

Abstract
Genomic and protein-coding transcriptomic data have suggested that germ cell tumours (GCTs) of childhood are biologically distinct from those of adulthood. Global messenger RNA profiles segregate malignant GCTs primarily by histology, but then also by age, with numerous transcripts showing age-related differential expression. Such differences are likely to account for the heterogeneous clinico-pathological behaviour of paediatric and adult malignant GCTs. In contrast, as global microRNA signatures of human tumours reflect their developmental lineage, we hypothesized that microRNA profiles would identify common biological abnormalities in all malignant GCTs owing to their presumed shared origin from primordial germ cells. MicroRNAs are short, non-protein-coding RNAs that regulate gene expression via translational repression and/or mRNA degradation. We showed that all malignant GCTs over-express the miR-371–373 and miR-302/367 clusters, regardless of patient age, histological subtype or anatomical tumour site. Furthermore, bioinformatic approaches and subsequent Gene Ontology analysis revealed that these two over-expressed microRNAs clusters co-ordinately down-regulated genes involved in biologically significant pathways in malignant GCTs. The translational potential of this finding has been demonstrated with the detection of elevated serum levels of miR-371–373 and miR-302/367 microRNAs at the time of malignant GCT diagnosis, with levels falling after treatment. The tumour-suppressor let-7 microRNA family has also been shown to be universally down-regulated in malignant GCTs, because of abundant expression of the regulatory gene LIN28. Low let-7 levels resulted in up-regulation of oncogenes including MYCN, AURKB and LIN28 itself, the latter through a direct feedback mechanism. Targeting LIN28, or restoring let-7 levels, both led to effective inhibition of this pathway. In summary, paediatric malignant GCTs show biological differences from their adult counterparts at a genomic and protein-coding transcriptome level, whereas they both display very similar microRNA expression profiles. These similarities and differences may be exploited for diagnostic and/or therapeutic purposes.
Keywords: biomarker, germ cell tumour, let-7, LIN28, microRNA, miR-302/367, miR-371–373, serum
Aims
The aim of this review was to focus on the biological studies of malignant germ cell tumours (GCTs) and highlight similarities and differences between tumours arising during childhood and those arising in adulthood. In addition to considering genomic and protein-coding transcriptomic changes, we will emphasize findings from expression profiling studies of short non-protein-coding RNAs, termed microRNAs, in GCTs. Our improving knowledge of the molecular mechanisms underlying the pathogenesis of GCTs is contributing to the identification of new biomarkers and therapeutic targets, and the development of clinico-biological algorithms for disease segmentation and risk stratification. Combined with the developing collaborations between international clinical trial groups, we can be cautiously optimistic that these approaches will, in the foreseeable future, improve the clinical management of the children, adolescents and young adults affected by this disease (Murray et al., 2009; Collinson et al., 2014; Stoneham et al., 2014).
Background
Germ cell tumours are clinically and pathologically complex neoplasms that occur from the neonatal period through to late adulthood (Murray & Nicholson, 2010). They show extensive clinico-pathological heterogeneity, for example in their incidence rate, presentation and histology. Only 50% of GCTs present at gonadal sites during childhood, whereas this figure increases to 95% in adulthood (Murray & Nicholson, 2010). GCTs display a characteristic bimodal age distribution in childhood, with a relatively high incidence rate in the first few years of life, which then declines to very low levels around 5 years of age, before increasing again in adolescence. As a result, their incidence in the paediatric (four per million) and older adult (14 per million) populations remains markedly lower than in teenagers and young adults (TYAs) (60 per million in males and eight per million in females) (Murray et al., 2009).
Despite these clinical differences, all GCTs are believed to originate from primordial germ cells (PGCs), regardless of patient age, tumour site and histological subtype (Teilum, 1965). Despite the common origin theory, the histological appearance of GCTs varies depending on the type and degree of subsequent differentiation. Those tumours that show extensive somatic differentiation are referred to as teratomas and are generally considered benign, particularly in paediatric practice. Malignant GCTs show varying degrees of differentiation and are classified into seminoma (undifferentiated) and non-seminomatous tumours [yolk sac tumour (YST), embryonal carcinoma (EC) and choriocarcinoma (CHC)]. The latter tumours show yolk sac, embryonal and trophoblastic/placental-like differentiation respectively (Murray & Nicholson, 2010). In this review, the term seminoma is used to collectively describe testicular seminomas, ovarian dysgerminomas and extragonadal germinomas. Mixed malignant GCTs also occur, which are composed of more than one histological subtype. Clinical differences between GCTs arising in children and adults, along with experimental data, such as the maturational stage and imprinting status of the originating PGC, led Oosterhuis and Looijenga to propose a classification system where GCTs are divided into five types (Oosterhuis & Looijenga, 2005). Relevant to this review, ‘Type I’ tumours occur in neonates and infants <5 years of age and predominantly comprise non-seminomatous teratomas and YSTs. ‘Type II’ tumours include both non-seminomatous and seminomatous GCTs and include testicular disease in patients >15 years and ovarian tumours in those aged >4 years (Oosterhuis & Looijenga, 2005).
Clinical perspective
Malignant GCTs rapidly became a highly curable disease, even when diagnosed at advanced clinical stages, with the advent of cisplatin-based chemotherapy in the 1970s (Einhorn & Donohue, 1977; Williams et al., 1987). However, up to 20% of patients may still eventually die of their disease. Although paediatric GCTs were initially treated with ‘adult’ type cisplatin-based regimes, the long-term sequelae of such schedules were substantial, including pulmonary fibrosis (Osanto et al., 1992), nephrotoxicity (Bosl et al., 1986), neuropathy (Glendenning et al., 2010) and ototoxicity (Bokemeyer et al., 1996a; Strumberg et al., 2002). Of particular concern are reports of increased risks of early onset cardiovascular disease (Huddart et al., 2003) and second malignancies (Travis et al., 1997, 2005) in survivors of malignant GCT disease. These side effect profiles resulted in the adoption of a carboplatin-based approach instead for affected children in the UK, with comparable survival for all tumour stages (Mann et al., 2000). However, carboplatin therapy has been shown to be inferior to cisplatin in adults with malignant testicular GCTs (TGCTs), although lower dose carboplatin regimes have typically been used in this age-group owing to the increased risk of myelosuppression (Bokemeyer et al., 1996b1996b; Horwich et al., 1997; Collinson et al., 2014). When attempting to identify the optimal treatment schedules for such patients however, the currently available clinical risk stratification systems are not sophisticated enough to truly distinguish those GCT patients who will have excellent outcomes from those destined to have poor outcomes. For example, within the International Germ Cell Consensus Classification (IGCCC) (1997) high-risk group it is not possible to identify upfront at the time of diagnosis the 55% of patients who will be chemotherapy resistant (and have either refractory disease or show subsequent relapse), from the 45% for whom four courses of standard cisplatin-based chemotherapy (BEP) will be sufficient for cure. Consequently, we need to extend our knowledge of GCT biology and use it to develop combined clinico-biological risk stratification algorithms, improved biomarkers and targets for the development of novel therapeutic agents for patients with malignant GCTs. This should ultimately lead to improved cure rates for those patients who currently have poor outcomes in the IGCCC (or equivalent risk stratification) intermediate- and high-risk disease groups and allow reduction of toxicity in those patient groups truly defined as low-risk.
Genomic changes
Atkin & Baker (1982) first described the detection of an isochromosome of the short arm of chromosome 12 [i(12p)] in TGCTs over three decades ago. This abnormality occurs in ∽80% of adult TGCTs, whereas more limited gain of 12p genomic material is almost invariably present in the remaining cases (Collinson et al., 2014). 12p gain is also a common cytogenetic event in ovarian GCTs (Kraggerud et al., 2000). Gain of 12p occurs regardless of histological subtype in adult gonadal malignant GCTs. In contrast, 12p gain was initially thought to occur rarely in paediatric malignant GCTs (Collinson et al., 2014), for example described in only 1/16 (6%) of cases in a small cohort of gonadal and extragonadal tumours that exclusively comprised YST histology (Perlman et al., 2000). A larger study of 33 paediatric gonadal and extragonadal malignant GCTs identified 12p gain in just three cases (8%; one testicular and two ovarian cases) (Schneider et al., 2001). Bussey et al. (2001) identified 12p gain in 6/53 (11%) paediatric GCT cases and identified increased prevalence in male (28%) compared with female (3%) patients. More recently, a paediatric study which included a large proportion of ovarian malignant GCTs and both seminoma and YST histology, revealed 12p gain in 44% of cases, with an increasing incidence with age: 53% in those aged 5–16 years compared with 29% in those <5 years (Palmer et al., 2007). These findings suggest that genomic copy number imbalances can distinguish GCT subgroups primarily by patient age, rather than by tumour site or histology.
As the only consistent structural chromosomal abnormality in malignant GCTs of adult patients, 12p gain is implicated in invasive malignant GCT development. The observation that the pre-invasive testicular lesion intratubular germ cell neoplasia unclassified demonstrates a similar pattern of overall genomic changes to those seen in invasive TGCTs, except for 12p gain, supports this theory (Rosenberg et al., 2000; Summersgill et al., 2001; Ottesen et al., 2003). Reports suggest that 12p gain in TGCTs results in the activation of key stem cell genes which promote cellular proliferation (Korkola et al., 2006). However, the identification of which specific genes on 12p are the fundamental universal ‘drivers’ of malignant GCT pathogenesis and which are merely ‘passengers’ has been difficult to achieve. For example, adult testicular seminomas and ECs over-express stem cell genes (e.g. NANOG) located at the common region of 12p gain (12p13.31), suggesting that they are likely to be important in tumorigenesis. However, other non-seminomatous tumours, for example YSTs, which also have 12p gain, do not over-express these genes, suggesting other mechanisms must be important.
Other genomic copy number imbalances in malignant GCTs have also been described in adults. Commonly observed imbalances include gains on chromosomes 1, 7, 8, 12, 21 and X, as well as losses on 4, 5, 11, 13 and 18. However, none of these reported abnormalities are as consistent as gain of 12p (Collinson et al., 2014). A wide range of copy number imbalances has also been described in paediatric malignant tumours, including gains on chromosomes 1q, 2p, 3, 7, 8, 13, 14, 20q, 21 and X, as well as losses on 1p36, 4q, 6q, 11, 13 and 18 (Collinson et al., 2014).
In the last few years, high-resolution approaches have become available to interrogate genomic abnormalities and regions of interest in far greater detail than has previously been possible. A small study of 25 adult malignant testicular seminomas has been published using this technique, which re-confirmed the presence of 12p gain (LeBron et al., 2011). Comparison of early- and late-stage disease identified copy number variations that correlated with progression, including 4q, 5p, 9q, 13q and 20p deletions and 9q and 13q amplifications (LeBron et al., 2011). Similar studies in childhood GCTs are ongoing and are likely to reveal many novel aberrations which may account for the observed differences in clinical behaviour and outcomes of these tumours.
Protein-coding gene expression
Protein-coding transcriptomic studies have predominantly been performed in adult TGCTs. A recent review article of 23 such studies highlighted common gene changes in TGCTs, elucidating transcriptional changes associated with malignant transformation and with differentiation patterns of malignant GCTs (Alagaratnam et al., 2011). This study implicated both known (KRAS, MYCN and TPD52) and novel (CCT6A, IGFBP3 and SALL2) cancer genes in TGCT pathogenesis. Gene expression patterns in malignant GCTs characteristic of embryonic stem cells (ESCs) were confirmed and a distinctive transcriptomic programme was identified for individual histological subtypes (Alagaratnam et al., 2011). For seminoma, this included LZTS1; for EC, DNMT3B, GAL and GPC4; for CHC CGA; and for YSTs AFP, APOA2, BMP2, VTN and OTX2 (Alagaratnam et al., 2011). Similarly, global mRNA gene expression profiles in paediatric malignant GCTs completely segregated the two main histological subtypes, YSTs and seminomas (Palmer et al., 2008). Within individual histological subtypes, however, tumours shared similar protein-coding transcriptomes, regardless of whether they were gonadal or extragonadal in origin (Palmer et al., 2008). As seen for adult seminomas (Korkola et al., 2006), paediatric seminomas were enriched for genes associated with pluripotency and the undifferentiated state [e.g. NANOG, POU5F1 (OCT3/4), TFAP2C and UTF] and paediatric YSTs were associated with genes such as AFP, those involved in differentiation (KRT8, KRT19), lipid metabolism (APOA1, APOA2) and proliferation pathways (Palmer et al., 2008). Interestingly, global mRNA profiles also segregated paediatric cases from adult malignant TGCTs of the same tumour subtype, suggesting that for mRNA expression, histological subtype is the main discriminator, and then patient age (Palmer et al., 2008) (Fig.1). Alterations in hormonal status that accompany puberty may partly account for the observed differences in mRNA expression profiles between paediatric and adult GCTs. Subsequent to these studies, a prognostic mRNA gene expression signature predictive of overall survival was identified and validated in adult males with non-seminomatous malignant GCTs (Korkola et al., 2009). Future studies of GCTs affecting patients in both childhood and adolescence will need to assess whether such an ‘adult’ mRNA signature is prognostic in these younger age-groups. If not, it will be important to identify which specific biomarkers are predictive of clinical outcome in paediatric and adolescent GCTs, to improve risk stratification and management.
Figure 1.
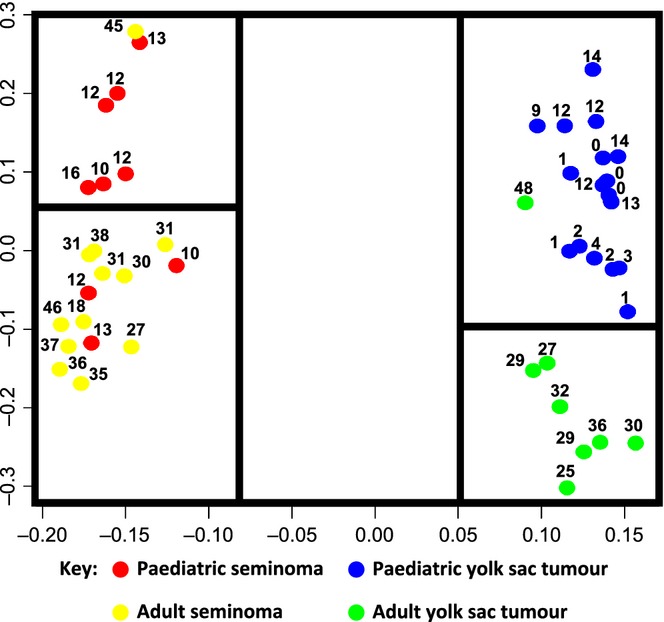
Principal component analysis of paediatric and adult malignant germ cell tumour (GCT) protein-coding gene expression data [adapted from (Palmer et al., 2008)]. Each malignant GCT sample is represented by a coloured circle; the number next to the circle shows the age of the relevant patient in years. The distance between samples represents biological (protein-coding transcriptome) differences. Consequently, those samples clustering close together demonstrate biological similarity, whereas those that are segregated are more dissimilar.
During the last decade, key insights into the molecular basis of cancer have been elucidated, resulting in a growing understanding of the complex cancer-associated signalling pathways that underlie tumour formation and progression. Among these pathways, Wnt, TGF-beta/BMP, PI3K/AKT/mTOR, RAS/RAF and VEGF signalling are of special interest, as they have driven the development of a new generation of anti-cancer drugs which target specific molecular events/mutations (Murray & Schönberger, 2014). Importantly, these pathways have also been identified as dysregulated in malignant GCTs, suggesting possible novel therapeutic targets (Murray & Schönberger, 2014). The generally more aggressive clinical behaviour of adult malignant GCTs in comparison with their paediatric counterparts is likely to be due, at least in part, to the differential expression of individual protein-coding genes within those pathways (Palmer et al., 2008). This differential gene expression may be owing to a number of factors, including genomic copy number alterations (Palmer et al., 2007) or differences in epigenetic mechanisms observed between the two age-groups of patients.
Differential expression of genes involved in cancer-associated signalling pathways has been shown between the two main pure histological subtypes of childhood malignant GCTs, namely YSTs and seminomas. An mRNA microarray analysis identified significant differential expression of genes involved in the Wnt pathway between these two subtypes (Fritsch et al., 2006). Besides WNT13, beta-catenin was also differentially expressed and showed nuclear translocation in over 50% of YSTs, in contrast to seminomas, where this occurred rarely. These differences suggested activation of Wnt signalling in YSTs. Subsequent gene expression analysis of intra- and extracellular regulators of the WNT pathway confirmed the differential expression of several genes between the two subtypes, for example SFRP2 and DKK1, predominantly owing to epigenetic mechanisms (Schönberger et al., 2010). Methylation of the DNA of these genes is therefore likely to account for the over-expression of Wnt/beta-catenin pathway genes seen in YSTs (Fritsch et al., 2006; Palmer et al., 2008). Similarly, differential protein-coding gene expression leads to activation of the TGF-beta/BMP pathway in YSTs, in contrast to undifferentiated tumours such as seminomas, where BMP pathway activity is absent (Fustino et al., 2011).
KIT and its ligand KITLG (steel factor) are not only involved in normal PGC development, resulting in oogenesis and spermatogenesis, but are also implicated in GCT development (Gilbert et al., 2011). They are known to activate the PI3K/AKT/mTOR and RAS/RAF pathway, and consequently, recent research has focused on KIT/KITLG in GCTs. Several immunohistochemical studies of adult GCTs detected KIT in seminomas in contrast to non-seminomatous tumours such as YSTs (Bokemeyer et al., 1996c1996c; Kemmer et al., 2004; Nakai et al., 2005; Biermann et al., 2007; Nikolaou et al., 2007). In addition, mutations in KIT in codon 816 (exon 17) are associated with the development of bilateral GCTs (Looijenga et al., 2003) and advanced stages of ovarian dysgerminoma (Cheng et al., 2011). Genetic and protein analysis identified different gain-of-function mutations in the KIT gene (D816V, D816H) in seminomas, resulting in phosphorylation of KIT and PI3K and therefore constitutive activation of the PI3K pathway, even in the absence of KITLG (Nakai et al., 2005). Unfortunately, although seminomas may harbour activating KIT mutations, responses in clinical studies of the KIT tyrosine kinase inhibitor imatinib mesylate have been disappointing, with no complete or even partial remissions (Einhorn et al., 2006). However, this is not entirely surprising as only those patients with mutations in exon 11 of KIT tend to respond to imatinib therapy. In vitro studies suggest that alternative tyrosine kinase inhibitors, such as dasatinib, which target exon 17 mutations, may be more promising as treatment options in vivo (Schittenhelm et al., 2006).
In many human cancers, mutations in the KRAS or BRAF gene lead to activation of the RAS/RAF pathway, resulting in over-expression of its target MAPK1 (ERK). Although MAPK1 is globally expressed in adult GCTs, mutations of KRAS and BRAF are rare events, pointing to the involvement of KIT as an upstream protein in activation of RAS/RAF signalling (McIntyre et al., 2005; Sommerer et al., 2005). Nevertheless, a European study identified that the BRAF mutation V600E is detectable in a subgroup of chemotherapy-resistant adult GCTs and correlates significantly with the presence of microsatellite instability (Honecker et al., 2009). Of note, however, this finding could not be verified in a recent US study of adult patients with malignant GCTs (Feldman et al., 2014), nor in a large cohort of paediatric GCTs (Masque-Soler et al., 2012).
Non-protein-coding gene (microRNA) expression
The identification of molecular abnormalities that are shared across the diverse spectrum of malignant GCTs is particularly important, as these are likely to be of fundamental significance in disease pathogenesis. MicroRNAs are short, non-protein-coding RNAs that regulate the expression of protein-coding genes through interactions with binding sites in the 3′ untranslated region (3′UTR) of target mRNAs, and whose expression profiles are dysregulated in cancer (Palmer et al., 2010). MicroRNAs may act directly as oncogenes or tumour suppressor genes through their interactions with mRNA targets (Palmer et al., 2010). As microRNA profiles reflect the developmental lineage of tumours (Lu et al., 2005), it was speculated that undertaking microRNA expression analysis in GCTs might identify such shared molecular abnormalities (Palmer et al., 2010).
Over-expressed tissue microRNAs
An early study revealed that the miR-371–373 microRNA cluster was highly expressed in adult TGCTs (Voorhoeve et al., 2006), suggesting that it may act as a potential novel oncogene in TGCTs via inhibition of the tumour suppressor gene LATS2 (Voorhoeve et al., 2006). Subsequently, it was confirmed that the miR-371–373 cluster was over-expressed in adult gonadal malignant GCTs, when compared with normal testis controls (Gillis et al., 2007).
Next, a global microRNA profiling study (n = 615 microRNAs) reported data from 48 paediatric samples, including gonadal and extragonadal (including intracranial) malignant GCTs (Palmer et al., 2010), and compared the profiles obtained with those from the adult gonadal GCT study described above (Gillis et al., 2007). The majority of differentially expressed microRNAs in paediatric GCTs were down-regulated (Palmer et al., 2010), consistent with observations in other malignancies (Lu et al., 2005). However, the most significant finding was that the miR-371–373 and miR-302/367 clusters were over-expressed in all malignant GCTs, independent of patient age (paediatric or adult), tumour histological subtype (YST, seminoma or EC) or anatomical site (gonadal or extragonadal) (Palmer et al., 2010). This finding was the first common biological abnormality identified in all malignant GCTs and was consistent with the presumed common origin of GCTs from PGCs (Palmer et al., 2010). Importantly for potential clinical use as highly sensitive and specific universal biomarkers of malignant GCTs, the expression levels of the eight main microRNA members from the miR-371–373 and miR-302/367 clusters accurately segregated malignant GCTs from the non-malignant group, comprising fetal and gonadal control samples and benign teratomas (Fig.2) (Palmer et al., 2010).
Figure 2.
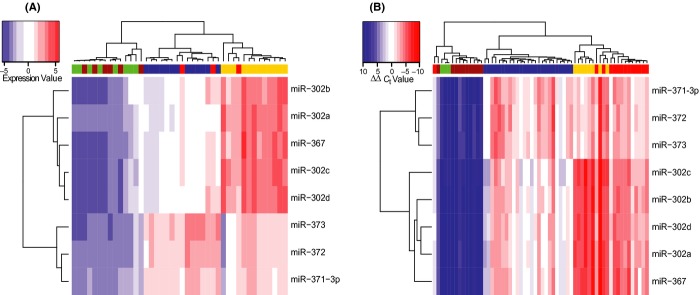
Differential expression of the miR-371–373 and miR-302/367 clusters in malignant germ cell tumours (GCTs) [adapted from (Palmer et al., 2010)]. Hierarchical clustering analysis based on the eight main microRNAs from the miR-371–373 and miR-302/367 clusters (rows) segregates (A) paediatric and (B) adult malignant GCT samples from non-malignant controls (comprising benign teratomas and normal gonadal controls) (columns). In the heatmap, red represents relative microRNA over-expression and blue represents under-expression. Green columns = normal gonadal controls; brown = teratoma; blue = seminoma; yellow = yolk sac tumour; red = embryonal carcinoma.
As both the miR-371–373 and miR-302/367 microRNA clusters are ESC-specific pluripotency markers (Suh et al., 2004; Thomson et al., 2006; Lakshmipathy et al., 2007; Barroso-del Jesus et al., 2008; Laurent, 2008), the co-ordinate expression of microRNAs from these clusters in malignant GCTs either represents the persistence of an embryonic pattern of microRNA expression that is not present in normal, somatically differentiated tissues, or acquired re-expression, regulated by an as yet undetermined mechanism(s) (Palmer et al., 2010). Of potential functional significance, six of the eight main microRNAs from the miR-371–373 clusters were noted to contain an identical ‘seed’ region at 5′ nucleotide positions 2–7 (2–7nt), critical for determining binding specificity to mRNA targets (Palmer et al., 2010). In addition to the universal miR-371–373 and miR-302/367 over-expression findings, each subtype of malignant GCT was additionally characterized by specific abnormalities of microRNA expression (Palmer et al., 2010). Interestingly, however, the striking differences in mRNA expression that were previously observed between paediatric and adult malignant GCTs (Palmer et al., 2008) were not reflected by similar differences in microRNA expression profiles (Palmer et al., 2010).
As microRNAs regulate gene expression via translational repression and mRNA destabilization (Gautier et al., 2004; Esquela-Kerscher & Slack, 2006; Giraldez et al., 2006), the latter resulting in mRNA expression changes (Lim et al., 2005; Calin & Croce, 2006), matched global mRNA profiles were ranked by expression change and then assessed by the Sylamer bioinformatic algorithm (van Dongen et al., 2008). This analysis showed that the identical seed region shared by microRNAs from the over-expressed miR-371–373 and miR-302/367 clusters resulted in a global down-regulation of target mRNAs in malignant GCTs and was therefore of functional significance (Palmer et al., 2010). Using Gene Ontology analysis, it was demonstrated that these mRNA targets mediated important cancer-associated cellular processes, such as signal transduction, cell cycle, development and morphogenesis (Palmer et al., 2010). Interestingly, the same microRNAs have been shown to be essential for regulating G1-S transition and promoting rapid proliferation in ESCs (Wang et al., 2008; Wang & Blelloch, 2009).
The profiles of the differentially expressed microRNAs that segregated the two main pure histological subtypes of malignant GCT, YST and seminoma, were very similar in both adult and paediatric patients, and may account for the observed differences in clinical outcome between the two tumour types (Murray et al., 2010). In particular, further over-expression of the miR-302/367 cluster was observed in YSTs compared with seminomas, which resulted in the down-regulation of cancer-associated protein-coding genes (Murray et al., 2010). Furthermore, in another study the relative miR-302/367 over-expression in YSTs was associated with increased bone morphogenetic protein (BMP) signalling activity in YSTs (compared with seminomas), presumably via multiple predicted mRNA targets in the transforming growth factor–beta/BMP pathway (Fustino et al., 2011).
Of note, the miR-371–373 cluster is involved in maintaining the pluripotent state in ESCs and germline stem cells, whereas miR-302/367 members are induced during the first stages of differentiation (Zovoilis et al., 2008). As miR-302/367 expression is lost in cells and tissues showing somatic differentiation (Suh et al., 2004; Barroso-del Jesus et al., 2008), it is likely that levels peak during early extraembryonic differentiation. If so, dynamic changes in miR-302/367 levels in normal embryonic development (Stadler et al., 2010) would be mirrored in GCTs showing equivalent differentiation states (Murray et al., 2010). Those GCTs with extraembryonic differentiation (i.e. YSTs and potentially CHC) would display high levels compared with undifferentiated tumours (seminomas), with a reduction to virtually undetectable levels in somatically differentiated tumours (teratomas) (Murray et al., 2010), in which microRNA profiles are almost identical to normal gonadal tissues (Palmer et al., 2010). Another study of childhood GCTs demonstrated DNA hypermethylation in YSTs compared with seminomas, coincident with higher levels of expression of the DNA methyltransferase, DNMT3B (Jeyapalan et al., 2011). Of particular interest, DNMT3B is a predicted target of the miR-29 family (miR-29a, -29b and -29c). Consistent with these observations, the miR-29 family is under-expressed in YSTs, when compared with seminomas (Palmer et al., 2010), and may account for clinical differences in behaviour.
In summary, miR-371–373 and miR-302/267 cluster over-expression occurs in all malignant GCTs, regardless of patient age, histological subtype and anatomical site of disease (Palmer et al., 2010). This is in contrast to mRNA profiles in malignant GCTs, where no such universal findings were described (Palmer et al., 2008). However, similar to mRNA profiles (Alagaratnam et al., 2011), microRNA expression differences between each individual histological subtype vs. control samples were identified, in addition to the key over-expressed miR-371–373 and miR-302/367 clusters (Palmer et al., 2010). Another similarity to protein-coding gene profiles are that there are microRNA expression differences between histological subtypes, for example seminoma and YST (Murray et al., 2010). Discordant with the findings for mRNA analysis (Fig.1), there do not appear to be any major microRNA expression differences within histological subtypes by patient age (i.e. in paediatric vs. adult samples) (Palmer et al., 2010).
Detection of serum microRNAs
Although the serum biomarkers alpha-fetoprotein (AFP) and human choriogonadotrophin (HCG) assist malignant GCT diagnosis, they have limitations in sensitivity and specificity (Murray & Nicholson, 2011). AFP is produced by YST components and HCG predominantly by CHC; consequently neither marker is raised in all cases of malignant GCT and both show elevations in non-malignant conditions (Murray & Nicholson, 2011). A biomarker that offered greater sensitivity and specificity for diagnosing or monitoring malignant GCTs would therefore be of considerable clinical value (Murray et al., 2011).
There are no reports demonstrating the clinical utility of serum levels of protein-coding mRNAs in malignant GCTs. Moreover, potential mRNA biomarkers can be subject to considerable variation in levels, for technical as well as biological reasons. In particular, mRNAs are inherently unstable at room temperature and rapidly degrade in blood-based samples that are not stored correctly (Rainen et al., 2002; Viprey et al., 2007). Furthermore, a small study describing non-protein-coding XIST transcripts in the plasma of male patients with TGCTs (Kawakami et al., 2004) had low sensitivity and has not been confirmed by others.
In contrast, microRNAs released from tumour cells into the bloodstream are stable because of protection from RNAse degradation by packaging within membrane-bound exosome particles (Caby et al., 2005; Valadi et al., 2007). MicroRNAs show stability in serum samples subjected to multiple freeze–thaw cycles (Chen et al., 2008; Mitchell et al., 2008) and in those left at room temperature prior to processing (Mitchell et al., 2008). Furthermore, a good correlation exists between levels of individual microRNAs in serum and plasma samples obtained from the same patient (Mitchell et al., 2008). As a result, blood-based microRNAs show considerable promise for cancer diagnosis and monitoring (Murray et al., 2011).
The first report of serum microRNA expression in malignant GCTs contained a detailed multiplexed qRT-PCR methodology and demonstrated elevated serum levels of all eight main members of the miR-371–373 and miR-302/367 clusters in the serum of a paediatric patient compared with pooled normal serum (Murray et al., 2011). Levels of miR-372 were over 700-fold higher at malignant GCT diagnosis and fell to normal levels during treatment and in uneventful clinical follow-up (Murray et al., 2011). Further study of malignant GCTs, including TGCTs, across a range of representative ages (paediatric and adult), anatomical sites (gonadal and extragonadal) and histological subtypes (YST, seminoma and EC) confirmed universal over-expression at diagnosis of serum levels of miR-372 and miR-367 (Fig.3) (Murray & Coleman, 2012). Importantly, the majority of the cases described were marker-negative by serum AFP and HCG estimation. There was a potential association between serum levels of both miR-372 and miR-367 and total tumour volume at diagnosis (Murray & Coleman, 2012).
Figure 3.
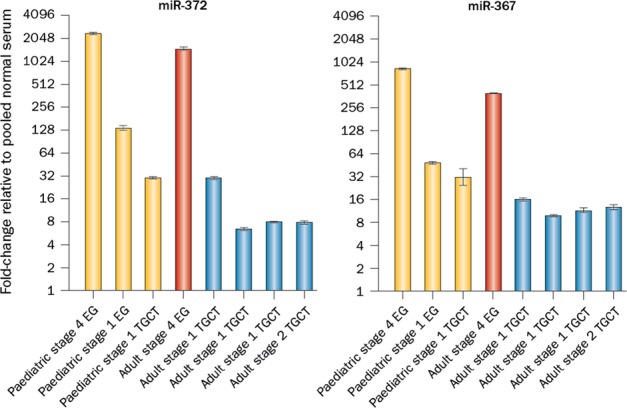
MicroRNAs from the miR-371–373 and miR-302/367 clusters as novel serum biomarkers of malignant germ cell tumours (GCTs) [adapted from (Murray & Coleman, 2012)]. Levels of miR-372 from the miR-371–373 cluster (left) and miR-367 from the miR-302/367 cluster (right) in the serum at the time of diagnosis in eight malignant GCTs of different patient age, anatomical site and histological subtype. Key: EG, extragonadal; TGCT, testicular germ cell tumour. Yellow columns = yolk sac tumour samples; red = embryonal carcinoma; blue = seminoma.
Using the same qRT-PCR methodology (Murray et al., 2011; Murray & Coleman, 2012), these initial findings have been extended. Elevated levels of the three main microRNAs from the miR-371–373 cluster were demonstrated in stage 1 TGCT patients at the time of diagnosis compared with control serum, and fell to normal levels within 5 days of orchidectomy (Belge et al., 2012; Dieckmann et al., 2012). The majority of patients described also had ‘marker-negative’ disease by serum AFP/HCG quantification (Belge et al., 2012; Dieckmann et al., 2012). In these studies, the highest serum levels were noted for miR-371a-3p, which also fell the most dramatically following treatment (Belge et al., 2012; Dieckmann et al., 2012). In addition, in advanced stage cases, serum levels of miR-371–373 microRNAs only reduced to the normal range after treatment with surgery and chemotherapy (Dieckmann et al., 2012). Importantly, for the benign teratoma cases included in the study, serum miR-371–373 levels at the time of diagnosis were in the range of the controls, demonstrating the potential clinical utility of these serum microRNAs to distinguish malignant disease from teratoma, for which the management approaches may be very different (Dieckmann et al., 2012).
Subsequently, a large collaborative serum microRNA qRT-PCR study employed a pipeline using non-human spike-in RNAs to control for technical variation in sample preparation (Gillis et al., 2013). Across all the datasets studied, a consistent, highly significant increase in the levels of four microRNAs (miR-371a-3p, miR-372, miR-373 and miR-367) was observed in serum from all patients with malignant GCTs at the time of diagnosis. Area under the curve values in receiver operator characteristic analyses were 0.88 (miR-371a-3p), 0.91 (miR-372), 0.96 (miR-373) and 0.94 (miR-367) (Gillis et al., 2013), significantly outperforming AFP and HCG. Combined use of these four microRNAs with AFP/HCG identified all diagnostic malignant GCT samples correctly. The sensitivity of each marker was between 80 and 90% at a specificity cut-off of 90% (Gillis et al., 2013).
Thus, microRNAs of the miR-371–373 and miR-302/367 clusters are emerging as promising bodyfluid biomarkers to improve clinical management of malignant GCTs. A further potential application has been identified by the recent demonstration of elevated levels of these microRNAs in the cerebrospinal fluid of patients with intracranial malignant GCTs (Terashima et al., 2013). Possible applications therefore include diagnosis of malignant GCTs in relatively inaccessible sites, such as the mediastinum, retroperitoneum or central nervous system, without the need for surgery, disease monitoring during chemotherapy and detection of subclinical tumour recurrence which may reduce the need for serial surveillance CT imaging and its inherent secondary cancer risks (Murray & Coleman, 2012). Together, the accumulating data strongly indicate that prospective studies of serum microRNAs in larger patient cohorts are warranted (Murray & Coleman, 2012).
Under-expressed tissue microRNAs
Members of the lethal-7 (let-7) family of microRNAs, which regulate cell proliferation (Johnson et al., 2007) and are important tumour suppressor genes (Takamizawa et al., 2004), were under-expressed in paediatric malignant GCTs when compared with non-malignant control tissues (Palmer et al., 2010). These microRNAs are specifically negatively regulated by the RNA binding protein LIN28 (Viswanathan et al., 2008, 2009), which is expressed at high levels in PGCs (West et al., 2009). Early studies used immunohistochemistry (Cao et al., 2011a,b; Xue et al., 2011) and RNA interference (Gillis et al., 2011) to investigate the expression and some aspects of LIN28 function in malignant GCTs. For example, LIN28 depletion in malignant GCT cells led to down-regulation of stem cell markers such as OCT4/POU5F1 and NANOG, and induction of cell differentiation (Gillis et al., 2011). However, LIN28 effects on let-7 expression were not assessed in this work (Gillis et al., 2011). Subsequently, it was shown that the low levels of let-7 observed in malignant GCTs were directly attributable to abundant LIN28 expression (Murray et al., 2013). As for miR-371–373 and miR-302/367 over-expression, let-7 under-expression in malignant GCTs was universal, occurring regardless of patient age, histological subtype or site of disease, thereby extending published reports describing predominantly or exclusively tumours from adult patients (Cao et al., 2011a,b; Gillis et al., 2011; Xue et al., 2011). Interestingly, all let-7 family members contained an identical 2–7nt seed region and the reduced abundance of this seed in malignant GCTs contributed to the subsequent up-regulation of important cancer-associated protein-coding genes, including MYCN (Murray et al., 2013) (Fig.4). Such post-transcriptional effects on MYCN levels are likely to explain why MYCN is frequently over-expressed in malignant GCTs (Alagaratnam et al., 2011) but shows copy number gain at the MYCN 2p23.4 locus in only one third of malignant GCTs from adult patients (Kraggerud et al., 2002) and less than one fifth of paediatric cases (Palmer et al., 2007).
Figure 4.
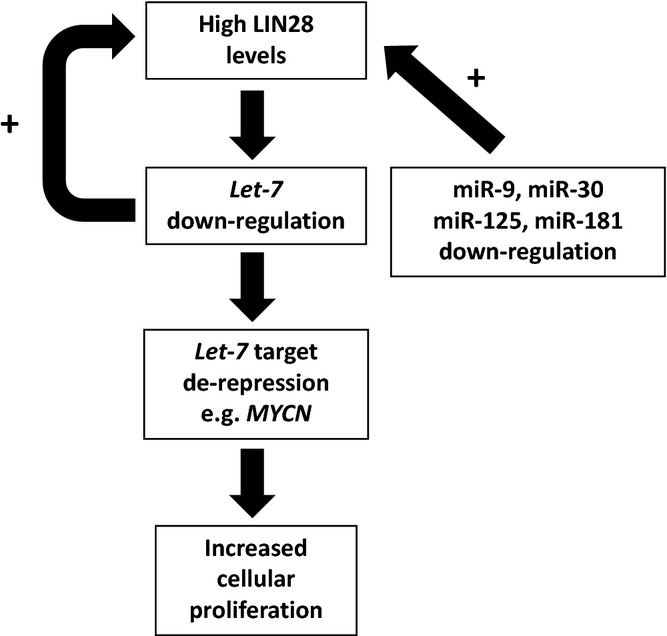
Schematic of the dysregulated LIN28/let-7 axis in malignant germ cell tumours (GCT)s. Let-7 down-regulation in malignant GCTs is attributable to abundant LIN28 expression (Murray et al., 2013). In turn, this leads to over-expression of let-7 targets (e.g. MYCN) and increased cellular proliferation. Loss of direct negative feedback from let-7 maintains high LIN28 levels and let-7 repression. Other contributions to abundant LIN28 expression include other microRNAs that bind to the 3′UTR of LIN28 transcripts, but which are down-regulated in malignant GCTs (Palmer et al., 2010). Taken together, these findings highlight the LIN28/let-7 axis as a novel therapeutic target in malignant GCTs.
In addition, low let-7 levels in malignant GCT cells resulted in increased LIN28 expression, through a binding site in the LIN28 3′UTR for the common let-7 seed region (Murray et al., 2013) (Fig.4). Other microRNAs, including miR-9 (Zhong et al., 2010), the miR-30 family (Zhong et al., 2010), miR-125 (Wu & Belasco, 2005; Zhong et al., 2010) and miR-181 (Li et al., 2012) have all been reported to down-regulate LIN28 in ESCs and cancer cells. Importantly, these microRNAs have previously been identified as being universally under-expressed in malignant GCTs (Murray et al., 2013). As copy number gain at the LIN28 locus (1p36.11) is not a feature of malignant GCTs (Palmer et al., 2007), down-regulation of these microRNAs is likely to be an important further contributor to LIN28 over-expression (Murray et al., 2013) (Fig.4). Interactions disrupting this LIN28/let-7 axis represent promising targets for novel therapies in malignant GCTs. As well as depleting LIN28, an alternative strategy is direct replacement of let-7 family members (Murray et al., 2013). Both approaches would provide a molecular ‘switch’ effect that should result in a sustained reversion of malignant GCT cell phenotype (Murray et al., 2013).
Conclusion
Our increasing knowledge of the biology of malignant GCTs needs to be harnessed to improve outcomes for both adult and paediatric patients. In particular, genomic and protein-coding transcriptomic data confirm that malignant GCTs of childhood are biologically distinct from those of adulthood and provide evidence supporting the different management approaches employed in patients of different ages. In contrast, all malignant GCTs over-express the miR-371–373 and miR-302/367 clusters regardless of patient age, histological subtype or anatomical tumour site. The detection of elevated serum levels of these microRNAs at the time of malignant GCT diagnosis, with levels falling after treatment, highlights the universal diagnostic potential of this finding. In addition, the dysregulation of the LIN28/let-7 axis in all malignant GCTs also suggests a pathway that may be a target for the development of novel therapeutic agents.
Further studies are now required, integrating genomic changes using high-resolution methods with combined analysis of mRNA and microRNA expression profiles in malignant GCTs from both adult and paediatric patients with good and adverse clinical outcomes. These approaches are likely to identify regulatory pathways and networks associated with treatment sensitivity and resistance. In particular, such studies should aim to define further the clinical relevance of age-specific biological characteristics of malignant GCTs. These insights may assist the optimal management of affected patients and identify biological targets suitable for the development of novel therapeutic agents. The ultimate aim of this approach will be to improve clinical outcomes for this under-investigated patient group, both through improved survival of patients with poor-risk disease and reduced late-effects of treatment for those with low-risk disease.
Acknowledgments
This work was funded by Cancer Research UK programme grant (NC), Medical Research Council Fellowship (MJM), Addenbrooke's Charitable Trust (MJM), SPARKS Charity (MJM/JN/NC), Children with Cancer UK/Great Ormond Street Hospital Children's Charity (MJM/JN/NC).
Conflicts of interest
None.
References
- Alagaratnam S, Lind GE, Kraggerud SM, Lothe RA. Skotheim RI. The testicular germ cell tumour transcriptome. Int J Androl. 2011;34:e133–e150. doi: 10.1111/j.1365-2605.2011.01169.x. discussion e150-131. [DOI] [PubMed] [Google Scholar]
- Atkin NB. Baker MC. Specific chromosome change, i(12p), in testicular tumours? Lancet. 1982;2:1349. doi: 10.1016/s0140-6736(82)91557-4. &. [DOI] [PubMed] [Google Scholar]
- Barroso-del Jesus A, Romero-Lopez C, Lucena-Aguilar G, Melen GJ, Sanchez L, Ligero G, Berzal-Herranz A. Menendez P. Embryonic stem cell-specific miR302-367 cluster: human gene structure and functional characterization of its core promoter. Mol Cell Biol. 2008;28:6609–6619. doi: 10.1128/MCB.00398-08. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belge G, Dieckmann KP, Spiekermann M, Balks T. Bullerdiek J. Serum levels of microRNAs miR-371-3: a novel class of serum biomarkers for testicular germ cell tumors? Eur Urol. 2012;61:1068–1069. doi: 10.1016/j.eururo.2012.02.037. &. [DOI] [PubMed] [Google Scholar]
- Biermann K, Heukamp LC, Steger K, Zhou H, Franke FE, Sonnack V, Brehm R, Berg J, Bastian PJ, Muller SC, Wang-Eckert L. Buettner R. Genome-wide expression profiling reveals new insights into pathogenesis and progression of testicular germ cell tumors. Cancer Genomics Proteomics. 2007;4:359–367. &. [PubMed] [Google Scholar]
- Bokemeyer C, Berger CC, Kuczyk MA. Schmoll HJ. Evaluation of long-term toxicity after chemotherapy for testicular cancer. J Clin Oncol. 1996a;14:2923–2932. doi: 10.1200/JCO.1996.14.11.2923. &. [DOI] [PubMed] [Google Scholar]
- Bokemeyer C, Kohrmann O, Tischler J, Weissbach L, Rath U, Haupt A, Schoffski P, Harstrick A. Schmoll HJ. A randomized trial of cisplatin, etoposide and bleomycin (PEB) versus carboplatin, etoposide and bleomycin (CEB) for patients with ‘good-risk’ metastatic non-seminomatous germ cell tumors. Ann Oncol. 1996b;7:1015–1021. doi: 10.1093/oxfordjournals.annonc.a010493. &. [DOI] [PubMed] [Google Scholar]
- Bokemeyer C, Kuczyk MA, Dunn T, Serth J, Hartmann K, Jonasson J, Pietsch T, Jonas U. Schmoll HJ. Expression of stem-cell factor and its receptor c-kit protein in normal testicular tissue and malignant germ-cell tumours. J Cancer Res Clin Oncol. 1996c;122:301–306. doi: 10.1007/BF01261407. &. [DOI] [PubMed] [Google Scholar]
- Bosl GJ, Leitner SP, Atlas SA, Sealey JE, Preibisz JJ. Scheiner E. Increased plasma renin and aldosterone in patients treated with cisplatin-based chemotherapy for metastatic germ-cell tumors. J Clin Oncol. 1986;4:1684–1689. doi: 10.1200/JCO.1986.4.11.1684. &. [DOI] [PubMed] [Google Scholar]
- Bussey KJ, Lawce HJ, Himoe E, Shu XO, Suijkerbuijk RF, Olson SB. Magenis RE. Chromosomes 1 and 12 abnormalities in pediatric germ cell tumors by interphase fluorescence in situ hybridization. Cancer Genet Cytogenet. 2001;125:112–118. doi: 10.1016/s0165-4608(00)00380-0. &. [DOI] [PubMed] [Google Scholar]
- Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G. Bonnerot C. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17:879–887. doi: 10.1093/intimm/dxh267. &. [DOI] [PubMed] [Google Scholar]
- Calin GA. Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. &. [DOI] [PubMed] [Google Scholar]
- Cao D, Allan RW, Cheng L, Peng Y, Guo CC, Dahiya N, Akhi S. Li J. RNA-binding protein LIN28 is a marker for testicular germ cell tumors. Hum Pathol. 2011a;42:710–718. doi: 10.1016/j.humpath.2010.09.007. &. [DOI] [PubMed] [Google Scholar]
- Cao D, Liu A, Wang F, Allan RW, Mei K, Peng Y, Du J, Guo S, Abel TW, Lane Z, Ma J, Rodriguez M, Akhi S, Dehiya N. Li J. RNA-binding protein LIN28 is a marker for primary extragonadal germ cell tumors: an immunohistochemical study of 131 cases. Mod Pathol. 2011b;24:288–296. doi: 10.1038/modpathol.2010.195. &. [DOI] [PubMed] [Google Scholar]
- Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, Guo J, Zhang Y, Chen J, Guo X, Li Q, Li X, Wang W, Zhang Y, Wang J, Jiang X, Xiang Y, Xu C, Zheng P, Zhang J, Li R, Zhang H, Shang X, Gong T, Ning G, Wang J, Zen K, Zhang J. Zhang CY. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006. doi: 10.1038/cr.2008.282. &. [DOI] [PubMed] [Google Scholar]
- Cheng L, Roth LM, Zhang S, Wang M, Morton MJ, Zheng W, Abdul Karim FW, Montironi R. Lopez-Beltran A. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer. 2011;117:2096–2103. doi: 10.1002/cncr.25794. &. [DOI] [PubMed] [Google Scholar]
- Collinson K, Murray MJ, Orsi NM, Cummings M, Shipley J, Joffe JK, Coleman N. Stark D. Age-related biological features of germ cell tumors. Genes Chromosomes Cancer. 2014;53:215–227. doi: 10.1002/gcc.22131. &. [DOI] [PubMed] [Google Scholar]
- Dieckmann KP, Spiekermann M, Balks T, Flor I, Loning T, Bullerdiek J. Belge G. MicroRNAs miR-371-3 in serum as diagnostic tools in the management of testicular germ cell tumours. Br J Cancer. 2012;107:1754–1760. doi: 10.1038/bjc.2012.469. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S, Abreu-Goodger C. Enright AJ. Detecting microRNA binding and siRNA off-target effects from expression data. Nat Methods. 2008;5:1023–1025. doi: 10.1038/nmeth.1267. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einhorn LH. Donohue JP. Improved chemotherapy in disseminated testicular cancer. J Urol. 1977;117:65–69. doi: 10.1016/s0022-5347(17)58338-x. &. [DOI] [PubMed] [Google Scholar]
- Einhorn LH, Brames MJ, Heinrich MC, Corless CL. Madani A. Phase II study of imatinib mesylate in chemotherapy refractory germ cell tumors expressing KIT. Am J Clin Oncol. 2006;29:12–13. doi: 10.1097/01.coc.0000195086.47548.ef. &. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A. Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. &. [DOI] [PubMed] [Google Scholar]
- Feldman DR, Iyer G, Van Alstine L, Patil S, Al-Ahmadie HA, Reuter VE, Bosl GJ, Chaganti RS. Solit DB. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin Cancer Res. 2014;20:3712–3720. doi: 10.1158/1078-0432.CCR-13-2868. &. [DOI] [PubMed] [Google Scholar]
- Fritsch MK, Schneider DT, Schuster AE, Murdoch FE. Perlman EJ. Activation of Wnt/beta-catenin signaling in distinct histologic subtypes of human germ cell tumors. Pediatr Dev Pathol. 2006;9:115–131. doi: 10.2350/08-05-0097.1. &. [DOI] [PubMed] [Google Scholar]
- Fustino N, Rakheja D, Ateek CS, Neumann JC. Amatruda JF. Bone morphogenetic protein signalling activity distinguishes histological subsets of paediatric germ cell tumours. Int J Androl. 2011;34:e218–e233. doi: 10.1111/j.1365-2605.2011.01186.x. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM. Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. &. [DOI] [PubMed] [Google Scholar]
- Gilbert D, Rapley E. Shipley J. Testicular germ cell tumours: predisposition genes and the male germ cell niche. Nat Rev Cancer. 2011;11:278–288. doi: 10.1038/nrc3021. &. [DOI] [PubMed] [Google Scholar]
- Gillis AJM, Stoop HJ, Hersmus R, Oosterhuis JW, Sun Y, Chen C, Guenther S, Sherlock J, Veltman I, Baeten J, van der Spek PJ, de Alarcon P. Looijenga LHJ. High-throughput microRNAome analysis in human germ cell tumours. J Pathol. 2007;213:319–328. doi: 10.1002/path.2230. &. [DOI] [PubMed] [Google Scholar]
- Gillis AJ, Stoop H, Biermann K, van Gurp RJ, Swartzman E, Cribbes S, Ferlinz A, Shannon M, Oosterhuis JW. Looijenga LH. Expression and interdependencies of pluripotency factors LIN28, OCT3/4, NANOG and SOX2 in human testicular germ cells and tumours of the testis. Int J Androl. 2011;34:e160–e174. doi: 10.1111/j.1365-2605.2011.01148.x. &. [DOI] [PubMed] [Google Scholar]
- Gillis AJ, Rijlaarsdam MA, Eini R, Dorssers LC, Biermann K, Murray MJ, Nicholson JC, Coleman N, Dieckmann KP, Belge G, Bullerdiek J, Xu T, Bernard N. Looijenga LH. Targeted serum miRNA (TSmiR) test for diagnosis and follow-up of (testicular) germ cell cancer patients: a proof of principle. Mol Oncol. 2013;7:1083–1092. doi: 10.1016/j.molonc.2013.08.002. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ. Schier AF. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–79. doi: 10.1126/science.1122689. &. [DOI] [PubMed] [Google Scholar]
- Glendenning JL, Barbachano Y, Norman AR, Dearnaley DP, Horwich A. Huddart RA. Long-term neurologic and peripheral vascular toxicity after chemotherapy treatment of testicular cancer. Cancer. 2010;116:2322–2331. doi: 10.1002/cncr.24981. &. [DOI] [PubMed] [Google Scholar]
- Honecker F, Wermann H, Mayer F, Gillis AJ, Stoop H, van Gurp RJ, Oechsle K, Steyerberg E, Hartmann JT, Dinjens WN, Oosterhuis JW, Bokemeyer C. Looijenga LH. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J Clin Oncol. 2009;27:2129–2136. doi: 10.1200/JCO.2008.18.8623. &. [DOI] [PubMed] [Google Scholar]
- Horwich A, Sleijfer DT, Fossa SD, Kaye SB, Oliver RT, Cullen MH, Mead GM, de Wit R, de Mulder PH, Dearnaley DP, Cook PA, Sylvester RJ. Stenning SP. Randomized trial of bleomycin, etoposide, and cisplatin compared with bleomycin, etoposide, and carboplatin in good-prognosis metastatic nonseminomatous germ cell cancer: a Multiinstitutional Medical Research Council/European Organization for Research and Treatment of Cancer Trial. J Clin Oncol. 1997;15:1844–1852. doi: 10.1200/JCO.1997.15.5.1844. &. [DOI] [PubMed] [Google Scholar]
- Huddart RA, Norman A, Shahidi M, Horwich A, Coward D, Nicholls J. Dearnaley DP. Cardiovascular disease as a long-term complication of treatment for testicular cancer. J Clin Oncol. 2003;21:1513–1523. doi: 10.1200/JCO.2003.04.173. &. [DOI] [PubMed] [Google Scholar]
- International Germ Cell Cancer Collaborative Group. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. J Clin Oncol. 1997;15:594–603. doi: 10.1200/JCO.1997.15.2.594. [DOI] [PubMed] [Google Scholar]
- Jeyapalan JN, Noor DA, Lee SH, Tan CL, Appleby VA, Kilday JP, Palmer RD, Schwalbe EC, Clifford SC, Walker DA, Murray MJ, Coleman N, Nicholson JC. Scotting PJ. Methylator phenotype of malignant germ cell tumours in children identifies strong candidates for chemotherapy resistance. Br J Cancer. 2011;105:575–585. doi: 10.1038/bjc.2011.218. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, Wilson M, Wang X, Shelton J, Shingara J, Chin L, Brown D. Slack FJ. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67:7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. &. [DOI] [PubMed] [Google Scholar]
- Kawakami T, Okamoto K, Ogawa O. Okada Y. XIST unmethylated DNA fragments in male-derived plasma as a tumour marker for testicular cancer. Lancet. 2004;363:40–42. doi: 10.1016/S0140-6736(03)15170-7. &. [DOI] [PubMed] [Google Scholar]
- Kemmer K, Corless CL, Fletcher JA, McGreevey L, Haley A, Griffith D, Cummings OW, Wait C, Town A. Heinrich MC. KIT mutations are common in testicular seminomas. Am J Pathol. 2004;164:305–313. doi: 10.1016/S0002-9440(10)63120-3. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkola JE, Houldsworth J, Chadalavada RS, Olshen AB, Dobrzynski D, Reuter VE, Bosl GJ. Chaganti RS. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66:820–827. doi: 10.1158/0008-5472.CAN-05-2445. &. [DOI] [PubMed] [Google Scholar]
- Korkola JE, Houldsworth J, Feldman DR, Olshen AB, Qin LX, Patil S, Reuter VE, Bosl GJ. Chaganti RS. Identification and validation of a gene expression signature that predicts outcome in adult men with germ cell tumors. J Clin Oncol. 2009;27:5240–5247. doi: 10.1200/JCO.2008.20.0386. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraggerud SM, Szymanska J, Abeler VM, Kaern J, Eknaes M, Heim S, Teixeira MR, Trope CG, Peltomaki P. Lothe RA. DNA copy number changes in malignant ovarian germ cell tumors. Cancer Res. 2000;60:3025–3030. &. [PubMed] [Google Scholar]
- Kraggerud SM, Skotheim RI, Szymanska J, Eknaes M, Fossa SD, Stenwig AE, Peltomaki P. Lothe RA. Genome profiles of familial/bilateral and sporadic testicular germ cell tumors. Genes Chromosom Cancer. 2002;34:168–174. doi: 10.1002/gcc.10058. &. [DOI] [PubMed] [Google Scholar]
- Lakshmipathy U, Love B, Goff LA, Jornsten R, Graichen R, Hart RP. Chesnut JD. MicroRNA expression pattern of undifferentiated and differentiated human embryonic stem cells. Stem Cells Dev. 2007;16:1003–1016. doi: 10.1089/scd.2007.0026. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent LC. MicroRNAs in embryonic stem cells and early embryonic development. J Cell Mol Med. 2008;12:2181–2188. doi: 10.1111/j.1582-4934.2008.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBron C, Pal P, Brait M, Dasgupta S, Guerrero-Preston R, Looijenga LH, Kowalski J, Netto G. Hoque MO. Genome-wide analysis of genetic alterations in testicular primary seminoma using high resolution single nucleotide polymorphism arrays. Genomics. 2011;97:341–349. doi: 10.1016/j.ygeno.2011.02.011. &. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang J, Gao L, McClellan S, Finan MA, Butler TW, Owen LB, Piazza GA. Xi Y. MiR-181 mediates cell differentiation by interrupting the Lin28 and let-7 feedback circuit. Cell Death Differ. 2012;19:378–386. doi: 10.1038/cdd.2011.127. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS. Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. &. [DOI] [PubMed] [Google Scholar]
- Looijenga LH, de Leeuw H, van Oorschot M, van Gurp RJ, Stoop H, Gillis AJ, de Gouveia Brazao CA, Weber RF, Kirkels WJ, van Dijk T, von Lindern M, Valk P, Lajos G, Olah E, Nesland JM, Fossa SD. Oosterhuis JW. Stem cell factor receptor (c-KIT) codon 816 mutations predict development of bilateral testicular germ-cell tumors. Cancer Res. 2003;63:7674–7678. &. [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR. Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. &. [DOI] [PubMed] [Google Scholar]
- Mann JR, Raafat F, Robinson K, Imeson J, Gornall P, Sokal M, Gray E, McKeever P, Hale J, Bailey S. Oakhill A. The United Kingdom Children's Cancer Study Group's second germ cell tumor study: carboplatin, etoposide, and bleomycin are effective treatment for children with malignant extracranial germ cell tumors, with acceptable toxicity. J Clin Oncol. 2000;18:3809–3818. doi: 10.1200/JCO.2000.18.22.3809. &. [DOI] [PubMed] [Google Scholar]
- Masque-Soler N, Szczepanowski M, Leuschner I, Vokuhl C, Haag J, Calaminus G. Klapper W. Absence of BRAF mutation in pediatric and adolescent germ cell tumors indicate biological differences to adult tumors. Pediatr Blood Cancer. 2012;59:732–735. doi: 10.1002/pbc.24005. &. [DOI] [PubMed] [Google Scholar]
- McIntyre A, Summersgill B, Spendlove HE, Huddart R, Houlston R. Shipley J. Activating mutations and/or expression levels of tyrosine kinase receptors GRB7, RAS, and BRAF in testicular germ cell tumors. Neoplasia. 2005;7:1047–1052. doi: 10.1593/neo.05514. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB. Tewari M. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray MJ. Coleman N. Testicular cancer: a new generation of biomarkers for malignant germ cell tumours. Nat Rev Urol. 2012;9:298–300. doi: 10.1038/nrurol.2012.86. &. [DOI] [PubMed] [Google Scholar]
- Murray MJ. Nicholson JC. Germ cell tumours in children and adolescents. Paediatrics and Child Health. 2010;20:109–116. &. [Google Scholar]
- Murray MJ. Nicholson JC. alpha-Fetoprotein. Arch Dis Child Educ Pract Ed. 2011;96:141–147. doi: 10.1136/adc.2011.213181. &. [DOI] [PubMed] [Google Scholar]
- Murray MJ. Schönberger S. Biology of germ cell tumors. In: Amatruda J, editor; Frazier A, editor. Pediatric Germ Cell Tumors. Berlin, Heidelberg: Springer; 2014. pp. 1–15. [Google Scholar]
- Murray MJ, Fern LA, Stark DP, Eden TO. Nicholson JC. Breaking down barriers: improving outcomes for teenagers and young adults with germ cell tumours. Oncol Rev. 2009;3:201–206. &. [Google Scholar]
- Murray MJ, Saini HK, van Dongen S, Palmer RD, Muralidhar B, Pett MR, Piipari M, Thornton CM, Nicholson JC, Enright AJ. Coleman N. The two most common histological subtypes of malignant germ cell tumour are distinguished by global microRNA profiles, associated with differential transcription factor expression. Mol Cancer. 2010;9:290. doi: 10.1186/1476-4598-9-290. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray MJ, Halsall DJ, Hook CE, Williams DM, Nicholson JC. Coleman N. Identification of microRNAs from the miR-371∽373 and miR-302 clusters as potential serum biomarkers of malignant germ cell tumors. Am J Clin Pathol. 2011;135:119–125. doi: 10.1309/AJCPOE11KEYZCJHT. &. [DOI] [PubMed] [Google Scholar]
- Murray MJ, Saini HK, Siegler CA, Hanning JE, Barker EM, van Dongen S, Ward DM, Raby KL, Groves IJ, Scarpini CG, Pett MR, Thornton CM, Enright AJ, Nicholson JC. Coleman N. LIN28 Expression in malignant germ cell tumors downregulates let-7 and increases oncogene levels. Cancer Res. 2013;73:4872–4884. doi: 10.1158/0008-5472.CAN-12-2085. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y, Nonomura N, Oka D, Shiba M, Arai Y, Nakayama M, Inoue H, Nishimura K, Aozasa K, Mizutani Y, Miki T. Okuyama A. KIT (c-kit oncogene product) pathway is constitutively activated in human testicular germ cell tumors. Biochem Biophys Res Commun. 2005;337:289–296. doi: 10.1016/j.bbrc.2005.09.042. &. [DOI] [PubMed] [Google Scholar]
- Nikolaou M, Valavanis C, Aravantinos G, Fountzilas G, Tamvakis N, Lekka I, Arapantoni-Dadioti P, Zizi A, Ghiconti I, Economopoulos T. Pectasides D. Kit expression in male germ cell tumors. Anticancer Res. 2007;27:1685–1688. &. [PubMed] [Google Scholar]
- Oosterhuis JW. Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer. 2005;5:210–222. doi: 10.1038/nrc1568. &. [DOI] [PubMed] [Google Scholar]
- Osanto S, Bukman A, Van Hoek F, Sterk PJ, De Laat JA. Hermans J. Long-term effects of chemotherapy in patients with testicular cancer. J Clin Oncol. 1992;10:574–579. doi: 10.1200/JCO.1992.10.4.574. &. [DOI] [PubMed] [Google Scholar]
- Ottesen AM, Skakkebaek NE, Lundsteen C, Leffers H, Larsen J. Rajpert-De Meyts E. High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosom Cancer. 2003;38:117–125. doi: 10.1002/gcc.10244. &. [DOI] [PubMed] [Google Scholar]
- Palmer RD, Foster NA, Vowler SL, Roberts I, Thornton CM, Hale JP, Schneider DT, Nicholson JC. Coleman N. Malignant germ cell tumours of childhood: new associations of genomic imbalance. Br J Cancer. 2007;96:667–676. doi: 10.1038/sj.bjc.6603602. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RD, Barbosa-Morais NL, Gooding EL, Muralidhar B, Thornton CM, Pett MR, Roberts I, Schneider DT, Thorne N, Tavare S, Nicholson JC. Coleman N. Pediatric malignant germ cell tumors show characteristic transcriptome profiles. Cancer Res. 2008;68:4239–4247. doi: 10.1158/0008-5472.CAN-07-5560. &. [DOI] [PubMed] [Google Scholar]
- Palmer RD, Murray MJ, Saini HK, van Dongen S, Abreu-Goodger C, Muralidhar B, Pett MR, Thornton CM, Nicholson JC, Enright AJ. Coleman N. Malignant germ cell tumors display common microRNA profiles resulting in global changes in expression of messenger RNA targets. Cancer Res. 2010;70:2911–2923. doi: 10.1158/0008-5472.CAN-09-3301. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman EJ, Hu J, Ho D, Cushing B, Lauer S. Castleberry RP. Genetic analysis of childhood endodermal sinus tumors by comparative genomic hybridization. J Pediatr Hematol Oncol. 2000;22:100–105. doi: 10.1097/00043426-200003000-00003. &. [DOI] [PubMed] [Google Scholar]
- Rainen L, Oelmueller U, Jurgensen S, Wyrich R, Ballas C, Schram J, Herdman C, Bankaitis-Davis D, Nicholls N, Trollinger D. Tryon V. Stabilization of mRNA expression in whole blood samples. Clin Chem. 2002;48:1883–1890. &. [PubMed] [Google Scholar]
- Rosenberg C, Van Gurp RJ, Geelen E, Oosterhuis JW. Looijenga LH. Overrepresentation of the short arm of chromosome 12 is related to invasive growth of human testicular seminomas and nonseminomas. Oncogene. 2000;19:5858–5862. doi: 10.1038/sj.onc.1203950. &. [DOI] [PubMed] [Google Scholar]
- Schittenhelm MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, Bokemeyer C, Deininger MW, Druker BJ. Heinrich MC. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–481. doi: 10.1158/0008-5472.CAN-05-2050. &. [DOI] [PubMed] [Google Scholar]
- Schneider DT, Schuster AE, Fritsch MK, Calaminus G, Harms D, Gobel U. Perlman EJ. Genetic analysis of childhood germ cell tumors with comparative genomic hybridization. Klin Padiatr. 2001;213:204–211. doi: 10.1055/s-2001-16852. &. [DOI] [PubMed] [Google Scholar]
- Schönberger S, Okpanyi V, Alemazkour K, Looijenga LH, Nicholson JC, Borkhardt A. Schneider DT. Extracellular regulators of the WNT signalling pathway in childhood germ cell tumors: methylation of the SFRP2 promoter leads to WNT activation and β-catenin accumulation. Pediatr Blood Cancer. 2010;55:804. &. [Google Scholar]
- Sommerer F, Hengge UR, Markwarth A, Vomschloss S, Stolzenburg JU, Wittekind C. Tannapfel A. Mutations of BRAF and RAS are rare events in germ cell tumours. Int J Cancer. 2005;113:329–335. doi: 10.1002/ijc.20567. &. [DOI] [PubMed] [Google Scholar]
- Stadler B, Ivanovska I, Mehta K, Song S, Nelson A, Tan Y, Mathieu J, Darby C, Blau CA, Ware C, Peters G, Miller DG, Shen L, Cleary MA. Ruohola-Baker H. Characterization of microRNAs involved in embryonic stem cell states. Stem Cells Dev. 2010;19:935–950. doi: 10.1089/scd.2009.0426. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoneham SJ, Hale JP, Rodriguez-Galindo C, Dang H, Olson T, Murray M, Amatruda JF, Thornton C, Arul GS, Billmire D, Krailo M, Stark D, Covens A, Hurteau J, Stenning S, Nicholson JC, Gershenson D. Frazier AL. Adolescents and young adults with a “rare” cancer: getting past semantics to optimal care for patients with germ cell tumors. Oncologist. 2014;19:689–692. doi: 10.1634/theoncologist.2014-0009. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumberg D, Brugge S, Korn MW, Koeppen S, Ranft J, Scheiber G, Reiners C, Mockel C, Seeber S. Scheulen ME. Evaluation of long-term toxicity in patients after cisplatin-based chemotherapy for non-seminomatous testicular cancer. Ann Oncol. 2002;13:229–236. doi: 10.1093/annonc/mdf058. &. [DOI] [PubMed] [Google Scholar]
- Suh MR, Lee Y, Kim JY, Kim SK, Moon SH, Lee JY, Cha KY, Chung HM, Yoon HS, Moon SY, Kim VN. Kim KS. Human embryonic stem cells express a unique set of microRNAs. Dev Biol. 2004;270:488–498. doi: 10.1016/j.ydbio.2004.02.019. &. [DOI] [PubMed] [Google Scholar]
- Summersgill B, Osin P, Lu YJ, Huddart R. Shipley J. Chromosomal imbalances associated with carcinoma in situ and associated testicular germ cell tumours of adolescents and adults. Br J Cancer. 2001;85:213–220. doi: 10.1054/bjoc.2001.1889. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T. Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. &. [DOI] [PubMed] [Google Scholar]
- Teilum G. Classification of endodermal sinus tumour (mesoblatoma vitellinum) and so-called “embryonal carcinoma” of the ovary. Acta Pathol Microbiol Scand. 1965;64:407–429. doi: 10.1111/apm.1965.64.4.407. [DOI] [PubMed] [Google Scholar]
- Terashima K, Shen J, Luan J, Yu A, Yamaguchi S, Suzuki T, Nishikawa R, Matsutani M, Liang Y, Man T-K. Lau C. MicroRNA 371–373 and 302a in cerebrospinal fluid are potential tumour-derived biomarkers for intracranial germ cell tumours. Br J Neurosurg. 2013;27:e1–e25. &. [Google Scholar]
- Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T. Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis LB, Curtis RE, Storm H, Hall P, Holowaty E, Van Leeuwen FE, Kohler BA, Pukkala E, Lynch CF, Andersson M, Bergfeldt K, Clarke EA, Wiklund T, Stoter G, Gospodarowicz M, Sturgeon J, Fraumeni JF., Jr Boice JD., Jr Risk of second malignant neoplasms among long-term survivors of testicular cancer. J Natl Cancer Inst. 1997;89:1429–1439. doi: 10.1093/jnci/89.19.1429. &. [DOI] [PubMed] [Google Scholar]
- Travis LB, Fossa SD, Schonfeld SJ, McMaster ML, Lynch CF, Storm H, Hall P, Holowaty E, Andersen A, Pukkala E, Andersson M, Kaijser M, Gospodarowicz M, Joensuu T, Cohen RJ, Boice JD, Jr, Dores GM. Gilbert ES. Second cancers among 40,576 testicular cancer patients: focus on long-term survivors. J Natl Cancer Inst. 2005;97:1354–1365. doi: 10.1093/jnci/dji278. &. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ. Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. &. [DOI] [PubMed] [Google Scholar]
- Viprey VF, Corrias MV, Kagedal B, Oltra S, Swerts K, Vicha A, Ladenstein R. Burchill SA. Standardisation of operating procedures for the detection of minimal disease by QRT-PCR in children with neuroblastoma: quality assurance on behalf of SIOPEN-R-NET. Eur J Cancer. 2007;43:341–350. doi: 10.1016/j.ejca.2006.08.007. &. [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ. Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O'Sullivan M, Lu J, Phillips LA, Lockhart VL, Shah SP, Tanwar PS, Mermel CH, Beroukhim R, Azam M, Teixeira J, Meyerson M, Hughes TP, Llovet JM, Radich J, Mullighan CG, Golub TR, Sorensen PH. Daley GQ. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–848. doi: 10.1038/ng.392. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhoeve PM, le Sage C, Schrier M, Gillis AJM, Stoop H, Nagel R, Liu Y-P, van Duijse J, Drost J, Griekspoor A, Zlotorynski E, Yabuta N, De Vita G, Nojima H, Looijenga LHJ. Agami R. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. &. [DOI] [PubMed] [Google Scholar]
- Wang Y. Blelloch R. Cell cycle regulation by microRNAs in embryonic stem cells. Cancer Res. 2009;69:4093–4096. doi: 10.1158/0008-5472.CAN-09-0309. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L. Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Viswanathan SR, Yabuuchi A, Cunniff K, Takeuchi A, Park IH, Sero JE, Zhu H, Perez-Atayde A, Frazier AL, Surani MA. Daley GQ. A role for Lin28 in primordial germ-cell development and germ-cell malignancy. Nature. 2009;460:909–913. doi: 10.1038/nature08210. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SD, Birch R, Einhorn LH, Irwin L, Greco FA. Loehrer PJ. Treatment of disseminated germ-cell tumors with cisplatin, bleomycin, and either vinblastine or etoposide. N Engl J Med. 1987;316:1435–1440. doi: 10.1056/NEJM198706043162302. &. [DOI] [PubMed] [Google Scholar]
- Wu L. Belasco JG. Micro-RNA regulation of the mammalian lin-28 gene during neuronal differentiation of embryonal carcinoma cells. Mol Cell Biol. 2005;25:9198–9208. doi: 10.1128/MCB.25.21.9198-9208.2005. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D, Peng Y, Wang F, Allan RW. Cao D. RNA-binding protein LIN28 is a sensitive marker of ovarian primitive germ cell tumours. Histopathology. 2011;59:452–459. doi: 10.1111/j.1365-2559.2011.03949.x. &. [DOI] [PubMed] [Google Scholar]
- Zhong X, Li N, Liang S, Huang Q, Coukos G. Zhang L. Identification of microRNAs regulating reprogramming factor LIN28 in embryonic stem cells and cancer cells. J Biol Chem. 2010;285:41961–41971. doi: 10.1074/jbc.M110.169607. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovoilis A, Nolte J, Drusenheimer N, Zechner U, Hada H, Guan K, Hasenfuss G, Nayernia K. Engel W. Multipotent adult germline stem cells and embryonic stem cells have similar microRNA profiles. Mol Hum Reprod. 2008;14:521–529. doi: 10.1093/molehr/gan044. &. [DOI] [PubMed] [Google Scholar]