

Abstract
Dishevelleds are modular proteins that lie at the crossroads of divergent Wnt signaling pathways. The DIX domain of dishevelleds modulates a β-catenin destruction complex, and thereby mediates cell fate decisions through differential activation of Tcf transcription factors. The DEP domain of dishevelleds mediates planar polarity of cells within a sheet through regulation of actin modulators. In Caenorhabditis elegans asymmetric cell fate decisions are regulated by asymmetric localization of signaling components in a pathway termed the Wnt/β-catenin asymmetry pathway. Which domain(s) of Disheveled regulate this pathway is unknown. We show that C. elegans embryos from dsh-2(or302) mutant mothers fail to successfully undergo morphogenesis, but transgenes containing either the DIX or the DEP domain of DSH-2 are sufficient to rescue the mutant phenotype. Embryos lacking zygotic function of SYS-1/β-catenin, WRM-1/β-catenin, or POP-1/Tcf show defects similar to dsh-2 mutants, including a loss of asymmetry in some cell fate decisions. Removal of two dishevelleds (dsh-2 and mig-5) leads to a global loss of POP-1 asymmetry, which can be rescued by addition of transgenes containing either the DIX or DEP domain of DSH-2. These results indicate that either the DIX or DEP domain of DSH-2 is capable of activating the Wnt/β-catenin asymmetry pathway and regulating anterior–posterior fate decisions required for proper morphogenesis.
Keywords: C. elegans, Dishevelled, Morphogenesis
Introduction
Throughout development, cells communicate with each other using intricate signal transduction pathways. One key group of such signaling pathways are those regulated by Wnt ligands, including the Wnt/β–catenin, planar cell polarity (PCP) and Wnt/Ca2+ pathways (reviewed in Copp et al., 2003; Herman and Wu, 2004; Kohn and Moon, 2005; Mlodzik, 2002; Sokol, 2000; Wallingford and Habas, 2005).
The activity of the Dishevelled (Dsh) proteins partially determines which Wnt pathway is activated. Dsh family proteins have three major conserved domains: an N-terminal DIX, a central PDZ, and a C-terminal DEP domain (reviewed in Wallingford and Habas, 2005; Wharton, 2003). The DIX and PDZ domains primarily interact with proteins that lead to activation of the Wnt/β–catenin pathway, which has many roles during development, including axis specification and cell fate determination (reviewed in Cadigan and Nusse, 1997; Moon et al., 1997; Weaver and Kimelman, 2004; Yamaguchi, 2001). In contrast, the DEP domain is responsible for both PCP and Wnt/Ca2+ signaling, which may act cooperatively through similar or identical pathways (reviewed in Wharton, 2003). These pathways control basic morphogenetic processes, such as the polarization of epithelial sheets and convergent extension movements (reviewed in Mlodzik, 2002; Veeman et al., 2003; Wallingford and Habas, 2005).
In Caenorhabditis elegans, Wnt signaling acts at many points throughout embryonic and larval stages of development to specify cell fate, to polarize cells in asymmetric divisions, and to control cell migrations (reviewed in Eisenmann, 2005; Herman and Wu, 2004; Korswagen, 2002). Blastomere polarity in the early embryo is controlled by a Wnt/spindle alignment pathway that does not require transcription (Schlesinger et al., 1999; Walston et al., 2004). Many asymmetric cell fate decisions in C. elegans are controlled through asymmetric localization of the Tcf transcription factor POP-1 and the β-catenins SYS-1 and WRM-1 in a pathway termed the Wnt/β-catenin asymmetry pathway (reviewed in Mizumoto and Sawa, 2007b). WRM-1, in association with the Nemo-like kinase LIT-1, targets POP-1 for nuclear export, leading to its asymmetric nuclear localization (Lo et al., 2004; Rocheleau et al., 1999). SYS-1 then functions as a limiting co-activator for POP-1 (Huang et al., 2007; Kidd et al., 2005; Phillips et al., 2007).
Although many developmental events in C. elegans require Wnt signaling, the roles of specific Dshs in these processes vary. C. elegans has three conserved Dsh genes, dsh-1, dsh-2, and mig-5. In the early embryo, all three Dshs function to varying degrees in orienting the mitotic spindle of the EMS blastomere in the four-cell embryo and the ABar blastomere in the eight-cell embryo (Walston et al., 2004). The Dshs also transduce the Wnt signal that specifies the fate of the EMS daughter cells, E (endoderm) and MS (mesoderm), and regulate myosin phosphorylation and contractility to drive the initial ingression of Ea and Ep during C. elegans gastrulation (Lee et al., 2006).
The Dshs also regulate several asymmetric cell divisions. These include those in the PHA neuronal lineage (Hawkins et al., 2005), the somatic gonad precursor lineage (Chang et al., 2005; Walston et al., 2006), the P11/P12 lineage, and the male-specific B cell lineages (Walston et al., 2006; Wu and Herman, 2007). In addition, MIG-5 controls polarity of the hermaphrodite B cell lineage through a PCP-like pathway involving RHO-1/RhoA and LET-502/ROCK (Wu and Herman, 2007). Asymmetric division in the B cell lineage and the PHA neuronal lineage requires the DEP but not the DIX domain (Hingwing et al., 2009; Wu and Herman, 2007). MIG-5 is also necessary for proper hypodermal morphogenesis (Walston et al., 2006). How the Dshs regulate hypodermal morphogenesis and how they activate the Wnt/β-catenin asymmetry pathway is unclear.
Here we describe the Wnt signaling pathways that act through DSH-2 to control hypodermal morphogenesis. Embryos from dsh-2 (or302) mutant mothers have disorganized hypodermal cells that migrate incorrectly. Removing conserved components of PCP signaling does not reveal a role for these proteins in hypodermal morphogenesis. However, removing zygotic function of POP-1, SYS-1, or WRM-1 using mutants characterized here for the first time results in defects in hypodermal morphogenesis similar to dsh-2 mutants. Removal of both DSH-2 and MIG-5 results in global loss of POP-1 asymmetry that can be rescued to wild-type levels by dsh-2 constructs lacking either the DIX or the DEP domain. These same constructs rescue morphogenetic defects in dsh-2 mutants. These results suggest that both the DIX and DEP domains of DSH-2 function in the Wnt/β-catenin asymmetry pathway to regulate cell fates prior to hypodermal morphogenesis.
Materials and methods
Strains and alleles
The Bristol strain N2 was used as wild-type. Strains were maintained and cultured as previously described (Brenner, 1974). A deletion allele of pop-1 was isolated from a UV-TMP mutagenized library of worms (Chase et al., 2001). Sequencing confirmed the presence of a 334 bp deletion extending from the first exon into the first intron, as well as a 22 bp insertion in the first intron of pop-1. The following mutations and transgenes were used for analysis or balancing. LGI: pop-1(q772), sys-1(q736) (Kidd et al., 2005), fog-1 (q325), fog-3(q520); LGII: dsh-2(or302) (Hawkins et al., 2005), mIn1 [dpy-10(e128); mIs14], eff-1(oj55) (Mohler et al., 2002); LGIII: frl-1 (ok460), wrm-1(ne1982ts) (Nakamura et al., 2005), wrm-1(ok738), dpy-1(e164), let-727(s2589), unc-32(e189); LGIV: jnk-1(gk7); LGX: mig-15(rh148), mig-15(rh80) (Poinat et al., 2002), mig-15(rh326), jkk-1(km2); transgenes: jcIs1[AJM-1::GFP] (Köppen et al., 2001), jcEx68 [nhr-73::gfp, rol-6(su1006)], jcEx76[nhr-73::gfp, rol-6(su1006)], qIs74 [pJK908(sys-1p::GFP::POP-1)] (Siegfried et al., 2004), jcIs26[pRF4(rol-6(su1006), pJS191(AJM-1::GFP), (CEH-16::GFP)].
For analyzing rescue of mutant phenotypes, the dsh-2 constructs (0.5 ng/μl) and the co-injection marker sur-5::gfp (80 ng/μl) (Gu et al., 1998) were injected into the gonads of NG3124 (dsh-2(or302)/mIn1(dpy-10(e128);mIs14)II) worms and maintained as extrachromosomal arrays in the following strains: full length dsh-2, SU302 (jcEx87), SU296 (jcEx82), SU297 (jcIs1, jcEx82); ΔDIX, SU303 (jcEx88), SU304 (jcEx89), SU321 (jcIs1, jcEx89); ΔPDZ, SU298 (jcEx83); ΔDEP, SU299 (jcEx84), SU300 (jcEx85), SU314 (jcIs1, jcEx85).
For analyzing rescue of POP-1 asymmetry, dsh-2 constructs (0.5 ng/μl) were transformed with rol-6(su1006D) (80ng/μl) as a co-injection marker into SU350 (dsh-2(or302)/mIn1II; qIs74III) and maintained as extrachromosomal arrays in the following strains: full length dsh-2, SU369 (jcEx100), ΔDIX, SU378 (jcEx103), SU379 (jcEx105), SU380 (jcEx106), SU381 (jcEx107), ΔDEP, SU371 (jcEx101), SU372 (jcEx108).
RNAi
For cDNAs, PCR amplification using T3 and T7 primers produced RNA templates. For cosmid DNA, nested gene-specific primers, with T3 or T7 added to the 5′ end of the inner pair, were amplified by PCR. Ambion Megascript T3 and T7 kits (Ambion, Austin, Texas) were used for in vitro transcription. Transcription products were purified, mixed in equimolar concentrations and double-stranded. Y. Kohara (Gene Network Lab, NIG, Japan) kindly provided the following cDNA clones: yk55h11 (dsh-2), yk216a12 (mig-5), yk291a11 (dsh-1), yk397b6 (dsh-1), yk653h6 (dsh-1), yk579a6 (mig-15), yk369b7 (jkk-1), yk1117g4 (jkk-1), yk1092h4 (jnk-1), yk385e6 (frl-1), yk539h3 (frl-1), yk626c12 (frl-1). RNA templates for fhod-2 were made from cosmid DNA (F56E10, obtained from Alan Coulson, Sanger Institute, England).
Double-stranded RNA was injected at a concentration of μg/μl into young adult hermaphrodites. Injected adults were allowed to lay embryos for at least 20 h prior to collection of embryos for imaging. RNAi efficacy was verified by comparison with published RNAi phenotypes available on Wormbase (www.wormbase.org).
Nomarski and fluorescent imaging of morphogenesis
Nomarski microscopy was used to collect 4D data sets as previously described (Raich et al., 1998). Typically, twenty optical sections with 1.5 μm spacing were collected at two- or three-minute intervals. Presence of the extrachromosomal array following 4D Nomarski imaging was determined by visualization of the coinjection marker SUR-5::GFP using epifluorescence microscopy.
For temperature shift experiments twenty-five optical sections with 1 μm spacing were collected at one-minute intervals on a custom made temperature controlled stage capable of shifting 10 °C/min. Embryos were shifted from the non-permissive temperature (25 °C) to the permissive temperature (15 °C; downshifts), or from the permissive temperature to the non-permissive temperature (upshift) at various times and analyzed for defects during morphogenesis.
Fluorescent images were collected with a Perkin-Elmer UltraVIEW spinning disk confocal microscope, using a Hamamatsu ORCA-ER CCD camera and Perkin Elmer UltraVIEW Imaging Suite software. For analysis of morphogenesis phenotypes twenty to thirty optical sections with 0.75 μm spacing were captured at two-minute intervals. Nomarski and fluorescent images were analyzed using macros and plugins for the public domain applications NIH Image and ImageJ (available at http://worms.zoology.wisc.edu/research/4d/4d.html). Pair-wise statistical tests between treatments were performed using Fisher’s Exact Test (Zar, 1999); p values represent two-tailed probabilities.
Imaging and analysis of POP-1 asymmetry
GFP::POP-1 asymmetry between sister cells was assessed by confocal microscopy using twenty-two optical sections with 0.5 μm spacing and one-minute intervals at the approximately 100-cell stage. Sister pairs were identified by direct observations of cell division during image acquisition. Relative levels of nuclear GFP::POP-1 were measured as the average pixel intensity in a 37 pixel2 circle at a middle focal plane of the nucleus using a modified version of ImageJ. The ratio of anterior to posterior fluorescence was calculated by dividing the average anterior pixel intensity minus the background pixel intensity by the average posterior pixel intensity minus the background pixel intensity. Histograms for each treatment were generated with the anterior to posterior ratio of GFP fluorescence entered into bins of 0.5, 0.67, 0.8, 0.91, 1.1, 1.25, 1.5 and 2.0. Each treatment was compared to wild-type and dsh-2, mig-5 double loss of function using a Dunn nonparametric post-hoc analysis (Zar, 1999).
Antibody staining
Gravid hermaphrodites were washed off plates, rinsed with water and incubated in a 0.71 M NaOH/4.4% NaOCl3 solution. Once the adults were dissolved, the remaining embryos were rinsed in water. The embryos were fixed and permeabilized using a procedure described by Guenther and Garriga (modified from Finney and Ruvkun, 1990; Guenther and Garriga, 1996). Overnight antibody incubations were at 4 °C. Primary antibodies were used at a 1:100 dilution of anti-DSH-2 antiserum, 1:200 dilution of MH27 and 1:1000 dilution of anti-GFP (Sigma, St. Louis, MO). Secondary antibodies were used at a 1:50 dilution.
dsh-2 deletion construct design
A dsh-2 minigene, containing the endogenous promoter and regulator regions fused with GFP (pNH77), was a template for deletion constructs (Hawkins et al., 2003). The deletion constructs removed the following regions: ΔDIX, bases 301–555 of the gene (aa 101–185) (pTW1); ΔPDZ, bases 826–1434 of the gene (aa 276–478) (pTW2), ΔDEP, bases 1555–1827 of the gene (aa 519–609) (pTW3). Deletion constructs were created as described (Wang and Wilkinson, 2001). Successful transformants were analyzed for presence of the deletion by gel electrophoresis. Cassettes containing the deleted regions were subcloned into the original vector and sequenced.
Results
Maternal effect embryonic lethality in dsh-2(or302) embryos results from defects in hypodermal morphogenesis
To better understand the requirements for Dshs during embryogenesis we characterized the phenotype of a dsh-2 mutant, dsh-2 (or302). Embryonic lethality in progeny from dsh-2(or302) heterozygous mothers is similar to wild-type, while animals from dsh-2 (or302) homozygous mothers display 69.7% embryonic lethality and 13.3% L1 larval lethality (Table 1). Of the remaining escapers, 11.5% of the larvae have body shape and other developmental defects; only 5.5% develop into viable adults. dsh-2(RNAi) results in lethality similar to embryos from dsh-2(or302) mutant mothers (54.0% embryonic lethality and 14.2% larval lethality; Table 1). Embryos from dsh-2 (or302) homozygous mothers also display defects in mitotic polarity of some blastomeres (Walston et al., 2004). While blastomere polarity is not paternally rescued (data not shown), embryonic lethality is reduced to 7.2% in embryos from dsh-2(or302) homozygous mothers mated to wild-type males (Table 1). This suggests that early defects are not causative of the embryonic lethality, and that dsh-2 function is required during later embryogenesis.
Table 1.
Embryonic lethality resulting from removal of the Dshs
| Genotype | Embryonic lethalitya | N |
|---|---|---|
| dsh-2(or302) (M− Z−)b | 69.7%e | 968 |
| dsh-2(RNAi) | 54.0% | 598 |
| dsh-2(or302) (M+ Z−)c | 0.9% | 2218 |
| dsh-2(or302) (M− Z+)d | 7.2% | 691 |
| mig-5(RNAi) | 3.6% | 2425 |
| dsh-2(or302) (M+ Z−); mig-5(RNAi) | 37.2% | 51 |
| dsh-1(RNAi) | 1.1% | 3326 |
| dsh-1(RNAi);dsh-2(RNAi) | 67.9%e | 1308 |
Embryos failing to hatch on NGM plates.
M− Z− are animals lacking both maternal and zygotic dsh-2 function.
M+ Z− are animals with maternal dsh-2 function but lacking zygotic dsh-2 function.
M− Z+ are animals with zygotic dsh-2 function but lacking maternal dsh-2 function.
Not significantly different; p=0.19.
4D Nomarski microscopy was used to more closely examine lethality of embryos from dsh-2(or302) mutant mothers. Following successful completion of gastrulation, 68.1% (n=51) of embryos displayed ruptures in the hypodermis at or prior to the 2-fold stage and 31.9% arrested at the 3- and 4-fold stages of elongation, suggesting defects in hypodermal morphogenesis.
dsh-2(or302) embryos display defects in dorsal intercalation
During dorsal intercalation two rows of ten cells intercalate to form a single row of twenty cells (Williams-Masson et al., 1998). Analysis of embryos from dsh-2(or302) homozygous hermaphrodites expressing AJM-1::GFP (an epithelial junctional protein; Köppen et al., 2001) reveals that all embryos (n=18) display dorsal intercalation defects in cells derived from both the AB (anterior) and C (posterior) lineages. Rather than migrating between their lateral neighbors, cells often migrate to the opposite seam boundary as pairs or threesomes (Fig. 1B). Cells within the dorsal array fail to migrate, resulting in regions of the dorsal hypodermis where individual cells do not span the space between the rows of seam cells (Fig. 1B). Just prior to migration during intercalation, dorsal cells of wild-type embryos adopt a wedge-shaped morphology, i.e., their apical profiles are triangular in shape (Williams-Masson et al., 1998). Often, in embryos from dsh-2(or302) homozygous hermaphrodites, laterally positioned cells adopt a similar morphology, even though at least some of these laterally positioned triangular cells express seam cell markers (Fig. 1D, H). This indicates that while some laterally positioned cells may be mispositioned dorsal cells attempting to intercalate, others are mispositioned seam cells that are either passively or actively rearranging.
Fig. 1.

Embryos from dsh-2(or302) homozygous mothers display hypodermal morphogenesis defects. (A, B) Dorsal intercalation in embryos expressing AJM-1::GFP. (A) Wild-type embryo. The posterior pointer pair (asterisks) are the last cells to intercalate. (B) Embryo from dsh-2(or302) homozygous mother. Many cells migrate in groups (arrows). Some cells fail to complete intercalation (arrowhead). (C, D) Dorsal views of embryos expressing nhr-73::gfp in the lateral (seam) cells. (C) During dorsal intercalation, the lateral cells remain in two linear rows in a wild-type embryo. (D) In an embryo from a dsh-2(or302) mother, a cell (arrow) expressing nhr-73::gfp intercalates between neighboring dorsal cells. (E, F) Ventral enclosure in embryos expressing AJM-1::GFP. (E) Wild-type embryo. The leading cells are the first to meet at the midline (asterisks). The pocket cells, posterior to the leading cells, migrate toward the midline following the leading cells. (F) Embryo from a dsh-2(or302) homozygous mother. The leading cells lag behind the pocket cells and often fail to meet at the ventral midline. Some pocket cells are disorganized and fail to migrate to the ventral midline (arrowheads). (G, H) Ventral views of embryos expressing nhr-73::gfp. (G) During ventral enclosure, the seam cells send out short filopodia in a wild-type embryo. (H) In an embryo from a dsh-2(or302) mother, a seam cell sends out a protrusion (arrow) to the ventral midline. Scale bar=10 μm. All embryos are oriented with anterior to the left.
Ventral enclosure and elongation are defective in dsh-2(or302) embryos
The majority of embryonic lethality in dsh-2(or302) is due to rupture along the ventral midline (64.3% n=28), indicating a failure of ventral enclosure (Raich et al., 1999; Williams-Masson et al., 1997), or rupture in the ventral anterior region (35.7% n=28), indicating a failure of head enclosure. The major cause of enclosure defects in embryos from dsh-2(or302) mothers results when leading cells (Williams-Masson et al., 1997) fail to lead and instead lag behind the migration of pocket cells (62.2% n=31 Fig. 1F). Ventral pocket cells are also disorganized in 37.8% (n=37) of embryos from dsh-2 (or302) mothers (Fig. 1F). This disorganization is due to individual pocket cells failing to migrate, and therefore, failing to meet their contralateral neighbor at the ventral midline. This appears to cause local weaknesses in the ventral hypodermis that can result in body shape defects during elongation (80.8% of 3-fold and older embryos, n=26; 67.7% of hatching larvae, n=164) in addition to ruptures.
Dshs in C. elegans are required redundantly throughout development
DSH-2 is expressed and cortically localized during morphogenesis (Fig. S1) similar to MIG-5 (Walston et al., 2006). We next addressed the possibility of redundancy between the Dshs during morphogenesis. Hypodermal morphogenesis defects were observed in both dsh-2 and mig-5 single mutants, although removal of mig-5 function by RNAi results in only 3.6% (n=2425) embryonic lethality (Table 1), similar to that published for mig-5 alleles (Walston et al., 2006). Simultaneous removal of the function of both dsh-2 and mig-5 by RNAi results in lethality during mid-gastrulation (Walston et al., 2004; our unpublished observations), precluding analysis of later requirements. To assess redundancy of dsh-2 and mig-5 during morphogenesis, mig-5 function was removed by RNAi in dsh-2(or302) heterozygotes. Embryonic lethality increased from 0.9% to 37.2%, with 78.9% of dead embryos displaying failure of ventral enclosure (n=51; Table 1). While removal of dsh-1 function by RNAi results in little embryonic lethality (1.1%; Table 1), enhancement of the dsh-2 (RNAi) phenotype was observed in dsh-1(RNAi); dsh-2(RNAi) embryos (from 54.0% for dsh-2(RNAi) to 67.9%). mig-5; dsh-1 double loss of function was similar to mig-5 single loss of function (data not shown). This demonstrates that in addition to redundant roles in early embryogenesis, the C. elegans Dshs also function at least semi-redundantly during hypodermal morphogenesis.
Hypodermal morphogenesis is rescued primarily by either the DIX or DEP domains of DSH-2
Using deletion constructs based on previous structure/function studies (Axelrod et al., 1998; Boutros et al., 1998; Heisenberg et al., 2000; Moriguchi et al., 1999; Penton et al., 2002; Rothbacher et al., 2000; Sheldahl et al., 2003; Wallingford et al., 2000), we next examined whether the DIX, the PDZ, or the DEP domains of DSH-2 are required to rescue morphogenesis defects in embryos from dsh-2 (or302) mothers. Rescue of morphogenetic phenotypes was analyzed in embryos from dsh-2(or302) mothers carrying the deletion constructs as extrachromosomal arrays. Embryos that did not display the co-injection marker showed similar lethality regardless of the deletion construct present in the mother (75.3–78.3% embryonic lethality, n>70 for each construct, vs. 86.3% lethality for embryos from dsh-2(or302) mothers; Table 2). Anti-GFP staining reported elsewhere confirms that the deletion constructs are expressed in embryos at comparable levels (Hingwing et al., 2009).
Table 2.
Rescue of dsh-2(or302) hypodermal morphogenesis defects with dsh-2 deletion constructs
| Array | Dorsal intercalation defectivea | Total embryonic lethalityb | Ventral ruptureb | Body shape defectsb | |
|---|---|---|---|---|---|
| dsh-2(or302) | 100% (n=38) | 86.3% (n=51) | 58.8% (n=51) | 80.8% (n=30)e | |
| Full length |
|
6.1% (n=33) | 14.9% (n=108) | 5.6% (n=108) | 14.9% (n=103) |
| ΔDIX |
|
57.1% (n=35)c | 29.4% (n=102) | 12.1% (n=102) | 31% (n=87) |
| ΔPDZ |
|
68.6% (n=35)c,d | 46% (n=87) | 20.7% (n=87) | 78.9% (n=71)e |
| ΔDEP |
|
81.5% (n=27)d | 59.5% (n=126) | 31.7% (n=126) | 58.8% (n=68) |
DSH-2 domains that are present in the constructs are as indicated.
Presence of intercalation defects as analyzed using both Nomarski imaging and confocal imaging with the junctional marker AJM-1::GFP.
Embryonic lethality, ventral rupture, and body shape defects observed in Nomarski movies of developing embryos.
Not significantly different, p=0.12.
Not significantly different, p=0.20.
Not significantly different, p=0.56.
Expression of full-length dsh-2 in embryos from dsh-2(or302) homozygous mothers reduced embryonic lethality (14.9% with only 5.6% ventral ruptures; n=108) and body shape defects (14.9%; n=103; Table 2). Dorsal intercalation defects were reduced to 6.1% as analyzed using AJM-1::GFP (n=33; Table 2). Therefore, expression of a full-length dsh-2 construct rescues many of the dsh-2 (or302) phenotypes.
A construct containing the PDZ and DEP domains should be sufficient to rescue PCP-like signaling, but should lack the ability to rescue the Wnt/β-catenin pathway. dsh-2(or302) offspring carrying the ΔDIX construct showed reduced embryonic lethality (29.4% with 12.1% ventral rupture; n=102), body shape defects (31.0%: n=87) and defects in dorsal intercalation (57.1%; n=15; Table 2).
The DEP domain is primarily responsible for PCP signaling. Therefore, a construct containing the DIX and PDZ domains would be expected to rescue Wnt/β-catenin signaling, but fail to rescue PCP-like signaling. Embryos from dsh-2(or302) homozygous mothers expressing the ΔDEP construct showed rescue of embryonic lethality (59.5% with 31.7% ventral rupture; n=126), body shape defects (58.8%; n=68) and dorsal intercalation defects (81.5%; n=27; Table 2).
The PDZ domain has been shown in some systems to be important for both Wnt/β-catenin and PCP signaling, whereas in other systems, it is dispensable for PCP signaling (reviewed in Boutros and Mlodzik, 1999; Wallingford and Habas, 2005; Wharton, 2003). We examined whether the PDZ domain was required to rescue dsh-2(or302) phenotypes during embryogenesis. dsh-2(or302) embryos expressing the ΔPDZ construct showed rescue of embryonic lethality (46.0% with 20.7% ventral rupture; n =87) and dorsal intercalation (68.8%, n=35), but the occurrence of body shape defects were similar to dsh-2(or302) mutants (78.9%; n=71; Table 2).
Our rescue experiments suggested that both a Wnt/β-catenin and a PCP-like pathway might regulate morphogenesis. To examine this possibility we examined the requirements for candidate genes in both pathways during morphogenesis.
Removal of conserved components of PCP signaling does not result in hypodermal morphogenesis defects
C. elegans has well conserved homologs of many genes implicated in PCP signaling downstream of Dsh, including components of the JNK pathway, Misshapen (Msn) (a Nck-Interacting Kinase (NIK) from the STE20 family of kinases), the formin homology domain protein DAAM-1, RHO-1/RhoA, and LET-502/ROCK (Kuhl, 2002; Noselli and Agnes, 1999; Paricio et al., 1999; Su et al., 2000).
Removing the function of jnk-1, the closest homologue of JNK in C. elegans, by RNAi or by the deletion allele gk7 resulted in no embryonic lethality or body shape defects (data not shown). The requirement for JNK signaling was further tested by removing the function of the only two Jnk-kinase (JKK) homologs, jkk-1 and mek-1. Levels of embryonic lethality in jkk-1 or mek-1 reduction of function embryos was comparable to wild-type (Table 3). Msn acts in several pathways, including upstream of Jnk and Jkk in PCP signaling during dorsal closure in Drosophila (Paricio et al., 1999; Su et al., 2000). A hypomorphic allele of mig-15(rh80), the C. elegans Msn homolog (Poinat et al., 2002; Su et al., 2000), gives a low level of embryonic lethality (10.2% with 0.2% enclosure defects; n=177) (Table 3). We were unable to use a stronger allele, mig-15(rh326), for this analysis, because in our hands the strain is barely viable, with brood sizes of <5 embryos per hermaphrodite, precluding a significant analysis of embryonic lethality. Although we cannot rule out a PCP-like role for mig-15 during morphogenesis, the low level of lethality of mig-15 (rh80) and the failure of mig-15(RNAi) to phenocopy the dsh-2 mutant suggest mig-15 is not a key regulator of morphogenesis in C. elegans.
Table 3.
Embryonic lethality in potential planar cell polarity and Wnt/β-catenin downstream pathway members
| Genotype | Embryonic lethalitya | n |
|---|---|---|
| WT | 0.9% | 2228 |
| PCP Homologs | ||
| jkk-1(RNAi) | 0.7% | 606 |
| mek-1(ks54) | 0.7% | 844 |
| mig-15(RNAi) | 1.7% | 2013 |
| mig-15(rh80) | 10.2% | 177 |
| fhod-2(RNAi) | 0.1% | 5006 |
| frl-1(RNAi) | 1.5% | 3366 |
| fhod-2(RNAi); frl-1(RNAi) | 0.7% | 1390 |
| Wnt/β-catenin | ||
| pop-1(q772)+/− | 26.0% | 2718 |
| sys-1(q736)+/− | 23.9% | 5236 |
| wrm-1(ok738)+/− | 23.9% | 3183 |
| pop-1(q772)+/− sys-1(q736)+/− | 48.3% | 499 |
| pop-1(q772)+/−;wrm-1(ok738)+/− | 44.0% | 1907 |
| sys-1(q736)+/−;wrm-1(ok738)+/− | 45.0% | 1015 |
Embryos failing to hatch on NGM plates.
C. elegans has two genes similar to Daam1, fhod-2 and frl-1. Removal of the function of either gene alone or in combination via RNAi does not result in embryonic lethality or body shape defects (fhod-2, 0.1% embryonic lethality, n=5006; frl-1, 1.5% embryonic lethality, n =3366; fhod-2; frl-1, 0.7% embryonic lethality, n=1390) (Table 3). Moreover, a deletion mutant for fhod-2, fhod-2(tm2133), does not yield obvious morphogenetic defects (our observations; note that the originally reported strain from the Mitani consortium carried a closely linked lethal mutation that has now been crossed away, yielding a fhod-2 homozygous line; I. Chin-Sang and A. Lam, pers. comm.). These results suggest that a Daam1 homologue does not act in a PCP-like pathway to regulate C. elegans morphogenesis.
Other potential PCP pathway members include RHO-1/RhoA and LET-502/ROCK, which act through MIG-5 to polarize the B cell in C. elegans (Wu and Herman, 2007). Weak let-502 mutants display embryonic lethality due to a failure of elongation (Piekny and Mains, 2002; Piekny et al., 2000; Wissmann et al., 1997), which is phenotypically different from defects observed in dsh-2 embryos. The involvement of rho-1 and let-502 could not be definitively determined, as removal of rho-1 or let-502 function results in cytokinesis defects in early embryos (Jantsch-Plunger et al., 2000; Spencer et al., 2001). Taken together, these results suggest that the signaling pathway(s) downstream of the DEP domain of DSH-2 must act through mechanisms that have yet to be implicated in PCP-like signaling.
Removal of Wnt/β–catenin asymmetry components yields defects similar to removal of DSH-2
There is no known embryonic role for the one β-catenin in C. elegans, BAR-1, that acts in a canonical Wnt pathway (Costa et al., 1998). We therefore examined whether components of the Wnt/β-catenin asymmetry pathway function downstream of dsh-2 during embryogenesis by conducting an in-depth characterization of hypodermal morphogenesis of deletion alleles for sys-1, wrm-1, and pop-1. Embryos homozygous for any of the various deletion alleles showed completely penetrant lethality during embryogenesis (Table 3). Intercalation was defective in all embryos examined (pop-1(q772), n=8; sys-1(q736), n=28; wrm-1(ok738), n=17). Similar to embryos from dsh-2(or302) mutant mothers, cells failed to complete intercalation and there were cells that intercalated as pairs or triplets (Fig. 2B–D). All three mutants also displayed ventral and head ruptures (pop-1 (q772) 37.5%, n =24; sys-1(q736) 89.3%, n=28; wrm-1(ok738) 90.6%, n=32). Unlike embryos from dsh-2(or302) mutant mothers, both sys-1(q736) and wrm-1(ok738) mutant embryos displayed a failure to complete gastrulation, preventing leading and pocket cells from completing ventral enclosure (sys-1(q736) 41.7%, n=24; wrm-1 (ok738) 26.7%, n=15). Intercalation and enclosure defects in pop-1 (q772) embryos were less severe than in sys-1(q736) and wrm-1 (ok738) mutants, with many elongating to the 1.5–2-fold stage prior to arresting with body shape defects (62.5% n=24). Transheterozygote analysis between the deletion mutants did not reveal additional genetic interactions during embryogenesis, but did produce the expected postembryonic Sys and Mig gonad defects (Table 3 and data not shown).
Fig. 2.

Morphogenesis defects in zygotic mutants for Wnt/β-catenin asymmetry pathway members. (A) Gene structure for sys-1, pop-1, and wrm-1 indicating the regions deleted in the alleles used for this study. Indicated protein domains are: red, armadillo repeats; green, β-catenin binding domain; and blue, HMG Box. (B–D) Dorsal and (E–G) ventral views of (B, E) sys-1(q736), (C, F) pop-1(q772) and (D, G) wrm-1(ok738) mutant embryos. Wnt/β-catenin asymmetry pathway members display variable defects in dorsal intercalation, including dorsal cells that fail to complete intercalation (arrowheads, B–D) and dorsal cells that migrate in groups (arrows, C). When gastrulation appears complete, leading cells (asterisks, E–G) in Wnt/β-catenin mutant embryos often migrate properly; however, pocket cells fail to meet at the ventral midline (arrow E–G). Head enclosure also fails in wrm-1(ok738) mutant embryos (arrowheads, G). Scale bar=10 μm.
Cell fate defects occur in lineages giving rise to hypodermal cells in sys-1, wrm-1, and dsh-2 mutants
While intercalation defects in dsh-2(or302), sys-1(q736), and wrm-1(ok738) mutant embryos were variable, we observed that a number of defects occurred near the position of the dorsal pointer cells and anterior deirid sheath (ADEsh) cells. These cells are derived from an asymmetric cell division. Given the well established role for Wnt/β-catenin asymmetry components in regulating asymmetric cell fates, we more closely examined the lineage giving rise to the dorsal pointer cells. We analyzed the ABarppaap lineage, which divides to give rise to the left dorsal pointer cell and a neuroblast in sys-1(q736) and wrm-1(ok738) embryos. We found that unlike wild-type controls the division was symmetric in size, with the posterior daughter remaining at the surface and showing wedging behavior, suggesting a posterior to anterior fate transformation (data not shown, n=3 each). To further study the ABarppaa lineage we examined dsh-2(or302), sys-1(q736), and wrm-1(ok738) mutant embryos expressing CEH-16::GFP and AJM-1::GFP. CEH-16::GFP is initially expressed in ABarppaa and expression continues in this lineage through the terminal division when it is down regulated in non-seam cells. Using this marker we observed posterior to anterior fate transformations, resulting in ABarppaapp generating an additional left dorsal pointer cell that wedged and fused with the dorsal syncytium (dsh-2 (or302) n=5/12, sys-1(q736) n= wrm-1 2/2, (ok738) n=6/7; Fig. 3). We were able to further correlate our CEH-16::GFP; AJM-1::GFP data with our AJM-1::GFP data and found that a large percentage of the intercalation defects in the middle region of the body correlate with the endogenous and the transformed left dorsal pointer failing to intercalate (dsh-2(or302) n=10/32, sys-1(q736) n=27/29, wmr-1 (ok738) n=11/13). Interestingly, the cell giving rise to the right dorsal pointer (ABarpppap) undergoes a similar asymmetric division which is strongly affected in sys-1(q736) mutants (n=25/31), but only weakly affected in dsh-2(or302) and wrm-1(ok738) mutants (n=2/36 and n=3/16 respectively). We conclude that dsh-2, wrm-1, and sys-1 function to regulate asymmetric cell fate decisions in some hypodermal lineages, and that defects in these lineages result in defects during morphogenesis.
Fig. 3.
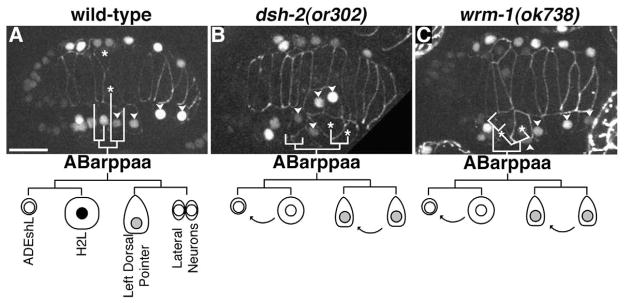
dsh-2 and Wnt/β-catenin asymmetry pathway mutants display asymmetric hypodermal lineage defects. (A–C) Dorsal views of embryos expressing CEH-16::GFP (nuclei) and AJM-1::GFP (cell borders). (A) Wild-type embryo. ABarppaa gives rise to (from anterior to posterior) the anterior deirid (ADEshL), a lateral (seam) cell (H2L), the left dorsal pointer cell, and a neuroblast. Only H2L retains strong expression of CEH-16::GFP (black nucleus in the schematic). (B) Embryo from dsh-2(or302) mutant mother. CEH-16::GFP levels become quickly diminished in all ABarppaa progeny. ABarppaap divides symmetrically, forming two dorsal pointer cells. (C) wrm-1(ok738) mutant embryo. ABarppaaa divides asymmetrically, but the larger posterior cell loses CEH-16::GFP expression and behaves like a second ADEshL. ABarppaap divides symmetrically, giving rise to two dorsal pointer cells. Asterisks = dorsal pointer cells; arrowheads = ABarppap derived seam cells. Scale bar=10 μm.
Global regulation of POP-1 asymmetry occurs via either the DIX or DEP domains of DSH-2
We examined whether dsh-2 functions upstream of POP-1 asymmetry by calculating the ratio of anterior to posterior GFP:: POP-1 in cells 5 to 10 min post-division at the 100 cell stage in wild-type, dsh-2(or302), mig-5(RNAi), and dsh-2(or302); mig-5(RNAi) embryos with or without either the full-length dsh-2 construct, the dsh-2(ΔDIX) construct, or the dsh-2(ΔDEP) construct. Anterior to posterior ratios of nuclear GFP were slightly but not significantly lower in embryos from dsh-2(or302) mutant mothers (mean ratio=1.72; n=3 embryos, 32 sister pairs) compared to wild-type (mean ratio=1.77; n=2 embryos, 30 sister pairs; Fig. 4). This is not surprising given the redundancy of Dshs during embryogenesis (Walston et al., 2004). Although the mean ratio was similar between wild-type and dsh-2(or302) mutant embryos, 93.3% of sister pairs in wild-type embryos showed a ratio greater than 1.5 between sister cells compared to 71.9% in dsh-2(or302) embryos. Thus, although globally anterior/posterior ratios of GFP::POP-1 are similar, a subset of cells display altered levels of POP-1 in dsh-2(or302) embryos.
Fig. 4.

ΔDIX and ΔDEP deletions of DSH-2 can each rescue POP-1 anterior/posterior (AP) asymmetry in dsh-2(or302); mig-5(RNAi) embryos. (A, B) Sister nuclei (connected by double-headed arrows) expressing GFP::POP-1. (A) In wild-type embryos, nuclear GFP::POP-1 levels are asymmetric, resulting in higher levels in the anterior sister than the posterior sister. The inset shows a representative sister A–P pair. (B) In dsh-2 (or302); mig-5(RNAi) embryos, overall nuclear levels of GFP::POP-1 are lower and nearly symmetric between sister cells. The inset shows a representative sister pair. (C) Histogram plots of the ratio of GFP::POP-1 between anterior and posterior sister nuclei. dsh-2(or302); mig-5(RNAi) embryos show a statistically significant difference in nuclear GFP::POP-1 ratios compared to wild-type (C, asterisk). In dsh-2(or302); mig-5 (RNAi) embryos, the presence of full-length dsh-2, dsh-2(ΔDIX) or dsh-2(ΔDEP) constructs yields a statistically significant rescue. Scale bar=10 μm.
To address whether the Dshs function redundantly to regulate POP-1 asymmetry in the embryo the functions of dsh-2 and mig-5 were simultaneously removed. dsh-2(or302); mig-5(RNAi) embryos displayed an overall reduction in GFP::POP-1 levels and a symmetric ratio of GFP::POP-1 between sister cells (mean ratio=1.02; n=4 embryos, 29 sister pairs, Fig. 4). While mig-5(RNAi) alone displayed a mean ratio=1.61 with 70% of sister pairs showing an anterior/posterior ratio of GFP::POP-1 greater than 1.5 (n=3 embryos, 31 sister pairs; Fig. 4). Significantly, when the function of both dsh-2 and mig-5 were removed most cell divisions were no longer oriented anterior to posterior. POP-1 asymmetry has previously been shown to require division along the anterior–posterior axis (Park and Priess, 2003). Thus it is difficult to determine if loss of POP-1 asymmetry is due to the altered division orientations in dsh-2(or302); mig-5(RNAi) embryos, or if the Dshs function directly upstream in regulating POP-1 asymmetry.
We next examined whether the full-length dsh-2, dsh-2(ΔDIX), or dsh-2(ΔDEP) constructs could rescue the division orientation and POP-1 asymmetry defects in dsh-2(or302); mig-5(RNAi) embryos. The constructs were maintained as extra-chromosomal arrays and sibling embryos not carrying the array showed POP-1 asymmetry defects consistent with dsh-2(or302); mig-5(RNAi) embryos, suggesting that mig-5(RNAi) was effective (data not shown). The full-length DSH-2:: GFP construct rescued division orientation and POP-1 asymmetry defects to near mig-5(RNAi) levels (mean ratio=1.52; n=3 embryos, 31 sister pairs) with 54.8% of sister pairs showing an anterior/posterior ratio of GFP::POP-1>1.5 (Fig. 4). Both the dsh-2(ΔDIX) (mean ratio=1.61; n=4 embryos, 50 sister pairs) and dsh-2(ΔDEP) (mean ratio 1.47; n=3 embryos, 39 sister pairs) constructs showed a similar POP-1 asymmetry rescue with 55.3% and 48.7% of sister pairs showing an anterior/posterior ratio of GFP::POP-1>1.5 (Fig. 4). The ability of either the N- or C-terminal fragments of DSH-2 to rescue division orientation and POP-1 asymmetry defects suggests that either fragment is sufficient to mediate signaling upstream of POP-1.
Discussion
Dshs are modular proteins that function at the branch-point of divergent Wnt signaling pathways. Rescue of dsh-2(or302) mutants with constructs lacking conserved Dsh domains reveals neither an absolute requirement for any one domain, nor a dispensability of any domain. The ability of both N- and C-terminal fragments of DSH-2 to rescue morphogenesis defects in dsh-2(or302) mutants differs from similar experiments examining asymmetric division of the B cell in the male tail as well as PHA neuronal lineages (Hingwing et al., 2009; Wu and Herman, 2007). In the B cell both the PDZ and the DEP domains of MIG-5/Dsh have been shown to be required to rescue B cell polarity in a mig-5 mutant (Wu and Herman, 2007). The DIX domain is not required for rescue of B cell polarity, which is consistent with polarity of this division being regulated by a PCP-like pathway (Herman, 2001; Herman and Wu, 2004; Wu and Herman, 2007). The DEP domain of DSH-2 is also required for asymmetric cell division in the PHA neuronal lineage (Hingwing et al., 2009). However, consistent with our results, DSH-2 deletion constructs lacking either the DIX or DEP domain are capable of rescuing asymmetric division of the somatic gonadal precursor in dsh-2(or302) mutants (Hingwing et al., 2009). This suggests that while polarity in some cells may be regulated by a β-catenin-independent PCP-like pathway, other cell divisions are regulated through a β-catenin-dependent asymmetry pathway that can be regulated through either the N- or C-terminal domains of DSH-2.
Does a PCP-like pathway regulate hypodermal morphogenesis?
Wnt/PCP signaling pathways regulate cell migration during morphogenesis in numerous systems (Wallingford and Habas, 2005). Therefore, a priori a PCP-like pathway seemed a likely candidate for regulating cell migration during dorsal intercalation and ventral enclosure in C. elegans, events that also involve directed cell migrations (Williams-Masson et al., 1998, 1997). This prediction is supported by the strong rescue of hypodermal defects in dsh-2(or302) embryos expressing a construct with the DEP domain. However, removal of candidate PCP homologs failed to demonstrate a role for these effectors in controlling hypodermal morphogenesis. It is possible that redundancy among known PCP components may have obscured the results. Alternatively, a novel PCP-like pathway may operate in C. elegans that may or may not be conserved among other organisms.
Another pathway by which the DEP domain may act is the Wnt/Ca2+ pathway. In Xenopus, constructs containing the DEP domain are sufficient to activate the Wnt/Ca2+ pathway (Sheldahl et al., 2003), and inhibit convergent extension movements (Kuhl et al., 2001). Additionally, pharmacological inhibition of inositol trisphosphate receptors (ITRs) produces phenotypes similar to Wnt mutants (Kohn and Moon, 2005). Although the Wnt/Ca2+ pathway has only been demonstrated to function in vertebrates, it is possible that a similar pathway acts during hypodermal morphogenesis in C. elegans. A hypomorphic allele of itr-1/ITR displays defects during dorsal intercalation and ventral enclosure (Thomas-Virnig et al., 2004), suggesting that Ca2+ signaling may function in regulating morphogenesis.
Cortical localization of Dishevelleds is a hallmark of PCP signaling (Axelrod et al., 1998). DSH-2 shows cortical localization throughout development, including during morphogenesis (Fig. S1). Cortical localization of both MIG-5 and DSH-2 has been shown to require the DEP domain, and consistent with this the DEP domain is required for β-catenin independent signaling in the B cell and PHA neuronal lineage (Hingwing et al., 2009-this issue; Wu and Herman, 2007). Intriguingly, a DSH-2 construct lacking the DEP domain is capable of rescuing morphogenesis, gonadogenesis, and POP-1 asymmetry defects despite failing to localize to the cortex, suggesting that cortical localization of Dshs may not be a requirement for Wnt/β-catenin asymmetry signaling.
A Wnt/β-catenin asymmetry pathway regulates morphogenesis in C. elegans
To determine whether the Wnt/β-catenin asymmetry pathway can account for defects observed in embryos from dsh-2(or302) mutant mothers, an in-depth characterization of morphogenesis defects in putative null alleles of pop-1, wrm-1 and sys-1 was conducted. Homozygotes for these alleles displayed dorsal intercalation defects similar to dsh-2(or302), including failure of cells to intercalate and cells that intercalated as pairs. As discussed below, cell fate defects are observed and may alter either cell–cell adhesion or morphogenetic programming to result in the observed defects.
Significantly, mutant embryos displayed posterior to anterior fate transformations, resulting in the H2L seam cell differentiating into a second anterior deirid and a neuroblast acting like a second left, dorsal pointer cell. These transformations are consistent with the well-established role of dsh-2, pop-1, sys-1, and wrm-1 in regulating cell fates (reviewed in Eisenmann, 2005). While the cell fate transformation produced two left dorsal pointer cells that wedged and fused with the dorsal syncytium, neither cell was fully capable of intercalating. This could be due to the often slight lateral mispositioning of these cells, an alteration in the size of these cells because the division is no longer asymmetric, or incomplete fate transformation.
Although we did identify A–P fate transformations, only a few asymmetric divisions during mid-embryogenesis showed obvious defects in dsh-2, pop-1, sys-1, and wrm-1 mutant backgrounds. This may reflect a difference in the signaling threshold required to specify these cells, or that their specification may be regulated through a separate mechanism. In addition, many of the defects we observed occurred in cells produced after symmetric divisions: most notably the C-derived dorsal hypodermal cells (Figs 1B and 2A) and ABarpp-derived seam cells (Fig. 3B). Wnt signaling may regulate the polarity of these cells during migration, or defects in these lineages may reflect subtle differences in cell fate between seemingly symmetric sister cells. These differences may be important for directing proper cell positioning and patterning prior to morphogenesis, which involves local cell–cell interactions termed “cell focusing” (Bischoff and Schnabel, 2006; Schnabel et al., 2006).
Although similar, the phenotypes observed in pop-1, sys-1, and wrm-1 mutants were not completely identical to those of embryos from dsh-2(or302) mutants. One explanation for this difference is that the Wnt/β-catenin asymmetry pathway may not be the only pathway downstream of DSH-2; a PCP-like pathway may also function during morphogenesis (see above). Alternatively, the differences we observed could be due to the zygotic requirement for pop-1, sys-1, and wrm-1 and redundancy of the Dshs.
POP-1 asymmetry is regulated by dishevelleds
dsh-2 function is required for regulating cell fate through the Wnt/β-catenin asymmetry pathway, as evidenced by reversal of POP-1 asymmetry in the somatic gonad (Chang et al., 2005). An analysis of POP-1 asymmetry during embryonic development in embryos from dsh-2(or302) mutant mothers reveals wild-type asymmetry. It is possible that although we were unable to find a significant change in POP-1 asymmetry, some cell fate decisions may be exquisitely sensitive to changes that were beyond our detection. Another possibility is that cell fate changes in a small subset of cells that showed POP-1 symmetry may be distinctively important for morphogenesis, even though these cells do not make a large numerical contribution to normal overall levels of POP-1 asymmetry. Consistent with this the percentage of sister pairs showing an anterior/posterior ratio of GFP::POP-1>1.5 was slightly lower in embryos from dsh-2 (or302) mutant mothers compared to wild-type. Alterations in POP-1 asymmetry in a small subset of cells is consistent with our cell fate and morphogenesis analysis, in which we only observed defects in a variable but small number of cells.
To examine functional redundancy between the Dshs in regulating POP-1 asymmetry, we removed the function of both dsh-2 and mig-5, resulting in POP-1 symmetry between sister cells. Our genetic analysis suggests that there is likely some redundancy/overlap in the function of the Dshs during morphogenesis, as we and others have already shown occurs in the early embryo during Wnt signaling and to orient blastomere divisions (Bei et al., 2002; Walston et al., 2004). While our analysis has examined domain-specific requirements for dsh-2 only, it is possible that there are similar or overlapping requirements for domains of mig-5 as well. Unfortunately, available mig-5 deletions (Wu and Herman, 2007) are in the mig-5(ok280) background. In addition to deleting a portion of mig-5, this allele deletes a piece of the neighboring gene (T05C12.7; cct-1), which RNAi screens indicate is essential for embryogenesis. Because we are addressing functions in the embryo, this confounds the experiment. Moreover, given the sensitivity of the Wnt signaling pathway to fluctuations in signaling component levels (e.g., see Phillips et al., 2007; Huang et al., 2007), analyzing the contribution of domains of various Dshs simultaneously is difficult and beyond the scope of this paper.
In our experiments, asymmetry was restored by the presence of a full-length dsh-2 construct, as well as constructs lacking either the DIX or DEP domains. This suggests that signals transduced through either the DIX or the DEP domain of DSH-2 is sufficient to achieve POP-1 asymmetry. The mechanisms by which each domain regulates POP-1 asymmetry remain unclear. POP-1 localization is regulated through phosphorylation by a complex of WRM-1/β-catenin and LIT-1/NLK, which triggers targeted nuclear export (Lo et al., 2004). Asymmetric WRM-1 nuclear localization is reciprocal to that of POP-1. Regulation of WRM-1 nuclear levels is mediated by selective nuclear retention through APR-1/APC (Mizumoto and Sawa, 2007a; Nakamura et al., 2005). Localization of DSH-2 and MIG-5 to the posterior cortex of cells undergoing asymmetric divisions suggests that they may function in regulating WRM-1 asymmetry (Mizumoto and Sawa, 2007a). However, localization of DSH-2 to the cortex may not be required for regulation in this pathway, since a DSH-2 construct lacking the DEP domain fails to localize to the cortex but is still capable of rescuing POP-1 asymmetry (Hingwing et al., 2009, and this study).
In order to interact with POP-1, WRM-1 requires LIT-1/NLK as a cofactor (Kaletta et al., 1997; Meneghini et al., 1999; Rocheleau et al., 1999; Shin et al., 1999). LIT-1 is independently activated by a MAPK cascade that includes MOM-4/Tak1, a MAPKKK (Ishitani et al., 1999; Meneghini et al., 1999; Shin et al., 1999). In mammalian tissue culture cells, increasing the levels of Wnt1 results in an increase in phosphorylation of C. elegans MOM-4, which demonstrates that Wnt-signaling can regulate the MAPK pathway that activates LIT-1 (Smit et al., 2004). The role of Dshs in activating the Wnt/Tak1 pathway has not been examined; however, the DEP domain of Dshs modulates MAPK pathways in other organisms (such as Msn in the JNK pathway in Drosophila; Paricio et al., 1999; Su et al., 2000). These data suggest an intriguing model, in which the DEP domain of DSH-2 may activate LIT-1 (and subsequently POP-1 in the Wnt/β-catenin asymmetry pathway) through a MAPK pathway.
In summary, hypodermal morphogenesis in C. elegans serves as a simple model for studying morphogenetic movements within the context of an epithelial sheet. We have shown that removal of DSH-2 results in hypodermal morphogenesis defects, which are also observed in mutants for downstream components of the Wnt/β-catenin asymmetry pathway. Moreover, our data suggest that either the DIX or the DEP domain of DSH-2 is sufficient to regulate POP-1 asymmetry prior to morphogenesis in C. elegans. Elucidating the mechanisms by which Dsh-dependent control of POP-1 asymmetry regulates morphogenetic events in C. elegans should yield further insights into this process.
Supplementary Material
Acknowledgments
This work was supported by NSF grant IOB0518081 to J.D.H., NIH Predoctoral Training Grant in Molecular Biosciences T32 GM07215 to R.S.K. and A.R.K., and NIH Predoctoral Training Grant in Genetics T32GM007133 to T.D.W. J.K. is an investigator of the HHMI. We would like to thank Elise Walck (Truman State University) for her assistance in analyzing the data. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR). Some mutant strains used in this work were created by the C. elegans Gene Knockout Consortium.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ydbio.2009.01.017.
References
- Axelrod JD, Miller JR, Shulman JM, Moon RT, Perrimon N. Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and Wingless signaling pathways. Genes Dev. 1998;12:2610–2622. doi: 10.1101/gad.12.16.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei Y, et al. SRC-1 and Wnt signaling act together to specify endoderm and to control cleavage orientation in early C. elegans embryos. Dev Cell. 2002;3:113–125. doi: 10.1016/s1534-5807(02)00185-5. [DOI] [PubMed] [Google Scholar]
- Bischoff M, Schnabel R. Global cell sorting is mediated by local cell–cell interactions in the C. elegans embryo. Dev Biol. 2006;294:432–444. doi: 10.1016/j.ydbio.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Boutros M, Mlodzik M. Dishevelled: at the crossroads of divergent intracellular signaling pathways. Mech Dev. 1999;83:27–37. doi: 10.1016/s0925-4773(99)00046-5. [DOI] [PubMed] [Google Scholar]
- Boutros M, Paricio N, Strutt DI, Mlodzik M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell. 1998;94:109–118. doi: 10.1016/s0092-8674(00)81226-x. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Chang W, Lloyd CE, Zarkower D. DSH-2 regulates asymmetric cell division in the early C. elegans somatic gonad. Mech Dev. 2005;122:781–789. doi: 10.1016/j.mod.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Chase DL, Patikoglou GA, Koelle MR. Two RGS proteins that inhibit Galpha(o) and Galpha(q) signaling in C. elegans neurons require a Gbeta(5)-like subunit for function. Curr Biol. 2001;11:222–231. doi: 10.1016/s0960-9822(01)00071-9. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Greene ND, Murdoch JN. Dishevelled: linking convergent extension with neural tube closure. Trends Neurosci. 2003;26:453–455. doi: 10.1016/S0166-2236(03)00212-1. [DOI] [PubMed] [Google Scholar]
- Costa M, Raich W, Agbunag C, Leung B, Hardin J, Priess JR. A putative catenin–cadherin system mediates morphogenesis of the Caenorhabditis elegans embryo. J Cell Biol. 1998;141:297–308. doi: 10.1083/jcb.141.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann DM Community, T.C.e.R, editor. Wnt signaling. WormBook. 2005 doi: 10.1895/wormbook.1.7.1. http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- Gu T, Orita S, Han M. Caenorhabditis elegans SUR-5, a novel but conserved protein, negatively regulates LET-60 Ras activity during vulval induction. Mol Cell Biol. 1998;18:4556–4564. doi: 10.1128/mcb.18.8.4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther C, Garriga G. Asymmetric distribution of the C. elegans HAM-1 protein in neuroblasts enables daughter cells to adopt distinct fates. Development. 1996;122:3509–3518. doi: 10.1242/dev.122.11.3509. [DOI] [PubMed] [Google Scholar]
- Hawkins NC, Ellis GC, Bowerman B, Garriga G. MOM-5 frizzled regulates the distribution of DSH-2 to control C. elegans asymmetric neuroblast divisions. Dev Biol. 2005;284:246–259. doi: 10.1016/j.ydbio.2005.05.024. [DOI] [PubMed] [Google Scholar]
- Hawkins NC, Garriga G, Beh CT. Creating precise GFP fusions in plasmids using yeast homologous recombination. Biotechniques. 2003;34:74–8. 80. doi: 10.2144/03341st03. [DOI] [PubMed] [Google Scholar]
- Heisenberg CP, Tada M, Rauch GJ, Saude L, Concha ML, Geisler R, Stemple DL, Smith JC, Wilson SW. Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature. 2000;405:76–81. doi: 10.1038/35011068. [DOI] [PubMed] [Google Scholar]
- Herman M. C. elegans POP-1/TCF functions in a canonical Wnt pathway that controls cell migration and in a noncanonical Wnt pathway that controls cell polarity. Development. 2001;128:581–590. doi: 10.1242/dev.128.4.581. [DOI] [PubMed] [Google Scholar]
- Herman MA, Wu M. Noncanonical Wnt signaling pathways in C. elegans converge on POP-1/TCF and control cell polarity. Front Biosci. 2004;9:1530–1539. doi: 10.2741/1306. [DOI] [PubMed] [Google Scholar]
- Hingwing K, Lee S, Nykilchuk L, Walston T, Hardin J, Hawkins N. CWN-1 functions with DSH-2 to regulate C. elegans asymmetric neuroblast division in a β-catenin independent Wnt pathway. Dev Biol. 2009;328:245–256. doi: 10.1016/j.ydbio.2009.01.025. [DOI] [PubMed] [Google Scholar]
- Huang S, Shetty P, Robertson SM, Lin R. Binary cell fate specification during C. elegans embryogenesis driven by reiterated reciprocal asymmetry of TCF POP-1 and its coactivator beta-catenin SYS-1. Development. 2007;134:2685–2695. doi: 10.1242/dev.008268. [DOI] [PubMed] [Google Scholar]
- Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N, Waterman M, Bowerman B, Clevers H, Shibuya H, Matsumoto K. The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta-catenin and transcription factor TCF. Nature. 1999;399:798–802. doi: 10.1038/21674. [DOI] [PubMed] [Google Scholar]
- Jantsch-Plunger V, Gonczy P, Romano A, Schnabel H, Hamill D, Schnabel R, Hyman AA, Glotzer M. CYK-4: a Rho family gtpase activating protein (GAP) required for central spindle formation and cytokinesis. J Cell Biol. 2000;149:1391–1404. doi: 10.1083/jcb.149.7.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaletta T, Schnabel H, Schnabel R. Binary specification of the embryonic lineage in Caenorhabditis elegans. Nature. 1997;390:294–298. doi: 10.1038/36869. [DOI] [PubMed] [Google Scholar]
- Kidd AR, III, Miskowski JA, Siegfried KR, Sawa H, Kimble J. A beta-catenin identified by functional rather than sequence criteria and its role in Wnt/MAPK signaling. Cell. 2005;121:761–772. doi: 10.1016/j.cell.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium. 2005;38:439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Köppen M, Simske JS, Sims PA, Firestein BL, Hall DH, Radice AD, Rongo C, Hardin JD. Cooperative regulation of AJM-1 controls junctional integrity in Caenorhabditis elegans epithelia. Nat Cell Biol. 2001;3:983–991. doi: 10.1038/ncb1101-983. [DOI] [PubMed] [Google Scholar]
- Korswagen HC. Canonical and non-canonical Wnt signaling pathways in Caenorhabditis elegans: variations on a common signaling theme. Bioessays. 2002;24:801–810. doi: 10.1002/bies.10145. [DOI] [PubMed] [Google Scholar]
- Kuhl M. Non-canonical Wnt signaling in Xenopus: regulation of axis formation and gastrulation. Semin Cell Dev Biol. 2002;13:243–249. doi: 10.1016/s1084-9521(02)00050-2. [DOI] [PubMed] [Google Scholar]
- Kuhl M, Geis K, Sheldahl LC, Pukrop T, Moon RT, Wedlich D. Antagonistic regulation of convergent extension movements in Xenopus by Wnt/beta-catenin and Wnt/Ca2+ signaling. Mech Dev. 2001;106:61–76. doi: 10.1016/s0925-4773(01)00416-6. [DOI] [PubMed] [Google Scholar]
- Lee JY, Marston DJ, Walston T, Hardin J, Halberstadt A, Goldstein B. Wnt/Frizzled signaling controls C. elegans gastrulation by activating actomyosin contractility. Curr Biol. 2006;16:1986–1997. doi: 10.1016/j.cub.2006.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo MC, Gay F, Odom R, Shi Y, Lin R. Phosphorylation by the beta-catenin/MAPK complex promotes 14-3-3-mediated nuclear export of TCF/POP-1 in signal-responsive cells in C. elegans. Cell. 2004;117:95–106. doi: 10.1016/s0092-8674(04)00203-x. [DOI] [PubMed] [Google Scholar]
- Meneghini MD, Ishitani T, Carter JC, Hisamoto N, Ninomiya-Tsuji J, Thorpe CJ, Hamill DR, Matsumoto K, Bowerman B. MAP kinase and Wnt pathways converge to downregulate an HMG-domain repressor in Caenorhabditis elegans. Nature. 1999;399:793–797. doi: 10.1038/21666. [DOI] [PubMed] [Google Scholar]
- Mizumoto K, Sawa H. Cortical beta-catenin and APC regulate asymmetric nuclear beta-catenin localization during asymmetric cell division in C. elegans. Dev Cell. 2007a;12:287–299. doi: 10.1016/j.devcel.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Mizumoto K, Sawa H. Two betas or not two betas: regulation of asymmetric division by beta-catenin. Trends Cell Biol. 2007b;17:465–473. doi: 10.1016/j.tcb.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Mlodzik M. Planar cell polarization: do the same mechanisms regulate Drosophila tissue polarity and vertebrate gastrulation? Trends Genet. 2002;18:564–571. doi: 10.1016/s0168-9525(02)02770-1. [DOI] [PubMed] [Google Scholar]
- Mohler WA, Shemer G, del Campo JJ, Valansi C, Opoku-Serebuoh E, Scranton V, Assaf N, White JG, Podbilewicz B. The type I membrane protein EFF-1 is essential for developmental cell fusion. Dev Cell. 2002;2:355–362. doi: 10.1016/s1534-5807(02)00129-6. [DOI] [PubMed] [Google Scholar]
- Moon RT, Brown JD, Torres M. WNTs modulate cell fate and behavior during vertebrate development. Trends Genet. 1997;13:157–162. doi: 10.1016/s0168-9525(97)01093-7. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Kawachi K, Kamakura S, Masuyama N, Yamanaka H, Matsumoto K, Kikuchi A, Nishida E. Distinct domains of mouse dishevelled are responsible for the c-Jun N-terminal kinase/stress-activated protein kinase activation and the axis formation in vertebrates. J Biol Chem. 1999;274(30):957–30. 962. doi: 10.1074/jbc.274.43.30957. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kim S, Ishidate T, Bei Y, Pang K, Shirayama M, Trzepacz C, Brownell DR, Mello CC. Wnt signaling drives WRM-1/beta-catenin asymmetries in early C. elegans embryos. Genes Dev. 2005;19:1749–1754. doi: 10.1101/gad.1323705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noselli S, Agnes F. Roles of the JNK signaling pathway in Drosophila morphogenesis. Curr Opin Genet Dev. 1999;9:466–472. doi: 10.1016/S0959-437X(99)80071-9. [DOI] [PubMed] [Google Scholar]
- Paricio N, Feiguin F, Boutros M, Eaton S, Mlodzik M. The Drosophila STE20-like kinase misshapen is required downstream of the Frizzled receptor in planar polarity signaling. EMBO J. 1999;18:4669–4678. doi: 10.1093/emboj/18.17.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park FD, Priess JR. Establishment of POP-1 asymmetry in early C. elegans embryos. Development. 2003;130:3547–3556. doi: 10.1242/dev.00563. [DOI] [PubMed] [Google Scholar]
- Penton A, Wodarz A, Nusse R. A mutational analysis of dishevelled in Drosophila defines novel domains in the dishevelled protein as well as novel suppressing alleles of axin. Genetics. 2002;161:747–762. doi: 10.1093/genetics/161.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BT, Kidd AR, III, King R, Hardin J, Kimble J. Reciprocal asymmetry of SYS-1/beta-catenin and POP-1/TCF controls asymmetric divisions in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2007;104:3231–3236. doi: 10.1073/pnas.0611507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekny AJ, Mains PE. Rho-binding kinase (LET-502) and myosin phosphatase (MEL-11) regulate cytokinesis in the early Caenorhabditis elegans embryo. J Cell Sci. 2002;115:2271–2282. doi: 10.1242/jcs.115.11.2271. [DOI] [PubMed] [Google Scholar]
- Piekny AJ, Wissmann A, Mains PE. Embryonic morphogenesis in Caenorhabditis elegans integrates the activity of LET-502 Rho-binding kinase, MEL-11 myosin phosphatase, DAF-2 insulin receptor and FEM-2 PP2c phosphatase. Genetics. 2000;156:1671–1689. doi: 10.1093/genetics/156.4.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poinat P, De Arcangelis A, Sookhareea S, Zhu X, Hedgecock EM, Labouesse M, Georges-Labouesse E. A conserved interaction between beta1 integrin/PAT-3 and Nck-interacting kinase/MIG-15 that mediates commissural axon navigation in C. elegans. Curr Biol. 2002;12:622–631. doi: 10.1016/s0960-9822(02)00764-9. [DOI] [PubMed] [Google Scholar]
- Raich WB, Agbunag C, Hardin J. Rapid epithelial-sheet sealing in the Caenorhabditis elegans embryo requires cadherin-dependent filopodial priming. Curr Biol. 1999;9:1139–1146. doi: 10.1016/S0960-9822(00)80015-9. [DOI] [PubMed] [Google Scholar]
- Raich WB, Moran AN, Rothman JH, Hardin J. Cytokinesis and midzone microtubule organization in Caenorhabditis elegans require the kinesin-like protein ZEN-4. Mol Biol Cell. 1998;9:2037–2049. doi: 10.1091/mbc.9.8.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau CE, Yasuda J, Shin TH, Lin R, Sawa H, Okano H, Priess JR, Davis RJ, Mello CC. WRM-1 activates the LIT-1 protein kinase to transduce anterior/posterior polarity signals in C. elegans. Cell. 1999;97:717–726. doi: 10.1016/s0092-8674(00)80784-9. [DOI] [PubMed] [Google Scholar]
- Rothbacher U, Laurent MN, Deardorff MA, Klein PS, Cho KW, Fraser SE. Dishevelled phosphorylation, subcellular localization and multimerization regulate its role in early embryogenesis. EMBO J. 2000;19:1010–1022. doi: 10.1093/emboj/19.5.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger A, Shelton CA, Maloof JN, Meneghini M, Bowerman B. Wnt pathway components orient a mitotic spindle in the early Caenorhabditis elegans embryo without requiring gene transcription in the responding cell. Genes Dev. 1999;13:2028–2038. doi: 10.1101/gad.13.15.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R, Bischoff M, Hintze A, Schulz AK, Hejnol A, Meinhardt H, Hutter H. Global cell sorting in the C. elegans embryo defines a new mechanism for pattern formation. Dev Biol. 2006;294:418–431. doi: 10.1016/j.ydbio.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Sheldahl LC, Slusarski DC, Pandur P, Miller JR, Kuhl M, Moon RT. Dishevelled activates Ca2+ flux, PKC, and CamKII in vertebrate embryos. J Cell Biol. 2003;161:769–777. doi: 10.1083/jcb.200211094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin TH, Yasuda J, Rocheleau CE, Lin R, Soto M, Bei Y, Davis RJ, Mello CC. MOM-4, a MAP kinase kinase kinase-related protein, activates WRM-1/LIT-1 kinase to transduce anterior/posterior polarity signals in C. elegans. Mol Cell. 1999;4:275–280. doi: 10.1016/s1097-2765(00)80375-5. [DOI] [PubMed] [Google Scholar]
- Siegfried KR, Kidd AR, III, Chesney MA, Kimble J. The sys-1 and sys-3 genes cooperate with Wnt signaling to establish the proximal–distal axis of the Caenorhabditis elegans gonad. Genetics. 2004;166:171–186. doi: 10.1534/genetics.166.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit L, Baas A, Kuipers J, Korswagen H, van de Wetering M, Clevers H. Wnt activates the Tak1/Nemo-like kinase pathway. J Biol Chem. 2004;279(17):232–17. 240. doi: 10.1074/jbc.M307801200. [DOI] [PubMed] [Google Scholar]
- Sokol S. A role for Wnts in morpho-genesis and tissue polarity. Nat Cell Biol. 2000;2:E124–E125. doi: 10.1038/35017136. [DOI] [PubMed] [Google Scholar]
- Spencer AG, Orita S, Malone CJ, Han M. A RHO GTPase-mediated pathway is required during P cell migration in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2001;98:13, 132–13,137. doi: 10.1073/pnas.241504098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YC, Maurel-Zaffran C, Treisman JE, Skolnik EY. The Ste20 kinase misshapen regulates both photoreceptor axon targeting and dorsal closure, acting downstream of distinct signals. Mol Cell Biol. 2000;20:4736–4744. doi: 10.1128/mcb.20.13.4736-4744.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Virnig CL, Sims PA, Simske JS, Hardin J. The inositol 1,4,5-trisphosphate receptor regulates epidermal cell migration in Caenorhabditis elegans. Curr Biol. 2004;14:1882–1887. doi: 10.1016/j.cub.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Habas R. The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development. 2005;132:4421–4436. doi: 10.1242/dev.02068. [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Rowning BA, Vogeli KM, Rothbacher U, Fraser SE, Harland RM. Dishevelled controls cell polarity during Xenopus gastrulation. Nature. 2000;405:81–85. doi: 10.1038/35011077. [DOI] [PubMed] [Google Scholar]
- Walston T, Guo C, Proenca R, Wu M, Herman M, Hardin J, Hedgecock E. mig-5/Dsh Controls Cell Fate Determination and Cell Migration in C. elegans. 2006 doi: 10.1016/j.ydbio.2006.06.053. in prep. [DOI] [PubMed] [Google Scholar]
- Walston T, Tuskey C, Edgar L, Hawkins N, Ellis G, Bowerman B, Wood W, Hardin J. Multiple Wnt signaling pathways converge to orient the mitotic spindle in early C. elegans embryos. Dev Cell. 2004;7:831–841. doi: 10.1016/j.devcel.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Wang J, Wilkinson MF. Deletion mutagenesis of large (12-kb) plasmids by a one-step PCR protocol. Biotechniques. 2001;31:722–724. doi: 10.2144/01314bm01. [DOI] [PubMed] [Google Scholar]
- Weaver C, Kimelman D. Move it or lose it: axis specification in Xenopus. Development. 2004;131:3491–3499. doi: 10.1242/dev.01284. [DOI] [PubMed] [Google Scholar]
- Wharton KA., Jr Runnin’ with the Dvl: proteins that associate with Dsh/Dvl and their significance to Wnt signal transduction. Dev Biol. 2003;253:1–17. doi: 10.1006/dbio.2002.0869. [DOI] [PubMed] [Google Scholar]
- Williams-Masson EM, Heid PJ, Lavin CA, Hardin J. The cellular mechanism of epithelial rearrangement during morphogenesis of the Caenorhabditis elegans dorsal hypodermis. Dev Biol. 1998;204:263–276. doi: 10.1006/dbio.1998.9048. [DOI] [PubMed] [Google Scholar]
- Williams-Masson EM, Malik AN, Hardin J. An actin-mediated two-step mechanism is required for ventral enclosure of the C. elegans hypodermis. Development. 1997;124:2889–2901. doi: 10.1242/dev.124.15.2889. [DOI] [PubMed] [Google Scholar]
- Wissmann A, Ingles J, McGhee JD, Mains PE. Caenorhabditis elegans LET-502 is related to Rho-binding kinases and human myotonic dystrophy kinase and interacts genetically with a homolog of the regulatory subunit of smooth muscle myosin phosphatase to affect cell shape. Genes Dev. 1997;11:409–422. doi: 10.1101/gad.11.4.409. [DOI] [PubMed] [Google Scholar]
- Wu M, Herman MA. Asymmetric localizations of LIN-17/Fz and MIG-5/Dsh are involved in the asymmetric B cell division in C. elegans. Dev Biol. 2007;303:650–662. doi: 10.1016/j.ydbio.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi TP. Heads or tails: Wnts and anterior–posterior patterning. Curr Biol. 2001;11:R713–R724. doi: 10.1016/s0960-9822(01)00417-1. [DOI] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis. Prentice Hall; Upper Saddle River, NJ: 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.