

Abstract
Cilnidipine is an L/N-type calcium channel blocker (CCB). The effects of cilnidipine on N-type channels give it unique organ-protective properties via the suppression of hyperactivity in the sympathetic nervous system (SNS) and renin-angiotensin-aldosterone system (RAAS). In the present study, we compared the effects of cilnidipine and amlodipine (an L-type CCB) on cardiac and renal functions in spontaneously-hypertensive rats injected with adriamycin (ADR). After the weekly administration of ADR for 3 weeks, spontaneously-hypertensive rats were orally administered cilnidipine (20 mg/kg per day), amlodipine (3 mg/kg per day), or vehicle once daily for 4 weeks. A control group received saline rather than ADR, followed by vehicle for 4 weeks. Cilnidipine and amlodipine produced similar reductions in blood pressure after 4 weeks. Cilnidipine ameliorated ADR-induced heart and kidney damage, whereas amlodipine slightly improved cardiac echocardiographic parameters, but did not protect against ADR-induced renal damage. Cilnidipine (but not amlodipine) suppressed the reflex SNS and RAAS hyperactivity caused by their antihypertensive effects. Furthermore, cilnidipine and amlodipine treatment decreased the urinary levels of adrenocortical hormones. The protective effects of cilnidipine against ADR-induced renal and cardiac dysfunction might be associated with its blockade of N-type calcium channels, in addition to its pleiotropic actions, which include the inhibition of the RAAS.
Keywords: adriamycin, calcium channel blockers, cilnidipine
Introduction
Calcium channel blockers (CCB) are a family of drugs commonly used for treating hypertension. In addition to their direct antihypertensive effects, CCB have been reported to ameliorate organ damage through their antioxidant and other beneficial properties. Cilnidipine is a CCB that blocks N-type calcium channels in addition to L-type channels, the traditional targets of CCB activity.1 Its effects on N-type channels give it a unique profile of beneficial actions that includes the suppression of sympathetic nervous system (SNS) hyperactivity.2,3 Furthermore, cilnidipine suppresses the elevation of plasma renin activity (PRA), angiotensin II (Ang II) levels, and aldosterone levels in plasma, which characterize the activation of the renin-angiotensin-aldosterone system (RAAS) in spontaneously-hypertensive rats (SHR) and in a canine model of chronic atrioventricular block with ventricular electrical remodelling.4,5 Conversely, the L-type CCB, amlodipine, has been shown to cause a reflex increase in PRA and plasma Ang II levels in response to its antihypertensive effect.6 Therefore, uniquely among CCB, cilnidipine has been reported to protect the cardiovascular system and kidneys.7–14 It remains unknown whether the primary mechanism underlying the beneficial effects of cilnidipine involves its effect on the heart or the kidneys.
The SHR is a commonly-used model of essential hypertension in humans. The administration of the cardiotoxic chemotherapeutic agent adriamycin (ADR) to SHR results in simultaneous cardiac and renal damage, mimicking human congestive heart failure and chronic renal failure.15–18 In the current study, ADR-treated SHR were used to investigate and compare the cardioprotective and renoprotective effects of the L/N-type CCB, cilnidipine, and the L-type CCB, amlodipine.
Results
Systolic blood pressure, body weight, and organ weight
SHR received no ADR (saline group, n = 10), ADR and vehicle (ADR group, n = 16), ADR and cilnidipine (20 mg/kg; ADR-Cil group, n = 16), or ADR and amlodipine (3 mg/kg; ADR-Aml group, n = 16) for 4 weeks. Systolic blood pressure (BP) and body weight were significantly lower, and the weights of the liver, lungs, and kidneys (but not the heart) were significantly higher in the ADR group than in the saline group (P < 0.001 for each) (Table1). A comparison of antihypertensive effects in the three ADR-treated groups revealed significantly lower systolic BP in the ADR-Cil and ADR-Aml groups than in the ADR group (P < 0.001 for each) (Table1). Liver weight was also significantly lower in the ADR-Cil and ADR-Aml groups than in the ADR group (P < 0.01 and P < 0.001, respectively) (Table1). Among the treatment groups, only the ADR-Cil group showed a significantly lower cardiac weight than that in the ADR group (P < 0.05) (Table1). The ADR-Cil group also showed a significantly lower cardiac weight than that shown by the ADR-Aml group (P < 0.001) (Table1).
Table 1.
Body weight, BP, heart rate, and relative organ weights in spontaneously-hypertensive rats
| Saline group (n = 10) | ADR group (n = 16) | ADR-Cil group (n = 16) | ADR-Aml group (n = 16) | |
|---|---|---|---|---|
| BW (g) | 380 ± 4 | 299 ± 7††† | 294 ± 3 | 294 ± 2 |
| Systolic BP (mmHg) | 205 ± 4 | 183 ± 2††† | 157 ± 3*** | 154 ± 3*** |
| Heart rate (bpm) | 389 ± 15 | 460 ± 8††† | 450 ± 14 | 474 ± 12 |
| Heart weight (mg/g BW) | 3.8 ± 0.0 | 3.6 ± 0.1 | 3.5 ± 0.0*§§§ | 3.7 ± 0.0 |
| Liver weight (mg/g BW) | 40.5 ± 0.2 | 57.7 ± 1.2††† | 53.4 ± 0.7** | 53.2 ± 0.6*** |
| Lung weight (mg/g BW) | 3.3 ± 0.1 | 4.1 ± 0.1††† | 4.0 ± 0.0 | 4.1 ± 0.0 |
| Kidney weight (mg/g BW) | 7.5 ± 0.1 | 14.9 ± 0.3††† | 15.5 ± 0.2 | 15.3 ± 0.1 |
P < 0.001 for the comparison with the saline group; *P < 0.05, **P < 0.01, ***P < 0.001 for the comparison with the ADR group; §§§P < 0.001 for the comparison with the ADR-Aml group.
Spontaneously-hypertensive rats were treated with ADR, followed by administration of vehicle (ADR group), 20 mg/kg per day cilnidipine (ADR-Cil group), or 3 mg/kg per day amlodipine (ADR-Aml group) for 4 weeks. Saline group received saline rather than ADR, followed by vehicle for 4 weeks.
ADR, adriamycin; Aml, amlodipine; BP, blood pressure; BW, body weight; Cil, cilnidipine.
Renal function analysis
The urinary albumin excretion (UAE) and uric protein excretion (UPE) in the ADR group (163.3 ± 20.0 mg/mg creatinine and 259.0 ± 24.5 mg/mg creatinine, respectively) were significantly higher than those in the saline group (UAE: 0.7 ± 0.2 mg/mg creatinine and UPE: 17.5 ± 1.4 mg/mg creatinine; P < 0.001 for each comparison) (Fig.1a,b). ADR-induced increases in UAE and UPE were significantly attenuated by cilnidipine treatment (UAE: 61.0 ± 3.4 mg/mg creatinine and UPE: 102.2 ± 2.6 mg/mg creatinine; P < 0.001 for the comparison with UAE or UPE in the ADR group) (Fig.1a,b). Amlodipine administration to ADR-treated rats resulted in relatively smaller decreases in UAE and UPE (106.7 ± 7.5 mg/mg creatinine and 194.3 ± 11.3 mg/mg creatinine, respectively; P > 0.05 for the comparison with UAE or UPE in the ADR group) (Fig.1a,b). UAE and UPE in the ADR-Cil group were significantly lower than those in the ADR-Aml group (P < 0.001 for each comparison) (Fig.1a,b). Plasma creatinine levels were not changed markedly by ADR treatment (saline: 0.20 ± 0.01 mg/dL, ADR: 0.28 ± 0.02 mg/dL, ADR-Cil: 0.28 ± 0.03 mg/dL, ADR-Aml: 0.34 ± 0.03 mg/dL). The mean glomerulosclerosis (GS) score of the ADR group (0.86 ± 0.04) was significantly higher than that of the saline group (0.32 ± 0.03, P < 0.001) (Fig.1c). The ADR-Cil group exhibited significantly lower GS scores (0.66 ± 0.05) than those shown by the ADR group (P < 0.001) (Fig.1c), but this effect was not observed in the ADR-Aml group. There was no significant difference in the GS scores of the ADR-Cil and ADR-Aml groups (P = 0.052, Student's t-test) (Fig.1c). Micrographs of the kidneys are shown in Fig.1d. ADR treatment induced tubulointerstitial injury, which improved in both the ADR-Cil and ADR-Aml groups.
Fig 1.
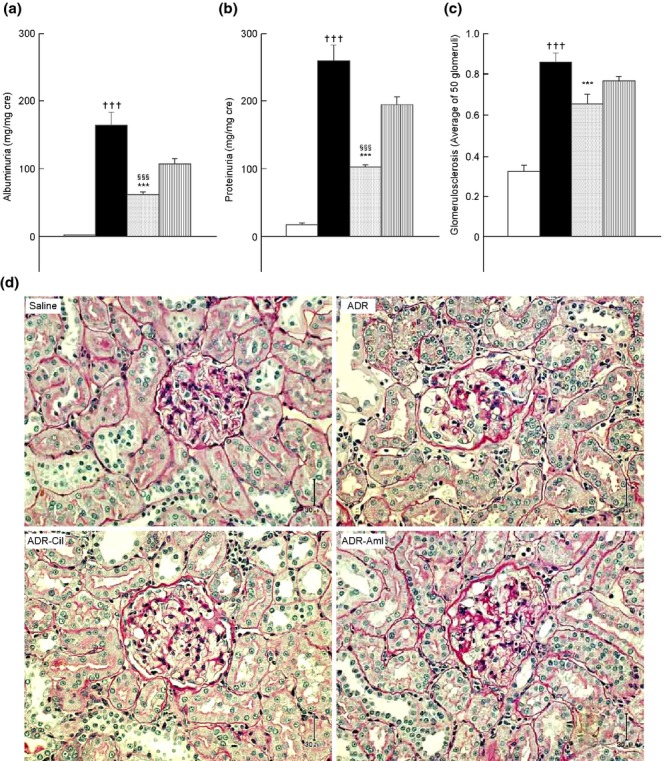
Urinary albumin levels (a), protein excretion levels (b), glomerulosclerosis scores (c), and kidney micrographs (d) in spontaneously-hypertensive rats (SHs) treated with adriamycin (ADR), followed by chronic administration of vehicle (ADR group), 20 mg/kg cilnidipine (ADR-Cil), or 3 mg/kg amlodipine (ADR-Aml) for 4 weeks. Saline group received saline rather than ADR, followed by vehicle for 4 weeks. Values are the mean ± standard error of the mean. †††P < 0.001 for the comparison with the saline group; ***P < 0.001 for the comparison with the ADR group; §§§P < 0.001 for the comparison with the ADR-Aml group.  , Saline;
, Saline;  , ADR;
, ADR;  , ADR-Cil;
, ADR-Cil;  , ADR-Aml.
, ADR-Aml.
Cardiac fibrosis and brain natriuretic peptide
The cardiac area affected by fibrosis was significantly larger in the ADR group than in the saline group (11.9 ± 0.7% and 5.5 ± 0.5%, respectively, P < 0.001) (Fig.2a). In addition, plasma brain natriuretic peptide (BNP) levels were significantly higher in the ADR group (91.8 ± 6.9 pg/mL) than in the saline group (66.3 ± 7.0 pg/mL, P < 0.05) (Fig.2b). In comparison to the ADR group, the ADR-Cil group showed significantly less cardiac fibrosis (9.1 ± 0.6%, P < 0.01) (Fig.2a) and a lower level of BNP (41.1 ± 1.5 pg/mL, P < 0.001) (Fig.2b). The ADR-Aml group showed comparatively weaker, but significant effects, on cardiac fibrosis area (9.5 ± 0.7%, P < 0.05 compared to the ADR group) (Fig.2a) and plasma BNP (58.1 ± 7.8 pg/mL, P < 0.01) (Fig.2b). When the ADR-Aml and ADR-Cil groups were compared using Student's t-test, the ADR-Cil group showed lower plasma BNP (P < 0.05) (Fig.2b), but there was no significant difference in cardiac fibrosis area between the groups (P = 0.611) (Fig.2a). Micrographs of cardiac tissue stained with Masson's trichrome are shown in Figure2c. The histological sections revealed interstitial fibrosis in the ADR group. Cardiac fibrosis in the ADR-Cil and ADR-Aml groups was attenuated.
Fig 2.
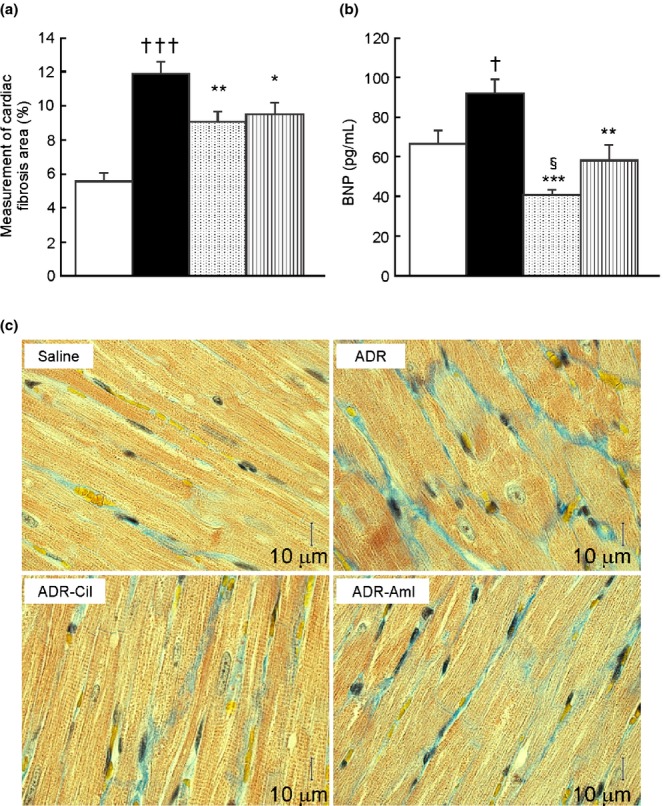
Cardiac fibrosis area (a) and plasma brain natriuretic peptide (BNP) levels (b), and micrographs of cardiac tissue with Masson's trichrome stain (c) in spontaneously-hypertensive rats (SHR) treated with adriamycin (ADR), followed by 4 weeks of vehicle (ADR group), 20 mg/kg cilnidipine (ADR-Cil), or 3 mg/kg amlodipine (ADR-Aml) administration. Saline group received saline rather than ADR, followed by vehicle for 4 weeks. Values are the mean ± standard error of the mean. †P < 0.05, †††P < 0.001 for the comparison with the saline group; *P < 0.05, **P < 0.01, ***P < 0.001 for the comparison with the ADR group; §P < 0.05 for the comparison with the ADR-Aml group.  , Saline;
, Saline;  , ADR;
, ADR;  , ADR-Cil;
, ADR-Cil;  , ADR-Aml.
, ADR-Aml.
Echocardiographic analysis
Following the last of the drug treatments, we performed an echocardiographic examination of myocardial function. Indices of left ventricular (LV) systolic function were found to be significantly worse in the ADR group (ejection fraction (EF): 49.7 ± 2.6%; fractional shortening (FS): 20.9 ± 1.5%) than in the saline group (EF: 72.0 ± 1.4%, P < 0.001; FS: 34.8 ± 1.2%, P < 0.001) (Table2). EF and FS in the ADR-Cil group (57.5 ± 2.2%, 25.1 ± 1.3%) were significantly higher than those recorded in the ADR group (P < 0.05, for each) (Table2). Additionally, LV mass was significantly lower in the ADR-Cil group (0.36 ± 0.01 g/g body weight) than in the ADR group (0.39 ± 0.01 g/g body weight, P < 0.05) (Table2). An analysis of the ADR-Cil and ADR-Aml group data using Student's t-test revealed no significant differences in the EF, FS, LV mass, and LV mass/body weight (P = 0.167, P = 0.171, P = 0.054 and P = 0.072, respectively) (Table2).
Table 2.
Echocardiography findings in spontaneously-hypertensive rats
| Saline group (n = 10) | ADR group (n = 16) | ADR-Cil group (n = 16) | ADR-Aml group (n = 16) | |
|---|---|---|---|---|
| Ejection fraction (%) | 72.0 ± 1.4 | 49.7 ± 2.6††† | 57.5 ± 2.2* | 52.6 ± 2.6 |
| Fractional shortening (%) | 34.8 ± 1.2 | 20.9 ± 1.5††† | 25.1 ± 1.3* | 22.4 ± 1.4 |
| Left ventricular mass (g) | 1.20 ± 0.1 | 1.14 ± 0.02 | 1.06 ± 0.02* | 1.13 ± 0.03 |
| Left ventricular mass/body weight (g/g) | 0.31 ± 0.0 | 0.39 ± 0.01††† | 0.36 ± 2.2* | 0.38 ± 0.01 |
P < 0.001 for the comparison with the saline group; *P < 0.05 for the comparison with the ADR group.
Spontaneously-hypertensive rats were treated with ADR, followed by administration of vehicle (ADR group), 20 mg/kg per day cilnidipine (ADR-Cil group), or 3 mg/kg per day amlodipine (ADR-Aml group) for 4 weeks. Saline group received saline rather than ADR, followed by vehicle for 4 weeks.
ADR, adriamycin; Aml, amlodipine; BP, blood pressure; BW, body weight; Cil, cilnidipine.
Plasma levels of noradrenaline and RAAS components
There was no significant difference in plasma noradrenaline (NA) levels between the saline group (0.56 ± 0.04 ng/mL) and the ADR group (0.63 ± 0.06 ng/mL) (Fig.3a). Conversely, the ADR-Aml group showed relatively higher plasma NA levels (1.05 ± 0.26 ng/mL); however, there was no significant difference compared to the ADR group levels (P = 0.186) (Fig.3a). Plasma NA levels did not differ significantly between the ADR-Aml and ADR-Cil groups (P = 0.129, Student's t-test) (Fig.3a).
Fig 3.
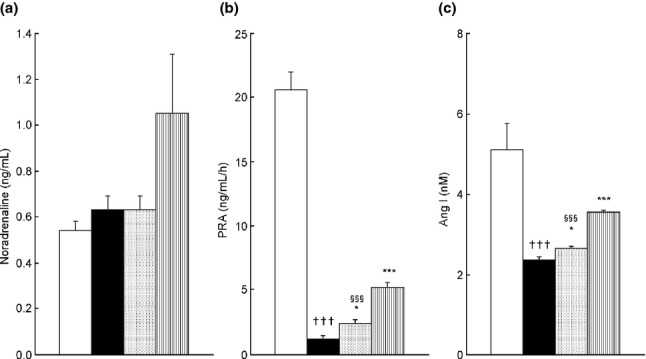
Plasma noradrenaline levels (Aa), plasma renin activity (PRA) (b), and plasma angiotensin I (Ang I) levels (c) in spontaneously-hypertensive rats (SHR) treated with adriamycin (ADR), followed by 4 weeks of vehicle (ADR group), 20 mg/kg cilnidipine (ADR-Cil), or 3 mg/kg amlodipine (ADR-Aml) administration. Saline group received saline rather than ADR, followed by vehicle for 4 weeks. Values are the mean ± standard error of the mean. †††P < 0.001 for the comparison with the saline group; *P < 0.05, ***P < 0.001 for the comparison with the ADR group; §§§P < 0.001 for the comparison with the ADR-Aml group.  , Saline;
, Saline;  , ADR;
, ADR;  , ADR-Cil;
, ADR-Cil;  , ADR-Aml.
, ADR-Aml.
PRA and plasma Ang I levels in the ADR group (1.1 ± 0.3 ng/mL per h and 2.4 ± 0.0 nmol/L, respectively) were significantly lower than those in the saline group (20.6 ± 1.4 ng/mL per h and 5.1 ± 0.7 nmol/L; P < 0.001 for the comparison of each parameter in the ADR and saline groups) (Fig.3b). Amlodipine treatment markedly increased PRA and plasma Ang I levels (5.2 ± 0.4 ng/mL per h and 3.6 ± 0.1 nmol/L, respectively; P < 0.001 for the comparison of each parameter in the ADR and ADR-Aml groups) (Fig.3c). Chronic treatment with cilnidipine induced relatively weak but significant increases in PRA (2.4 ± 0.3 ng/mL per h, P < 0.05 compared to the ADR group) and plasma Ang I levels (2.7 ± 0.0 nmol/L, P < 0.05 compared to the ADR group) (Fig.3c). PRA and plasma Ang I levels were significantly lower in the ADR-Cil group than in the ADR-Aml group (P < 0.001 for each comparison) (Fig.3b,c).
Urinary levels of adrenocortical hormones
Urinary corticosterone levels were significantly elevated in the ADR group (139.0 ± 13.7 ng/mg creatinine) compared to those in the saline group (41.1 ± 2.3 ng/mg creatinine, P < 0.001) (Fig.4a). Cilnidipine treatment suppressed urinary corticosterone levels (74.7 ± 3.4 ng/mg creatinine, P < 0.001 compared to the ADR group) (Fig.4a) more effectively than amlodipine treatment (84.0 ± 3.2 ng/mg creatinine, P < 0.01 compared to the ADR group) (Fig.4a). Among all the treatment groups, only the ADR-Cil group showed a decrease in aldosterone levels (78.5 ± 13.0 pg/mg creatinine) compared to those in the ADR group (159.1 ± 24.2 pg/mg creatinine, P < 0.05) (Fig.4b). Comparison of the ADR-Aml and ADR-Cil groups using Student's t-test did not reveal any significant differences in the levels of corticosterone or aldosterone between the groups (P = 0.057 and P = 0.337, respectively) (Fig.4a,b).
Fig 4.
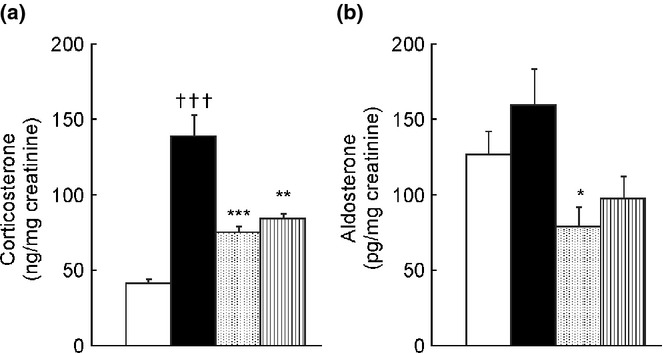
Urinary levels of corticosterone (a) and aldosterone (b) in spontaneously-hypertensive rats (SHR) treated with adriamycin (ADR) followed by 4 weeks of vehicle (ADR group), 20 mg/kg cilnidipine (ADR-Cil), or 3 mg/kg amlodipine (ADR-Aml) administration. Saline group received saline rather than ADR, followed by vehicle for 4 weeks. Values are the mean ± standard error of the mean. †††P < 0.001 for the comparison with the saline group; *P < 0.05, **P < 0.01, ***P < 0.001 for the comparison with the ADR group.  , Saline;
, Saline;  , ADR;
, ADR;  , ADR-Cil;
, ADR-Cil;  , ADR-Aml.
, ADR-Aml.
Discussion
In the present study, the effects of ADR on the heart and kidney were consistent with those reported previously.18,19 Cilnidipine ameliorated ADR-induced heart and kidney damage. Although both CCB tested in this study effectively reduced BP, amlodipine elicited only slight improvements in cardiac function, as evaluated by echocardiography, and did not ameliorate ADR-induced renal damage.
Strong clinical evidence supports the beneficial effects of SNS and RAAS blockade in the management of heart failure, and the efficacy of RAAS blockers in slowing the progression of chronic renal disease.20–25 In this study, ADR-treated SHR developed congestive heart failure and progressive renal disease. Despite producing similar effects on BP, cilnidipine was more effective than amlodipine in ameliorating ADR-induced cardiac and renal dysfunction. The variation in cardioprotective efficacy might reflect a difference in the capacity of individual CCB to suppress the SNS and RAAS. However, animals in the ADR-Aml group showed higher plasma NA levels than those in the ADR group. PRA and plasma Ang I levels in the ADR group were significantly lower than those in the saline group. Our evaluation of the outcomes in ADR-treated animals that were administered CCB showed that amlodipine treatment induced an increase in NA, PRA, and plasma Ang I levels. In comparison, cilnidipine treatment induced a relatively small increase in these parameters.
The strong antihypertensive effect of CCB has been reported to cause reflex activation of the SNS and RAAS.26 Furthermore, excess calcium levels have been reported to inhibit renin expression in juxtaglomerular cells by the direct inhibition of gene transcription and destabilization of renin mRNA.27 L-type CCB might therefore increase renin transcription in the juxtaglomerular cells. However, the blockade of T- and N-type calcium channels did not affect calcium influx in these cells.28 Cilnidipine, an L/N-type CCB, has been reported to suppress the SNS over-activation associated with RAAS activation by blocking N-type calcium channels and to inhibit renin transcription in juxtaglomerular cells.1,29 The results of the present study are in agreement with those of past reports describing the mechanism of action of cilnidipine and typical CCB, suggesting that cilnidipine could attenuate the SNS and RAAS activation induced by its own blockade of L-type calcium channels.
Cilnidipine significantly reduced urinary corticosteroid excretion, whereas amlodipine suppressed the urinary excretion of corticosterone. By inhibiting the influx of calcium into adrenocortical cells, dihydropyridine CCB reduce corticosteroid production in response to a variety of stimulating factors, including increased levels of plasma potassium, adrenocorticotropic hormone, and Ang II.30 Our previous studies suggested that L-type CCB suppressed the release of cortisol more effectively than they suppressed the release of aldosterone, whereas N-type CCB suppressed the release of both glucocorticoids.31 Therefore, the differences in corticosteroid release observed in this study might reflect the calcium channel subtype-selectivity of cilnidipine and amlodipine.
Although the renoprotective properties of cilnidipine are well known, its effects on cardiac function have not been studied in detail. The guidelines of the Japanese Society of Hypertension do not recommend the use of CCB for the treatment of hypertension in patients with heart failure, despite indications that cilnidipine might exhibit cardioprotective activity, in addition to its positive effects on kidney function. Our data suggest that the calcium channel subtype specificity of CCB influences their applicability to patients with cardiac failure. Specifically, N-type CCB might be useful in the treatment of patients with heart failure, possibly because of their suppression of SNS hyperactivity and corticosteroid secretion. Excessive stimulation of the SNS is a recognized feature of congestive heart failure and has been reported in rats with ADR-induced cardiac dysfunction.32–34 In contrast to these previous findings, we observed no significant increase in plasma NA levels following ADR treatment. The lack of effect of ADR treatment or cilnidipine administration on circulating NA levels found in this study, however, supports previous studies suggesting that modulation of SNS activation by cilnidipine occurs via effects on local tissue rather than systemic changes.27–30 Because tissue NA levels are tightly regulated, minimizing the spill-over of excess NA to the systemic circulation, it is feasible that the severity of the disease is reflected in differences in tissue NA levels. Cilnidipine was found to elicit little or no increase in plasma NA levels through the suppression of SNS neurotransmitter release by N-type calcium channel blockade, in contrast to the reflex sympathetic hyperactivity caused by the antihypertensive effect of amlodipine.35 These differences in the SNS effects of CCB are in agreement with previous clinical findings.36 Additionally, elevated plasma corticosteroid concentrations are associated with an increased risk of a cardiac event.37–39 Our evaluation of the effects of CCB on ADR-induced cardiac fibrosis and BNP elevation suggested that both amlodipine and cilnidipine exhibited cardioprotective effects. Taking into consideration the recognized association between glucocorticoid-induced morphological changes and cardiovascular risk factors, the cardioprotective effects of CCBs could, at least in part, be attributed to their antihypertensive and glucocorticoid-lowering effects.40,41 Further study on this issue is required, because it is important to identify fibrosis-related molecules by a multifaceted approach, such as immunohistology of the extracellular matrix, to investigate the mechanisms underlying these suppressive effects on fibrosis and BNP.
In the present study, ADR-treated SHRs showed lower BP and levels of RAAS components (PRA and Ang I) than those shown by saline-treated SHR. ADR toxicity is characterized by hypotension, tachycardia, and various arrhythmias, in addition to reduced food intake and inhibition of protein synthesis, resulting in a loss of body weight.42,43 Previous work demonstrated that ADR-treated SHR exhibited lower BP than that observed in untreated SHR.44 Because heart failure is generally associated with increased RAAS, the ADR-induced decrease in plasma levels of RAAS components observed both in this study and in our preliminary investigations (data not shown) might reflect pathophysiological differences between ADR-induced cardiac dysfunction and clinical congestive heart failure. Several previous studies detected RAAS activation following ADR treatment, and indicated that RAAS inhibitors could prevent ADR-induced tissue damage.45–48 Additional investigations are warranted to elucidate the exact effect of ADR administration on RAAS activity, in addition to the mechanisms underlying these effects.
In addition, cilnidipine has been reported to possess pleiotropic effects, including an antioxidant effect that might be critical for protection against ADR-induced cytotoxicity. Therefore, in addition to N-type calcium channel blockade, the pleiotropic effects of cilnidipine could play a role in its protection against ADR-induced injury.49
In conclusion, the L/N-type CCB, cilnidipine, effectively ameliorated both renal and cardiac dysfunction induced by repeated administration of ADR. Because these effects of cilnidipine were more pronounced than those of the L-type CCB, amlodipine, the blockade of N-type calcium channels might play an important role in the protection of renal and cardiac functions in the presence of ADR. In contrast to amlodipine, cilnidipine suppressed SNS and RAAS hyperactivity caused by its antihypertensive action. The renoprotective and cardioprotective effects of cilnidipine might involve the inhibition of adrenocortical hormone release.
Methods
Animals
Male SHR (11 weeks of age) were purchased from Japan SLC (Shizuoka, Japan). The rats were fed standard laboratory chow (CRF-1, Charles River, Kanagawa, Japan) and tap water ad libitum for the duration of the experiments. All procedures involving the care and use of animals were approved by the Institutional Animal Care and Use Committee of the Pharmaceutical Research Laboratories of Ajinomoto Pharmaceuticals (Tokyo, Japan) before they were performed.
Materials
Cilnidipine and amlodipine were purchased from Ajinomoto and Moehs Catalana, SL (Barcelona, Spain), respectively.
Drug administration
ADR (1.5 mg/kg; Kyowa Hakko Kirin, Tokyo, Japan) was administered intravenously to SHR once a week for 3 weeks, corresponding to a cumulative dose of 4.5 mg/kg. One week after the last ADR administration, the rats were divided into three groups. The ADR group (n = 16) was administered with vehicle, the ADR-Cil group (n = 16) received 20 mg/kg cilnidipine, and the ADR-Aml group (n = 16) received 3 mg/kg amlodipine for 4 weeks. Control SHR (saline group, n = 10) were treated identically to the ADR-treated animals, except that they received intravenous saline for the first 3 weeks, followed by daily oral administration of drug vehicle for 4 weeks. Cilnidipine and amlodipine were suspended in 0.5% hydroxypropyl methylcellulose (Sigma-Aldrich, St Louis, MO, USA) immediately prior to oral administration to the animals. The dosages used were selected based on preliminary studies (data not shown). Rats were assigned to treatment groups by stratified randomization based on the measurements of BP, body weight, and UAE. At the end of the study period (3–4 h after the last administration), the rats were killed, a sample of blood was collected, and the organs of interest were quickly removed.
Measurement of systolic BP
Systolic BP was measured by the tail-cuff method using a Softron BP-98A BP meter (Softron, Tokyo, Japan). The rats were introduced into a plastic wire holder and placed in a thermostatically-warmed tube, which was maintained at 34–36°C during the measurements. After environmental acclimatization, three measurements were performed for each animal, and the mean values were recorded.50
Echocardiographic analysis
After the drug treatments had been completed and immediately before killing, the rats were anesthetized by intraperitoneal administration of pentobarbital and subjected to transthoracic echocardiography. M-mode echocardiography was performed with a 12.5-MHz transducer (Xario SSA-660A; Toshiba Medical Systems, Tochigi, Japan). LV end-diastolic and end-systolic dimensions (LVDd and LVD, respectively), interventricular septum thickness (IVST) and LV posterior wall thickness (LVPWT) were measured. LVFS, LV mass, end-diastolic volume, and end-systolic volume were calculated using the following formulae:
LV EF was calculated using the formula of Teichholz.51
Measurements of biochemical parameters
Blood samples collected from the postcaval vein at the time of euthanasia were centrifuged (3000 g) for 15 min. Plasma NA levels were measured using high-performance liquid chromatography, as described previously.50 Plasma BNP concentration was measured using an enzyme-linked immunosorbent assay (ELISA) kit (Assay Max Rat BNP-32; AssayPro, St Charles, MO, USA). PRA was determined using a radioimmunoassay system (Yamasa Shoyu, Chiba, Japan), and Ang I levels were determined by liquid chromatography-tandem mass spectrometry after solid-phase extraction, as described previously.52
Collection and analysis of urine
The rats were individually placed in metabolic cages, and urine samples were collected for 24 h on the last week of the study to quantify UAE levels. Albumin concentrations were measured using an ELISA kit (Nephrat II; Exocell, Philadelphia, PA, USA). Protein concentrations were measured using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Kanagawa, Japan). Urinary and plasma creatinine concentrations were measured with an enzymatic Creatinine Plus version 2 reagent kit, using an automatic analyzer (COBAS INTEGRA 400 plus; Roche, Basel, Switzerland). Urinary corticosterone and aldosterone levels were measured using an enzyme immunoassay system (Cayman Chemical, Ann Arbor, MI, USA). UAE, UPE, urinary corticosterone and aldosterone levels were normalized to the urinary creatinine concentration.
Histological studies
The left kidney of each rat was fixed in fresh 10% formaldehyde and embedded in paraffin. Sections (3 μm thick) were stained with periodic acid-Schiff to assess GS. Fifty glomeruli in each specimen were examined. Lesions were observed under a light microscope (BX50; Olympus, Tokyo, Japan) and scored based on the percentage of glomerular involvement by an observer blinded to the sample treatment group. Lesion scores were assigned according to the following scale: 0, no lesions; 1+, less than 25% glomerular involvement; 2+, 26% to 50%; 3+, 51% to 75%; and 4+, more than 76%.53
Immediately following killing, hearts were removed and fixed in 10% formalin. Sections of the myocardium (3 μm thick) were stained with haematoxylin–eosin or Masson's trichrome stain to quantify the myocyte cross-sectional dimensions and interstitial fibrosis. Analysis of cardiac sections by light microscope (BX50) was performed by an observer blinded to the treatment group of the sample. The relative volume occupied by each tissue element of the ventricle (myocardial fibres and fibrous tissue) was quantified using image processing software (winroof version 3.5; Mitani, Fukui, Japan).
Statistical analysis
Data were expressed as mean ± the standard error of the mean (SEM). The differences between the ADR group and the CCB-treated animal groups were compared using Dunnett's test. ADR versus saline groups and ADR-Cil versus ADR-Aml groups were compared using Student's unpaired t-test. These analyses were performed using exsus Ver. 7.7.1 (CAC, Tokyo, Japan). Differences were considered statistically significant when P < 0.05.
References
- 1.Takahara A. Cilnidipine: A new generation Ca2+ channel blocker with inhibitory action on sympathetic neurotransmitter release. Cardiovasc. Ther. 2009;27:124–39. doi: 10.1111/j.1755-5922.2009.00079.x. [DOI] [PubMed] [Google Scholar]
- 2.Takahara A, Koganei H, Takeda T, Iwata S. Antisympathetic and hemodynamic property of a dual L/N-type Ca(2+) channel blocker cilnidipine in rats. Eur. J. Pharmacol. 2002;434:43–7. doi: 10.1016/s0014-2999(01)01521-7. [DOI] [PubMed] [Google Scholar]
- 3.Shiga T, Yamada Y, Matsuda N, et al. Influence of cilnidipine or nisoldipine on sympathetic activity in healthy male subjects. Heart Vessels. 2007;22:404–9. doi: 10.1007/s00380-007-0984-y. [DOI] [PubMed] [Google Scholar]
- 4.Konda T, Enomoto A, Aritomi S, et al. Different effects of L/N-type and L-type calcium channel blockers on the renin-angiotensin-aldosterone system in SHR/Izm. Am. J. Nephrol. 2009;30:155–61. doi: 10.1159/000210396. [DOI] [PubMed] [Google Scholar]
- 5.Takahara A, Nakamura Y, Wagatsuma H, et al. Long-term blockade of L/N-type Ca(2+) channels by cilnidipine ameliorates repolarization abnormality of the canine hypertrophied heart. Br. J. Pharmacol. 2009;158:1366–74. doi: 10.1111/j.1476-5381.2009.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fournier A, Oprisiu-Fournier R, Serot J-M, et al. Prevention of dementia by antihypertensive drugs: How AT1-receptor-blockers and dihydropyridines better prevent dementia in hypertensive patients than thiazides and ACE-inhibitors. Expert Rev. Neurother. 2009;9:1413–31. doi: 10.1586/ern.09.89. [DOI] [PubMed] [Google Scholar]
- 7.Varagic J, Susic D, Frohlich ED. Cilnidipine improves spontaneously hypertensive rat coronary hemodynamics without altering cardiovascular mass and collagen. J. Hypertens. 2002;20:317–22. doi: 10.1097/00004872-200202000-00023. [DOI] [PubMed] [Google Scholar]
- 8.Sakata K, Yoshida H, Tamekiyo H, et al. Comparative effect of clinidipine and quinapril on left ventricular mass in mild essential hypertension. Drugs Exp. Clin. Res. 2003;29:117–23. [PubMed] [Google Scholar]
- 9.Nagai H, Minatoguchi S, Chen X-H, et al. Cilnidipine, an N+L-type dihydropyridine Ca channel blocker, suppresses the occurrence of ischemia/reperfusion arrhythmia in a rabbit model of myocardial infarction. Hypertens. Res. 2005;28:361–8. doi: 10.1291/hypres.28.361. [DOI] [PubMed] [Google Scholar]
- 10.Takatsu M, Hattori T, Murase T, et al. Comparison of the effects of cilnidipine and amlodipine on cardiac remodeling and diastolic dysfunction in Dahl salt-sensitive rats. J. Hypertens. 2012;30:1845–55. doi: 10.1097/HJH.0b013e3283567645. [DOI] [PubMed] [Google Scholar]
- 11.Takai S, Jin D, Aritomi S, Niinuma K, Miyazaki M. Powerful vascular protection by combining cilnidipine with valsartan in stroke-prone, spontaneously hypertensive rats. Hypertens. Res. 2013;36:342–8. doi: 10.1038/hr.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou X, Ono H, Ono Y, Frohlich ED. N-and L-type calcium channel antagonist improves glomerular dynamics, reverses severe nephrosclerosis, and inhibits apoptosis and proliferation in an l-NAME/SHR model. J. Hypertens. 2002;20:993–1000. doi: 10.1097/00004872-200205000-00035. [DOI] [PubMed] [Google Scholar]
- 13.Tsuchihashi T, Ueno M, Tominaga M, et al. Anti-proteinuric effect of an N-type calcium channel blocker, cilnidipine. Clin. Exp. Hypertens. 2005;27:583–91. doi: 10.1080/10641960500298558. [DOI] [PubMed] [Google Scholar]
- 14.Kojima S, Shida M, Yokoyama H. Comparison between cilnidipine and amlodipine besilate with respect to proteinuria in hypertensive patients with renal diseases. Hypertens. Res. 2004;27:379–85. doi: 10.1291/hypres.27.379. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Clark JR, Herman EH, Ferrans VJ. Doxorubicin-induced apoptosis in spontaneously hypertensive rats: Differential effects in heart, kidney and intestine, and inhibition by ICRF-187. J. Mol. Cell. Cardiol. 1996;28:1931–43. doi: 10.1006/jmcc.1996.0186. [DOI] [PubMed] [Google Scholar]
- 16.Guerra J, De Jesus A, Santiago-Borrero P, Roman-Franco A, Rodriguez E, Crespo MJ. Plasma nitric oxide levels used as an indicator of doxorubicin-induced cardiotoxicity in rats. Hematol. J. 2005;5:584–8. doi: 10.1038/sj.thj.6200573. [DOI] [PubMed] [Google Scholar]
- 17.Hazari MS, Haykal-Coates N, Winsett DW, Costa DL, Farraj AK. Continuous electrocardiogram reveals differences in the short-term cardiotoxic response of Wistar-Kyoto and spontaneously hypertensive rats to doxorubicin. Toxicol. Sci. 2009;110:224–34. doi: 10.1093/toxsci/kfp092. [DOI] [PubMed] [Google Scholar]
- 18.Mihailović-Stanojević N, Jovović D, Miloradović Z, Grujić-Milanović J, Jerkić M, Marković-Lipkovski J. Reduced progression of adriamycin nephropathy in spontaneously hypertensive rats treated by losartan. Nephrol. Dial. Transplant. 2009;24:1142–50. doi: 10.1093/ndt/gfn596. [DOI] [PubMed] [Google Scholar]
- 19.Okuda S, Oh Y, Tsuruda H, Onoyama K, et al. Adriamycin-induced nephropathy as a model of chronicprogresive glomerular disease. Kidney Int. 1986;29:502–10. doi: 10.1038/ki.1986.28. [DOI] [PubMed] [Google Scholar]
- 20.Braunwald E. ACE inhibitors–a cornerstone of the treatment of heart failure. N. Engl. J. Med. 1991;325:351–3. doi: 10.1056/NEJM199108013250508. [DOI] [PubMed] [Google Scholar]
- 21.The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N. Engl. J. Med. 1991;325:293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 22.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999;341:709–17. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 23.Ruggenenti P, Perna A, Gherardi G, et al. Renoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet. 1999;354:359–64. doi: 10.1016/S0140-6736(98)10363-X. [DOI] [PubMed] [Google Scholar]
- 24.Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: The candesartan and lisinopril microalbuminuria (CALM) study. BMJ. 2000;321:1440–4. doi: 10.1136/bmj.321.7274.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ibrahim HN, Hostetter TH. Aldosterone in renal disease. Curr. Opin. Nephrol. Hypertens. 2003;12:159–64. doi: 10.1097/00041552-200303000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Tsutamoto T, Tsutsui T, Maeda K, et al. Effects of long-acting calcium channel antagonists on neurohumoral factors: Comparison of nifedipine coat-core with amlodipine. J. Cardiovasc. Pharmacol. 2003;41(Suppl. 1):77–81. [PubMed] [Google Scholar]
- 27.Klar J, Sigl M, Obermayer B, Schweda F, Krämer BK, Kurtz A. Calcium inhibits renin gene expression by transcriptional and posttranscriptional mechanisms. Hypertension. 2005;46:1340–6. doi: 10.1161/01.HYP.0000192025.86189.46. [DOI] [PubMed] [Google Scholar]
- 28.Lai EY, Wang Y, Persson AEG, Manning RD, Liu R. Pressure induces intracellular calcium changes in juxtaglomerular cells in perfused afferent arterioles. Hypertens. Res. 2011;34:942–8. doi: 10.1038/hr.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konda T, Enomoto A, Matsushita J, Takahara A, Moriyama T. The N- and L-type calcium channel blocker cilnidipine suppresses renal injury in Dahl rats fed a high-sucrose diet, an experimental model of metabolic syndrome. Nephron Physiol. 2005;101:1–13. doi: 10.1159/000085713. [DOI] [PubMed] [Google Scholar]
- 30.Lisurek M, Bernhardt R. Modulation of aldosterone and cortisol synthesis on the molecular level. Mol. Cell. Endocrinol. 2004;215:149–59. doi: 10.1016/j.mce.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Aritomi S, Wagatsuma H, Numata T, et al. Expression of N-type calcium channels in human adrenocortical cells and their contribution to corticosteroid synthesis. Hypertens. Res. 2011;34:193–201. doi: 10.1038/hr.2010.191. [DOI] [PubMed] [Google Scholar]
- 32.Gu R, Lu W, Xie J, Bai J, Xu B. Renalase deficiency in heart failure model of rats–a potential mechanism underlying circulating norepinephrine accumulation. PLoS ONE. 2011;6:e14633. doi: 10.1371/journal.pone.0014633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zucker IH, Pliquett RU. Novel mechanisms of sympatho-excitation in chronic heart failure. Heart Fail. Monit. 2002;3:2–7. [PubMed] [Google Scholar]
- 34.Zhang S, Zhang F, Sun H, Zhou Y, Han Y. Enhanced sympathetic activity and cardiac sympathetic afferent reflex in rats with heart failure induced by adriamycin. J. Biomed. Res. 2012;26:425–31. doi: 10.7555/JBR.26.20120035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hosono M, Fujii S, Hiruma T, et al. Inhibitory effect of cilnidipine on vascular sympathetic neurotransmission and subsequent vasoconstriction in spontaneously hypertensive rats. Jpn. J. Pharmacol. 1995;69:127–34. doi: 10.1254/jjp.69.127. [DOI] [PubMed] [Google Scholar]
- 36.Sakata K, Shirotani M, Yoshida H, et al. Effects of amlodipine and cilnidipine on cardiac sympathetic nervous system and neurohormonal status in essential hypertension. Hypertension. 1999;33:1447–52. doi: 10.1161/01.hyp.33.6.1447. [DOI] [PubMed] [Google Scholar]
- 37.Yamaji M, Tsutamoto T, Kawahara C, et al. Serum cortisol as a useful predictor of cardiac events in patients with chronic heart failure: The impact of oxidative stress. Circ. Heart Fail. 2009;2:608–15. doi: 10.1161/CIRCHEARTFAILURE.109.868513. [DOI] [PubMed] [Google Scholar]
- 38.Girerd N, Pang PS, Swedberg K, et al. Serum aldosterone is associated with mortality and re-hospitalization in patients with reduced ejection fraction hospitalized for acute heart failure: Analysis from the EVEREST trial. Eur. J. Heart Fail. 2013;15:1228–35. doi: 10.1093/eurjhf/hft100. [DOI] [PubMed] [Google Scholar]
- 39.Weir RAP, Tsorlalis IK, Steedman T, et al. Aldosterone and cortisol predict medium-term left ventricular remodelling following myocardial infarction. Eur. J. Heart Fail. 2011;13:1305–13. doi: 10.1093/eurjhf/hfr129. [DOI] [PubMed] [Google Scholar]
- 40.Fardet L, Cabane J, Kettaneh A, Lebbé C, Flahault A. Corticosteroid-induced lipodystrophy is associated with features of the metabolic syndrome. Rheumatology. 2007;46:1102–6. doi: 10.1093/rheumatology/kem062. [DOI] [PubMed] [Google Scholar]
- 41.Souverein P, Berard A, Van Staa T, et al. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case–control study. Heart. 2004;90:859–65. doi: 10.1136/hrt.2003.020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gnanapragasam A, Ebenezar KK, Sathish V, Govindaraju P, Devaki T. Protective effect of Centella asiatica on antioxidant tissue defense system against adriamycin induced cardiomyopathy in rats. Life Sci. 2004;76:585–97. doi: 10.1016/j.lfs.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Tong J, Ganguly PK, Singal PK. Myocardial adrenergic changes at two stages of heart failure due to adriamycin treatment in rats. Am. J. Physiol. 1991;260:909–16. doi: 10.1152/ajpheart.1991.260.3.H909. [DOI] [PubMed] [Google Scholar]
- 44.Jovanovic D, Jovovic D, Mihailovic-Stanojevic N, et al. Effect of carvedilol on pulse pressure and left ventricular hypertrophy in spontaneously hypertensive rats with adriamycin nephropathy. Biomed. Pharmacother. 2009;63:571–6. doi: 10.1016/j.biopha.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 45.Rashikh A, Pillai KK, Ahmad SJ, Akhtar M, Najmi AK. Aliskiren alleviates doxorubicin-induced nephrotoxicity by inhibiting oxidative stress and podocyte injury. J. Renin Angiotensin Aldosterone Syst. 2013;14:14–22. doi: 10.1177/1470320312459980. [DOI] [PubMed] [Google Scholar]
- 46.Alves de Souza RC, Camacho AA. Neurohormonal, hemodynamic, and electrocardiographic evaluations of healthy dogs receiving long-term administration of doxorubicin. Am. J. Vet. Res. 2006;67:1319–25. doi: 10.2460/ajvr.67.8.1319. [DOI] [PubMed] [Google Scholar]
- 47.Muñoz M, Rincón J, Pedreañez A, Viera N, Hernández-Fonseca JP, Mosquera J. Proinflammatory role of angiotensin II in a rat nephrosis model induced by adriamycin. J. Renin Angiotensin Aldosterone Syst. 2011;12:404–12. doi: 10.1177/1470320311410092. [DOI] [PubMed] [Google Scholar]
- 48.Rashikh A, Ahmad SJ, Pillai KK, Kohli K, Najmi AK. Aliskiren attenuates myocardial apoptosis and oxidative stress in chronic murine model of cardiomyopathy. Biomed. Pharmacother. 2012;66:138–43. doi: 10.1016/j.biopha.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 49.Harada E, Uchida H, Sugino K, et al. Effects of L/N-type Ca2+ channel blocker, cilnidipine on release of reactive oxygen species from leukocytes. Jpn. Pharmacol. Ther. 2014;42:413–8. [Google Scholar]
- 50.Aritomi S, Niinuma K, Ogawa T, Konda T, Nitta K. Effects of an N-type calcium antagonist on angiotensin II-renin feedback. Am. J. Nephrol. 2011;33:168–75. doi: 10.1159/000323969. [DOI] [PubMed] [Google Scholar]
- 51.Teichholz LE, Kreulen T, Herman MV, et al. Problems in echocardiographic volume determinations: Echocardiographic-angiographic correlations in the presence of absence of asynergy. Am. J. Cardiol. 1976;37:7–11. doi: 10.1016/0002-9149(76)90491-4. [DOI] [PubMed] [Google Scholar]
- 52.Aritomi S, Niinuma K, Kawakami M, et al. Cilnidipine, an L-/N-type calcium channel blocker, changes the circulating angiotensin–(1-7)/angiotensin II ratio. J. Hypertens. 2012;1:102. Open Access. [Google Scholar]
- 53.Aritomi S, Koganei H, Wagatsuma H, et al. The N-type and L-type calcium channel blocker cilnidipine suppresses renal injury in Dahl rats fed a high-salt diet. Heart Vessels. 2010;25:549–55. doi: 10.1007/s00380-010-0005-4. [DOI] [PubMed] [Google Scholar]