

ABSTRACT
Prioritizing missense variants for further experimental investigation is a key challenge in current sequencing studies for exploring complex and Mendelian diseases. A large number of in silico tools have been employed for the task of pathogenicity prediction, including PolyPhen‐2, SIFT, FatHMM, MutationTaster‐2, MutationAssessor, Combined Annotation Dependent Depletion, LRT, phyloP, and GERP++, as well as optimized methods of combining tool scores, such as Condel and Logit. Due to the wealth of these methods, an important practical question to answer is which of these tools generalize best, that is, correctly predict the pathogenic character of new variants. We here demonstrate in a study of 10 tools on five datasets that such a comparative evaluation of these tools is hindered by two types of circularity: they arise due to (1) the same variants or (2) different variants from the same protein occurring both in the datasets used for training and for evaluation of these tools, which may lead to overly optimistic results. We show that comparative evaluations of predictors that do not address these types of circularity may erroneously conclude that circularity confounded tools are most accurate among all tools, and may even outperform optimized combinations of tools.
Keywords: pathogenicity prediction tools, exome sequencing
Introduction
Current high‐throughput techniques to investigate the genetic basis of inherited diseases yield large numbers of potentially pathogenic sequence alterations [Tennessen et al., 2012; Purcell et al., 2014]. Conducting further in‐depth functional analyses on these large numbers of candidates is generally impractical. Reliable strategies that allow investigators to decide which of these variants to prioritize are therefore imperative. Researchers will often focus on nonsynonymous single‐nucleotide variants (nsSNVs), which are disproportionately deleterious compared with synonymous variants [Hindorff et al., 2009; Kiezun et al., 2012; MacArthur et al., 2012], and filter out common variants, which are presumed to be more likely to be neutral. However, in many cases, tens of thousands of candidates still remain after this step. Computational tools that can be used to identify those missense variants most likely to have a pathogenic effect, that is, most likely to contribute to a disease, are therefore of high‐practical value.
A number of such tools are already available, such as MutationTaster‐2 (MT2) [Schwarz et al., 2014], LRT [Chun and Fay, 2009], PolyPhen‐2 (PP2) [Adzhubei et al., 2010], SIFT [Ng and Henikoff, 2003], MutationAssessor (MASS) [Reva et al., 2011], FatHMM weighted (FatHMM‐W) and unweighted (FatHMM‐U) [Shihab et al., 2013], Combined Annotation Dependent Depletion (CADD) [Kircher et al., 2014], phyloP [Cooper and Shendure, 2011], and GERP++ [Davydov et al., 2010]. They are widely used for separating pathogenic variants from neutral variants in sequencing studies [Leongamornlert et al., 2012; Rudin et al., 2012; Kim et al., 2013; Thevenon et al., 2014; Weinreb et al., 2014; Zhao et al., 2015]. While these tools are all commonly applied to the problem of pathogenicity prediction, the original purposes they were designed for varies (see Table 1). Some measure sequence conservation (phyloP [Cooper and Shendure, 2011], GERP++ [Davydov et al., 2010], and SIFT [Ng and Henikoff, 2003]), others try to assess the impact of variants on protein structure or function (e.g., PP2 [Adzhubei et al., 2010]) or to quantify the overall pathogenic potential of a variant based on diverse types of genomic information (e.g., CADD [Kircher et al. 2014]). Note that SIFT is both a measure of sequence conservation and provides an analytically derived threshold for predicting whether or not protein function will be affected [Ng and Henikoff, 2003]. Furthermore, popular benchmark datasets for pathogenicity prediction differ in the way they define the pathogenic or neutral character of a variant (see Table 2). For instance, neutral variants are supposed to have a minor allele frequency larger than 1% in HumVar [Adzhubei et al., 2010], of less than 1% in ExoVar [Li et al., 2013], and of more than 40% in VariBench [Thusberg et al., 2011; Nair and Vihinen, 2013].
Table 1.
Overview of the Prediction Tools Used in This Study
| Tool (abbreviation) | Version | N | AA | Purpose, as stated by developers |
|---|---|---|---|---|
| PolyPhen‐2 (PP2) | 2.2.2 | Yes | Yes | “Predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations”a |
| MutationTaster‐2 (MT2) | 2 | Yes | No | “Evaluation of the disease‐causing potential of DNA sequence alterations”b |
| MutationAssessor (MASS) | 2 | Yes | Yes | “Predicts the functional impact of amino acid substitutions in proteins, such as mutations discovered in cancer or missense polymorphisms”c |
| LRT | ‐ | Yes | No | “Identify a subset of deleterious mutations that disrupt highly conserved amino acids within protein‐coding sequences, which are likely to be unconditionally deleterious”d |
| SIFT | 1.03 | Yes | Yes | “Predicts whether an amino acid substitution affects protein function”e |
| GERP++ | – | Yes | No | “Identifies constrained elements in multiple alignments by quantifying substitution deficits. These deficits represent substitutions that would have occurred if the element were neutral DNA, but did not occur because the element has been under functional constraint. We refer to these deficits as “rejected substitutions.” Rejected substitutions are a natural measure of constraint that reflects the strength of past purifying selection on the element”f |
| phyloP | – | Yes | No | “Compute conservation or acceleration P values based on an alignment and a model of neutral evolution”g |
| FatHMM unweighted (FatHMM‐U) | 2.2–2.3 | No | Yes | Predicts “functional consequences of both coding variants, that is, nonsynonymous single‐nucleotide variants, and noncoding variants”h |
| FatHMM weighted (FatHMM‐W) | 2.2–2.3 | No | Yes | Predicts “functional consequences of both coding variants, that is, nonsynonymous single‐nucleotide variants, and noncoding variants” and its weighting scheme attributes higher tolerance scores to SNVs in proteins, related proteins, or domains that already include a high fraction of pathogenic variantsh |
| Combined Annotation Dependent Depletion (CADD) | 1.0 | Yes | No | “CADD is a tool for scoring the deleteriousness of single‐nucleotide variants as well as insertion/deletions variants in the human genome”i |
For each tool, the first column shows the version of the tool, the second column (N) shows whether it accepts nucleotide changes as input, the third column (AA) shows whether it accepts amino acid changes as input. The last column provides a description of the tool, as stated by the developers.
Table 2.
Purpose of Each Dataset, as Described by Dataset Creators
| Dataset | Purpose | Positive control: damaging/deleterious/disease causing/pathogenic | Negative control: neutral/benign/nondamaging/tolerated |
|---|---|---|---|
| HumVar | Mendelian disease variant identification | “All disease‐causing mutations from UniProtKB”a | “Common human nsSNPs (MAF > 1%) without annotated involvement in disease…treated as nondamaging”a |
| ExoVar | “Dataset composed of pathogenic nsSNVs and nearly nonpathogenic rare nsSNVs”b | “5,340 alleles with known effects on the molecular function causing human Mendelian diseases from the UniProt database…positive control variants.” “Pathogenic nsSNVs”b | “4,752 rare (alternative/derived allele frequency <1%) nsSNVs with at least one homozygous genotype for the alternative/derived allele in the 1000 Genomes Project…negative control variants.” “Other rare variants”b |
| VariBench | “Variation datasets affecting protein tolerance”c | “The pathogenic dataset of 19,335 missense mutations obtained from the PhenCode database downloaded in June 2009), IDbases and from 18 individual LSDBs. For this dataset, the variations along with the variant position mappings to RefSeq protein (> = 99% match), RefSeq mRNA, and RefSeq genomic sequences are available for download.”c | “This is the neutral dataset or nonsynonymous coding SNP dataset comprising 21,170 human nonsynonymous coding SNPs with allele frequency 40.01 and chromosome sample count 449 from the dbSNP database build 131. This dataset was filtered for the disease‐associated SNPs. The variant position mapping for this dataset was extracted from dbSNP database.”c |
| predictSNP | “Benchmark dataset used for the evaluation of…prediction tools and training of consensus classifier PredictSNP”d | Disease‐causing and deleterious variants from SwissProt, HGMD, HumVar, Humsavar, dbSNP, PhenCode, IDbases, and 16 individual locus‐specific databases. | Neutral variants from SwissProt, HGMD, HumVar, Humsavar, dbSNP, PhenCode, IDbases, and 16 individual locus‐specific databases. |
| SwissVar | “Comprehensive collection of single amino acid polymorphisms (SAPs) and diseases in the UniProtKB/Swiss‐Prot knowledgebase”e | “A variant is classified as disease when it is found in patients and disease association is reported in literature. However, this classification is not a definitive assessment of pathogenicity”f | “A variant is classified as polymorphism if no disease association has been reported”f |
For each dataset, the first column shows the general purpose. The last two columns describe the positive and negative control categories of variants.
Given this wealth of different methods and benchmarks that can be used for pathogenicity prediction, an important practical question to answer is whether one or several tools systematically outperform all others in prediction accuracy. To address this question, we comprehensively assess the performance of 10 tools that are widely used for pathogenicity prediction: MT2 [Schwarz et al., 2014], LRT [Chun and Fay, 2009], PP2 [Adzhubei et al., 2010], SIFT [Ng and Henikoff, 2003], MASS [Reva et al., 2011], FatHMM‐W and FatHMM‐U [Shihab et al., 2013], CADD [Kircher et al., 2014], phyloP [Cooper and Shendure, 2011], and GERP++ [Davydov et al., 2010]. We evaluate performance across major public databases previously used to test these tools [Adzhubei et al., 2010; Mottaz et al., 2010; Thusberg et al., 2011; Li et al., 2013; Nair and Vihinen, 2013; Bendl et al., 2014] and show that two types of circularity severely affect the interpretation of the results. Here, we use the term “circularity” to describe the phenomenon that predictors are evaluated on variants or proteins that were used to train their prediction models. While a number of authors have acknowledged the existence of one particular form of circularity before (stemming specifically from overlap between data used to develop the tools and data upon which those tools are tested) [Adzhubei et al., 2010; Thusberg et al., 2011; Nair and Vihinen, 2013; Vihinen, 2013], our study is the first to provide a clear picture of the extent and impact of this phenomenon in pathogenicity prediction.
The first type of circularity we encounter is due to overlaps between datasets that were used for training and evaluation of the models. Tools such as MT2 [Schwarz et al., 2014], PP2 [Adzhubei et al., 2010], MASS [Reva et al., 2011], and CADD [Kircher et al., 2014], which require a training dataset to determine the parameters of the model, run the risk of capturing idiosyncratic characteristics of their training set, leading to poor generalization when applied on new data. To prevent the phenomenon of overfitting [Hastie et al., 2009], it is imperative that tools be evaluated on variants that were not used for the training of these tools [Vihinen, 2013]. This is particularly true when evaluating combinations of tool scores, as different tools have been trained on different datasets, increasing the likelihood that variants in the evaluation set appear in at least one of these datasets [González‐Pérez and López‐Bigas, 2011; Capriotti et al., 2013; Li et al., 2013; Bendl et al., 2014]. Notably, this type of circularity, which we refer to as type 1 circularity, could cause spurious increases in prediction accuracy for both single tools and combinations of tool scores.
The second type of circularity, which we refer to as type 2 circularity, is closely linked to a statistical property of current variant databases: often, all variants from the same gene are jointly labeled as being pathogenic or neutral. As a consequence, a classifier that predicts pathogenicity based on known information about specific variants in the same gene will achieve excellent results, while being unable to detect novel risk genes, for which no variants have been annotated before. Further, it will not be able to perform another critical function: discrimination of pathogenic variants from neutral ones within a given protein.
Furthermore, we evaluate the performance of two tools that combine scores across methods, Condel [González‐Pérez and López‐Bigas, 2011] and Logit [Li et al., 2013], and examine whether these tools are affected by circularity as well. These tools are based on the expectation that individual predictors have complementary strengths, because they rely on diverse types of information, such as sequence conservation or modifications at the protein level. Combining them hence has the potential to boost their discriminative power, as reported in a number of studies [González‐Pérez and López‐Bigas, 2011; Capriotti et al., 2013; Li et al., 2013; Bendl et al., 2014]. The problem of circularity, however, could be exacerbated when combining several tools. First, consider the case where the data that are used to learn the weights assigned to each individual predictor in the combination also overlaps with the training data of one or more of the tools. Here, tools that have been fitted to the data already will appear to perform better and may receive artificially inflated weights. Second, consider the case where the data used to assess the combination of tools overlaps with the data on which the tools have been trained. Here, the tools themselves are biased toward performing well on the evaluation data, which can make their combination appear to perform better than it actually does.
Materials and Methods
Datasets and Data Preprocessing
In this study, we used five different datasets to assess the performance of available prediction tools and their combinations. We used publicly available and commonly used benchmark datasets: HumVar [Adzhubei et al., 2010], ExoVar [Li et al., 2013], VariBench [Thusberg et al., 2011; Nair and Vihinen, 2013], predictSNP [Bendl et al., 2014], and the latest SwissVar (December 2014) database [Mottaz et al. 2010] (Table 3). As these tools can require either nucleotide or amino acid substitutions as input, we used Variant Effect Predictor (VEP) [McLaren et al., 2010] to convert between both formats. We excluded all variants for which we could not determine an unambiguous nucleotide or amino acid change. Note that by contrast, analyses such as that of Thusberg et al. (2011) only assess tools that require amino acid changes as input. As the intersection of the training data from the tool CADD [Kircher et al., 2014] and that of all other datasets is small (fewer than a hundred variants), we systematically excluded all variants overlapping with the CADD training data from all other data sets. The VariBench dataset (benchmark database for variations) was created [Thusberg et al., 2011; Nair and Vihinenm 2013] to address the problem of type 1 circularity. However, while the pathogenic variants of this dataset were new, its neutral variants may have been present in the training data of other tools. VariBench has an overlap of approximately 50% with both HumVar and ExoVar (Supp. Fig. S1). We kept the nonoverlapping variants to build an independent evaluation dataset, which we called VariBenchSelected and make available along with this manuscript (Supp. Data S1). From the predictSNP benchmark dataset, we systematically excluded all variants that overlap with HumVar, ExoVar, and VariBench and called the resulting dataset predictSNPSelected. Eventually, we created a fifth dataset, SwissVarSelected. Here, we excluded from the latest SwissVar database (December 2014) all variants overlapping with the other four datasets — HumVar, ExoVar, VariBench, and predictSNP. Thus, SwissVarSelected should be the dataset containing the newest variants across all datasets. With one possible exception, none of the prediction tools or conservation scores we investigated in this manuscript were trained on VariBenchSelected, predictSNPSelected, or SwissVarSelected. The exception is that some variants in the selected datasets may overlap partially with variants used to train MT2 [Schwarz et al., 2014] because MT2 was trained on private data (a large collection of disease variants from HGMD Professional [Stenson et al., 2014]).
Table 3.
All Datasets Used in This Study
| Datasets | Deleterious variants (D) | Neutral variants (N) | Total | Ratio (D:Total) | Tools potentially trained on data (fully or partly) | Removed variants overlapping with: |
|---|---|---|---|---|---|---|
| HumVar | 21,090 | 19,299 | 40,389 | 0.52 | MT2, MASS, PP2, FatHMM‐W | CADD training data |
| ExoVar | 5,156 | 3,694 | 8,850 | 0.58 | MT2, MASS, PP2, FatHMM‐W | CADD training data |
| VariBenchSelected | 4,309 | 5,957 | 10,266 | 0.42 | MT2 | CADD training data, HumVar, ExoVar |
| predictSNPSelected | 10,000 | 6,098 | 16,098 | 0.62 | MT2 | CADD training data, HumVar, ExoVar, VariBench |
| SwissVarSelected | 4,526 | 8,203 | 12,729 | 0.36 | MT2 | CADD training data, HumVar, ExoVar, VariBench, predictSNP |
These preprocessed and filtered datasets are used to evaluate the performance of different prediction tools.
Pathogenicity Prediction Score Sources and Conservation Scores
For any given variant, we obtained scores and prediction labels for each tool directly from their respective Web servers or standalone tools (Table 1). The pathogenicity score of a missense variant may depend on which transcript of the corresponding gene is considered. For this reason, we standardized our analyses by examining the same transcript across all tools; if available, we chose the canonical transcript [Hubbard et al., 2009]. In contrast, ANNOVAR [Wang et al., 2010] and dbNSFP 2.0 [Liu et al., 2013] use the transcript that yields the worst (e.g., most damaging) score, which means that different tools may select different transcripts for the same variant.
Data Availability and Reproducibility of Results
For each dataset, we compiled comma‐separated files containing all obtained tool scores and predicted labels as well as information about the variant (true label, nucleotide and amino acid changes, minor allele frequencies, UniProt accession IDs [Magrane and Consortium, 2011], Ensembl gene, transcript and protein IDs [Flicek et al. 2014], and dbSNP IDs (rs#) if available [Sherry et al., 2001]). All these datasets including the tool predictions can be found at the VariBench Website (http://structure.bmc.lu.se/VariBench/GrimmDatasets.php) as well as a single Excel file at the journals Website (Supp. Data S2).
Further, we provide all Python scripts used to generate all tables and figures in this study along with all datasets, compressed as a single ZIP file (Supp. Data S1). All data and Python scripts can also be downloaded from: http://www.bsse.ethz.ch/mlcb/research/bioinformatics‐and‐computational‐biology/pathogenicity‐predi‐ction.html.
Performance Evaluation
To evaluate the performance of all the tools in this study, we used a collection of statistics derived from a confusion matrix. To this end, we counted a correctly classified test point as a true positive (TP) if and only if the test point corresponds to the positive class (pathogenic or damaging) and as a true negative (TN) if and only if the test point corresponded to the negative class (neutral or benign). Accordingly, a false positive (FP) is a negative test point that is classified to be positive and a false negative (FN) a positive test point classified as a negative one. Since a few datasets are slightly unbalanced, we assessed the performance of the single tools by computing receiver operating characteristic curves (ROC‐curves). Furthermore, we computed Precision‐Recall curves (ROC‐PR‐curves) [Davis and Goadrich 2006]. The ROC‐curve is the fraction of the TP over all positives TP+FN (sensitivity or TP rate) against the fraction of the FP over all negatives TN+FP (1‐specificity or FP rate), whereas the ROC‐PR curve is the fraction of the TP over all positives TP+FN (recall or sensitivity) against the fraction of the TP over all TP+FP (precision). To measure the performance, we computed the area under the ROC and ROC‐PR curves (AUC and AUC‐PR, respectively). The area under the curve can take values between 0 and 1. A perfect classifier has an AUC and AUC‐PR of 1. The AUC of a random classifier is 0.5. Additionally, we assessed the performance with seven commonly used parameters as described in the Human Mutation guidelines [Vihinen, 2012 , 2013] and reported the results in the Supp. Information:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
Evaluation of the Weighting Scheme of FatHMM‐W
Given the superior performance of FatHMM‐W on VariBenchSelected and predictSNPSelected (see Results and Fig. 1), we examined this prediction tool in more detail. This section provides relevant details about FatHMM's weighted (FatHMM‐W) and unweighted (FatHMM‐U) versions and our evaluation of the weighting of FatHMM, which accounts for its superior performance. To evaluate the role of the weighting scheme of FatHMM‐W [Shihab et al., 2013] (Supp. Text S1), we compared the original FatHMM‐W method with an L1‐regularized logistic regression [Lee et al., 2006] over the log‐transformed features ln(Wn) and ln(Wd). These features are used by FatHMM to reweight the FatHMM‐U score and construct FatHMM‐W, the weighted version of FatHMM (Supp. Text S1). We performed a 10‐fold cross‐validation on the five datasets HumVar [Adzhubei et al., 2010], ExoVar [Li et al., 2013], VariBenchSelected [Thusberg et al., 2011; Nair and Vihinen, 2013], predictSNPSelected [Bendl et al., 2014], and SwissVarSelected [Mottaz et al., 2010] (see Table 3 and the Methods section). For this purpose, we randomly split the dataset of interest into 10 subsets of equal size (to the extent possible). Simultaneously, we kept the ratio of neutral to pathogenic variants the same across all subsets. This avoids generating a biased subset containing only variants of the same kind. We then combined nine subsets to train the model and used the remaining one to assess the performance (testing). We repeated this cross‐validation procedure 10 times. Because we were using a regularization term, we had to find a trade‐off between the matching model on the training set and the best generalization. For this purpose, we had to select a reasonable value C. This we did by performing an internal line‐search for each fold to find the C j that leads to the best AUC from a set of C values, C = (1e‐4, 1e‐3, 1e‐2, 1e‐1, 1, 1e1, 1e2). The overall performance of the model was the average across all 10 AUC values. We then computed AUC, AUC‐PR, and the seven commonly used parameters as described in the Human Mutation guidelines [Vihinen, 2012, 2013]. For our experiments, we used custom Python scripts (see Supp. Data S1) and scikit‐learn [Pedregosa et al., 2011], an efficient machine learning library for Python that includes an L1‐regularized logistic regression using the LIBLINEAR library [Fan et al., 2008].
Figure 1.
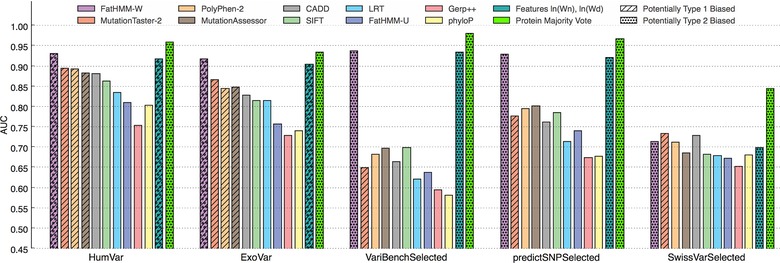
Evaluation of the 10 different pathogenicity prediction tools (by AUC) over five datasets. The hatched bars indicate potentially biased results, due to the overlap (or possible overlap) between the evaluation data and the data used (by tool developers) for training the prediction tool. The dotted bars indicate that the tool is biased due to type 2 circularity. The protein MV predictor and the logistic regression (over the features used in the weighting scheme of FatHMM‐W) are discussed in the second part of the Results section.
Protein Majority Vote
To analyze how protein‐related features can influence the performance of the prediction tools, we performed a protein majority vote (MV). For each of the five evaluation datasets, we split the benchmark into 10 subsets, and for each of the subsets, used the union of the nine other subsets as training data. Within that framework, we scored a variant by the pathogenic‐to‐neutral ratio, in the training data, of the protein that variant belongs to. If the protein did not appear in the training data, we assigned a score of 0.5. This strategy, while statistically effective on the currently existing databases, is not appropriate, as it cannot discriminate between neutral and pathogenic variants within the same protein.
Results
Evaluation of 10 Pathogenicity Prediction Tools on Five Variant Datasets
We evaluated the performance of eight different prediction tools: MT2 [Schwarz et al., 2014], LRT [Chun and Fay, 2009], PP2 [Adzhubei et al., 2010], SIFT [Ng and Henikoff, 2003], MASS [Reva et al., 2011], FatHMM‐W and FatHMM‐U [Shihab et al., 2013], and CADD [Kircher et al., 2014] as well as two conservation scores: phyloP [Cooper and Shendure, 2011] and GERP++ [Davydov et al., 2010]. An overview of these tools and conservation scores can be found in Table 1. Details on how these scores were obtained can be found in the Methods section.
We evaluated these tools using a range of preprocessed public datasets and subsets of VariBench, predictSNP, and SwissVar (see Methods), resulting in five evaluation datasets [Adzhubei et al., 2010; Mottaz et al., 2010; Thusberg et al., 2011; Li et al., 2013; Nair and Vihinen, 2013; Bendl et al., 2014] (see Methods; Table 3; Supp. Fig. S1). Importantly, two of these datasets (HumVar [Adzhubei et al., 2010] and ExoVar [Li et al., 2013]) overlap with at least one of the training sets used to train the individual tools FatHMM‐W, MT2, MASS, and PP2 (Supp. Fig. S1; Table 3). The selected datasets can be considered to be truly independent evaluation datasets, which are free of type 1 circularity (see Methods).
We report AUC values per tool and per dataset in Figure 1 and Supp. Table S1 (corresponding ROC, PR curves, AUC/AUC‐PR values as well as other evaluation metrics can be found in Supp. Figs. S2–S6 and Supp. Table S1). Hatched bars in Figure 1 indicate that the evaluation data were used in part or entirely to train the corresponding tool; these results may suffer from overfitting. Dotted bars indicate that the tools are biased, due to type 2 circularity (see section “Type 2 Circularity as an Explanation of the Good Performance of FatHMM Weighted”).
Five central observations can be made in Figure 1: first, on the two benchmarks HumVar and ExoVar, the four best performing methods were fully or partly trained on these datasets. Second, while MT2, PP2, and MASS outperform CADD and SIFT on benchmarks that include some of their training data (HumVar, ExoVar), this is not the case on the independent VariBenchSelected and predictSNPSelected datasets. A potential explanation is that type 1 circularity — that is overlap between training and evaluation sets — might lead to overly optimistic results on the first two datasets. Third, across the first four datasets, FatHMM‐W outperforms all other tools (Supp. Table S1; Fig. 1). All measured evaluation criteria support these findings on the VariBenchSelected and predictSNPSelected datasets (Supp. Table S1), even though FatHMM‐W has no type 1 bias on VariBenchSelected and predictSNPSelected. However, it is confounded by type 2 circularity. Fourth, FatHMM‐W shows a severe drop in performance on the SwissVarSelected dataset. Finally, we observed across all datasets that trained predictors generally outperform untrained conservation scores.
Type 2 Circularity as an Explanation of the Good Performance of FatHMM Weighted
The superiority of FatHMM‐W's [Shihab et al., 2013] predictions on VariBenchSelected and predictSNPSelected and the severe drop in performance on SwissVarSelected made us investigate its underlying model to find the reason for its superior performance on all but one dataset. FatHMM‐W's weighting scheme attributes higher scores to amino acid substitutions in proteins, related proteins, or domains that already include a high fraction of pathogenic variants (see Supp. Text S1). A key element of this weighting scheme is the use of the two parameters Wn and Wd, which represent the relative frequency of neutral variants (Wn) and pathogenic variants (Wd) in the relevant protein family, defined through SUPERFAMILY [Gough et al., 2001] or Pfam [Sonnhammer et al., 1997]. To further evaluate the role of this weighting in the performance of FatHMM‐W, we compared the original FatHMM‐W with a logistic regression over the features (ln(Wn) and ln(Wd)) in a 10‐fold cross‐validation on the selected datasets (see Methods and Supp. Text S1). The use of these features alone was sufficient to achieve approximately the same predictive performance as FatHMM‐W (see Supp. Table S2 and Fig. 1).
Given that the ratio of neutral and pathogenic variants in the same protein family is the key feature used by FatHMM‐W, we further analyzed how an even simpler statistic — the fraction of pathogenic variants in the same protein — performs as a predictor. We refer to this predictor as a protein MV (see Methods). MV systematically outperforms FatHMM‐W (Supp. Table S2; Fig. 1). The pathogenicity of neighboring variants within the same protein is therefore the best predictor of pathogenicity across these datasets. This strategy, while statistically effective on the currently existing databases, is not appropriate. Indeed, it assigns the same label to all variants in the same protein, based on information likely obtained at the protein level (i.e., that it is associated with a disease), and cannot distinguish between pathogenic and neutral variants within the same protein.
To better understand the outstanding performance of FatHMM‐W and the protein‐based MV, we examined the relative frequency of pathogenic variants across proteins in all our datasets. In the independent evaluation dataset VariBenchSelected, we found that more than 98% of all proteins (4,425 out of 4,490; Table 4) contain variants from a single class, that is, either “pathogenic” or “neutral” (Fig. 2A). For the remainder of the manuscript, we shall refer to proteins with only one class of variant as “pure” proteins (divided in “pathogenic‐only” proteins and “neutral‐only” proteins). The existence of such “pure” proteins — while theoretically possible — should not be interpreted as a biological phenomenon. Rather, these designations are based on current knowledge, and are at least partially an artifact of how these particular datasets are populated.
Table 4.
Protein Categories and Variants Per Category
| Datasets | “Pure” pathogenic proteins | Pathogenic variants in “pure” proteins | “Pure” neutral proteins | Neutral variants in “pure” proteins | Mixed proteins | Variants in mixed proteins | Total number of proteins |
|---|---|---|---|---|---|---|---|
| HumVar | 1,277 | 10,484 | 8,400 | 17,140 | 911 | 12,765 | 10,588 |
| ExoVar | 891 | 4,336 | 2,794 | 3,478 | 165 | 1,036 | 3,850 |
| VariBenchSelected | 286 | 3,865 | 4,139 | 5,869 | 65 | 532 | 4,490 |
| predictSNPSelected | 855 | 7,090 | 3,738 | 5,649 | 228 | 3,359 | 4,821 |
| SwissVarSelected | 1,444 | 2,749 | 3,614 | 6,568 | 540 | 3,412 | 5,598 |
Overview about the total number of proteins per dataset and the composition of these datasets.
Figure 2.
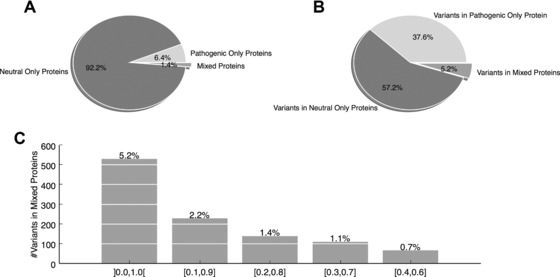
In the VariBenchSelected dataset, most SNPs are in genes with only neutral or only pathogenic variants. A: Protein perspective: proportion of proteins containing only neutral variants (“neutral‐only”), only pathogenic variants (“pathogenic‐only”), and both types of variants (“mixed”). Only 1.4% of the proteins are mixed. B: Variant perspective: proportions, of variants in each of the three categories of proteins. Only 5.2% of variants are in mixed proteins. C: Fractions of variants, in the VariBenchSelected dataset, containing various ratios of pathogenic‐to‐neutral variants, binned into increasingly narrow bins, approaching balanced proteins. The open interval ]0.0, 1.0[ contains all mixed proteins (as in B). Only 0.7% of all variants belong to almost perfectly balanced proteins (closed interval [0.4, 0.6]).
Nearly all (94.8%) variants in VariBenchSelected are located in pure proteins with 57.2% in neutral‐only proteins and 37.6% in pathogenic‐only proteins (Fig. 2B). On such a dataset, excellent accuracies can be achieved by predicting the status of a variant based on the other variants in the same protein. We refer to this phenomenon as type 2 circularity. The remaining 5.2% of VariBenchSelected variants are located in “mixed” proteins (Fig. 2B and C), which contain both pathogenic and neutral variants (pathogenic‐to‐neutral ratio in the open interval ]0.0, 1.0[ (in Fig. 2C). While the MV approach will necessarily misclassify some of these variants, it will still perform well on proteins containing primarily neutral or primarily pathogenic variants, and overall, only 0.7% of all variants are in proteins containing an almost balanced ratio of pathogenic and neutral variants (pathogenic‐to‐neutral ratio in the interval [0.4, 0.6] in Fig. 2C). Similar dataset compositions can be observed in the other three datasets HumVar, ExoVar, and predictSNPSelected (Supp. Figs. S7–S9). A striking property of SwissVarSelected is its much larger fraction of proteins with almost balanced pathogenic‐to‐neutral ratio: 6.5% of all variants (832 out of 12,729) can be found in the most balanced category of mixed proteins [0.4, 0.6] (see Supp. Fig. S10), compared with an average of 1.5% in the other four datasets.
To further understand FatHMM‐W's performance, we evaluated it separately on mixed proteins. As shown in Figure 3, Supp. Figures S11 and S12, FatHMM‐W performs well on pure proteins but loses much of its predictive power on the mixed proteins, as it is misled by its weighting scheme. On almost‐balanced proteins, FatHMM‐W is therefore outperformed by all other tools but phyloP (Fig. 3). This may also be the first reason why FatHMM‐W performs worse on SwissVarSelected than on all other datasets: SwissVarSelected contains many more variants in the most mixed categories (Fig. S10). FatHMM‐W even performs poorly on mixed proteins from its own training dataset (Supp. Fig. S11). We observed that PolyPhen‐2 outperforms all other tools in the mixed categories for the datasets predictSNPSelected and SwissVarSelected (Supp. Fig. S12). For the VariBenchSelected dataset, no clear winner can be determined (Supp. Fig. S12).
Figure 3.
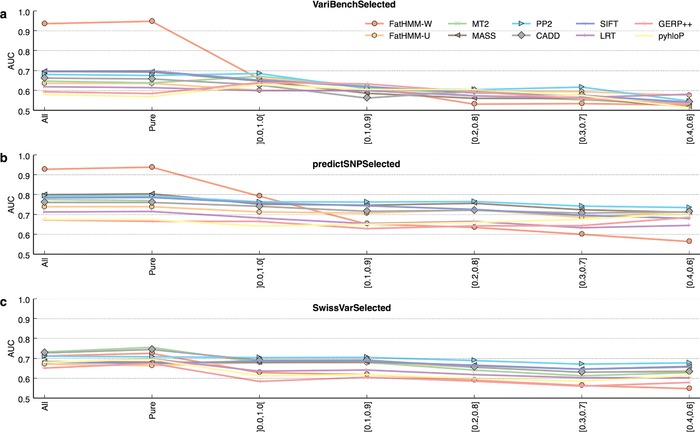
Performance of 10 pathogenicity prediction tools according to protein pathogenic‐to‐neutral variant ratio. Evaluation of tool performance on subsets of VariBenchSelected, predictSNPSelected, and SwissVarSelected, defined according to the relative proportions of pathogenic and neutral variants in the proteins they contain. “Pure” indicates variants belonging to proteins containing only one class of variant. (x and y) indicate variants belonging to mixed proteins, containing a ratio of pathogenic‐to‐neutral variants between x and y. ]0.0, 1.0[ therefore indicate all mixed proteins (the ratios of 0.0 and 1.0 being excluded by the reversed brackets). While FatHMM‐W performs well or excellently on variants belonging to pure proteins (VariBenchSelected and predictSNPSelected), it performs poorly on those belonging to mixed proteins.
The second reason for the drop in performance is the presence of “new” proteins in SwissVarSelected that are unknown to the FatHMM‐W weighting database. To show this, we used the HumVar and ExoVar datasets as a proxy for the training data among all our tools (FatHMM's training data is not fully publicly available). We observed that approximately 91% of all pathogenic and approximately 68% of all neutral variants in VariBenchSelected are located in proteins that also occur in HumVar/ExoVar (Supp. Fig. S13 and Supp. Table S3). As FatHMM‐W makes use of information from protein families, we computed pairwise BLASTP [Camacho et al., 2009] alignments between all proteins in our selected datasets and proteins in HumVar/ExoVar. Approximately 99% of all pathogenic variants in VariBenchSelected are located in proteins from HumVar/Exovar or proteins with more than 70% sequence similarity to a protein in HumVar/Exovar. Similar statistics can be observed for predictSNPSelected (Supp. Fig. S13 and Supp. Table S4). However, for SwissVarSelected, we observed that only approximately 61% of all pathogenic and approximately 56% of all neutral variants belong to proteins from HumVar/ExoVar (Supp. Fig. S13 and Supp. Table S5). Approximately 78% of all pathogenic and approximately 77% of all neutral variants in SwissVarSelected are located in proteins from HumVar/ExoVar or in proteins with high‐sequence similarity (70% sequence similarity) to a protein from HumVar/ExoVar (Supp. Fig. S13 and Supp. Table S5). Hence, a significant proportion of SwissVarSelected variants cannot be found in proteins from the proxy training dataset or proteins with high‐sequence similarity. All of these findings lead to the conclusion that FatHMM‐W's good performance on VariBenchSelected and predictSNPSelected is largely due to type 2 circularity.
Evaluation of Two Combined Predictors
After studying in silico tools for pathogenicity prediction, we compared the performance of two methods that combine individual tools into a joint prediction, the metapredictors Condel [González‐Pérez and López‐Bigas, 2011] and Logit [Li et al., 2013]. Based on our previous findings, we were interested in how their performance compares when evaluated on datasets that avoid type 1 circularity. Furthermore, we wanted to compare the performance of metapredictors that include FatHMM‐W and may suffer from type 2 circularity, to metapredictors that do not include FatHMM‐W.
Condel's Web server provides two combinations of tool scores: the older (original) version combines PP2 [Adzhubei et al., 2010], SIFT [Ng and Henikoff, 2003], and MASS [Reva et al., 2011] and the latest one adds FatHMM‐W [Shihab et al., 2013]. We refer to these two combinations as Condel and Condel+, respectively (Supp. Table S6). To provide a fair comparison, we used the same sets of tools for the Logit (and Logit+) model by Li et al. (2013) (Logit and Logit+, see Supp. Table S6). Thus, in our manuscript, Logit combines PP2, SIFT, and MASS; Logit+ combines these three tools and FatHMM‐W.
To avoid type 1 circularity, we chose the selected datasets as our evaluation datasets, as they do not overlap with the training dataset of any individual tool or metapredictor. Our results using Logit on all selected datasets confirm those reported by Li et al. (2013): Logit outperforms all individual tools and Condel in terms of AUC (Fig. 4; Supp. Figs. S14–S18). Condel's performance (AUC = 0.70) is on par with SIFT (AUC = 0.70) for VariBenchSelected, the best performing of the tools it combines. We then evaluated the combination of tools on the pure and mixed proteins on VariBenchSelected, predictSNPSelected, and SwissVarSelected (Supp. Fig. S19). While Logit performs well on the pure proteins, Condel performs at least as well as Logit on variants in mixed proteins.
Figure 4.
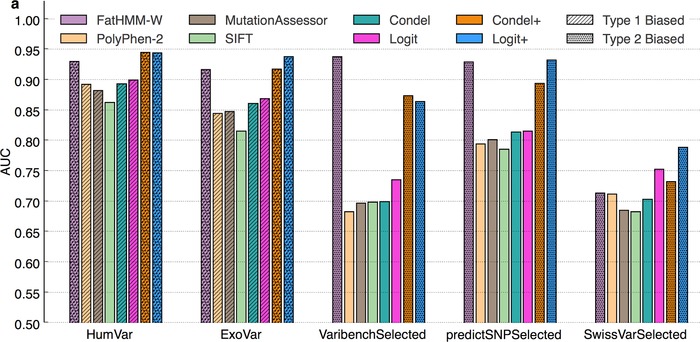
Comparison of the performance of two metapredictors (Logit and Condel) and their component tools, across five datasets. Bar heights reflect AUC for each tool and tool combination. Logit and Condel are metapredictors combining MASS, PP2, and SIFT. The “+” versions of Logit and Condel also include FatHMM‐W. While effective in prediction, FATHMM‐W (alone and in the Logit+ and Condel+ metapredictors) is optimistically biased due to type 2 circularity (see Results section). In the “Selected” datasets, Logit provides the best unbiased performance. SIFT has the lowest performance in the HumVar and ExoVar datasets, but it is also the only predictor that is unbiased in these two datasets.
The evaluation of Condel+ and Logit+ may be optimistically biased by type 2 circularity, given the inclusion of FatHMM‐W. Across all datasets, we observed that adding FatHMM‐W to either tool score combination (Condel+ or Logit+) led to a performance boost (Fig. 4; Supp. Figs. S14–S18). However, this did not hold for mixed proteins, providing strong evidence for type 2 circularity. For mixed proteins, we observed a significant drop in performance for both Logit+ and Condel+ on all datasets but SwissVarSelected (see Supp. Fig. S19).
Discussion
The wealth of pathogenicity prediction tools proposed in the literature raises the question whether there are systematic differences in the quality of their predictions when evaluated on a large number of variant databases. In an attempt to answer this question, we performed a comparative evaluation of pathogenicity prediction tools and demonstrated the existence of two types of circularity that meaningfully impair comparison of in silico pathogenicity prediction tools. We showed how ignoring these effects could lead to overly optimistic assessments of tool performance. One severe consequence of this phenomenon is that it may hinder the discovery of novel disease risk genes, as these tools are widely used to choose variants for further functional investigation.
In this manuscript, we have described and demonstrated “type 1” and “type 2” circularity. Type 1 circularity occurs because of an overlap between training and evaluation datasets, possibly resulting in overfitting [Hastie et al., 2009], meaning that a tool is highly tailored to a given dataset, but will perform worse on novel variants. Type 1 circularity is a well‐known and studied phenomenon in machine learning [Hastie et al., 2009] and there are guidelines on how it can be avoided [Vihinen, 2013]. To avoid type 1 circularity, we built the Selected datasets, in which none of the variants have previously been seen by any of the tools (with the possible and unavoidable exception of MT2). This makes them the most appropriate datasets on which to draw conclusions regarding the relative performance of the tools. Figure 1 and Supp. Table S1 suggest that MT2, PP2, and MASS overfit on their training data and have rather weak generalization abilities.
Our efforts to understand the outstanding performance of FatHMM‐W on four out of five datasets led to additional insights about type 2 circularity. Our findings about type 2 circularity demonstrate that predicting the pathogenicity of a variant — based on the pathogenicity of all other known variants in the same protein — is a statistically successful, but ultimately inappropriate strategy. It is inappropriate because it will often fail to correctly classify variants in proteins that contain both pathogenic and neutral variants. Further, it will often fail to discover pathogenic variants in unannotated proteins.
The apparent success of this strategy is due to the fact that, in variant databases, it is frequently the case that all the variants of the same protein are annotated with the same status. Furthermore, pathogenic‐only proteins contain many more labeled variants than neutral‐only proteins. In these databases, pathogenic amino acid substitutions are heavily concentrated in a few key genes (Fig. 2; Supp. Figs. S7–S9). These observations regarding the distribution of variants in our datasets likely result — in part — from research practices relevant to the way variant databases are populated. Often, an initial discovery of a pathogenic variant in a gene (for a given disease) leads to additional discoveries of pathogenic variants in the same gene, in part because a given gene will be more heavily investigated once it has been identified as harboring pathogenic variants.
These properties of variant databases explain why we observe that pathogenicity can be predicted from the annotation of variants within the same protein by a MV with excellent accuracies (Fig. 1; Supp. Table S2). They also explain why FatHMM‐W, whose predictive power is driven by the pathogenic‐to‐neutral ratio of variants in the same protein, performs so well on VariBenchSelected and predictSNPSelected (Fig. 1; Supp. Table S2). This approach, however, will often fail to correctly classify amino acid substitutions in proteins that contain both pathogenic and neutral variants (Fig. 3; Supp. Figs. S11 and S12). This partially explains why FatHMM‐W performs worse on SwissVarSelected than on all other datasets, as it contains many more variants in the most mixed categories (Fig. 3; Fig. S10). The same phenomenon also occurs when building metapredictors: the performance of Logit+ and Condel+ are similarly optimistically biased because they contain FathHMM‐W.
The pervasiveness of circularity makes it difficult to draw definitive conclusions regarding the relative performance of these prediction tools. We do nevertheless observe a reassuring trend for tools trained for the purpose of predicting pathogenicity to outperform conservation scores. In addition, the Logit combination of SIFT, PP2, and MASS is a slightly better predictor of pathogenicity than any of these tools taken individually. Also, for the mixed proteins in particular, Condel performs better than, or is on par with, Logit across the datasets (Supp. Fig. S19).
It is important to note that the drop in performance that we observe when applying a method to a dataset (that it was not trained on) could also be due to the different definitions of pathogenicity and neutrality used in the different benchmark datasets (see Table 2). It should be an important goal of future studies to quantify this impact. However, as long as circularity exists in a comparative study, it will mask the effect of these differences in the definition of pathogenicity: in Figure 1, FatHMM‐W seems to provide excellent prediction results across four different benchmark datasets, irrespective of the different definitions of pathogenicity and neutrality. However, our analysis shows that the true origin of this superior performance is type 2 circularity and not robustness to different definitions of pathogenicity and neutrality.
Therefore, a key step in future studies examining this problem will be to avoid any type of circularity. The existence of these types of circularity has immediate implications for the further development and evaluation of pathogenicity prediction tools: at the very least, and as recommended previously [Vihinen, 2013], prediction tools should only be compared on benchmarks that do not overlap with any of the datasets used to train the tool. We provide such datasets, VariBenchSelected, predictSNPSelected, and SwissVarSelected (see Supp. Data S1) for this kind of independent evaluation. All prediction tools should make their training datasets public, as type 1 circularity cannot be excluded if any portion of a training dataset is kept private.
A more rigorous strategy would be to retrain all predictors on the same dataset, in order to truly evaluate the predictors and not the quality of their training datasets. However, this is only possible if the raw variant descriptors (variant features) — from which the tools derive their predictions — are made available. We investigated all tools presented here and only for PP2 was it straightforward to obtain these descriptors [Adzhubei et al., 2010].
To address the problem of type 2 circularity, it is imperative that future studies report prediction accuracy as a function of the pathogenic‐to‐neutral ratio, as in Figure 3. An even better strategy would be to stratify training and test datasets such that variants from the same protein only occur in either the training or the test dataset, completely removing the possibility of classification within the same protein [Adzhubei et al., 2010]. Furthermore, one could construct predictors for different classes of variants, defined by the pathogenic‐to‐neutral ratio in the proteins, to address the problem that none of the existing tools achieves constantly good results across these classes on VariBenchSelected. Both these alternative approaches, however, rely critically on the availability of raw descriptors used by the corresponding tools.
Finally, there is another potential source of circularity to beware of in the future: The novel variants entered in databases may be annotated using existing pathogenicity prediction tools. These tools will therefore appear to perform well on “new” data (from later versions of mutation databases), whereas in fact they will only be recovering labels that they have themselves provided. We therefore advocate documentation of the sources of evidence that were used to assign labels to variants when they are entered into a variant database. This is akin to standard practice for gene function databases, which record whether the annotation of a gene with a particular function was biologically validated and/or computationally predicted.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Table S1. Performance evaluation of 10 state‐of‐the‐art pathogenicity prediction tools over 5 datasets
Table S2. Performance evaluation of FatHMM‐W, a logistic regression over the features used to weight FatHMM‐W, and a protein majority vote (MV), over 5 datasets
Table S3. Number of pathogenic and neutral variants from VariBenchSelected in identical or similar proteins in HumVar and ExoVar
Table S4. Number of pathogenic and neutral variants from predictSNPSelected in identical or similar proteins in HumVar and ExoVar
Table S5. Number of pathogenic and neutral variants from SwissVarSelected in identical or similar proteins in HumVar and ExoVar
Table S6. Overview of all combined predictors
Figure S1. Venn diagram showing the overlap between five datasets used in this study.
Figure S2. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the HumVar dataset.
Figure S3. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the ExoVar dataset.
Figure S4. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the VariBenchSelected dataset.
Figure S5. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the predictSNPSelected dataset.
Figure S6. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the SwissVarSelected dataset.
Figure S7. Composition of the HumVar dataset.
Figure S8. Composition of the ExoVar dataset.
Figure S9. Composition of the dataset predictSNPSelected.
Figure S10. Composition of the dataset SwissVarSelected.
Figure S11. Evaluation of FatHMM‐W according to pathogenic‐to‐neutral ratios.
Figure S12. Performance of ten pathogenicity prediction tools according to protein pathogenic‐to‐neutral variant ratio in mixed proteins only.
Figure S13. Variants from the Selected datasets that are in identical or similar proteins in the proxy training datasets HumVar/ExoVar
Figure S14. Performance of meta‐predictors on HumVar.
Figure S15. Performance of meta‐predictos on ExoVar.
Figure S16. Performance of meta‐predictors on VariBenchSelected.
Figure S17. Performance of meta‐predictors on predictSNPSelected.
Figure S18. Performance of meta‐predictors on SwissVarSelected.
Figure S19. Performance of 4 meta‐predictors according to protein pathogenic‐to‐neutral variant ratio.
Data
Acknowledgments
We thank Shaun Purcell for helpful feedback and advice throughout the project and Barbara Rakitsch and Dean Bodenham for fruitful discussion. We thank our anonymous reviewers for their helpful feedback.
Communicated by Mauno Vihinen
Contract grant sponsors: The research of Professor Dr. Karsten Borgwardt was supported by the Alfried Krupp Prize for Young University Teachers of the Alfried Krupp von Bohlen und Halbach‐Stiftung. Dr. Smoller is supported in part by NIH/NIMH grant K24MH094614.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J. 2014. PredictSNP: robust and accurate consensus classifier for prediction of disease‐related mutations. PLoS Comput Biol 10:e1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Altman RB, Bromberg Y. 2013. Collective judgment predicts disease‐associated single nucleotide variants. BMC Genomics 14:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC. 2009. Identification of deleterious mutations within three human genomes. Genome Res 19:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Shendure J. 2011. Needles in stacks of needles: finding disease‐causal variants in a wealth of genomic data. Nat Rev Genet 12:628–640. [DOI] [PubMed] [Google Scholar]
- Davis J, Goadrich M. 2006. The relationship between precision‐recall and ROC curves. In Proceedings of the 23rd international conference on Machine learning (ICML '06), p 233–240.
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R‐E, Chang K‐W, Hsieh C‐J, Wang X‐R, Lin C‐J. 2008. LIBLINEAR: a library for large linear classification. J Mach Learn Res 9:1871–1874. [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho‐Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Girón CG, et al. 2014. Ensembl 2014. Nucleic Acids Res 42:D749–D755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Pérez A, López‐Bigas N. 2011. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough J, Karplus K, Hughey R, Chothia C. 2001. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol 313:903–919. [DOI] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R, Friedman J. 2009. Model assessment and selection In The elements of statistical learning. New York: Springer; p 219–259. [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. 2009. Potential etiologic and functional implications of genome‐wide association loci for human diseases and traits. Proc Natl Acad Sci 106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TJP, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Clarke L, Coates G, Fairley S, et al. 2009. Ensembl 2009. Nucleic Acids Res 37:D690–D697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, Gupta N, Sklar P, Sullivan PF, Moran JL, Hultman CM, Lichtenstein P, et al. 2012. Exome sequencing and the genetic basis of complex traits. Nat Genet 44:623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Mendiratta S, Kim J, Pecot CV, Larsen JE, Zubovych I, Seo BY, Kim J, Eskiocak B, Chung H, McMillan E, Wu S, et al. 2013. Systematic identification of molecular subtype‐selective vulnerabilities in non‐small‐cell lung cancer. Cell 155:552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee H, Abbeel P, Ng AY. 2006. Efficient L1 regularized logistic regression. In Proceedings of the 21st national conference on Artificial intelligence — Volume 1 (AAAI'06), Anthony Cohn (Ed.), Vol. 1. AAAI Press 401–408.
- Leongamornlert D, Mahmud N, Tymrakiewicz M, Saunders E, Dadaev T, Castro E, Goh C, Govindasami K, Guy M, O'Brien L, Sawyer E, Hall A, et al. 2012. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer 106:1697–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M‐X, Kwan JSH, Bao S‐Y, Yang W, Ho S‐L, Song Y‐Q, Sham PC. 2013. Predicting Mendelian disease‐causing non‐synonymous single nucleotide variants in exome sequencing studies. PLoS Genet 9:e1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. 2013. dbNSFP v2.0: a database of human non‐synonymous SNVs and their functional predictions and annotations. Hum Mutat 34:E2393–E2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZD, et al. 2012. A systematic survey of loss‐of‐function variants in human protein‐coding genes. Science 335:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M, Consortium U. 2011. UniProt knowledgebase: a hub of integrated protein data. Database:bar009–bar009. [DOI] [PMC free article] [PubMed]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. 2010. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics 26:2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottaz A, David FPA, Veuthey A‐L, Yip YL. 2010. Easy retrieval of single amino‐acid polymorphisms and phenotype information using SwissVar. Bioinforma Oxf Engl 26:851–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair PS, Vihinen M. 2013. VariBench: a benchmark database for variations. Hum Mutat 34:42–49. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2003. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, et al. 2011. Scikit‐learn: machine learning in python. J Mach Learn Res 12:2825–2830. [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, et al. 2014. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, Sander C. 2011. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39:e118–e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, et al. 2012. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small‐cell lung cancer. Nat Genet 44:1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D. 2014. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods 11:361–362. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward M‐H, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, Day INM, Gaunt TR. 2013. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer ELL, Eddy SR, Durbin R. 1997. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins Struct Funct Genet 28:405–420. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN. 2014. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, et al. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenon J, Milh M, Feillet F, St‐Onge J, Duffourd Y, Jugé C, Roubertie A, Héron D, Mignot C, Raffo E, Isidor B, Wahlen S, et al. 2014. Mutations in SLC13A5 cause autosomal‐recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet 95:113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J, Olatubosun A, Vihinen M. 2011. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat 32:358–368. [DOI] [PubMed] [Google Scholar]
- Vihinen M. 2012. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genomics 13:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M. 2013. Guidelines for reporting and using prediction tools for genetic variation analysis. Hum Mutat 34:275–282. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 38:e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb I, Piscuoglio S, Martelotto LG, Waggott D, Ng CKY, Perez‐Ordonez B, Harding NJ, Alfaro J, Chu KC, Viale A, Fusco N, Cruz Paula A da, et al. 2014. Hotspot activating PRKD1 somatic mutations in polymorphous low‐grade adenocarcinomas of the salivary glands. Nat. Genet 46:1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang F, Wang H, Li Y, Alexander S, Wang K, Willoughby CE, Zaneveld JE, Jiang L, Soens ZT, Earle P, Simpson D, et al. 2015. Next‐generation sequencing‐based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum Genet 134:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Table S1. Performance evaluation of 10 state‐of‐the‐art pathogenicity prediction tools over 5 datasets
Table S2. Performance evaluation of FatHMM‐W, a logistic regression over the features used to weight FatHMM‐W, and a protein majority vote (MV), over 5 datasets
Table S3. Number of pathogenic and neutral variants from VariBenchSelected in identical or similar proteins in HumVar and ExoVar
Table S4. Number of pathogenic and neutral variants from predictSNPSelected in identical or similar proteins in HumVar and ExoVar
Table S5. Number of pathogenic and neutral variants from SwissVarSelected in identical or similar proteins in HumVar and ExoVar
Table S6. Overview of all combined predictors
Figure S1. Venn diagram showing the overlap between five datasets used in this study.
Figure S2. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the HumVar dataset.
Figure S3. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the ExoVar dataset.
Figure S4. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the VariBenchSelected dataset.
Figure S5. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the predictSNPSelected dataset.
Figure S6. Predictive performance of 10 state‐of‐the‐art pathogenicity prediction tools on the SwissVarSelected dataset.
Figure S7. Composition of the HumVar dataset.
Figure S8. Composition of the ExoVar dataset.
Figure S9. Composition of the dataset predictSNPSelected.
Figure S10. Composition of the dataset SwissVarSelected.
Figure S11. Evaluation of FatHMM‐W according to pathogenic‐to‐neutral ratios.
Figure S12. Performance of ten pathogenicity prediction tools according to protein pathogenic‐to‐neutral variant ratio in mixed proteins only.
Figure S13. Variants from the Selected datasets that are in identical or similar proteins in the proxy training datasets HumVar/ExoVar
Figure S14. Performance of meta‐predictors on HumVar.
Figure S15. Performance of meta‐predictos on ExoVar.
Figure S16. Performance of meta‐predictors on VariBenchSelected.
Figure S17. Performance of meta‐predictors on predictSNPSelected.
Figure S18. Performance of meta‐predictors on SwissVarSelected.
Figure S19. Performance of 4 meta‐predictors according to protein pathogenic‐to‐neutral variant ratio.
Data
Data Availability Statement
For each dataset, we compiled comma‐separated files containing all obtained tool scores and predicted labels as well as information about the variant (true label, nucleotide and amino acid changes, minor allele frequencies, UniProt accession IDs [Magrane and Consortium, 2011], Ensembl gene, transcript and protein IDs [Flicek et al. 2014], and dbSNP IDs (rs#) if available [Sherry et al., 2001]). All these datasets including the tool predictions can be found at the VariBench Website (http://structure.bmc.lu.se/VariBench/GrimmDatasets.php) as well as a single Excel file at the journals Website (Supp. Data S2).
Further, we provide all Python scripts used to generate all tables and figures in this study along with all datasets, compressed as a single ZIP file (Supp. Data S1). All data and Python scripts can also be downloaded from: http://www.bsse.ethz.ch/mlcb/research/bioinformatics‐and‐computational‐biology/pathogenicity‐predi‐ction.html.