PubMed] [Google Scholar]
- 19. Meyer M. B., Benkusky N. A., Pike J. W. (2014) The RUNX2 cistrome in osteoblasts: characterization, down-regulation following differentiation, and relationship to gene expression. J. Biol. Chem. 289, 16016–16031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stein G. S., Stein J. L., Van Wijnen A. J., Lian J. B., Montecino M., Croce C. M., Choi J. Y., Ali S. A., Pande S., Hassan M. Q., Zaidi S. K., Young D. W. (2010) Transcription factor-mediated epigenetic regulation of cell growth and phenotype for biological control and cancer. Adv. Enzyme Regul. 50, 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zaidi S. K., Young D. W., Montecino M. A., Lian J. B., van Wijnen A. J., Stein J. L., Stein G. S. (2010) Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat. Rev. Genet. 11, 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grumbles R. M., Shao L., Jeffrey J. J., Howell D. S. (1996) Regulation of rat interstitial collagenase gene expression in growth cartilage and chondrocytes by vitamin D3, interleukin-1β, and okadaic acid. J. Cell. Biochem. 63, 395–409 [DOI] [PubMed] [Google Scholar]
- 23. Uchida M., Shima M., Chikazu D., Fujieda A., Obara K., Suzuki H., Nagai Y., Yamato H., Kawaguchi H. (2001) Transcriptional induction of matrix metalloproteinase-13 (collagenase-3) by 1α,25-dihydroxyvitamin D3 in mouse osteoblastic MC3T3-E1 cells. J. Bone Miner. Res. 16, 221–230 [DOI] [PubMed] [Google Scholar]
- 24. Meyer M. B., Benkusky N. A., Lee C. H., Pike J. W. (2014) Genomic determinants of gene regulation by 1,25-dihydroxyvitamin D3 during osteoblast-lineage cell differentiation. J. Biol. Chem. 289, 19539–19554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng Y., Ma Z., Kim B. H., Wu W., Cayting P., Boyle A. P., Sundaram V., Xing X., Dogan N., Li J., Euskirchen G., Lin S., Lin Y., Visel A., Kawli T., Yang X., Patacsil D., Keller C. A., Giardine B., Mouse ENCODE Consortium, Kundaje A., Wang T., Pennacchio L. A., Weng Z., Hardison R. C., Snyder M. P. (2014) Principles of regulatory information conservation between mouse and human. Nature 515, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meyer M. B., Benkusky N. A., Pike J. W. (2014) 1,25-Dihydroxyvitamin D3 induced histone profiles guide discovery of VDR action sites. J. Steroid Biochem. Mol. Biol. 144, 19–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sudo H., Kodama H. A., Amagai Y., Yamamoto S., Kasai S. (1983) In vitro differentiation and calcification in a new clonal osteogenic cell line derived from newborn mouse calvaria. J. Cell Biol. 96, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Brien C. A., Gubrij I., Lin S. C., Saylors R. L., Manolagas S. C. (1999) STAT3 activation in stromal/osteoblastic cells is required for induction of the receptor activator of NF-κB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D3 or parathyroid hormone. J. Biol. Chem. 274, 19301–19308 [DOI] [PubMed] [Google Scholar]
- 29. Fu Q., Manolagas S. C., O'Brien C. A. (2006) Parathyroid hormone controls receptor activator of NF-κB ligand gene expression via a distant transcriptional enhancer. Mol. Cell. Biol. 26, 6453–6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim S., Yamazaki M., Zella L. A., Shevde N. K., Pike J. W. (2006) Activation of receptor activator of NF-κB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers. Mol. Cell. Biol. 26, 6469–6486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zella L. A., Kim S., Shevde N. K., Pike J. W. (2006) Enhancers located within two introns of the vitamin D receptor gene mediate transcriptional autoregulation by 1,25-dihydroxyvitamin D3. Mol. Endocrinol. 20, 1231–1247 [DOI] [PubMed] [Google Scholar]
- 32. Meyer M. B., Goetsch P. D., Pike J. W. (2010) A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1α,25-dihydroxyvitamin D3. J. Biol. Chem. 285, 15599–15610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A., 3rd, Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 35. Westendorf J. J., Zaidi S. K., Cascino J. E., Kahler R., van Wijnen A. J., Lian J. B., Yoshida M., Stein G. S., Li X. (2002) Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell. Biol. 22, 7982–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weber K., Bartsch U., Stocking C., Fehse B. (2008) A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. 16, 698–706 [DOI] [PubMed] [Google Scholar]
- 37. Yamamoto H., Shevde N. K., Warrier A., Plum L. A., DeLuca H. F., Pike J. W. (2003) 2-Methylene-19-nor-(20S)-1,25-dihydroxyvitamin D3 potently stimulates gene-specific DNA binding of the vitamin D receptor in osteoblasts. J. Biol. Chem. 278, 31756–31765 [DOI] [PubMed] [Google Scholar]
- 38. Meyer M. B., Goetsch P. D., Pike J. W. (2012) VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: impact on c-FOS and c-MYC gene expression. Mol. Endocrinol. 26, 37–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyer M. B., Watanuki M., Kim S., Shevde N. K., Pike J. W. (2006) The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells. Mol. Endocrinol. 20, 1447–1461 [DOI] [PubMed] [Google Scholar]
- 40. Meyer M. B., Zella L. A., Nerenz R. D., Pike J. W. (2007) Characterizing early events associated with the activation of target genes by 1,25-dihydroxyvitamin D3 in mouse kidney and intestine in vivo. J. Biol. Chem. 282, 22344–22352 [DOI] [PubMed] [Google Scholar]
- 41. Cartharius K., Frech K., Grote K., Klocke B., Haltmeier M., Klingenhoff A., Frisch M., Bayerlein M., Werner T. (2005) MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21, 2933–2942 [DOI] [PubMed] [Google Scholar]
- 42. Quandt K., Frech K., Karas H., Wingender E., Werner T. (1995) MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 23, 4878–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Selvamurugan N., Jefcoat S. C., Kwok S., Kowalewski R., Tamasi J. A., Partridge N. C. (2006) Overexpression of Runx2 directed by the matrix metalloproteinase-13 promoter containing the AP-1 and Runx/RD/Cbfa sites alters bone remodeling in vivo. J. Cell. Biochem. 99, 545–557 [DOI] [PubMed] [Google Scholar]
- 44. Walling H. W., Chan P. T., Omura T. H., Barmina O. Y., Fiacco G. J., Jeffrey J. J., Partridge N. C. (1998) Regulation of the collagenase-3 receptor and its role in intracellular ligand processing in rat osteoblastic cells. J. Cell. Physiol. 177, 563–574 [DOI] [PubMed] [Google Scholar]
- 45. Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J. M., Behringer R. R., de Crombrugghe B. (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29 [DOI] [PubMed] [Google Scholar]
- 46. Paredes R., Arriagada G., Cruzat F., Villagra A., Olate J., Zaidi K., van Wijnen A., Lian J. B., Stein G. S., Stein J. L., Montecino M. (2004) Bone-specific transcription factor Runx2 interacts with the 1α,25-dihydroxyvitamin D3 receptor to up-regulate rat osteocalcin gene expression in osteoblastic cells. Mol. Cell. Biol. 24, 8847–8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tsuji K., Kraut N., Groudine M., Noda M. (2001) Vitamin D3 enhances the expression of I-mfa, an inhibitor of the MyoD family, in osteoblasts. Biochim. Biophys. Acta 1539, 122–130 [DOI] [PubMed] [Google Scholar]
- 48. Chimge N. O., Baniwal S. K., Luo J., Coetzee S., Khalid O., Berman B. P., Tripathy D., Ellis M. J., Frenkel B. (2012) Opposing effects of Runx2 and estradiol on breast cancer cell proliferation: in vitro identification of reciprocally regulated gene signature related to clinical letrozole responsiveness. Clin. Cancer Res. 18, 901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zanotti S., Stadmeyer L., Smerdel-Ramoya A., Durant D., Canalis E. (2009) Misexpression of CCAAT/enhancer binding protein β causes osteopenia. J. Endocrinol. 201, 263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hirata M., Kugimiya F., Fukai A., Saito T., Yano F., Ikeda T., Mabuchi A., Sapkota B. R., Akune T., Nishida N., Yoshimura N., Nakagawa T., Tokunaga K., Nakamura K., Chung U. I., Kawaguchi H. (2012) C/EBPβ and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2α as the inducer in chondrocytes. Hum. Mol. Genet. 21, 1111–1123 [DOI] [PubMed] [Google Scholar]
- 51. Tominaga H., Maeda S., Hayashi M., Takeda S., Akira S., Komiya S., Nakamura T., Akiyama H., Imamura T. (2008) CCAAT/enhancer-binding protein β promotes osteoblast differentiation by enhancing Runx2 activity with ATF4. Mol. Biol. Cell 19, 5373–5386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Benderdour M., Tardif G., Pelletier J. P., Dupuis M., Geng C., Martel-Pelletier J. (2002) A novel negative regulatory element in the human collagenase-3 proximal promoter region. Biochem. Biophys. Res. Commun. 291, 1151–1159 [DOI] [PubMed] [Google Scholar]
- 53. Hashimoto K., Otero M., Imagawa K., de Andrés M. C., Coico J. M., Roach H. I., Oreffo R. O., Marcu K. B., Goldring M. B. (2013) Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J. Biol. Chem. 288, 10061–10072 [DOI] [PMC free article] [PubMed] [Google Scholar]