

Background: The mechanisms of transcriptional regulation of TNFRSF10A and TNFRSF10B are not well described.
Results: DDIT3 and KAT2A cooperatively up-regulated TNFRSF10A and TNFRSF10B.
Conclusion: DDIT3 and KAT2A promote the transcription of TNFRSF10A and TNFRSF10B via the AP-1 and DDIT3 binding sites, respectively.
Significance: Our findings offer new insights on the mechanisms of ER stress-mediated apoptosis.
Keywords: Apoptosis, Endoplasmic Reticulum Stress (ER Stress), Histone Acetylase, Lung Cancer, Transcription Factor, TNFRSF10A, TNFRSF10B, DIT3, KAT2A, Apoptosis
Abstract
TNFRSF10A and TNFRSF10B are cell surface receptors that bind to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and mediate the extrinsic pathway of apoptosis. However, the mechanisms of transcriptional regulation of TNFRSF10A and TNFRSF10B remain largely uncharacterized. In this study, two putative DDIT3 binding sites (−1636/−1625; −374/−364) and a putative AP-1 binding site (−304/−298) were identified in the TNFRSF10A promoter region. We found that DDIT3 interacts with phospho-JUN, and the DDIT3·phospho-JUN complex binds to the AP-1 binding site (−304/−298) within the TNFRSF10A promoter region. In addition, we confirmed that KAT2A physically interacts with the N-terminal region (amino acids 1–26) of DDIT3. Importantly, knockdown of KAT2A down-regulated TNFRSF10A and TNFRSF10B and dramatically decreased promoter activity of cells transfected with luciferase reporter plasmid containing the AP-1 binding site (−304/−298) of the TNFRSF10A promoter, as well as cells transfected with luciferase reporter plasmid containing DDIT3 binding site (−276/−264) of the TNFRSF10B promoter. ChIP results suggest that KAT2A may participate in a KAT2A·DDIT3·phospho-JUN complex, or may participate in a KAT2A·DDIT3 complex and acetylate H3K9/K14, respectively. Moreover, we verified that TNFRSF10A mediates apoptosis triggered by endoplasmic reticulum stress in human lung cancer cells. Collectively, we demonstrate that DDIT3 and KAT2A cooperatively up-regulate TNFRSF10A and TNFRSF10B. Our findings highlight novel mechanisms underlying endoplasmic reticulum stress-induced TNFRSF10A and TNFRSF10B expressions and apoptosis. These findings will be helpful for elucidating mechanisms related to anticancer drugs in mediating apoptosis.
Introduction
TNFRSF10A (also known as DR4 or TRAIL-R1) and TNFRSF10B (also known as DR5 or TRAIL-R2) are two cell surface receptors that bind to TRAIL3 and mediate the extrinsic pathway of apoptosis (1, 2). TNFRSF10A and TNFRSF10B mediate TRAIL-induced apoptosis and contribute to enhancement of apoptosis induced by anticancer drugs (3–8). In this process, the trimerized TNFRSF10A and TNFRSF10B interact specifically with the adaptor protein FADD, which subsequently recruits the pro-CASP8 through the death effector domain. Aggregation of pro-CASP8 leads to auto-cleavage and activation of CASP8 and activates effector caspases such as CASP3 and then induces apoptosis (9).
The up-regulation of TNFRSF10A expression usually occurs at the transcriptional level via transcriptional factors AP-1 (10), TP53 (11), or NF-κB (12). Transcription of TNFRSF10B is regulated by TP53 (13, 14), NF-κB (15, 16), YY1 (17), or DDIT3 (18, 19). Further insights into the transcriptional regulation of TNFRSF10A and TNFRSF10B may help understand the molecular mechanisms underlying TNFRSF10A/10B-mediated apoptosis.
Endoplasmic reticulum (ER) stress plays an important role in anticancer drug-induced apoptosis (7). Four members of BH3-only family, BBC3, PMAIP1, BID, and BCL2L11, mediate apoptosis triggered by ER stress (18, 20–22). In addition, BBC3, PMAIP1, BCL2L11, and TNFRSF10B are regulated by DDIT3 (18, 21, 23), which is an ER stress-inducible gene and a key mediator of ER stress-induced apoptosis in many cell types including murine fibroblast cells (24), lymphocyte cells (25), and pancreatic β cells (26).
DDIT3 is a member of the CCAAT/enhancer-binding proteins (C/EBPs), which consist of six members: C/EBPα, C/EBPβ, C/EBPγ, C/EBPδ, C/EBPϵ, and C/EBP homologous protein (CHOP, also called DDIT3 or GADD153) (27). The classical C/EBP contains a transcriptional activation domain and a basic region-leucine zipper (bZIP) region for DNA binding and dimerization. DDIT3 has an atypical basic region as compared with other C/EBP family proteins, and therefore DDIT3 lacks DNA binding activity at the C/EBP binding site, but DDIT3 does bind to a unique DNA sequence and acts as a transactivator (28). DDIT3 acts as a transcription factor to enhance TNFRSF10B expression and trigger ER stress-induced apoptosis (29). The accumulation of TNFRSF10B provides a DISC (death-inducing signaling complex)-like intracellular platform for caspase-8 recruitment and apoptosis initiation (30). However, details underlying these mechanisms are lacking. In addition, whether TNFRSF10A is also regulated by DDIT3 and mediates ER stress-induced apoptosis remains unknown.
Histone acetyltransferases (HATs) act as transcription co-activators. They are directly recruited by transcriptional activators to gene promoters and enhance the transcription activity by adding acetyl groups to lysine residues within the N-terminal tails of histones, which facilitates the transcription complex formation (31). HATs contain five families, including the KAT2A/KAT2B family, MYST (MOZ, Ybf2 (Sas3), Sas2, and Tip60) family, TAFII250 family, CREBBP/EP300 family, and SRC family (32). KAT2A/KAT2B and CREBBP/TP300 give rise to histone acetylation and lead to transcriptional activation (33–35). DDIT3 interacts with TP300 through the N-terminal region (36). However, whether other HATs interact with DDIT3 and act as co-activators to enhance transcription activity has not been explored.
In the present study, we found that DDIT3 and TNFRSF10A are induced by two ER stress inducers, thapsigargin and tunicamycin, in human non-small cell lung cancer (NSCLC) cells. We verified that DDIT3 enhances TNFRSF10A transcription via interaction with phospho-JUN of AP-1 complex at the AP-1 binding site located at −304/−298 bp in the TNFRSF10A promoter region. Moreover, we confirmed that KAT2A interacts with the N-terminal region of DDIT3 and acts as a transcription co-activator of DDIT3, leading to H3K9/K14 acetylation, and further enhances TNFRSF10A and TNFRSF10B transcription. In addition, we found that TNFRSF10A mediated ER stress-induced apoptosis in a DDIT3-dependent manner in NSCLC cells. Our findings shed light on the molecular mechanism underlying TNFRSF10A and TNFRSF10A-mediated apoptosis in human lung cancer cells.
EXPERIMENTAL PROCEDURES
Reagents
RPMI 1640 medium (R6504), Dulbecco's modified Eagle's medium (D5648), FBS (12003C), thapsigargin (T9033), anti-TNFRSF10A antibody (SAB3500428), and anti-ACTB antibody (SAB1403520) were purchased from Sigma-Aldrich. Tunicamycin (TF1129) was purchased from Sangon Biotech Co., Ltd. (Shanghai, China). Primary antibodies against DDIT3 (2895), KAT2A (3305), CASP8 (9746), PARP1 (9542), JUN (9165), phospho-JUN (3270), FOS (4384), phospho-FOS (5348), and acetylated lysine (9441) were purchased from Cell Signaling Technology (Danvers, MA). Anti-DDIT3 (sc-7351), anti-phospho-JUN (sc-822), anti-FOS (sc-52), isotype mouse IgG1 (sc-3877), and the normal rabbit isotype IgG (sc-3888) were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-acetyl histone H3K9/K14 polyclonal antibody (A-4021) was purchased from Epigentek Group (Farmingdale, NY). X-tremeGENE HP DNA transfection reagent (06366546001) and X-tremeGENE siRNA transfection reagent (4476115001) were purchased from Roche Diagnostics (Mannheim, Germany). Luciferase Assay System with reporter lysis buffer (E4030) was purchased from Promega (Madison, WI). ChIP assay kit (17-295) was purchased from EMD Millipore (Billerica, MA). Universal DNA purification kit (DP214) was purchased from TIANGEN Biotech Co., Ltd. (Beijing, China). Annexin V:PE/7-AAD apoptosis detection kit I (559763) was purchased from BD Biosciences.
Cell Lines and Cell Culture
All cell lines used in this study were obtained from the American Type Culture Collection (Manassas, VA). A549 and H1792 cell lines were recently authenticated by Microread gene technology (Beijing Microread Genetics, Beijing, China) by short tandem repeat analysis. A549, H1792, H460, and H157 cells were maintained in RPMI 1640 medium supplemented with 5% (v/v) FBS at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
siRNA Transfections
siRNAs were synthesized by GenePharma (Shanghai, China). The transfection of siRNA was conducted as described previously (29). DDIT3 #1 and #2 siRNAs target the sequences 5′-GCCTGGTATGAGGACCTGC-3′ and 5′-AAGAACCAGCAGAGGUCACAA-3′, respectively. TNFRSF10A #1 and TNFRSF10A #2 siRNAs target the sequences 5′-TACACCAATGCTTCCAACAAT-3′ and 5′-AATGAGATCGATGTGGTCAGA-3′, respectively. KAT2A#1 and #2 siRNAs target the sequences 5′-GAAGCUGAUUGAGCGCAAA-3′, and 5′-GGAAAUGCAUCCUGCAGAU-3′, respectively.
Western Blot Analysis
Preparation of whole-cell protein lysates and the procedures for the Western blot were described previously (29).
Immunoprecipitation Assay
H1792 cells were treated with DMSO, thapsigargin (1 μm), and tunicamycin (1 μm), respectively, for 12 h, and the cells were lysed in precipitation lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, and 1 μg/ml leupeptin) supplemented with protease inhibitors. Cell lysates were immunoprecipitated and analyzed by Western blot.
ChIP Assay
ChIP assay was conducted using the ChIP assay kit following the manufacturer's instructions. Briefly, H1792 cells were treated with DMSO, thapsigargin (1 μm), and tunicamycin (1 μm). After 12 h, genomic DNA and protein were cross-linked by formaldehyde (1% final concentration) for 10 min at 37 °C. Cells then were collected and lysed in 200 μl of SDS lysis buffer supplemented with protease inhibitors for 10 min, and then the cell lysates were sonicated to generate 200–1000-bp DNA fragments. After centrifugation, the cleared supernatant was diluted with 10-fold ChIP dilution buffer and incubated at 4 °C overnight with anti-DDIT3, anti-phospho-JUN, anti-H3K9/K14ac, and isotype moue IgG1 or rabbit normal IgG antibodies, respectively. On the second day, the immune complexes were precipitated, washed, and eluted as recommended. After DNA-protein cross-links were reversed at 65 °C for 4 h, DNA was extracted using the Universal DNA purification kit. The input and immunoprecipitated samples were used as template for PCR amplification of fragments containing the AP-1 binding site in the TNFRSF10A promoter region using specific primers as follows: sense, 5′-CGAGCTTGACTCCATCTCAAAAA-3′; antisense, 5′-CTCAGCCTTTCTGTGACCCAG-3′; they amplify a 193-bp region of the human TNFRSF10A promoter, which contains an AP-1 binding site: TGAATCA (10). The fragments containing the DDIT3 binding site in TNFRSF10B promoter region used specific primers as follows: sense, 5′-AGGTTAGTTCCGGTCCCTTC-3′; antisense, 5′-CAACTGCAAATTCCACCACA-3′; they amplify a 111-bp region of the human TNFRSF10B promoter, which contains a DDIT3 binding site: GAGGATTGCGTTG. GAPDH was used as an internal control, using primers as follows: sense, 5′-TACTAGCGGTTTTACGGGCG-3′; antisense, 5′-TCGAACAGGAGGAGCAGAGAGCGA-3′; they amplify a 166-bp region of the human GAPDH promoter.
Luciferase Activity Assay
H1792 cells were transfected with plasmids containing luciferase reporter gene for 24 h, with plasmids containing luciferase reporter gene and other indicated plasmids for 24 h, with plasmids containing luciferase reporter gene and KAT2A siRNA for 24 h, or with plasmids containing luciferase reporter gene in combination with treatment with thapsigargin (TG) (1 μm) and tunicamycin (Tm) (1 μm) for 12 h. Cell lysates were prepared and luciferase assays were performed according to the manufacturer's instructions (Promega). The transfection efficiency was normalized by co-transfection with pCH110 (β-gal) or by Dual-Luciferase reporter assay system using the pRL-SV40 vector (catalog number E2231, Promega). All experiments were performed three times independently, and the values obtained were used to calculate means and standard deviations.
Plasmid Constructions
The TNFRSF10A promoter region (−1640/−1 bp) was amplified from A549 cell genomic DNA and cloned into the pGL3-Basic luciferase reporter vector by using MluI and BglII restriction sites, and the resulting plasmid was designated as pGL3-WT. Based on this plasmid, five plasmids were constructed using the PCR mutagenesis method. The plasmid pGL3-BS1mt contains the DDIT3-BS1 (−1636/−1625) mutant, the plasmid pGL3-BS2mt contains the DDIT3-BS2(−374/−364) mutant, the plasmid pGL3-BS1/2mt contains the DDIT3-BS1 and BS2 mutant, the plasmid pGL3-AP-1BSmt contains the AP-1(−304/−298) mutant, and the plasmid pGL3-3mt contains the combination of all three mutant sites. PCR primers were described as follows: for pGL3-WT, The sense and antisense primers are: 5′-CACGCGTCAATGCAATCCCAATTAAAATTCC; and 5′-CAGATCTTGCGGCTCCTGTTGGCTAAC-3′, respectively. The putative DDIT3-BS1 (−1636–1625) and mutant sequence are: 5′-CAATGCAATCCC-3′ and 5′-CAATacgatCCC-3′, respectively. The putative DDIT3-BS2(−374/−364) and mutant sequence are: 5′-CACTGCACTCC-3′ and 5′-CAacattaTCC-3′, respectively. The AP-1BS and mutant sequence are: 5′-TGAATCA-3′ and 5′-acaacaa-3′, respectively.
In addition, we also constructed two pGL3-Basic plasmids that contained AP-1BS and AP-1BS mutant, respectively. The primers used are: TNFRSF10A/AP-1(−304/−298) sense, 5′-CACGCGTCAGGCTGAATCACTCGCCCGGTAGCTCGCC-3′; TNFRSF10A/AP-1(−304/−298)(mutant)-BS sense, 5′-CACGCGTCAGGCacaacaaCTCGCCCGGTAGCTCGCC-3′. Their antisense primer was the same as the pGL3-WT one. The TNFRSF10A promoter region (−304/−1 bp) was amplified from A549 cell genomic DNA and cloned into the pGL3-Basic luciferase reporter vector by using MluI and BglII restriction sites, and the resulting plasmid was designated as pB-TNFRSF10A/AP-1-BS(−304/−298)-luc and pB-TNFRSF10A/AP-1-BS(−304/−298)(mt)-luc. The TNFRSF10B reporter constructs pB-TNFRSF10B/DDIT3(−276/−264)-BS-luc and pB-TNFRSF10B/DDIT3(−276/−264)(mt)-BS-luc used in this study were described previously (37). Briefly, pB-TNFRSF10B/DDIT3(−276/−264)-BS-luc is a pGL3-Basic plasmid containing (−522/−1 bp) TNFRSF10B promoter region with a wild type DDIT3 binding site, and pB-TNFRSF10B/DDIT3(−276/−264)(mt)-BS-luc is the same except that it contains the mutant DDIT3 binding site.
The DDIT3 was amplified from A549 cells cDNA with PCR amplification using the primers described as follows: sense, 5′-CCTCGAGATGCTTGGTGCAGATTCACC-3′; antisense, 5′-CGGGCCCTCATTGGTCTTCCTCCTCTTCCT-3′. The fragments were then cloned into the pcDNA3.1 (+) vector by using XhoI and ApaI restriction sites. The different fragments of DDIT3 (aa 1–99), (aa 27–99), and (aa 100–162) and the full-length DDIT3 (aa 1–169) were amplified with PCR using the primers described as follows: (aa 1–99) sense, 5′-CGCGGCCGCTCATTGGTCTTCCTCCTCTTCCT-3′; antisense, 5′-AAGGAAAAAAGCGGCCGCAAAAGGAAAATCATTGGTCTTCCTCCTCTTCCT; (aa 27–99) sense, 5′-CGGATCCGAGGTCCTGTCTTCAGATGA-3′; antisense, 5′-AAGGAAAAAAGCGGCCGCAAAAGGAAAATCATTGGTCTTCCTCCTCTTCCT; (aa 100–162) sense, 5′-CGGATCCGGGAGAACCAGGAAACGGA-3′; antisense, 5′-CGCGGCCGCTCATCGGTCAATCAGAGCTCGG-3′; (aa 1–169) sense, 5′-CGCGGCCGCTCATTGGTCTTCCTCCTCTTCCT-3′; antisense, 5′-AAGGAAAAAAGCGGCCGCAAAAGGAAAATCATGCTTGGTGCAGATTCAC-3′. The fragments then were cloned into the pEBG vector by using BamHI and NotI restriction sites.
Apoptosis Assays
Apoptosis was evaluated by annexin V:PE/7-AAD apoptosis detection kit (BD Biosciences) following the manufacturer's instructions. Caspase activation was detected by Western blot.
Statistical Analysis
The percentage of total apoptotic cells was calculated and expressed as the mean ± S.D., and differences between groups were evaluated by Student's t test. In all statistical analyses, results were considered to be statistically significant when p value was less than 0.05.
RESULTS
DDIT3 and TNFRSF10A Expressions Are Up-regulated by ER Stress Inducers in Human NSCLC Cells
To explore whether TNFRSF10A is induced during ER stress in human NSCLC cells, the cell lines A549, H1792, and H460 were treated with ER stress inducers TG or Tm at 0.5, 1, and 2 μm for 24 h. Western blot analysis revealed that TNFRSF10A was induced in a concentration-dependent manner (Fig. 1A). Tm alters TNFRSF10A protein size via inhibition of glycosylation (38). Tm treatment generated two forms of TNFRSF10A: TNFRSF10A (large) and TNFRSF10A (small) (Fig. 1A). We treated A549, H1792, and H460 cells with TG (1 μm) or Tm (1 μm) for 4, 8, 12, and 24 h. We found that TNFRSF10A expression was up-regulated by TG and Tm in a time-dependent manner (Fig. 1B). Given that DDIT3 plays an important role in ER stress-induced apoptosis (18, 21, 23), we also investigated DDIT3 protein levels after treatment with TG or Tm. Western blot analysis indicated that DDIT3 induction preceded TNFRSF10A induction. Moreover, we examined the TNFRSF10A mRNA levels after TG or Tm treatment. The RT-PCR results indicated that TNFRSF10A was up-regulated at the transcriptional level (Fig. 1C). These data suggest that DDIT3 and TNFRSF10A are induced by ER stress inducers in a concentration- and time-dependent manner in human lung cancer cells.
FIGURE 1.

ER stress induces TNFRSF10A expression in human NSCLC cells. A, A549, H1792, and H460 cells were treated with 0.5, 1, and 2 μm TG or Tm for 24 h. Protein expression was analyzed by Western blot using antibodies against DDIT3, TNFRSF10A, and ACTB. B, A549, H1792, and H460 cells were treated with 1 μm TG or Tm for the indicated times. Protein expression was analyzed by Western blot using antibodies against DDIT3, TNFRSF10A, and ACTB. C, A549, H1792, and H460 cells were treated with 0.5, 1, and 2 μm TG or Tm for 24 h. RT-PCR was performed to examine the mRNA levels.
TNFRSF10A Expression Is Up-regulated by ER Stress Inducers in a DDIT3-dependent Manner
To investigate whether DDIT3 regulates induction of TNFRSF10A, we transfected pcDNA3.1-DDIT3 plasmid into A549, H1792, H460, and H157 cell lines and evaluated the effects. As shown in Fig. 2A, DDIT3 overexpression induced TNFRSF10A in the four human lung cancer cell lines. In addition, we also suppressed the expression of DDIT3 using the specific siRNAs against DDIT3 (#1 and #2) and then detected the TNFRSF10A protein levels after treatment with TG or Tm for 12 h in A549, H1792, and H460 cells. The results show that TNFRSF10A induction was inhibited in DDIT3 knockdown cells after treatment with ER stress inducers (Fig. 2B). Thus, these results suggest that TNFRSF10A expression is regulated in a DDIT3-dependent manner.
FIGURE 2.

ER stress up-regulates TNFRSF10A expression through a DDIT3-dependent mechanism. A, A549, H1792, H460, and H157 cells were transfected with pcDNA3.1-DDIT3 plasmid for 24 h, and samples were analyzed by Western blot using antibodies against DDIT3, TNFRSF10A, and ACTB. B, A549, H1792, and H460 cells were transfected with DDIT3 siRNAs (#1 and #2). Forty-eight hours later, the cells were treated with DMSO or 1 μm TG for another 12 h, and samples were analyzed by Western blot. Ctrl, control. C, A549, H1792, and H460 cells were transfected with DDIT3 siRNAs (#1 and #2). After 48 h, the cells were treated with DMSO or 1 μm Tm for another12 h, and samples were analyzed by Western blot using antibodies against DDIT3, TNFRSF10A, and ACTB.
The AP-1 Binding Site on the TNFRSF10A Promoter Plays a Critical Role in Mediating DDIT3-dependent Transcription
DDIT3 acts as a transcription factor and induces TNFRSF10B expression via TNFRSF10B promoter binding (27). We evaluated whether DDIT3 directly up-regulates TNFRSF10A transcription. To determine whether putative DDIT3 binding sites exist in the TNFRSF10A promoter, we compared the sequences between DDIT3 binding site sequence PuPuPuTGCAAT(A/C)CCC (Pu, purine) and the TNFRSF10A promoter. We identified two potential DDIT3 binding sites located at −1636/−1625 and −374/−364 bp in the TNFRSF10A promoter. In addition, we found an AP-1 binding site located at −304/−398 bp (Fig. 3A). To explore whether TNFRSF10A/DDIT3-BS and AP-1BS mediate DDIT3-dependent TNFRSF10A gene expression, we designed and constructed six plasmids based on the pGL3-Basic luciferase reporter vector as shown in Fig. 3A. The six constructs are designated as: pGL3-WT, pGL3-BS1mt, PGL3-BS2mt, pGL3-BS1/2mt, pGL3-AP-1BSmt, and pGL3-3mt. We transfected plasmids into H1792 cells for 24 h and then incubated them with TG (1 μm) or Tm (1 μm) for an additional 12 h. We found that the luciferase activities of pGL3-WT, pGL3-BS1mt, pGL3-BS2mt, and pGL3-BS1/2mt increased significantly after TG or Tm treatment as compared with the control treatment. However, the luciferase activities of pGL3-AP-1BSmt and pGL3-3mt did not increase after treatment with TG or Tm (Fig. 3B). These data suggest that the AP-1 binding site on the TNFRSF10A promoter is necessary to mediate its ER stress inducer-induced transcription.
FIGURE 3.

DDIT3 enhances TNFRSF10A transcription via AP-1 binding site. A, schematic of putative DDIT3 and AP-1 binding sites and the design of the mutations in the TNFRSF10A promoter region (−1640 bp to 1 bp). Position +1 designates the initiation site for the TNFRSF10A transcription. The DNA fragments were amplified by PCR and cloned into the pGL3-Basic luciferase reporter vector. The mutations were made by PCR mutagenesis. B, H1792 cells were co-transfected with the pRL-SV40 vector (0.1 μg/well) and the pGL3-Basic constructs, respectively, (0.25 μg/well) for 24 h, and the cells were treated with TG (1 μm) or Tm (1 μm) for another 12 h. The lysates were prepared and luciferase assays were performed using Dual-Luciferase reporter assay system according to the manufacturer's instructions (Promega). Ctrl, control. C, the H1792 cells were seeded into 24-well cell culture plates and incubated overnight. Cells were co-transfected with pcDNA3.1 (0.25 μg/well) or pcDNA3.1-DDIT3 (0.25 μg/well) and pB-TNFRSF10A/AP-1(−304/−298)-BS-luc (0.25 μg/well) or pB-TNFRSF10A/AP-1(−304/−298)(mt)-BS-luc (0.25 μg/well) for 24 h, cell lysates were prepared, and luciferase assays were performed according to the manufacturer's instructions (Promega). The transfection efficiency was normalized by co-transfection with pCH110 (β-gal) (0.1 μg/well). D, the H1792 cells were seeded into 24-well cell culture plates and incubated overnight. Next the cells were co-transfected with DDIT3 siRNA and pB-TNFRSF10A/AP-1(−304/−298)-BS-luc (0.25 μg/well) or pB-TNFRSF10A/AP-1(−304/−298)(mt)-BS-luc (0.25 μg/well) for 24 h, and the cells were then treated with TG (1 μm) or Tm (1 μm) for another 12 h. Cell lysates were prepared, and luciferase assays were performed according to the manufacturer's instructions. The transfection efficiency was normalized by co-transfection with pCH110 (β-gal) (0.1 μg/well). Columns, mean of triplicate treatments; bars, ± S.D. The statistical differences between the two treatments were analyzed by two-sided unpaired Student's t tests (**, p < 0.01; ***, p < 0.001; ns, no significant difference).
To further determine whether the AP-1 binding site at −304/−298 bp is critical for DDIT3-dependent TNFRSF10A transcription, we cloned the fragment of the TNFRSF10A promoter region (−304/−1 bp), containing the AP-1 binding site, into the pGL3-Basic vector. Based on this plasmid, we constructed a plasmid with a mutation in the AP-1 binding site. These two constructs were designated pB-TNFRSF10A/AP-1-BS(−304/−298)-luc and pB-TNFRSF10A/AP-1-BS(−304/−298)(mt)-luc, as shown under “Experimental Procedures.” First, we co-transfected with pcDNA3.1 or pcDNA3.1-DDIT3 and pB-TNFRSF10A/AP-1-BS(−304/−298)-luc or pB-TNFRSF10A/AP-1-BS(−304/−298)(mt)-luc plasmid into H1792 cells for 24 h, respectively. We found that transfection of pcDNA3.1-DDIT3 dramatically increased the luciferase activity of reporter plasmid carrying TNFRSF10A/AP-1-BS(−304/−298)-luc. However, the luciferase activity of pB-TNFRSF10A/AP-1-BS(−304/−298)(mt)-BS-luc was at a minimum level (Fig. 3C). Second, we co-transfected with control or DDIT3 siRNA and pB-TNFRSF10A/AP-1(−304/−298)-BS-luc or pB-TNFRSF10A/AP-1(−304/−298)(mt)-BS-luc plasmid into H1792 cells for 24 h, respectively. The cells were then treated with TG (1 μm) or Tm (1 μm) for an additional 12 h. The results indicate that the luciferase activity of pB-TNFRSF10A/AP-1(−304/−298)-BS-luc was dramatically decreased in DDIT3 knockdown cells (Fig. 3D). Altogether, these data demonstrate that the AP-1 binding site located at −304/−298 bp on the TNFRSF10A promoter plays a critical role in mediating DDIT3-dependent transcription.
DDIT3 Enhances TNFRSF10A Transcription via Interaction with Phospho-JUN
To investigate whether DDIT3 enhances TNFRSF10A transcription via interaction with members of the AP-1 family of transcription factors, we performed co-immunoprecipitation experiments to determine whether endogenous DDIT3 interacts with active phospho-JUN and FOS. We treated H1792 cells with TG (1 μm) or Tm (1 μm) for 12 h and found that immunoprecipitation of endogenous DDIT3 co-precipitated phospho-JUN. However, there is no significant phospho-FOS pulled down relative to the negative control (Fig. 4A). This suggests DDIT3 interacts with phospho-JUN to form a complex in lung cancer cells. To further verify whether DDIT3·p-JUN complex binds to the AP-1 binding site located at −304/−298 bp within the TNFRSF10A promoter region (Fig. 3A), we performed ChIP analysis using DDIT3 or phospho-JUN antibody in the H1792 cell lysate after treatment with TG (1 μm) or Tm (1 μm) for 12 h. The results demonstrate that both DDIT3 and phospho-JUN antibodies captured the AP-1 binding site region of human TNFRSF10A promoter, and the quantity of PCR product containing the AP-1 binding site region was increased in cell lysate prepared from TG- or Tm-treated H1792 cells (Fig. 4B). These data show that DDIT3 regulates TNFRSF10A transcription through DDIT3·p-JUN transcriptional complex that binds to the AP-1 binding site (−304/−298).
FIGURE 4.

DDIT3 enhances TNFRSF10A transcription via DDIT3·p-JUN complex. A, H1792 cells were treated with DMSO, TG (1 μm), or Tm (1 μm) for 12 h, and cell lysates were immunoprecipitated (IP) using the anti-DDIT3 antibody. The isotype IgG1 antibody served as control. The indicated proteins were detected using Western blot analysis. p-JUN, phospho-Jun; p-FOS, phospho-FOS. B, H1792 cells were treated with DMSO, TG (1 μm), or Tm (1 μm) for 12 h, and then the cell lysates were prepared and ChIP assays were performed using the anti-DDIT3 and anti-phospho-JUN antibodies and using the isotype IgG antibody as negative control (data not show). The input and immunoprecipitated samples were used as template for PCR amplification of fragments containing the AP-1 binding site, and GAPDH was used as a loading control.
KAT2A Interacts with DDIT3 by Binding to Its N-terminal Region
HATs KAT2A/KAT2B and CREBBP/EP300 are transcription co-activators that regulate gene activation (32). DDIT3 interacts with EP300 through the N-terminal region (36). However, whether other HATs also interact with DDIT3 and act as co-activators to enhance TNFRSF10A transcription activity has not been studied. We treated H1792 cells with TG (1 μm) or Tm (1 μm) for 12 h and harvested the cell lysates, and then performed co-immunoprecipitation experiments. Western blot analysis indicates that KAT2A co-immunoprecipitated with DDIT3 antibody (Fig. 5A). Given that the DDIT3 N-terminal domain is critical for its transactivity (36), we speculated that KAT2A interacts with the N-terminal region of DDIT3 and enhances its transcriptional activity. To verify this hypothesis, we generated a series of DDIT3 truncation fragments based on previous studies (28, 36, 39) and transfected into 293FT cells for 24 h, and then tested their interaction with endogenous KAT2A by Western blot analysis. The results show that KAT2A binds to the amino acid residues 1–26 of DDIT3 (Fig. 5B), indicating that KAT2A enhances DDIT3 transcriptional activity through interaction with its N-terminal transactivation domain.
FIGURE 5.
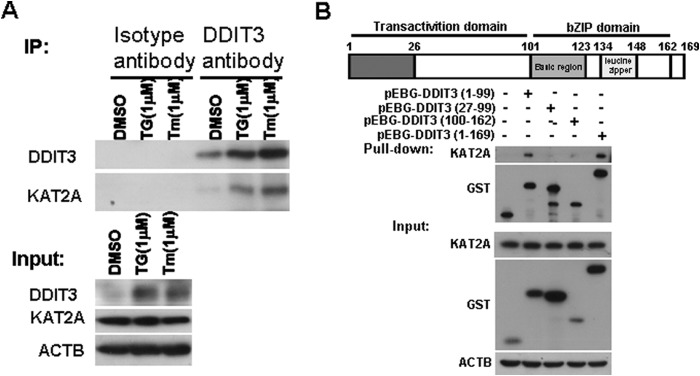
KAT2A interacts with DDIT3 by binding to its N-terminal region. A, H1792 cells were treated with DMSO, TG (1 μm), or Tm (1 μm), for 12 h, and cell lysates were immunoprecipitated (IP) using the anti-DDIT3 antibody. The isotype IgG1 antibody served as control. The proteins were detected using Western blot analysis. B, GST-tagged DDIT3 (1–99), (27–99), and (100–162) and full-length DDIT3 (1–169) were transfected into 293FT cells for 24 h, followed by glutathione-Sepharose 4B pulldown and Western blot analysis. bZIP, basic region-leucine zipper.
KAT2A Is a Co-activator of DDIT3 and Enhances TNFRSF10A Transcription by Acetylation of H3K9/K14
To further clarify whether KAT2A regulates the expression of TNFRSF10A, KAT2A siRNAs were transfected in A549, H1792, and H460 cells. We found that TNFRSF10A protein levels decreased in KAT2A knockdown cells (Fig. 6A). KAT2A acetylates the IFH1 transcription factor and regulates its transcription activity (40). To test whether KAT2A interacts with and acetylates DDIT3 to affect its regulation of TNFRSF10A, we overexpressed DDIT3 in H1792 cells and evaluated its acetylation level with immunoprecipitation assay. We detected no acetylation modification in DDIT3 (data not shown), suggesting that KAT2A acts as a co-activator of DDIT3 to regulate TNFRSF10A transcription. To validate this hypothesis, we performed a luciferase activity assay. The results show that the luciferase activity of pB-TNFRSF10A/AP-1(−304/−298)-BS-luc was dramatically decreased in KAT2A knockdown cells as compared with control cells (Fig. 6B).
FIGURE 6.
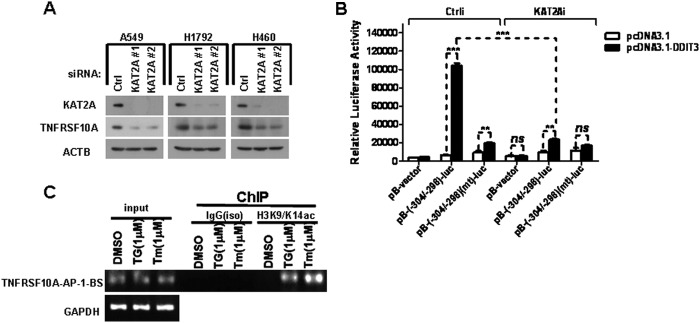
KAT2A is a co-activator of DDIT3 and enhance TNFRSF10A transcription by mediating acetylation of H3K9/K14. A, A549, H1792, and H460 cells were transfected with KAT2A siRNAs (#1 and #2) for 24 h, and then analyzed by Western blot. Levels of protein expression were detected using antibodies against KAT2A, TNFRSF10A, and ACTB. B, H1792 cells were co-transfected with KAT2A siRNA and pB-TNFRSF10A/AP-1(−304/−298)-BS-luc (0.25 μg/well) or pB-TNFRSF10A/AP-1(−304/−298)(mt)-BS-luc (0.25 μg/well) for 24 h. Cell lysates were prepared, and luciferase assays were performed according to the manufacturer's instructions. The transfection efficiency was normalized by co-transfection with pCH110 (β-gal) (0.1 μg/well). All luciferase activity detection experiments were performed three times independently, and the values obtained were used to calculate means and standard deviations. Ctrl, control. C, H1792 cells were treated with DMSO, TG (1 μm), or Tm (1 μm) for 12 h, and then the cell lysates were prepared and ChIP assays were performed using the anti-H3K9/K14ac antibody and the normal IgG antibody as negative control. The input and immunoprecipitated samples were used as template for PCR amplification of fragments containing the AP-1 binding site, and GAPDH was used as a loading control. Columns, mean of triplicate treatments; bars, ± S.D. The statistical differences between the two treatments were analyzed by two-sided unpaired Student's t tests (**, p < 0.01; ***, p < 0.001; ns, no significant difference).
Given that KAT2A increases histone H3K9/K14 acetylation (H3K9/K14ac) and accessibility of factors promoting transcription (33, 41, 42), we speculated that KAT2A is recruited to DDIT3·phospho-JUN complex via physical interaction with DDIT3 at the AP-1 binding site (−304/−298) in the TNFRSF10A promoter region, leading to acetylation of H3K9/K14 and further enhancement of TNFRSF10A transcription. To verify this hypothesis, we performed ChIP experiments using anti-H3K9/K14ac antibody in the H1792 cell lysates after treatment with TG (1 μm) or Tm (1 μm) for 12 h. We found that the antibody against H3K9/K14ac captured the AP-1 binding site region of human TNFRSF10A promoter, and the quantity of PCR product containing the AP-1 binding site was increased in cell lysate prepared from TG- or Tm-treated H1792 cells, respectively (Fig. 6C). Taken together, these results suggest that KAT2A interacts with DDIT3·phospho-JUN complex and acts as a co-activator to further enhance TNFRSF10A transcription via acetylation of H3K9/K14.
KAT2A Is a Co-activator of DDIT3 and Enhances TNFRSF10B Transcription by Acetylation of H3K9/K14
DDIT3 transcriptionally regulates TNFRSF10B expression during ER stress-induced apoptosis by binding to the TNFRSF10B promoter (−276/−264 bp) region (18). To explore whether KAT2A also regulates TNFRSF10B transcription as a co-activator of DDIT3, we transfected KAT2A siRNA in A549, H1792, and H460 cells, and then analyzed by Western blot. The results show that TNFRSF10B protein levels were decreased in KAT2A knockdown cells (Fig. 7A). Moreover, we transfected H1792 cells with control siRNA or KAT2A siRNA for 24 h, and then co-transfected with pcDNA3.1 or pcDNA3.1-DDIT3 and pB-TNFRSF10B/DDIT3(−276/−264)-BS-luc or pB-TNFRSF10B/DDIT3(−276/−264)(mt)-BS-luc plasmid. The results show that luciferase activity of pB-TNFRSF10B/DDIT3(−276/−264)-BS-luc was dramatically decreased in KAT2A knockdown cells (Fig. 7B). We performed ChIP assays using anti-H3K9/K14ac antibody and cell lysates from H1792 cells treated with TG (1 μm) for 12 h. We found that antibody against H3K9/K14ac captured the DDIT3 binding site region located at (−276/−264 bp) in TNFRSF10B promoter, and the quantity of PCR product containing DDIT3 binding site sequence was increased in cell lysate prepared from TG-treated H1792 cells (Fig. 7C). Taken together, these results show that KAT2A also acts as a co-activator of DDIT3 to regulate TNFRSF10B transcription via acetylation of H3K9/K14.
FIGURE 7.
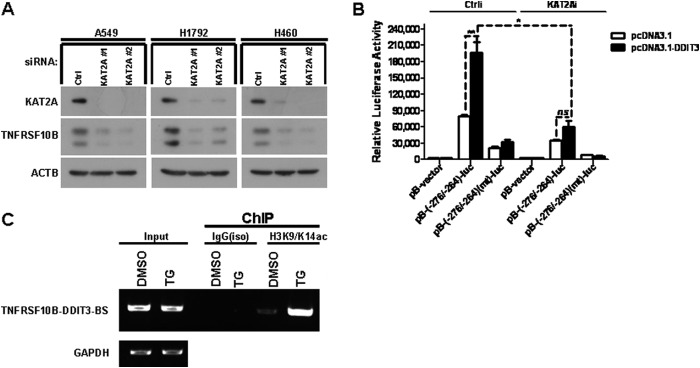
KAT2A is a co-activator of DDIT3 and enhances TNFRSF10B transcription by mediating acetylation of H3K9/K14. A, A549, H1792, and H460 cells were transfected with KAT2A siRNAs (#1 and #2) for 24 h, and samples were analyzed by Western blot using antibodies against KAT2A, TNFRSF10B, and ACTB. Ctrl, control. B, H1792 cells were co-transfected with KAT2A siRNA and pB-TNFRSF10B/DDIT3(−276/−264)-BS-luc (0.25 μg/well) or pB-TNFRSF10B/DDIT3(−276/−264)(mt)-BS-luc (0.25 μg/well) for 24 h. Cell lysates were prepared, and luciferase assays were performed according to the manufacturer's instructions. The transfection efficiency was normalized by co-transfection with pCH110 (β-gal) (0.1 μg/well). All luciferase activity detection experiments were performed three times independently, and the values obtained were used to calculate means and standard deviations. C, H1792 cells were treated with DMSO and TG (1 μm) for 12 h, and then the cell lysates were prepared and ChIP assays were performed using the anti-H3K9/K14ac antibody and the normal IgG antibody as negative control. The input and immunoprecipitated samples were used as template for PCR amplification of fragments containing the DDIT3 binding site, and GAPDH was used as a loading control. Columns, mean of triplicate treatments; bars, ± S.D. The statistical differences between the two treatments were analyzed by two-sided unpaired Student's t tests (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, no significant difference).
TNFRSF10A Mediates ER Stress-induced Apoptosis in a DDIT3-dependent Manner
To investigate whether TNFRSF10A mediates ER stress-induced apoptosis in lung cancer cells, we transfected A549 cells with TNFRSF10A siRNAs (#1 and #2) to block the expression of TNFRSF10A. We then measured the cleavage of CASP3 and PARP1 after treatment with 1 μm TG or Tm for 48 h. The results show a decrease in CASP3 and PARP1 cleavage in TNFRSF10A knockdown cells after treatment with TG or Tm as compared with control cells (Fig. 8A). To explore whether TNFRSF10A mediates ER stress-induced apoptosis via a DDIT3-dependent mechanism in human lung cancer cells, we performed several knockdown experiments in A549 cells using DDIT3 and TNRSF10A siRNAs. We then detected the CASP8, CASP3, and PARP1 protein levels using Western blot analysis. The results show that the cleavage of CASP8, CASP3, and PARP1 were decreased significantly in DDIT3 or TNFRSF10A knockdown cells (Fig. 8B). Furthermore, TG treatment induced 13.8% apoptosis in DDIT3 knockdown cells, 11.5% apoptosis in TNFRSF10A knockdown cells, and 10.6% apoptosis in DDIT3 and TNFRSF10A knockdown cells, whereas it caused 24% apoptosis in control knockdown cells. Tm treatment induced 10.5% apoptosis in DDIT3 knockdown cells, 9.35% apoptosis in TNFRSF10A knockdown cells, and 8.25% apoptosis in DDIT3/TNFRSF10A knockdown cells, whereas it caused 20.45% apoptosis in control knockdown cells when evaluated by annexin V-PE/7-AAD staining and flow cytometry analysis (Fig. 8C).
FIGURE 8.

TNFRSF10A mediates ER stress-induced apoptosis in a DDIT3-dependent mechanism. A, A549 cells were transfected with control (Ctrl) siRNA or TNFRSF10A siRNA #1 and #2. Forty-eight hours later, the cells were treated with DMSO, TG (1 μm), or Tm (1 μm) for another 24 h, and samples were analyzed by Western blot. B, A549 cells were transfected with control siRNA, DDIT3 siRNA, or TNFRSF10A siRNA #2 or co-transfected with DDIT3 and TNFRSF10A siRNA. After 48 h, the cells were treated with DMSO, TG (1 μm), or Tm (1 μm) for another 24 h, and samples were analyzed by Western blot using antibodies against DDIT3, TNFRSF10A, CASP8, CASP3, PARP1, and ACTB. C, A549 cells were transfected with Control siRNA, DDIT3 siRNA, or TNFRSF10A siRNA #2 or co-transfected with DDIT3 and TNFRSF10A siRNA. After 48 h, the cells were treated with DMSO, TG (1 μm), or Tm (1 μm) for another 24 h, and then cells were stained with annexin V-PE/7-AAD, and apoptosis was detected by flow cytometry analysis. Columns, mean of triplicate treatments; bars, ± S.D. The statistical differences between the two treatments were analyzed by two-sided unpaired Student's t tests (*, p < 0.05; **, p < 0.01).
DISCUSSION
ER stress plays an important role in anticancer drug-induced apoptosis (39). BBC3, PMAIP1, BID, BCL2L11, and TNFRSF10B mediate apoptosis triggered by ER stress (18, 20–22). Our previous study confirmed that DDIT3, an important ER stress-inducible protein (24), mediates ER stress-induced apoptosis via ATF4-ATF3-DDIT3-TNFRSF10B signaling axis in human lung cancer cells (43). However, details underlying the regulation of TNFRSF10B by DDIT3 have not been explored. In addition, the details of the role of TNFRSF10A, a key death receptor, in DDIT3-mediated ER stress-induced apoptosis have not been well elucidated. Our study demonstrates that thapsigargin and tunicamycin, two well known strong ER stress inducers (44), up-regulated TNFRSF10A protein levels rapidly in human lung cancer cell lines (Fig. 1). We show that TNFRSF10A protein level was up-regulated in transient DDIT3-overexpressed cells, whereas it was down-regulated in DDIT3 knockdown cells (Fig. 2). These data indicate that DDIT3 is involved in regulation of TNFRSF10A gene expression in human NSCLC cells during ER stress. This is the first report on DDIT3 regulation of TNFRSF10A expression.
DDIT3 transcriptionally regulates the expression of BCL2L11, BBC3, and TNFRSF10B during ER stress-induced apoptosis (18, 21, 23). In this study, we explored whether DDIT3 also regulates TNFRSF10A transcription. The analysis of TNFRSF10A promoter sequence revealed two putative DDIT3 binding sites located at −1636/−1625 and −374/−364, respectively. There is also an AP-1 binding site located at −304/−298, which is important for the regulation of TNFRSF10A transcription (10). We constructed six plasmids to test which sites are important to the TNFRSF10A transcription. We note that mutation of the two putative DDIT3 binding sites affects transcription according to the -fold change of luciferase activity between the pGL3-WT and pGL3-BS1/2mt transfection (Fig. 3B). However, it is clear that the AP-1 binding site on the TNFRSF10A promoter is necessary to mediate TG/Tm-induced transcription. The further analysis of luciferase reporter assay verified that the AP-1 binding site located at −304/−298 bp on the TNFRSF10A promoter plays a critical role in mediating DDIT3-dependent transcription (Fig. 3, C and D).
AP-1 is composed of homodimeric or heterodimeric basic region-leucine zipper (bZIP) proteins belonging to the Jun (JUN, JUNB, and JUND) and Fos (FOS, FOSB, FOSL1, and FOSL2) sub-families (45). DDIT3 interacts with members of the AP-1 transcription factor family and enhances transcription of AP-1 target genes (23). Given the co-up-regulation of DDIT3 and TNFRSF10A by ER stress, as well as the essential role of the AP-1 binding site for DDIT3-induced transcription activity of TNFRSF10A, we speculated that DDIT3 and AP-1 cooperate to activate TNFRSF10A transcription. This hypothesis is supported by our finding that DDIT3 up-regulation by ER stress inducers increased the interaction between DDIT3 and phospho-JUN. DDIT3 up-regulation by ER stress inducers significantly increased the amount of fragment containing the AP-1 binding site region captured by DDIT3 and phospho-JUN antibodies. These results suggest that ER stress-induced TNFRSF10A up-regulation requires cooperation between DDIT3 and phospho-JUN.
KAT2A is one of the best characterized HATs and the catalytic subunit of the SAGA complex that acetylates lysines primarily on histones H3 and H2B as a transcriptional co-activator that mediates the transcription of many genes (46–48). However, we do not know whether KAT2A also regulates the transcription of TNFRSF10A or TNFRSF10B. DDIT3 interacts with TP300 through the N-terminal region (36). Whether other HATs also interact with DDIT3 and act as co-activators to enhance TNFRSF10A or TNFRSF10B transcription activity has not been explored. In the present study, we report that KAT2A interacts with the N-terminal region (aa 1–26) of DDIT3 and functions as a co-activator of DDIT3 in the transcriptional regulation of TNFRSF10A and TNFRSF10B. Our findings support the hypothesis that KAT2A is recruited to DDIT3·phospho-JUN complex via physical interaction with DDIT3 at the AP-1 binding site located at −304/−298 bp in the TNFRSF10A promoter region and mediates H3K9/K14 acetylation to further enhance TNFRSF10A transcription. Moreover, we verified that KAT2A is recruited by DDIT3 at the DDIT3 binding site located at −276/−264 bp in TNFRSF10B promoter region and mediates H3K9/K14 acetylation to further enhance TNFRSF10B transcription.
TNFRSF10A and TNFRSF10B are two important TRAIL receptors belonging to the TNF family, and both death receptors contribute to apoptosis induced by anticancer drugs (3–8). Therefore, TNFRSF10A and TNFRSF10B have been considered therapeutic targets for cancer therapy (49). TNFRSF10B mediates ER stress-induced apoptosis (18), but whether TNFRSF10A contributes to apoptosis triggered by ER stress has not been well explored. In this study, we show that TNFRSF10A expression is up-regulated by ER stress inducers and is dependent on the induction of DDIT3 in human lung cancer cells. Moreover, the results of apoptosis assays demonstrate that TNFRSF10A mediates ER stress-induced apoptosis in a DDIT3 activation-dependent manner. We note that DDIT3 regulates both TNFRSF10A and TNFRSF10B, whereas TNFRSF10A knockdown almost protects cells from apoptosis thoroughly (Fig. 8C). Anyway, TNFRSF10A and TNFRSF10B can form a heterodimer (50). However, the role of their interaction has not been elucidated. We also found TNFRSF10A knockdown reduced the level of TNFRSF10B (51), which may be due to the destruction of the interaction between them. This may explain why TNFRSF10A knockdown protected cells from apoptosis. Nonetheless, this finding warrants further study.
In summary, the present study has revealed two novel mechanisms where DDIT3 cooperates with KAT2A to modulate TNFRSF10A and TNFRSF10B expression during ER stress in human lung cancer cells. One pathway suggests that the induced DDIT3 is involved in DDIT3·p-JUN complex at the AP-1 binding site located at −304/−298 bp in the TNFRSF10A promoter region and recruits KAT2A for acetylation of H3K9/K14 to cooperatively promote the transcription of TNFRSF10A. The other suggests that the induced DDIT3 is located at −276/−264 bp in the TNFRSF10B promoter region and recruits KAT2A for acetylation of H3K9/K14 to cooperatively promote the transcription of TNFRSF10B. Moreover, TNFRSF10A mediates apoptosis triggered by ER stress inducers in a DDIT3-dependent manner in human lung cancer cells. Therefore, our findings highlight the different mechanisms underlying ER stress-induced TNFRSF10A/10B expression and apoptosis, which will be helpful for elucidating the mechanism by which anticancer drugs induce apoptosis.
Acknowledgment
We thank Dr. Austin Cape for careful reading and comments.
This work was supported by grants from the National Natural Science Foundation of China (31371402, 31171332, 81472686, 30971479), National Key Basic Research Program of China (2013CB910900, project 3).
- TRAIL
- tumor necrosis factor-related apoptosis-inducing ligand
- FADD
- Fas-associated protein with death domain
- ER
- endoplasmic reticulum
- Tm
- tunicamycin
- aa
- amino acids
- PE
- phycoerythrin
- 7-AAD
- 7-amino-actinomycin D
- C/EBP
- CCAAT/enhancer-binding protein
- CREBBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- HAT
- histone acetyltransferase
- NSCLC
- non-small cell lung cancer
- DMSO
- dimethyl sulfoxide
- mt
- mutant
- luc
- luciferase.
REFERENCES
- 1. Ashkenazi A. (2008) Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat. Rev. Drug Discov. 7, 1001–1012 [DOI] [PubMed] [Google Scholar]
- 2. Ashkenazi A., Dixit V. M. (1998) Death receptors: signaling and modulation. Science 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 3. Tsai J. Y., Hung C. M., Bai S. T., Huang C. H., Chen W. C., Chung J. G., Kuo S. C., Way T. D., Huang L. J. (2010) Induction of apoptosis by HAC-Y6, a novel microtubule inhibitor, through activation of the death receptor 4 signaling pathway in human hepatocellular carcinoma cells. Oncol. Rep. 24, 1169–1178 [DOI] [PubMed] [Google Scholar]
- 4. Jin X., Wu X. X., Abdel-Muneem Nouh M. A., Kakehi Y. (2007) Enhancement of death receptor 4 mediated apoptosis and cytotoxicity in renal cell carcinoma cells by subtoxic concentrations of doxorubicin. J. Urol. 177, 1894–1899 [DOI] [PubMed] [Google Scholar]
- 5. Lee B. S., Cha H. Y., Shin Y. S., Kim Y. S., Kim C. H. (2013) AY4, an agonistic anti-death receptor 4 MAB, induces apoptotic cell death in anaplastic thyroid cancer cells via downregulation of Bcl-xL with reactive oxygen species generation. Endocr. Relat. Cancer 20, 283–291 [DOI] [PubMed] [Google Scholar]
- 6. Lim J. H., Park J. W., Choi K. S., Park Y. B., Kwon T. K. (2009) Rottlerin induces apoptosis via death receptor 5 (DR5) upregulation through CHOP-dependent and PKC δ-independent mechanism in human malignant tumor cells. Carcinogenesis 30, 729–736 [DOI] [PubMed] [Google Scholar]
- 7. Elrod H. A., Sun S. Y. (2008) Modulation of death receptors by cancer therapeutic agents. Cancer Biol. Ther. 7, 163–173 [DOI] [PubMed] [Google Scholar]
- 8. Sun S. Y. (2005) Chemopreventive agent-induced modulation of death receptors. Apoptosis 10, 1203–1210 [DOI] [PubMed] [Google Scholar]
- 9. Thorburn A. (2004) Death receptor-induced cell killing. Cell. Signal. 16, 139–144 [DOI] [PubMed] [Google Scholar]
- 10. Guan B., Yue P., Lotan R., Sun S. Y. (2002) Evidence that the human death receptor 4 is regulated by activator protein 1. Oncogene 21, 3121–3129 [DOI] [PubMed] [Google Scholar]
- 11. Liu X., Yue P., Khuri F. R., Sun S. Y. (2004) p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res. 64, 5078–5083 [DOI] [PubMed] [Google Scholar]
- 12. Mendoza F. J., Ishdorj G., Hu X., Gibson S. B. (2008) Death receptor-4 (DR4) expression is regulated by transcription factor NF-κB in response to etoposide treatment. Apoptosis 13, 756–770 [DOI] [PubMed] [Google Scholar]
- 13. Wu G. S., Burns T. F., McDonald E. R., 3rd, Jiang W., Meng R., Krantz I. D., Kao G., Gan D. D., Zhou J. Y., Muschel R., Hamilton S. R., Spinner N. B., Markowitz S., Wu G., el-Deiry W. S. (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 17, 141–143 [DOI] [PubMed] [Google Scholar]
- 14. Takimoto R., El-Deiry W. S. (2000) Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 19, 1735–1743 [DOI] [PubMed] [Google Scholar]
- 15. Ravi R., Bedi G. C., Engstrom L. W., Zeng Q., Mookerjee B., Gélinas C., Fuchs E. J., Bedi A. (2001) Regulation of death receptor expression and TRAIL/Apo2L-induced apoptosis by NF-κB. Nat. Cell Biol. 3, 409–416 [DOI] [PubMed] [Google Scholar]
- 16. Shetty S., Graham B. A., Brown J. G., Hu X., Vegh-Yarema N., Harding G., Paul J. T., Gibson S. B. (2005) Transcription factor NF-κB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol. Cell. Biol. 25, 5404–5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baritaki S., Katsman A., Chatterjee D., Yeung K. C., Spandidos D. A., Bonavida B. (2007) Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J. Immunol. 179, 5441–5453 [DOI] [PubMed] [Google Scholar]
- 18. Yamaguchi H., Wang H. G. (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279, 45495–45502 [DOI] [PubMed] [Google Scholar]
- 19. Yoshida T., Shiraishi T., Nakata S., Horinaka M., Wakada M., Mizutani Y., Miki T., Sakai T. (2005) Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res. 65, 5662–5667 [DOI] [PubMed] [Google Scholar]
- 20. Li J., Lee B., Lee A. S. (2006) Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 281, 7260–7270 [DOI] [PubMed] [Google Scholar]
- 21. Puthalakath H., O'Reilly L. A., Gunn P., Lee L., Kelly P. N., Huntington N. D., Hughes P. D., Michalak E. M., McKimm-Breschkin J., Motoyama N., Gotoh T., Akira S., Bouillet P., Strasser A. (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 [DOI] [PubMed] [Google Scholar]
- 22. Upton J. P., Austgen K., Nishino M., Coakley K. M., Hagen A., Han D., Papa F. R., Oakes S. A. (2008) Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell. Biol. 28, 3943–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cazanave S. C., Elmi N. A., Akazawa Y., Bronk S. F., Mott J. L., Gores G. J. (2010) CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G236–G243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., Ron D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pino S. C., O'Sullivan-Murphy B., Lidstone E. A., Yang C., Lipson K. L., Jurczyk A., diIorio P., Brehm M. A., Mordes J. P., Greiner D. L., Rossini A. A., Bortell R. (2009) CHOP mediates endoplasmic reticulum stress-induced apoptosis in Gimap5-deficient T cells. PLoS One 4, e5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fonseca S. G., Gromada J., Urano F. (2011) Endoplasmic reticulum stress and pancreatic beta cell death. Trends Endocrinol. Metab. 22, 266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Averous J., Bruhat A., Jousse C., Carraro V., Thiel G., Fafournoux P. (2004) Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J. Biol. Chem. 279, 5288–5297 [DOI] [PubMed] [Google Scholar]
- 28. Ubeda M., Wang X. Z., Zinszner H., Wu I., Habener J. F., Ron D. (1996) Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol. Cell. Biol. 16, 1479–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu X., Yue P., Zhou Z., Khuri F. R., Sun S. Y. (2004) Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J. Natl. Cancer Inst. 96, 1769–1780 [DOI] [PubMed] [Google Scholar]
- 30. Lu M., Lawrence D. A., Marsters S., Acosta-Alvear D., Kimmig P., Mendez A. S., Paton A. W., Paton J. C., Walter P., Ashkenazi A. (2014) Cell death: opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345, 98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marmorstein R., Roth S. Y. (2001) Histone acetyltransferases: function, structure, and catalysis. Curr. Opin. Genet. Dev. 11, 155–161 [DOI] [PubMed] [Google Scholar]
- 32. Miyagi S., Mishima Y., Iwama A. (2011) [Structure and function of MYST family histone acetyltransferases]. Rinsho ketsueki 52, 490–496 [PubMed] [Google Scholar]
- 33. Jin Q., Yu L. R., Wang L., Zhang Z., Kasper L. H., Lee J. E., Wang C., Brindle P. K., Dent S. Y., Ge K. (2011) Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gunderson F. Q., Johnson T. L. (2009) Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet. 5, e1000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chérasse Y., Maurin A. C., Chaveroux C., Jousse C., Carraro V., Parry L., Deval C., Chambon C., Fafournoux P., Bruhat A. (2007) The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 35, 5954–5965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohoka N., Hattori T., Kitagawa M., Onozaki K., Hayashi H. (2007) Critical and functional regulation of CHOP (C/EBP homologous protein) through the N-terminal portion. J. Biol. Chem. 282, 35687–35694 [DOI] [PubMed] [Google Scholar]
- 37. Sun S. Y., Liu X., Zou W., Yue P., Marcus A. I., Khuri F. R. (2007) The farnesyltransferase inhibitor lonafarnib induces CCAAT/enhancer-binding protein homologous protein-dependent expression of death receptor 5, leading to induction of apoptosis in human cancer cells. J. Biol. Chem. 282, 18800–18809 [DOI] [PubMed] [Google Scholar]
- 38. Yoshida T., Shiraishi T., Horinaka M., Wakada M., Sakai T. (2007) Glycosylation modulates TRAIL-R1/death receptor 4 protein: different regulations of two pro-apoptotic receptors for TRAIL by tunicamycin. Oncol. Rep. 18, 1239–1242 [PubMed] [Google Scholar]
- 39. Verfaillie T., Garg A. D., Agostinis P. (2013) Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 332, 249–264 [DOI] [PubMed] [Google Scholar]
- 40. Downey M., Knight B., Vashisht A. A., Seller C. A., Wohlschlegel J. A., Shore D., Toczyski D. P. (2013) Gcn5 and sirtuins regulate acetylation of the ribosomal protein transcription factor Ifh1. Curr. Biol. 23, 1638–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sterner D. E., Berger S. L. (2000) Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnsson A., Durand-Dubief M., Xue-Franzén Y., Rönnerblad M., Ekwall K., Wright A. (2009) HAT-HDAC interplay modulates global histone H3K14 acetylation in gene-coding regions during stress. EMBO Rep. 10, 1009–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu G., Su L., Hao X., Zhong N., Zhong D., Singhal S., Liu X. (2012) Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J. Cell. Mol. Med. 16, 1618–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 [DOI] [PubMed] [Google Scholar]
- 45. Shaulian E., Karin M. (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136 [DOI] [PubMed] [Google Scholar]
- 46. Zhang W., Bone J. R., Edmondson D. G., Turner B. M., Roth S. Y. (1998) Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J. 17, 3155–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suka N., Suka Y., Carmen A. A., Wu J., Grunstein M. (2001) Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol. Cell 8, 473–479 [DOI] [PubMed] [Google Scholar]
- 48. Marmorstein R. (2001) Structure of histone acetyltransferases. J. Mol. Biol. 311, 433–444 [DOI] [PubMed] [Google Scholar]
- 49. Yang A., Wilson N. S., Ashkenazi A. (2010) Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical translation. Curr. Opin. Cell Biol. 22, 837–844 [DOI] [PubMed] [Google Scholar]
- 50. Kimberley F. C., Screaton G. R. (2004) Following a TRAIL: update on a ligand and its five receptors. Cell Res. 14, 359–372 [DOI] [PubMed] [Google Scholar]
- 51. Liu X., Yue P., Chen S., Hu L., Lonial S., Khuri F. R., Sun S. Y. (2007) The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 67, 4981–4988 [DOI] [PubMed] [Google Scholar]