

Abstract
A two-week study in rats identified target organs of oxfendazole toxicity to be bone marrow, epididymis, liver, spleen, testis, and thymus. Female rats had greater oxfendazole exposure and exhibited toxicities at lower doses than did males. Decreased WBC levels, a class effect of benzimidazole anthelminthics, returned to normal during the recovery period. The NOAEL was determined to be >5 but < 25 mg/kg/d and the MTD 100 mg/kg/d. The highest dose, 200 mg/kg/d resulted in significant toxicity and mortality, leading to euthanization of the main study animals in this group after seven days. Oxfendazole did not exhibit genetic toxicology signals in standard Ames bacterial, mouse lymphoma or rat micronucleus assays, nor did it provoke safety concerns when evaluated for behavioral effects in rats or cardiovascular safety effects in dogs. These results support the transition of oxfendazole to First in Human safety studies preliminary to its evaluation in human helminth diseases.
Keywords: benzimidazole, anthelminthic, soil transmitted helminths, cysticercosis
Introduction
The majority of benzimidazole (BZ) carbamate anthelminthics in veterinary and human clinical use were approved over 30 years ago. Despite their extensive use for soil transmitted helminths (STH) and other parasitic diseases, the BZs albendazole (ALB) and mebendazole (MEB) are far from perfect. Their efficacy against some human helminth species is clearly sub-optimal,1,2 and if submitted today would probably not obtain approval for some of these indications. A very limited number of treatments are available for some zoonotic helminth infections, such as neurocysticercosis and echinococcosis, and all available treatments for these diseases are less than ideal in terms of efficacy or safety, and not appropriate for use in control programs.3
In addition, widespread use of benzimidazoles in animal health practice has led to drug resistance in helminths of domestic animals (sheep, cattle, horses, goats etc.).4 While there has been little evidence of resistance in helminth species that affect humans (due in part to the differing approach to treatment of human helminths, with, until recently, single annual dose treatment being the norm), attempts at control of a number of human helminth species such as lymphatic filariasis and STH has resulted in a massive increase in drug use, with whole communities being treated. As a response to the potential threat of drug resistance,5 as well as to increase the drug armamentarium available for most parasitic diseases (particularly tissue-dwelling larval helminths) there is a pressing clinical need for alternative drugs to be developed.
A number of BZ anthelminthics used in veterinary medicine have been considered for transition to human use, including oxibendazole and oxfendazole (OXF). In addition, flubendazole, already approved for use as an intestinal anthelminthic, is also being developed as a treatment against adult filarial worms causing river blindness and elephantiasis6 in humans. Oxfendazole has long been under pre-clinical investigation as a potential new treatment for neurocysticercosis and echinococcosis. The initial placebo controlled study of Taenia solium cysticercosis in pigs (the normal host for larval forms of the cestode) showed that a single high dose of OXF (30 mg/kg) but not praziquantel (PZQ) (50 mg/kg PZQ) could eliminate the encysted larvae7 as observed in necropsy 10-12 weeks after drug administration. A follow up study demonstrated the dose-dependent cysticidal effects of OXF in several tissues, with no viable cysts being observed at the highest dose of OXF tested, 30 mg/kg.8 A third study confirmed that cysts do not die immediately, the decrease in viable cysts becoming more pronounced across the 12 week time frame of the study;9 12 weeks after the administration of a single dose of OXF, viable cysts had been fully eliminated from muscle, heart and tongue and fully cleared from the brains of three out of four pigs studied. Importantly, the pharmacokinetic study of OXF disposition in pigs revealed that the metabolic half-life of OXF is long, 21.6 hr,10 a value comparable to its half-life in ruminants.
Although there is considerable experience with BZ anthelminthics, both individually and as a class, many of the available data come from experience post registration rather than from extensive preclinical testing. Since the original work on most BZ anthelminthics was conducted some 30-40 years ago, the toxicological data used for the original approvals are limited, often unpublished, and do not comply with current regulatory standards. The lack of a requirement to update basic toxicological data or to conduct new studies once a compound is approved means that there has been little incentive to undertake new studies and even less to place any data gathered into the public domain. Thus, in order to transition any BZ from animal to human use, it is necessary to fill the current data gaps and to bring the dated toxicology package up to current regulatory standards. Typically, the areas lacking in these earlier toxicological evaluations are genetic toxicology, pharmacokinetics and toxicokinetics, safety pharmacology, and the species and numbers of animals studied. Using the available published data on OXF,11,12 the data from the original toxicology package13 and knowledge of BZ class toxicology,14 a series of OXF studies in rat and dog was devised to provide missing data and to bring the toxicology package up to date in support of an IND for a First in Human (FIH) study. Toxicokinetic (TK) evaluation is also a key component of the present studies, as TK data for OXF have not been available in the public domain.
Methods
Animal care
Animal care and housing were in accordance with the National Research Council (NRC) Guide for the Care and Use of Laboratory Animals (1996) and the Animal Welfare Standards incorporated in 9 CFR Part 3, 1991. All use of animals was approved by the Institutional Animal Care and Use Committee (IACUC) and conducted at SRI International (Menlo Park, CA) in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Rat pharmacokinetics
Three groups of Sprague-Dawley rats, each consisting of three males and three females, were administered OXF, one group at each of the following pharmacologic doses: 5 mg/kg i.v., 15 mg/kg p.o. or 60 mg/kg p.o. Oral dose administration was by gavage. Blood samples were drawn into EDTA containing tubes for determination of OXF concentration pre-dose and at 5, 15, 30 min; 1, 2, 4, 8, 12, 24, 32, 48 hr (i.v. group) or 0.5, 1, 2, 4, 6, 8, 12, 24, 32, 48 hr (p.o. group).
Plasma was separated from the blood samples by centrifugation within 30 min of collection. Rat plasma (80 μl samples) was treated with 350 μl of acetonitrile (containing 25 ng/ml mebendazole as an internal standard) to precipitate plasma proteins. These mixtures were then vortexed for 15 min and suspensions were then clarified by centrifugation (18000 g, 15 min); 300 μl of the resulting supernatants were transferred to a polypropylene container for solvent evaporation at 40°C. The dried samples were reconstituted with 150 μl 25% (v/v) acetonitrile in Milli-Q water. The samples were sonicated for 5 min and then vortexed for an additional 10 min. Samples were analyzed by LC-MS/MS analysis using a Waters 2795 Alliance Integrated System with a Gemini C6-Phenyl column (3 μm, 50 × 2.0 mm 110A). Mobile phase was: A= 0.1% (v/v) acetic acid in water; B= 0.1% (v/v) acetic acid in acetonitrile. Study samples were quantified using a set of calibration standards prepared in blank matrix that were processed in parallel. This analytical method had a LLQ of 5 ng/ml. Pharmacokinetic parameters were calculated from the resulting data with WinNonlin® Professional.
Rat toxicokinetics
Groups of Sprague-Dawley rats (3 male and 3 female per dose) were orally administered 5, 25, 100, or 200 mg/kg of OXF for 14 days, and blood samples were drawn for determination of plasma OXF levels on days 1 and 14 at 0.25, 1, 3, 6, 12, and 24 hr following OXF administration. Samples were processed and analyzed as described above for the rat PK study.
Rat functional observational battery (FOB)
The FOB was conducted on the toxicology study rats (5 male and 5 female per group) at 3-5 hr following the oral administration of 0, 5, 25, 100, or 200 mg/kg of OXF on day one of dosing. The FOB components, designed to fulfill the requirements of the FDA Guidance,15 included home-cage observations, handling observations, open-field observations, and reflexes and physiological measurements. Each component had several endpoints that were evaluated to yield approximately 30 endpoints in total. Parametric data included rearing, defecation, urination, landing foot splay, fore- and hind-limb grip strength, body weight and body temperature. Positive control groups of rats were administered amphetamine (5 mg/kg, males) or carbaryl (50 mg/kg, females).
Rat two week toxicology study
Groups of Sprague-Dawley rats (10 male and 10 female per dose) were orally administered 5, 25, 100, or 200 mg/kg of OXF for 14 days, doses spanning those studied in previously conducted rat toxicology studies.13 Half the animals (5 males and 5 females) were sacrificed 24 hr after the last dose administered, and the other half was kept on study for an additional two weeks without dosing for evaluation of recovery from any changes resulting from OXF administration. Clinical observations were recorded once daily (approximately 2-4 hr postdose) on days of treatment, or more often as clinical signs warranted, and on the day of sacrifice. Animals were examined for any altered clinical signs, including gross motor and behavioral activity, and observable changes in appearance. Body weights were determined before dosing and weekly thereafter. Food consumption was recorded at 24 hr intervals once a week throughout the study. Blood samples were taken 24 hr after administration of the last dose (day 15 or earlier when animals in the high dose group were euthanized) and on Day 28 for the recovery group. Measurements included hematology and clinical chemistry measurements. Urine for urinalysis (protein, glucose, bilirubin, urobilinogen and cell content) was collected during the week prior to each scheduled sacrifice. At necropsy, major organ systems and tissues were collected and weighed and then examined for gross lesions as well as for microscopic changes.
Dog CV safety and tolerability study
Three male and three female beagle dogs were orally administered vehicle followed by rising doses of OXF (5, 25 or 100 mg/kg), with a two week interval between doses. Post-dose evaluations included clinical observations (4–8 hr postdose when possible, or more often as clinical signs warranted), body weight, body temperature, clinical pathology (hematology, serum chemistry, and coagulation), and cardiology parameters (blood pressure, heart rate, and electrocardiogram).
Genetic toxicology evaluation
Ames test
The potential mutagenic activity of OXF was examined in the Salmonella typhimurium-Escherichia coli/microsome plate incorporation assay.16-18 A range-finding experiment was conducted with strain TA100 over doses ranging from 125 to 4000 μg/plate in the presence and absence of a metabolic activation (MA) system containing 5% Aroclor 1254-induced rat-liver microsomes (S9). The maximum test concentration was based on the limit of OXF solubility. Mutagenicity was then studied in two experiments with five tester strains TA1535, TA1537, TA98, and TA100 and E. coli strain WP2 (uvrA) at doses ranging from 125 to 4000 μg/plate, in both the presence and absence of MA containing 5% S9 (first experiment) or 10% S9 (second experiment).
Mouse lymphoma gene mutation assay
The ability of OXF to induce mutations at the thymidine kinase locus (tk) in the L5178Y Mouse Lymphoma Cell tk +/- → tk-/- Gene Mutation Assay was examined using a standard procedure.19,20 Cells were exposed to OXF for 4 and 24 hr in the absence of metabolic activation (MA), and for 4 hr in the presence of MA. Additional details on cell growth, positive control treatments, data collection and statistical analysis were published earlier.21 The end points evaluated were cell growth during expression periods, relative suspension growth (RSG), relative total growth (RTG), relative cloning efficiency (RCE), mutation frequency (MF), and numbers of small (≤ 0.6 mm in diameter) colonies from trifluorothymidine-resistant (TFT1) cells.
Micronucleus evaluation
Bone marrow smears were prepared from the left femur of each rat (5/sex/group) taken on Day 15 in the main group of rats in the two-week toxicology study. All the main group slides and positive control (cyclophosphamide) slides, the latter prepared under a separate GLP study and randomized in with the study slides, were stained, coded and evaluated for the presence of micronuclei using standard procedures.22-24
Results
Rat pharmacokinetics
Rats administered OXF by the i.v. or the p.o. route exhibited plasma levels of the drug (Table 1), with higher exposures seen at the 60 mg/kg than at the 15 mg/kg, p.o. dose. The T½ for OXF was approximately 2.5 hr. The PK parameters obtained following iv administration indicated that OXF distributed to extravascular locations and was eliminated rather quickly (only 10% remained at about 8 hr post dose). There was a possible trend for higher systemic exposure, extravascular distribution and longer elimination time in females compared to males, and these sex-dependent differences were more apparent at the higher oral doses.
Table 1. Calculated parameters from rat PK study.
| Administration Route | Dose (mg/g) | Sex | Cp (μg/ml) | Tmax (hr) | AUClast (hr.μg/ml) | MRTlast (hr) | T1/2 (hr) | V (L/kg) | CL (ml/hr/kg) |
|---|---|---|---|---|---|---|---|---|---|
| i.v. | 5 | M | 10.75 ± 0.73 | 0.14 ± 0.10 | 35.08 ± 3.48 | 3.4 ± 0.3 | 2.4 ± 0.1 | 0.48 ± 0.04 | 142 ± 17 |
| F | 11.20 ± 0.44 | 0.14 ± 0.10 | 35.42 ± 1.25 | 3.1 ± 0.0 | 2.5 ± 0.1 | 0.5 ± 0.03 | 136 ± 4 | ||
|
| |||||||||
| Administration Route | Dose (mg/g) | Sex | Cmax (μg/ml) | Tmax (hr) | AUClast (hr.μg/ml) | MRTlast (hr) | T1/2 (hr) | V (L/kg) | CL (ml/hr/kg) |
|
| |||||||||
| p.o. | 15 | M | 4.63 ± 0.20 | 4.0 ± 0.0 | 34.7 ± 3.67 | 5.9 ± 0.4 | 2.2 ± 0.3 | 0.44 ± 0.06 | 142 ± 1 |
| F | 5.84 ± 2.52 | 4.0 ± 0.0 | 57.12 ± 29.45 | 6.7 ± 0.5 | 3.1 ± 0.9 | 0.60 ± 0.16 | 136 ± 1 | ||
| 60 | M | 9.14 ± 1.76 | 5.3 ± 1.2 | 87.75 ± 35.23 | 6.9 ± 1.0 | 2.2 ± 0.3 | 0.45 ± 0.08 | 142 ± 5 | |
| F | 10.79 ± 5.03 | 5.3 ± 1.2 | 198.70 ± 142.46 | 12.5 ± 1.7 | 4.5 ± 0.8 | 0.87 ± 0.16 | 135 ± 0 | ||
Rat toxicokinetics
Rats orally administered OXF exhibited dose related, although not fully dose proportional, exposure to OXF, as expressed in the Cmax and AUC (Tables 1 and 2 and Fig. 1). Exposures were higher in females than in males, findings paralleled by higher Vd and T½ values. Note, for example, that females administered a 100 mg/kg oral dose of OXF had Cmax and AUC values similar to the values in males receiving 200 mg/kg (Table 2 and Fig. 1).
Table 2. Toxicokinetic Parameters of Oxfendazole on Day 1 and Day 14.
| Day | Dose (mg/kg) | Sex | Cmax (ng/ml) | Tmax (hr) | AUClast (hr·ng/ml) | t1/2 (hr) |
|---|---|---|---|---|---|---|
| 1 | 5 | M | 653 ± 187 | 3 | 4529 ± 555 | 3.0 |
| F | 1197 ± 124 | 3 | 7738 ± 399 | 2.7 | ||
| Mean | 925 | 3 | 6134 | 2.9 | ||
| 25a | M | 3817 ± 175 | 3 | 35833 ± 3612 | 3.9 | |
| F | 5653 ± 776 | 3 | 61842 ± 4477 | 3.0 | ||
| Mean | 4735 | 3 | 48838 | 3.5 | ||
| 100 | M | 7620 ± 797 | 3 | 107448 ± 12412 | 5.9 | |
| F | 13067 ± 1525 | 6 | 159757 ± 13257 | 2.9 | ||
| Mean | 10344 | 4.5 | 133603 | 4.4 | ||
| 200 | M | 10033 ± 722 | 3 | 149249 ± 9745 | 9.6 | |
| F | 13020 ± 971 | 3 | 226117 ± 19107 | 12.9 | ||
| Mean | 11527 | 3 | 187683 | 11.3 | ||
| 14 | 5 | M | 792 ± 90 | 3 | 5173 ± 611 | 2.7 |
| F | 856 ±113 | 1 | 5109 ± 793 | 4.6 | ||
| Mean | 824 | 2 | 5141 | 3.7 | ||
| 25 | M | 3430 ± 267 | 3 | 34427 ± 5099 | 6.8 | |
| F | 6523 ± 721 | 3 | 71454 ± 3686 | 5.4 | ||
| Mean | 4977 | 3 | 52941 | 6.1 | ||
| 100 | M | 6767 ± 1003 | 3 | 106684 ± 7626 | 9.9 | |
| F | 11225 ± 2675 | 6 | 180364 ± 31729 | NRc | ||
| Mean | 8996 | 4.5 | 143524 | 9.9 | ||
| 200b | M | 12445 ± 2035 | 3 | 26370 ± 4295 | NDd | |
| F | 12500 ± 3203 | 1 | 26910 ± 5845 | ND | ||
| Mean | 12473 | 2 | 26640 |
Animals # 218-223 (6 of 9 males) received 22.35 mg/kg on Day 1. TK analysis used the nominal 25 mg/kg for dose level.
Males and females in the 200 mg/kg groups were terminated on Day 10 and Day 7, respectively, due to toxicity of oxfendazole.
NR = not reported. t1/2 was not reported as the goodness of fit parameter (r2) was < 0.8.
ND = no data. There were only two time points in which plasma was collected.
Figure 1.
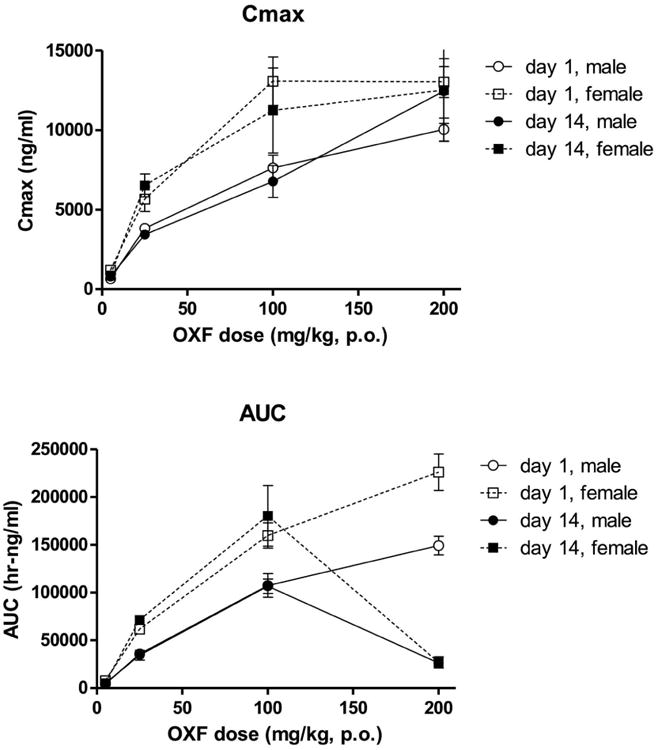
Exposure to oxfendaxole in rat toxicity study. Shown are the Cmax (upper panel) and AUC (lower panel) values obtained in the rat two week toxicity study. The symbols represent the calculated oxfendazole values (±SEM) determined in the plasma of rats administered the stated oxfendazole doses. Data were obtained from three rats per gender per dose. Note: males and females in the 200 mg/kg group were terminated on day 10 and day 7 respectively because of toxicity.
Rat functional observational battery (FOB)
No changes at any FOB endpoint (home-cage, handling, open field, reflexes and physiology components) were observed in male rats treated with OXF compared with rats administered vehicle. Female rats treated with OXF exhibited a statistically significant reduction in landing foot splay, but in the absence of a dose dependent relationship, similar observations in males, or significant findings for related endpoints, this finding was not considered to be toxicologically relevant. In contrast, treatment of rats with the positive control substances, amphetamine or carbaryl (data not shown), resulted in significant changes in behavior and physiology.
Rat two-week toxicology study
Following repeated oral administration of OXF (5, 25, 100 or 200 mg/kg/day), clinical changes were seen at only the two highest dose levels, 100 and 200 mg/kg. Observations included hunched posture; swelling (soft or hard) in the chin, muzzle, throat, and genitalia; paraphimosis; red discharge in the nose, mouth, and head; ruffled fur; hypoactivity; and yellow discolored stool and/or diarrhea. Mortality was seen in some females treated with 200 mg/kg OXF after 6-8 days of repeated dose administration. Of the 19 females on test, 6 were found dead between days 7-9, and 6 were sacrificed in moribund condition on Day 7. This high dose group was terminated before the scheduled sacrifice, and the recovery group was allowed to continue, without treatment, after 7 days of dosing. In general, the OXF related treatment mortality and clinical findings were more severe in females than in males at the same dose levels.
A significant decrease in body weight gain in the high dose group (compared with the control group) was observed in the first week of treatment and in the recovery phase after the last dose administration. On Day 8, the high dose group was 5 and 10% lower than control body weights in males and females, respectively. Mean weight gain in the high dose group during the Day 1-8 interval was 28 g vs 44.3 g in controls in males, and 0 g vs. 16.5 g in females. While sporadic changes in food consumption, both increases and decreases, were observed throughout the study, there was no clear association between food consumption and body weight changes in any of the treatment groups.
A dose-dependent decrease in white blood cell count (WBC) was seen at day 15 in females, with corresponding decreases in white blood cell subtypes (lymphocytes, neutrophils, monocytes, eosinophils, and basophils). The effect on WBC was severe in females, with decreases relative to controls of 8%, 13%, 51% and 75% at 5, 25, 100 (on Day 15), and 200 mg/kg (on Day 7), respectively. Levels returned to normal historical ranges by the end of the recovery period (Day 22 or 28). Effects seen in males were much less severe, with decreases in WBC seen in only the 200 mg/kg group (36% decrease in Day 8 males vs Day 15 control male level). WBC recovered in surviving animals once OXF administration was suspended. Minor effects on red blood cell count (RBC) were seen, but these were less significant than the WBC effects. None of the clinical chemistry changes were considered toxicologically relevant.
The primary target organs of toxicity identified microscopically were bone marrow, epididymis, liver, spleen, testis, and thymus. Bone marrow cell line (myeloid megakaryocytes and neutrophils) depletion was present on Day 8 in male rats given 200 mg/kg OXF and on Days 15 and 8 in female rats administered 100 and 200 mg/kg, respectively. This finding is consistent with the pronounced effects on WBC seen in these animals. The presence of epididymal multinucleated sperm precursors in rats given 25, 100, and 200 mg/kg OXF was noted on study Day 15 (25 and 100 mg/kg) and Day 8 (200 mg/kg), respectively. This finding was still present in the recovery groups in male rats treated with OXF at 100 mg/kg (Day 28) and 200 mg/kg (Day 22). Bilateral tubular atrophy was present on Day 8 in the testes of 2 rats given 200 mg/kg OXF, and it was present unilaterally on Day 28 in the testes of one rat given 5 and one given 100 mg/kg OXF. These effects, consistent with the decreased testis weight noted at necropsy, were not seen on Day 22 in the recovery animals from the 200 mg/kg/day treatment group, indicating that these effects are reversible following cessation of treatment.
Hepatic midzonal fatty change was present in male and female rats given 25 and 100 mg/kg OXF (Day 15) and those given 200 mg/kg OXF (Day 8). After a 14 day recovery, for females in the 25, 100, and 200 mg/kg groups and males in the 100 and 200 mg/kg groups, the changes were still present. This effect is consistent with mottled, pale, dark, or discolored livers seen at necropsy. However, no effects on liver enzymes were seen in the clinical chemistry results, suggesting that the fatty change did not result in overt hepatotoxicity or compromised hepatic function; therefore, the toxicologic significance of this observation is unclear. Splenic and thymic lymphoid depletion was present on Day 8 in male and female rats given 200 mg/kg OXF and on Day 15 in female rats given 100 mg/kg OXF. On recovery Day 28, splenic lymphoid depletion was still present in one female animal in the 100 mg/kg group, and thymic lymphoid depletion was still seen in 3 females in the 100 mg/kg and 1 female in the 200 mg/kg groups.
Thus, repeat daily oral administration of the highest dose of OXF, 200 mg/kg/day, resulted in significant toxicity and mortality in Sprague-Dawley rats. The main study animals in this group were euthanized after 7 consecutive daily oral dose administrations. Hematology, necropsy, organ weight, and histopathological results identified bone marrow, epididymis, liver, spleen, testis, and thymus as potential target organs of toxicity. On the basis of the toxicology endpoints evaluated in this study, the maximum tolerated dose (MTD) of OXF is approximately 100 mg/kg/day for a 14-day oral dose administration in rats. The NOAEL is considered to be greater than 5 mg/kg/day but less than 25 mg/kg/day for a 14-day oral dose administration in rats.
Dog CV safety and tolerability study
Following the administration of single oral OXF doses of 5, 50 and 100 mg/kg to three conscious dogs of each sex, no changes were observed in the cardiovascular parameters measured (blood pressure, heart rate or electrocardiogram). Neither were there any measured changes in clinical observations, body weight, postdose body temperature or clinical pathology (hematology, serum chemistry, and coagulation). Thus, based on the parameters evaluated in this study, the maximum tolerated dose was not reached, but appears to be greater than 100 mg/kg for administration of a single oral dose of OXF to dog. The no observed adverse effect level (NOAEL) is at least 100 mg/kg for a single oral dose administration of OXF in beagle dogs.
Genetic toxicology evaluation
Ames test
No cytotoxicity was seen at any dose level in the range-finding experiment conducted with strain TA100. In the follow up experiments, precipitate was seen on the plates at OXF doses of 2000 μg/plate, but no statistically significant increase in the number of revertant colonies was seen under any test condition. Control values were consistent with historical values obtained in this test and laboratory. OXF was judged to be nonmutagenic under the test conditions used in this study; therefore, OXF was determined to be negative in the bacterial reverse mutation assay.
Mouse lymphoma gene mutation assay
In the gene mutation assay using L5178Y mouse lymphoma cells, cytotoxicity (< 20% relative suspension growth) was observed following exposure to OXF levels ≥ 200 μg/ml for 4 hr in the presence of MA. In an initial mutagenicity experiment conducted at dose levels of 20, 40, 80, 160, and 320 μg/ml in the presence of MA for 4 hr, clones were obtained at all but the highest dose; inadequate cell growth precluded cloning at this dose. Neither statistically significant increases in mutation frequency (MF) compared with controls nor statistically significant dose-related increases in MF were found in the clones obtained from the treated cultures. In the follow up experiment at OXF dose levels of 10, 20, 40, 80, and 160 μg/ml for 4 hr in the presence of MA, data from the highest dose level was not included in the statistical analysis since its relative total growth (RTG) was less than 10%. Neither statistically significant increases in MF compared with controls nor statistically significant dose-related increases in MF were observed in the treated cultures.
In the absence of MA, cytotoxicity was observed at 400 μg/ml for 4 hr exposure and at ≥ 25 μg/ml for 24 hr exposure. In the initial mutagenicity experiment conducted at dose levels of 10, 20, 40, 80, 160 and 320 μg/ml in the absence of MA for 4 hr exposure, clones were obtained from cells treated with all doses. Because inadequate RTG was observed at 320 μg/ml, these data were not included in the statistical analysis. Neither statistically significant increases in MF compared with controls nor statistically significant dose-related increases in MF were observed in the treated cultures. In the follow up experiment, conducted at dose levels of 1.25, 2.5, 5, 10, and 20 μg/ml for 24 hr, clones were obtained from cells treated at all dose levels. Because inadequate RTG was seen at the 10 and 20 μg/ml dose levels, these results were not included in the statistical analysis. Neither statistically significant increases in MF compared with controls nor statistically significant dose-related increases in MF were found in the treated cultures. Under the test conditions, OXF met the criteria for a negative response as defined for this study.
Micronucleus evaluation
No significant suppression of polychromatic erythrocytes (PCE) occurred among red blood cells, and no statistically significant increase in the frequency of micronucleated PCE were seen in rats treated with OXF for two weeks. Thus, OXF was considered negative for the induction of micronuclei under the test conditions used in this mammalian bone marrow micronucleus assay.
Discussion
Pharmaco- and toxicokinetics
The metabolic half-life observed here for OXF in rat is shorter than that found in any other mono-gastric [dog,6 pig,10 horse25] or multi-gastric [cattle,25 sheep25] species previously reported. In fact, the 2 hr T 1/2 observed for rat is one-tenth of that observed in most other species studied; of the other species studied, only dog exhibited a half-life less than 20 hr, namely 5.5 hr.
In the present study, exposure to OXF in female rats exceeded that measured in males. The gender difference increased with OXF dosage through the 100 mg/kg level and was reflected in higher Cmax (ng/ml) and AUC (hr-ng/ml) values in females than in males.
Although measurement of OXF metabolites was not part of the present study, a previously conducted rat PK study25 found that OXF sulfone was the predominant plasma species across the 24 hr following OXF administration, accounting for 71% of the OXF related moieties quantified. In contrast, in most other species studied, including cattle,25 sheep,25 dog6 and pig,10 OXF itself was the predominant 0-24 hr plasma analyate. In horse, as in rat, an OXF metabolite predominated, but in horse that metabolite was fenbendazole25 rather than oxfendazole sulfone as found in rat.25
In contrast, following oral administration of the anthelminthic BZ ALB, the parent compound is barely detectable in the plasma. Rather, the analyates present are ALB sufoxide and a secondary metabolite, ALB sulfone,26 the former considered generally responsible for the drug's therapeutic and toxic effects. Another BZ anthelminthic, MEB, is itself considered the active moiety; its metabolism proceeds via inactivation pathways, reduction and conjugation.26
Toxicology
Oxfendazole
In the present rat studies, significant decreases in body weight at the higher OXF doses were accompanied by lowered WBC levels and a corresponding decrease in white blood cell types, including neutrophils. The observed target organs of toxicity, bone marrow, epididymis, liver, spleen, testis and thymus, corroborate those stated in the Review Summary of the NADA approval.13 In two-week toxicology studies in rat, OXF reduced neutrophil levels at the higher dose levels, 33 mg/kg in females and 100 mg/kg in both females and males. In those NADA supporting studies, doses of 33 and 100 mg/kg were also associated with reduced activity in lymphoid, testicular and bone marrow tissues. Importantly, in both the NADA supporting study13 and in the present investigation, WBC levels returned to normal during the study recovery phase.
The gender difference in toxicity observed in the present study is probably due to differences in OXF exposure, being higher in female than in male rats administered the same oral doses. It is not known whether female rats were also exposed to higher levels of the metabolite OXF sulfone or whether female rats biotransformed OXF more slowly to its sulfone, leaving higher levels of the parent compound. Indeed, in the present PK study, the T½ for OXF is longer in female than in male rats, especially at the higher oral dosage. Females also had a higher OXF volume of distribution following the drug's oral administration. In any case, the greater toxicity in female rats is probably due to greater exposure to OXF, to an OXF metabolite or both.
The target organs of toxicity in dog13 appear to be similar to those observed here and previously in rat: in two-week toxicology studies in dog,13 some animals in all OXF treated groups (11, 33 or 100 mg/kg) exhibited reduced myeloid maturation in bone marrow, and most OXF treated dogs exhibited splenic tissue and thymic atrophy. These supportive findings are particularly interesting given the difference in the predominant OXF-related analyate in the two species, OXF in dog and OXF sulfone in rat, suggesting that both OXF and OXF sulfone have similar toxicologic profiles.
Because the toxicities observed in in the present rat studies may have been induced by moieties other than OXF itself, the predictive value of the present toxicology findings to those that will be observed in human is presently unknown, as the metabolic profile of OXF in humans has not yet been determined. However, rat and dog are the two most widely used animal species for assessment of human risk of small molecules. Differences in metabolism and pharmacokinetics aside, these species are reasonable surrogates for the estimation of potential human risk, and data from these species will ultimately be used to set conservative, safe starting doses for initial human clinical trials.
Other benzimidazoles
Only limited data from mouse, rat and dog toxicology studies on other BZs exist in the public domain; summaries of these data are available in published reviews from the regulatory agencies, many of which have been complied by Dayan.26 From these data, the toxicology profiles of ALB and MEB appear similar to that of OXF. For example, repeated administration of high doses of ALB to rats led to leukopenia, enlargement of the liver and testicular hyperplasia.26 Similarly, administration of MEB in the diet to rats for 13 weeks led to an inhibition of body weight gain, anemia, hepatocyte vacuolation, liver weight gain and testicular damage.26 Most probably, the testicular effects of ALB derive from its disruption of microtubules,27 a mechanism shared by other BZs, including OXF and MEB.
Despite limited publically available non-clinical metabolism and toxicology data, the BZ ALB and MEB are widely used as anthelminthic agents in both veterinary practice and human treatment.26,28 Undoubtedly, ALB's safety profile and ease of administration has contributed to its use in the treatment of whole communities.28 The similar preclinical toxicologic profile of OXF to those of ALB and MEB hold promise for a suitable clinical safety profile for OXF.
Genetic toxicology
OXF was not mutagenic in a five tester strain Ames assay, with or without metabolic activation, a finding similar to that obtained for ALB.26 Further, OXF did not induce mutations in a mouse lymphoma cell line, with or without metabolic activation. OXF was also negative for the induction of micronuclei in a mammalian (rat) micronucleus assay, whereas ALB gave a positive mouse lymphoma cell response.26 Indeed, ALB as well as its metabolite ALB sulfoxide induced nondisjunction in cultured human lymphocytes29 at doses lower than those at which micronuclei were observed. And importantly, the lymphocytes of patients with hepatic hydatid disease showed increased sister chromatid exchange and micronuclei following ALB treatment.30 Thus, with a cleaner preclinical genetic toxicology profile, OXF may have a more favorable clinical profile in this regard.
While the current study demonstrated lack of genotoxicity of OXF in the three most widely used genotoxicity assays, previously published reports have presented somewhat conflicting results. El-Makawy et al.31 reported OXF to be both genotoxic and teratogenic in mice, whereas Holden et al.32 reported no chromosomal effects of OXF in human lymphocytes. Based on the current data, conducted in full compliance with current FDA testing requirements, genotoxicity does not appear to be a significant risk factor for OXF, giving it a more favorable profile than other BZs.
Toward FIH
Earlier work on OXF includes chronic toxicity studies in rat and dog.13 In those one year studies, rat was more sensitive to the adverse effects of OXF than was dog, the no effect dose for rat being 0.7 mg/kg/d and for dog being 13.5 mg/kg/d. Extrapolating (using a body surface area calculation) from the results of those chronic oxfendazole toxicology studies, the most conservative estimate of a no-effect chronic dose for humans (based on a NOEL derived from a 1 year study) is 0.13 mg/kg. A conservative dose of 0.3 mg/kg has been proposed as the starting point for a single dose Phase I OXF study. Furthermore, since the principal toxicology target organs appear to be the bone marrow and liver, safety monitoring for the proposed Phase I study includes clinical chemistry indicators of bone marrow and liver function. Coupled with OXF's lack of induction of behavioral effects in rat, cardiovascular effects in dog or genetic toxicology signals in standard Ames bacterial, mouse lymphoma or rat micronucleus assays, the results of the present rat pharmacokinetic, toxicokinetic, toxicology and safety studies support the investigation of OXF as a potential anthelminthic medication against human helminth species.
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN266200600011C / N01-AI-60011. H. H. G. is supported by a Wellcome Trust Senior International Research Fellowship in Public Health and Tropical Medicine.
References
- 1.Espadero B. Efficacy of current drugs against soil-transmitted helminth infections: systematic review and meta-analysis. Evidencias en Pediatria. 2009:53. doi: 10.1001/jama.299.16.1937. [DOI] [PubMed] [Google Scholar]
- 2.Geary TG, Woo K, McCarthy JS, Mackenzie CD, Horton J, Prichard RK, de Silva NR, Olliaro PL, Lazdins-Helds JK, Engels DA, Bundy DA. Unresolved issues in anthelmintic pharmacology for helminthiases of humans. International Journal for Parasitology. 2010;40:1–13. doi: 10.1016/j.ijpara.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 3.García HH, Gonzalez AE, Evans CA, Gilman RH. Taenia solium cysticercosis. The Lancet. 2003;362:547–56. doi: 10.1016/S0140-6736(03)14117-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan RM. Drug resistance in nematodes of veterinary importance: a status report. Trends in Parasitology. 2004;20:477–81. doi: 10.1016/j.pt.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Geary TG. Are new anthelmintics needed to eliminate human helminthiases? Current Opinion in Infectious Diseases. 2012;25:709–17. doi: 10.1097/QCO.0b013e328359f04a. [DOI] [PubMed] [Google Scholar]
- 6.Gokbulut C, Bilgili A, Hanedan B, McKellar Q. Comparative plasma disposition of fenbendazole, oxfendazole and albendazole in dogs. Veterinary Parasitology. 2009;148:279–87. doi: 10.1016/j.vetpar.2007.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Gonzales AE, Garcia HH, Gilman RH, Gavidia CM, Tsang VC, Bernal T, Falcon N, Romero M, Lopez-Urbina MT. Effective, single-dose treatment or porcine cysticercosis with oxfendazole. American Journal of Tropical Medicine and Hygiene. 1996;54:391–4. doi: 10.4269/ajtmh.1996.54.391. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez AE, Falcon N, Gavidia C, Garcia HH, Tsang VC, Bernal T, Romero M, Gilman RH. Treatment of porcine cysticercosis with oxfendazole: a dose-response trial. The Veterinary Record. 1997;141:420–2. doi: 10.1136/vr.141.16.420. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez AE, Falcon N, Gavidia C, Garcia HH, Tsang VC, Bernal T, Romero M, Gilman RH. Time-response curve of oxfendazole in the treatment of swine cysticercosis. American Journal of Tropical Medicine and Hygiene. 1998;59:832–6. doi: 10.4269/ajtmh.1998.59.832. [DOI] [PubMed] [Google Scholar]
- 10.Moreno L, Lopez-Urbina M, Farias C, Domingue G, Donadeu M, Dungu B, García H, Gomez-Puerta L, Lanusse C, González A. A high oxfendazole dose to control porcine cysticercosis: pharmacokinetics and tissue residue profiles. Food and Chemical Toxicology. 2012;50:3819–25. doi: 10.1016/j.fct.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Oxfendazole, Fenbendazole, Febantel, Summary Report: Committee for Veterinary Medicinal Products. EMEA. 1997 [Google Scholar]
- 12.Oxfendazole Summary Report (3): Committee for Veterinary Medicinal Products. EMEA. 1997 [Google Scholar]
- 13. [Accessed November 7, 2011];Freedom of Information Summary, NADA 140-854 Synanthic-original approval. 1990 at http://www.fda.gov/AnimalVeterinary/Products/ApprovedAnimalDrugProducts/FOIADrugSummaries/ucm049983.html.
- 14.Albenza (albendazle) 2009 at http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/020666s005s006lbl.pdf.
- 15.CDER, editor. FDA. Safety Pharmacology Studies for Human Pharmaceuticals – Central Nervous System. 2000. [Google Scholar]
- 16.Mortelmans K, Riccio ES. The bacterial tryptophan reverse mutation assay with Escherichia coli WP2. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2000;455:61–9. doi: 10.1016/s0027-5107(00)00076-2. [DOI] [PubMed] [Google Scholar]
- 17.Mortelmans K, Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2000;455:29–60. doi: 10.1016/s0027-5107(00)00064-6. [DOI] [PubMed] [Google Scholar]
- 18.Maron DM, Ames BN. Revised methods for the Salmonella mutagenicity test. Mutation Research/Environmental Mutagenesis and Related Subjects. 1983;113:173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- 19.Clive D, Johnson K, Spector J, Batson A, Brown M. Validation and characterization of the L5178Y/T+/- mouse lymphoma mutagen assay system. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 1979;59:61–108. doi: 10.1016/0027-5107(79)90195-7. [DOI] [PubMed] [Google Scholar]
- 20.Moore MM, Honma M, Clements J, Harrington-Brock K, Awogi T, Bolcsfoldi G, Cifone M, Collard D, Fellows M, Flanders K. Mouse lymphoma thymidine kinase gene mutation assay: Follow-up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, April 2000. Environmental and Molecular Mutagenesis. 2002;40:292–9. doi: 10.1002/em.10122. [DOI] [PubMed] [Google Scholar]
- 21.Doppalapudi RS, Riccio ES, Rausch LL, Shimon JA, Lee PS, Mortelmans KE, Kapetanovic IM, Crowell JA, Mirsalis JC. Evaluation of chemopreventive agents for genotoxic activity. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2007;629:148–60. doi: 10.1016/j.mrgentox.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi M, Sofuni T, Ishidate M., Jr An application of acridine orange fluorescent staining to the micronucleus test. Mutation Research Letters. 1983;120:241–7. doi: 10.1016/0165-7992(83)90096-9. [DOI] [PubMed] [Google Scholar]
- 23.MacGregor JT, Heddle JA, Hite M, Margolin BH, Ramel C, Salamone MF, Tice RR, Wild D. Guidelines for the conduct of micronucleus assays in mammalian bone marrow erythrocytes. Mutation Research/Genetic Toxicology. 1987;189:103–12. doi: 10.1016/0165-1218(87)90016-4. [DOI] [PubMed] [Google Scholar]
- 24.Schmid W. The micronucleus test for cytogenetic analysis. In: Hollander A, editor. Chemical Mutagens. New York: Plenum Press; 1976. pp. 31–53. [Google Scholar]
- 25. [Accessed September 3, 2014];41-4-Oxfendazole. 2003 at ftp://ftp.fao.org/ag/agn/jecfa/vetdrug/41-4-oxfendazole.pdf)
- 26.Dayan A. Albendazole, mebendazole and praziquantel. Review of non-clinical toxicity and pharmacokinetics. Acta Tropica. 2003;86:141–59. doi: 10.1016/s0001-706x(03)00031-7. [DOI] [PubMed] [Google Scholar]
- 27.Okamura M, Watanabe T, Kashida Y, Machida N, Mitsumori K. Possible mechanisms underlying the testicular toxicity of oxfendazole in rats. Toxicologic Pathology. 2004;32:1–8. doi: 10.1080/01926230490260655. [DOI] [PubMed] [Google Scholar]
- 28.Horton J. Albendazole: a review of anthelmintic efficacy and safety in humans. Parasitology. 2000;121:S113–S32. doi: 10.1017/s0031182000007290. [DOI] [PubMed] [Google Scholar]
- 29.Ramírez T, Eastmond DA, Herrera LA. Non-disjunction events induced by albendazole in human cells. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2007;626:191–5. doi: 10.1016/j.mrgentox.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Oztas S, Salman AB, Tatar A, Yigiter M, Yazgi H, Ertek M, Yesilyurt A, Ocak Z, Kursad H. Genotoxic effect of albendazole in pediatric patients with hepatic hydatid disease. International Journal of Infectious Diseases. 2007;11:446–9. doi: 10.1016/j.ijid.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 31.El-Makawy A, Radwan HA, Ghaly IS, El-Raouf AA. Genotoxical, teratological and biochemical effects of anthelmintic drug oxfendazole Maximum Residue Limit (MRL) in male and female mice. Reproduction Nutrition Development. 2006;46:139–56. doi: 10.1051/rnd:2006007. [DOI] [PubMed] [Google Scholar]
- 32.Holden HE, Crider PA, Wahrenburg MG. Mitotic arrest by benzimidazole analogs in human lymphocyte cultures. Environmental Mutagenesis. 1980;2:67–73. doi: 10.1002/em.2860020110. [DOI] [PubMed] [Google Scholar]