

Abstract
The public health and economic burden of heart failure (HF) is staggering and the need for relevant pathophysiologic and clinical biomarkers to advance the field and improve HF therapy remains high. Renal dysfunction is common among HF patients and is associated with increased HF hospitalization and mortality. It is widely recognized that mechanisms contributing to HF pathogenesis include a complex bidirectional interaction between the kidney and heart, encompassed by the term cardiorenal syndrome (CRS). Among a new wave of urinary biomarkers germane to CRS, C-type natriuretic peptide (CNP) has emerged as an innovative biomarker of renal structural and functional impairment in HF and chronic renal disease states. CNP is a hormone, synthesized in the kidney, and is an important regulator of cell proliferation and organ fibrosis. Hypoxia, cytokines and fibrotic growth factors, which are inherent to both cardiac and renal remodeling processes, are among the recognized stimuli for CNP production and release. In this review we aim to highlight current knowledge regarding the biology and pathophysiologic correlates of urinary CNP, and its potential clinical utility as a diagnostic and prognostic biomarker in HF and renal disease states.
Keywords: C-type natriuretic peptide, biomarker, heart failure, cardiorenal syndrome, renal remodeling
1. Introduction
There are an estimated 23 million patients with heart failure (HF) worldwide and, as the elderly population increases, this prevalence is projected to rise [1, 2]. Despite greater utilization of current HF therapies and modest survival gains, the absolute mortality remains sobering: 50% of HF patients die within 5 years of diagnosis [3]. Thus, there remains a critical need for additional pathophysiologic and clinical insights to identify unresolved issues, improve the application of existing therapies, and inform the development of novel HF management strategies.
2. The cardiorenal axis in heart failure
Renal dysfunction is extremely common among HF patients and is associated with increased HF hospitalization and mortality [4, 5]. The term cardiorenal syndrome (CRS) has been used to describe the complex interaction whereby acute or chronic cardiac dysfunction can precipitate acute kidney injury (type I CRS) or chronic kidney disease (type II CRS) respectively [6]. Subsequent development of moderate to severe renal dysfunction marks an advanced stage of HF. Importantly however, worsening renal function and chronic kidney disease may also promote cardiac remodeling (types III and IV CRS) and increase the risk of adverse events [6]. Therefore, alterations in renal structure and function become relevant to all aspects of HF including pathogenesis, progression, decompensation and ensuing complications.
Timely recognition and optimal treatment of CRS have been identified as key evidence gaps in contemporary HF management guidelines [7]. Challenges arise because renal dysfunction may involve a combination of lesions within glomerular, tubulointerstitial, and vascular compartments of the kidney, while frequently only parameters of glomerular function are measured. Worsening renal function is generally defined by an increase in serum creatinine or reduction in glomerular filtration rate (GFR), which reflects a late decline in renal function and precludes early identification. Likewise, treatment of CRS is hampered by limited differentiation between transient but potentially cardio- and reno-protective increases in serum creatinine, related to diuretics (hemoconcentration) [8], angiotensin-converting enzyme (ACE) inhibitors, or angiotensin receptor blockers (ARBs) [9, 10], versus a deleterious increase in creatinine due to progressive renal remodeling and fibrosis.
Direct measurement of proteins in the urine, as compared to serological assessment, has the potential to offer earlier and more specific insight into intrinsic renal injury and reparative processes. A number of novel urinary biomarkers have been proposed to detect renal tubular damage, including kidney injury molecule 1 (KIM-1), neutrophil gelatinase-associated lipocalin (NGAL), and N-acetyl-β-D-glucosaminidase (NAG), which are elevated in the urine of HF patients before a rise in serum creatinine. However variable and modest correlations have been observed with clinical outcomes [11-14]. Given the prominent role of cardiac natriuretic peptides in the serologic diagnosis of HF and the sensitivity of NT-pro-B type natriuretic peptide (NT-proBNP) for cardiac stress, injury and remodeling, there has been increasing interest in a role for renal-derived C-type natriuretic peptide (CNP) as a urinary biomarker of renal dysfunction and chronic renal remodeling in HF and CRS. In this review we aim to highlight current knowledge regarding the biology and pathophysiologic correlates of urinary CNP, and its potential clinical utility in the diagnosis and management of HF and CRS.
3. C-type natriuretic peptide biology
3.1. Discovery and processing of CNP
CNP was first isolated from porcine brain in 1990 [15]; though subsequent studies have demonstrated the highest levels of CNP expression and production in the kidney [16-18]. CNP expression has also been detected in cardiomyocytes [19], vascular endothelium [20], and bone [21]. Among the family of natriuretic peptides, CNP is the most highly conserved between species and may in fact represent the ancestral precursor from which atrial (ANP) and B-type (BNP) natriuretic peptides evolved [22, 23]. However, whereas ANP and BNP are predominantly expressed in atrial and ventricular myocardium respectively, cardiac expression of CNP is typically low [24]. Furthermore, in the absence of disease, plasma levels of CNP are significantly lower than those of ANP and BNP, giving rise to the notion that CNP principally operates as an autocrine or paracrine factor [25-27] (Figure 1).
Figure 1.
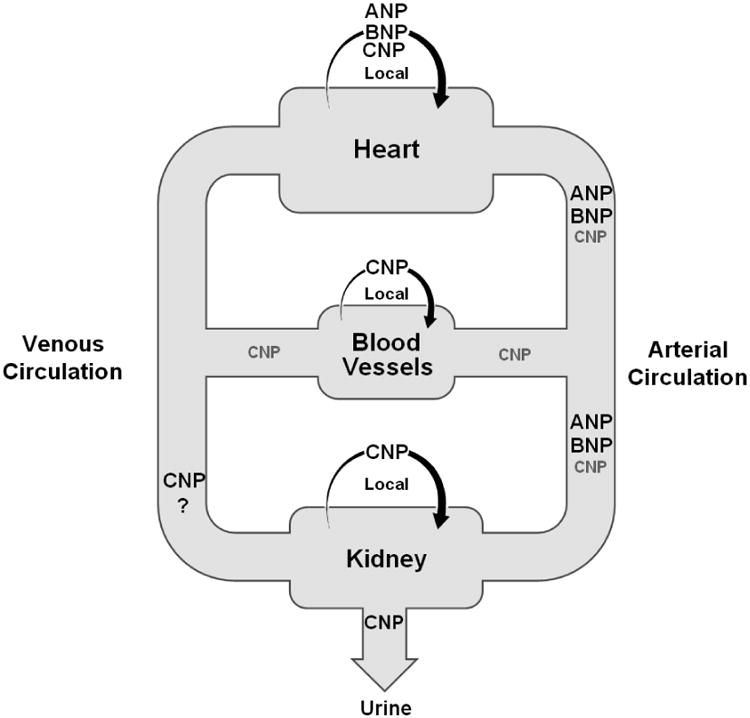
Major sites of C-type natriuretic peptide (CNP) production include the vascular endothelium, kidney and heart, where it is believed to principally operate as an autocrine or paracrine factor. In patients with heart failure, circulating levels of ANP and BNP are higher than those of CNP; however urinary excretion of CNP is high.
The CNP gene is located on chromosome 2, versus chromosome 1 for ANP and BNP [22, 28]. CNP is initially synthesized as a 103 amino acid (AA) prohormone, proCNP (AA 1-103), which is then cleaved into NT-proCNP (AA 1-50) and CNP53 (AA 51-103) by the ubiquitous convertase, furin [29]. At this stage CNP53 may be secreted [20] or undergo further biological processing resulting in two additional products: CNP22 (AA 82-103) and NT-CNP53 (AA 51-81, which may otherwise be referred to as NT-proCNP; Figure 2). Theoretically, CNP22 may be directly derived by alternative cleavage of proCNP, by an unidentified mechanism, which concurrently produces an accessory 81AA NT-proCNP (AA1-81; Figure 2). However this pathway has not previously been described and requires further validation.
Figure 2.
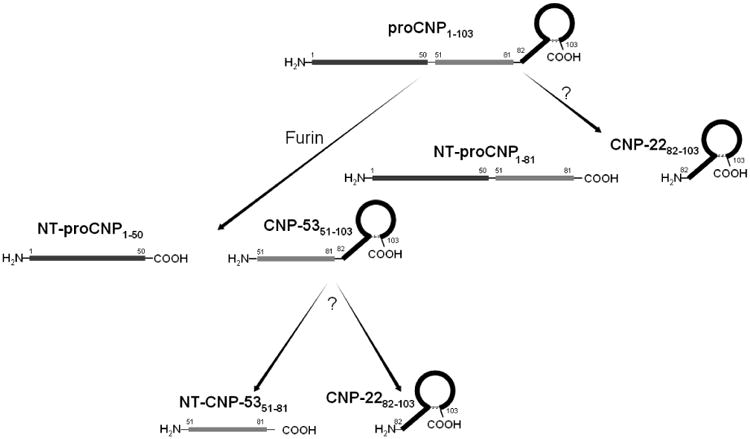
Schematic diagram of the biological processing pathways of pro C-type natriuretic peptide (proCNP) to downstream CNP molecular forms.
Among the products of CNP processing, it is thought that CNP22 is the mature and biologically active form and has therefore been most extensively studied. However, CNP22 has a short half-life (2-6 minutes) [30], and thus detected levels may not accurately reflect CNP synthesis. CNP53 [14, 17] and NT-CNP53 [14] have been detected in urine from patients with HF and healthy controls. Under physiological conditions, approximately 70% of urinary protein content originates from the kidney and urinary tract, while only 30% from plasma [31]. In view of the low circulating levels of CNP, urinary CNP is predominantly thought to derive from local renal production and the CNP urinary excretion rate reflective of renal structural integrity and function.
3.2. Biological effects and role of CNP
The biological effects of CNP result from its selective activation of the cell surface particulate guanylyl cyclase receptor B (GC-B), which contrasts the selective activation of GC-A by ANP and BNP [32]. Binding of CNP to the GC-B receptor catalyzes the conversion of GTP to the downstream second messenger, cyclic guanosine monophosphate (cGMP). The bioavailability of CNP is modulated by three mechanisms: the transmembrane clearance receptor, NPR-C, which binds and sequesters ANP, BNP, and CNP from the circulation; enzymatic hydrolysis by the zinc metallopeptidase, neutral endopeptidase (NEP), which is found in heart, lung, kidney and endothelial tissue; and finally urinary excretion. CNP is the most rapidly degraded of the natriuretic peptides by NEP [33], suggesting that enzymatic hydrolysis and/or urinary excretion may be more important for CNP regulation than receptor-mediated clearance.
Our understanding of the biological role of CNP continues to evolve. While ANP and BNP exhibit a number of homeostatic actions which are beneficial in HF, including diuresis, natriuresis, arterial vasodilation and renin-angiotensin-aldosterone system inhibition [34, 35], CNP lacks significant diuretic or natriuretic effects at physiologic concentrations [30]. However, unique among the natriuretic peptides, CNP has demonstrated significant anti-proliferative and anti-fibrotic properties. These include potent suppression of fibroblast proliferation and collagen production [36-39], inhibition of vascular smooth muscle cell proliferation [40, 41], and accelerated regeneration of endothelial cells [42, 43]. Moreover, CNP is a recognized vasodilator [34,35,36] and particularly potent venodilator [37], which appears to play an important role in the local regulation of vascular tone. Of relevance to HF and CRS, hypoxia [39] that may occur with renal congestion, and cytokines and fibrotic growth factors [25, 40], inherent to cardiac and renal remodeling, are all recognized stimuli for CNP production and release. Thus it is proposed that CNP represents a marker of reparative and restorative physiology as well as structural alterations, as will be outlined below.
3.3 Measurement of CNP
CNP molecular forms in the urine and plasma are currently assessed using proprietary radio- or sandwich immunoassays [14] [38] [44-46]. Reproducibility and a high degree of sensitivity and specificity have been reported within individual pre-clinical and clinical research studies, particularly with the use of the radioimmunoassays [14, 44, 47]. However a clinical laboratory platform for CNP measurement in clinical practice has yet to be developed. Important next steps towards translation of recent research findings to the clinical arena will include the development of a scalable platform, generalizable and consistent reporting of urinary CNP excretion rates adjusted for urinary creatinine, and the establishment of age and gender-specific normal reference ranges.
4. C-type natriuretic peptide in aging and disease
4.1. Aging
Physiologic aging has been associated with a progressive decline in renal function, which is not entirely due to age-related comorbidities [48, 49]. Reductions in glomerular filtration, renal perfusion and altered glomerular basement membrane (GBM) permeability may accompany advanced age, with eventual contributions from glomerulosclerosis and tubulointerstitial fibrosis [49]. Importantly, a concurrent reduction in muscle mass can maintain near-constancy of serum creatinine and initially mask the decline, as urinary creatinine excretion is proportional to muscle mass. By contrast, urinary CNP excretion has been shown to increase with normal aging and further exhibits a robust correlation with renal fibrosis in aging rodents, before the onset of significant proteinuria or blood pressure elevation [44]. In human kidney biopsy specimens, the localization of CNP was similar to aging rodents in both young and older kidney donors [44]. As GFR and plasma CNP also declined with age in this study, renal-derived, rather than filtered, CNP was suggested to correlate with renal fibrosis [44]. It follows, therefore, that elevated urinary CNP excretion may represent a sensitive marker of renal structural remodeling prior to the development of overt symptoms and disease. This association with aging extends the reno-protection and reparative hypothesis, by suggesting that increased CNP expression and its subsequent excretion in the urine may be sustained over many months and years, in contrast to more fluctuant levels observed with other urinary biomarkers [50].
4.2. Heart failure
A potential role for urinary CNP in HF was first proposed in 1994 when Mattingly et al. [17] reported elevated urinary excretion of CNP53 and CNP22 in patients with stable HF. Subsequently, in the experimental setting, Borgeson et al. [51] observed a marked increase in urinary, but interestingly not plasma, CNP in a canine HF model of acute intravascular volume overload with increased left ventricular filling pressures. More recently significantly elevated levels of CNP molecular forms were detected in urine from patients with acute decompensated HF (ADHF) versus healthy controls, and exhibited poor correlation with plasma CNP levels [14, 52]. These data suggest activation of the renal natriuretic peptide system in HF, wherein elevated urinary excretion of CNP is purported to reflect increased renal interstitial pressure, renal tubular injury, hypoxia, and potentially renal fibrosis. Importantly, in the setting of ADHF, urinary NT-CNP53 excretion was significantly associated with prognosis and conveyed incremental prognostic information than was obtained from patient age, GFR, proteinuria, or plasma NT-proBNP, with respect to all-cause mortality and a combined endpoint of HF hospitalization and death [14]. Furthermore, the fact that urinary tubulospecific biomarkers KIM-1 and NGAL were not associated with clinical outcomes in the same study highlights a broader scope for urinary CNP as a biomarker of global renal processes beyond renal tubular injury alone.
Intrinsic renal damage in HF may result from kidney hypoperfusion due to arterial underfilling or increased renal outflow impedance due to venous congestion. In some cases renal hypoxia and tubular necrosis may occur, and subclinical or overt renal dysfunction persist despite decongestion. Whereas tubulospecific biomarkers such as KIM-1, NAG, and NGAL, have been shown to rise early and markedly in the course of renal tubular dysfunction in HF, their levels have been shown to fall with diuretic therapy [50]. Serum creatinine is a late marker of overt renal dysfunction. CNP, on the other hand, has been localized to the renal tubules and may represent an additional marker of renal tubular integrity. Moreover its positive association with prognosis in ADHF [14] suggests a greater sensitivity or even detection of a different type of renal dysfunction in HF, thus better characterizing the evolution of CRS. However, further prospective studies are needed to evaluate the specific type of kidney injury detectable by urinary CNP and its response to HF therapies.
Remarkably, the adverse prognosis associated with increased (ostensibly renal-derived) urinary CNP in HF appears to conflict with a postulated renoprotective effect of CNP, which would be expected to improve overall prognosis. It is reasonable to speculate that CNP activation in the kidney is initially compensatory and reparative, but in HF and CRS these homeostatic mechanisms are exceeded. Thus, an increase in urinary NT-CNP53 may represent a failure or a deficiency of the renal natriuretic peptide system's counter-regulatory mechanisms in HF [14]. Notably, the biological activity and role of different CNP molecular forms remain unclear and further investigation may provide relevant pathophysiologic and prognostic insights. These may be particularly important in HF syndromes strongly associated with comorbidities such as HF with preserved ejection fraction (HFpEF). In the proof of concept study by Zakeri et al. [14], consecutive patients hospitalized with acute decompensated HF and reduced ejection fraction (HFrEF) were assessed. To our knowledge, the clinical utility of urinary CNP testing in HFpEF, compared with either HFrEF or healthy controls has not been directly examined.
4.3. Renal disease states
Examination of urinary CNP in experimental and human nephropathy has advanced our understanding of its relationship to renal structure and function. In experimental nephropathy secondary to ureteral obstruction, Hu et al. [53] reported an increase in urinary CNP excretion well before changes were observed in urinary protein, albumin, blood urea nitrogen, and creatinine. Importantly, experimental nephropathy was associated with histological evidence of tubulointerstitial fibrosis, higher urine than plasma concentrations of CNP, and no significant arteriovenous CNP concentration gradient. Taken together, these findings suggest enhanced renal expression of CNP and subsequent elevation in urinary excretion of CNP may serve as an early marker of tubulointerstitial fibrosis in obstructive nephropathy [53].
Diabetes is a recognized cause of cardiac and renal remodeling. In rats with streptozotocin-induced diabetes, increased renal CNP synthesis (via mRNA expression) and a parallel increase in urinary CNP excretion was observed by Shin et al. [54]. Notably renal CNP synthesis was attenuated by dietary salt-restriction, which highlights a potential for dynamic alterations in CNP expression. Conceivably decreased renal interstitial pressure may reduce mechanical stimulation of CNP production [54].
Moreover in normotensive patients with nephrotic syndrome, which exhibits some similarities to diabetic nephropathy, urinary CNP excretion was elevated compared to healthy control subjects of similar age and body mass [55]. After administration of a low protein diet, urinary, but not plasma, CNP levels were reduced without a detectable change in creatinine clearance. This corroborates a role for urinary CNP in the early detection of renal dysfunction. Although a concurrent improvement in urinary albumin was also observed in this study, decreases in CNP were of greater magnitude [55], suggesting that urinary CNP exhibits heightened sensitivity to changes in glomerular function and/or detects extra-glomerular renal disease.
Finally, patients with cirrhosis and concomitant renal dysfunction were reported to have higher urinary CNP excretion compared to patients with cirrhosis and normal renal function, or healthy controls [47]. Urinary CNP demonstrated an inverse correlation with urinary sodium excretion and following interventions, which improved renal perfusion and pressures (transjugular intrahepatic portosystemic shunt insertion or ornipressin infusion), urinary CNP excretion declined. Plasma CNP concentrations were not affected and no arteriovenous (femoral artery-renal vein) CNP concentration gradient was observed, excluding major renal extraction of circulating CNP as a potential cause [47]. In sum, these observations are compatible with increased and dynamic renal CNP activation resulting in greater urinary CNP excretion in conditions of renal stress. It is possible that more advanced renal remodeling and fibrosis may be reflected in constancy and plasticity of urinary CNP excretion, however this hypothesis requires further validation.
5. Future directions and conclusion
Renal remodeling and fibrosis are key pathological processes underlying progression to chronic kidney disease, which adversely impacts prognosis in HF and CRS. Unique among the family of natriuretic peptides, CNP is synthesized in the kidney, where it acts in a predominantly autocrine/paracrine fashion, and appears to be activated in HF and renal disease states. CNP and its various molecular forms are detectable in the urine and may enable early diagnosis of renal congestion, renal injury and structural remodeling due to disease or physiologic aging. Evidence for a reno-protective effect of CNP is also mounting and deserves further exploration as a potential therapeutic mechanism. At present there is no specific therapy for renal fibrosis or CRS. Current data suggest that elevated urinary CNP may predict the evolution and progression of renal fibrosis and may therefore be of clinical utility as a pragmatic endpoint for trials of novel anti-fibrotic agents, where existing clinical endpoints mandate long and expensive follow-up. Further investigation is also needed to elucidate the relative biological significance and relationships between urinary CNP molecular forms in HF with reduced and preserved ejection fraction and in CRS. Evidence to date suggests that urinary CNP may offer relevant, non-invasive insight into intrinsic renal processes, which are incompletely described by existing parameters of renal function. Among the new wave of urinary biomarkers, CNP holds great promise for translation to new diagnostic and therapeutic strategies, which are urgently needed to improve outcomes for patients with HF and CRS.
Highlights.
C-type natriuretic peptide (CNP) is synthesized in the kidney, where it acts in a predominantly autocrine/paracrine fashion, and is an important regulator of cell proliferation and organ fibrosis.
CNP molecular forms can be detected in urine and are elevated in patients with heart failure and in renal disease states.
Urinary CNP excretion may enable early diagnosis of renal congestion, renal injury and structural remodeling and predict the evolution and progression of renal fibrosis.
Acknowledgments
This work was supported by a Scientist Development Grant from the American Heart Association (13SDG16910051 to Dr. Sangaralingham), grants from the National Institutes of Health (R01 HL036634, P01 HL076611 and R01 HL083231 to Dr. Burnett), a Career Development Award in Cardiovascular Research - St. Jude Foundation (to Dr. Sangaralingham) and the Mayo Foundation. This work was also supported by the Mayo Clinic Center for Clinical and Translational Science (TL1 TR000137 and TL1RR02415 from the National Center for Advancing Translational Science [NCATS]; Drs. Sangaralingham and Zakeri). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest: Drs. Burnett and Sangaralingham are named as co-inventors on a patent application relating to the use of CNP as a biomarker.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McMurray JJ, Petrie MC, Murdoch DR, Davie AP. Clinical epidemiology of heart failure: public and private health burden. Eur Heart J. 1998;19(Suppl P):P9–16. [PubMed] [Google Scholar]
- 2.Curtis LH, Whellan DJ, Hammill BG, Hernandez AF, Anstrom KJ, Shea AM, et al. Incidence and prevalence of heart failure in elderly persons, 1994-2003. Arch Intern Med. 2008;168:418–24. doi: 10.1001/archinternmed.2007.80. [DOI] [PubMed] [Google Scholar]
- 3.Roger VL, Weston SA, Redfield MM, Hellermann-Homan JP, Killian J, Yawn BP, et al. Trends in heart failure incidence and survival in a community-based population. Jama. 2004;292:344–50. doi: 10.1001/jama.292.3.344. [DOI] [PubMed] [Google Scholar]
- 4.Campbell RC, Sui X, Filippatos G, Love TE, Wahle C, Sanders PW, et al. Association of chronic kidney disease with outcomes in chronic heart failure: a propensity-matched study. Nephrol Dial Transplant. 2009;24:186–93. doi: 10.1093/ndt/gfn445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McAlister FA, Ezekowitz J, Tarantini L, Squire I, Komajda M, Bayes-Genis A, et al. Renal dysfunction in patients with heart failure with preserved versus reduced ejection fraction: impact of the new Chronic Kidney Disease-Epidemiology Collaboration Group formula. Circ Heart Fail. 2012;5:309–14. doi: 10.1161/CIRCHEARTFAILURE.111.966242. [DOI] [PubMed] [Google Scholar]
- 6.Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527–39. doi: 10.1016/j.jacc.2008.07.051. [DOI] [PubMed] [Google Scholar]
- 7.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:e240–327. doi: 10.1161/CIR.0b013e31829e8776. [DOI] [PubMed] [Google Scholar]
- 8.Testani JM, Chen J, McCauley BD, Kimmel SE, Shannon RP. Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation. 2010;122:265–72. doi: 10.1161/CIRCULATIONAHA.109.933275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ljungman S, Kjekshus J, Swedberg K. Renal function in severe congestive heart failure during treatment with enalapril (the Cooperative North Scandinavian Enalapril Survival Study [CONSENSUS Trial]) Am J Cardiol. 1992;70:479–87. doi: 10.1016/0002-9149(92)91194-9. [DOI] [PubMed] [Google Scholar]
- 10.Ruggenenti P, Remuzzi G. Worsening kidney function in decompensated heart failure: treat the heart, don't mind the kidney. Eur Heart J. 2011;32:2476–8. doi: 10.1093/eurheartj/ehr242. [DOI] [PubMed] [Google Scholar]
- 11.Damman K, Van Veldhuisen DJ, Navis G, Vaidya VS, Smilde TD, Westenbrink BD, et al. Tubular damage in chronic systolic heart failure is associated with reduced survival independent of glomerular filtration rate. Heart. 2010;96:1297–302. doi: 10.1136/hrt.2010.194878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jungbauer CG, Birner C, Jung B, Buchner S, Lubnow M, von Bary C, et al. Kidney injury molecule-1 and N-acetyl-beta-D-glucosaminidase in chronic heart failure: possible biomarkers of cardiorenal syndrome. Eur J Heart Fail. 2011;13:1104–10. doi: 10.1093/eurjhf/hfr102. [DOI] [PubMed] [Google Scholar]
- 13.Sarnak MJ, Katz R, Newman A, Harris T, Peralta CA, Devarajan P, et al. Association of urinary injury biomarkers with mortality and cardiovascular events. J Am Soc Nephrol. 2014;25:1545–53. doi: 10.1681/ASN.2013070713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zakeri R, Sangaralingham SJ, Sandberg SM, Heublein DM, Scott CG, Burnett JC., Jr Urinary C-type natriuretic peptide: a new heart failure biomarker. JACC Heart Fail. 2013;1:170–7. doi: 10.1016/j.jchf.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sudoh T, Minamino N, Kangawa K, Matsuo H. C-type natriuretic peptide (CNP): a new member of natriuretic peptide family identified in porcine brain. Biochem Biophys Res Commun. 1990;168:863–70. doi: 10.1016/0006-291x(90)92401-k. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki E, Hirata Y, Hayakawa H, Omata M, Kojima M, Kangawa K, et al. Evidence for C-type natriuretic peptide production in the rat kidney. Biochem Biophys Res Commun. 1993;192:532–8. doi: 10.1006/bbrc.1993.1448. [DOI] [PubMed] [Google Scholar]
- 17.Mattingly MT, Brandt RR, Heublein DM, Wei CM, Nir A, Burnett JC., Jr Presence of C-type natriuretic peptide in human kidney and urine. Kidney Int. 1994;46:744–7. doi: 10.1038/ki.1994.329. [DOI] [PubMed] [Google Scholar]
- 18.Dean AD, Vehaskari VM, Greenwald JE. Synthesis and localization of C-type natriuretic peptide in mammalian kidney. Am J Physiol. 1994;266:F491–6. doi: 10.1152/ajprenal.1994.266.3.F491. [DOI] [PubMed] [Google Scholar]
- 19.Del Ry S, Cabiati M, Vozzi F, Battolla B, Caselli C, Forini F, et al. Expression of C-type natriuretic peptide and its receptor NPR-B in cardiomyocytes. Peptides. 2011;32:1713–8. doi: 10.1016/j.peptides.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Stingo AJ, Clavell AL, Heublein DM, Wei CM, Pittelkow MR, Burnett JC., Jr Presence of C-type natriuretic peptide in cultured human endothelial cells and plasma. Am J Physiol. 1992;263:H1318–21. doi: 10.1152/ajpheart.1992.263.4.H1318. [DOI] [PubMed] [Google Scholar]
- 21.Inoue A, Hiruma Y, Hirose S, Yamaguchi A, Furuya M, Tanaka S, et al. Stimulation by C-type natriuretic peptide of the differentiation of clonal osteoblastic MC3T3-E1 cells. Biochem Biophys Res Commun. 1996;221:703–7. doi: 10.1006/bbrc.1996.0660. [DOI] [PubMed] [Google Scholar]
- 22.Tawaragi Y, Fuchimura K, Tanaka S, Minamino N, Kangawa K, Matsuo H. Gene and precursor structures of human C-type natriuretic peptide. Biochem Biophys Res Commun. 1991;175:645–51. doi: 10.1016/0006-291x(91)91614-i. [DOI] [PubMed] [Google Scholar]
- 23.Inoue K, Naruse K, Yamagami S, Mitani H, Suzuki N, Takei Y. Four functionally distinct C-type natriuretic peptides found in fish reveal evolutionary history of the natriuretic peptide system. Proc Natl Acad Sci U S A. 2003;100:10079–84. doi: 10.1073/pnas.1632368100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vollmar AM, Gerbes AL, Nemer M, Schulz R. Detection of C-type natriuretic peptide (CNP) transcript in the rat heart and immune organs. Endocrinology. 1993;132:1872–4. doi: 10.1210/endo.132.4.8462485. [DOI] [PubMed] [Google Scholar]
- 25.Wei CM, Heublein DM, Perrella MA, Lerman A, Rodeheffer RJ, McGregor CG, et al. Natriuretic peptide system in human heart failure. Circulation. 1993;88:1004–9. doi: 10.1161/01.cir.88.3.1004. [DOI] [PubMed] [Google Scholar]
- 26.Igaki T, Itoh H, Suga S, Hama N, Ogawa Y, Komatsu Y, et al. C-type natriuretic peptide in chronic renal failure and its action in humans. Kidney Int Suppl. 1996;55:S144–7. [PubMed] [Google Scholar]
- 27.Kalra PR, Clague JR, Bolger AP, Anker SD, Poole-Wilson PA, Struthers AD, et al. Myocardial production of C-type natriuretic peptide in chronic heart failure. Circulation. 2003;107:571–3. doi: 10.1161/01.cir.0000047280.15244.eb. [DOI] [PubMed] [Google Scholar]
- 28.Ogawa Y, Itoh H, Yoshitake Y, Inoue M, Yoshimasa T, Serikawa T, et al. Molecular cloning and chromosomal assignment of the mouse C-type natriuretic peptide (CNP) gene (Nppc): comparison with the human CNP gene (NPPC) Genomics. 1994;24:383–7. doi: 10.1006/geno.1994.1633. [DOI] [PubMed] [Google Scholar]
- 29.Wu C, Wu F, Pan J, Morser J, Wu Q. Furin-mediated processing of Pro-C-type natriuretic peptide. J Biol Chem. 2003;278:25847–52. doi: 10.1074/jbc.M301223200. [DOI] [PubMed] [Google Scholar]
- 30.Hunt PJ, Richards AM, Espiner EA, Nicholls MG, Yandle TG. Bioactivity and metabolism of C-type natriuretic peptide in normal man. J Clin Endocrinol Metab. 1994;78:1428–35. doi: 10.1210/jcem.78.6.8200946. [DOI] [PubMed] [Google Scholar]
- 31.Pieper R, Gatlin CL, McGrath AM, Makusky AJ, Mondal M, Seonarain M, et al. Characterization of the human urinary proteome: a method for high-resolution display of urinary proteins on two-dimensional electrophoresis gels with a yield of nearly 1400 distinct protein spots. Proteomics. 2004;4:1159–74. doi: 10.1002/pmic.200300661. [DOI] [PubMed] [Google Scholar]
- 32.Suga S, Nakao K, Hosoda K, Mukoyama M, Ogawa Y, Shirakami G, et al. Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinology. 1992;130:229–39. doi: 10.1210/endo.130.1.1309330. [DOI] [PubMed] [Google Scholar]
- 33.Kenny AJ, Bourne A, Ingram J. Hydrolysis of human and pig brain natriuretic peptides, urodilatin, C-type natriuretic peptide and some C-receptor ligands by endopeptidase-24.11. Biochem J. 1993;291(Pt 1):83–8. doi: 10.1042/bj2910083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhn M. Molecular physiology of natriuretic peptide signalling. Basic research in cardiology. 2004;99:76–82. doi: 10.1007/s00395-004-0460-0. [DOI] [PubMed] [Google Scholar]
- 35.Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocrine reviews. 2006;27:47–72. doi: 10.1210/er.2005-0014. [DOI] [PubMed] [Google Scholar]
- 36.Horio T, Tokudome T, Maki T, Yoshihara F, Suga S, Nishikimi T, et al. Gene expression, secretion, and autocrine action of C-type natriuretic peptide in cultured adult rat cardiac fibroblasts. Endocrinology. 2003;144:2279–84. doi: 10.1210/en.2003-0128. [DOI] [PubMed] [Google Scholar]
- 37.Peltonen TO, Taskinen P, Soini Y, Rysa J, Ronkainen J, Ohtonen P, et al. Distinct downregulation of C-type natriuretic peptide system in human aortic valve stenosis. Circulation. 2007;116:1283–9. doi: 10.1161/CIRCULATIONAHA.106.685743. [DOI] [PubMed] [Google Scholar]
- 38.Sangaralingham SJ, Huntley BK, Martin FL, McKie PM, Bellavia D, Ichiki T, et al. The aging heart, myocardial fibrosis, and its relationship to circulating C-type natriuretic Peptide. Hypertension. 2011;57:201–7. doi: 10.1161/HYPERTENSIONAHA.110.160796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soeki T, Kishimoto I, Okumura H, Tokudome T, Horio T, Mori K, et al. C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. Journal of the American College of Cardiology. 2005;45:608–16. doi: 10.1016/j.jacc.2004.10.067. [DOI] [PubMed] [Google Scholar]
- 40.Furuya M, Yoshida M, Hayashi Y, Ohnuma N, Minamino N, Kangawa K, et al. C-type natriuretic peptide is a growth inhibitor of rat vascular smooth muscle cells. Biochemical and biophysical research communications. 1991;177:927–31. doi: 10.1016/0006-291x(91)90627-j. [DOI] [PubMed] [Google Scholar]
- 41.Porter JG, Catalano R, McEnroe G, Lewicki JA, Protter AA. C-type natriuretic peptide inhibits growth factor-dependent DNA synthesis in smooth muscle cells. The American journal of physiology. 1992;263:C1001–6. doi: 10.1152/ajpcell.1992.263.5.C1001. [DOI] [PubMed] [Google Scholar]
- 42.Doi K, Ikeda T, Itoh H, Ueyama K, Hosoda K, Ogawa Y, et al. C-type natriuretic peptide induces redifferentiation of vascular smooth muscle cells with accelerated reendothelialization. Arteriosclerosis, thrombosis, and vascular biology. 2001;21:930–6. doi: 10.1161/01.atv.21.6.930. [DOI] [PubMed] [Google Scholar]
- 43.Yamahara K, Itoh H, Chun TH, Ogawa Y, Yamashita J, Sawada N, et al. Significance and therapeutic potential of the natriuretic peptides/cGMP/cGMP-dependent protein kinase pathway in vascular regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3404–9. doi: 10.1073/pnas.0538059100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sangaralingham SJ, Heublein DM, Grande JP, Cataliotti A, Rule AD, McKie PM, et al. Urinary C-type natriuretic peptide excretion: a potential novel biomarker for renal fibrosis during aging. Am J Physiol Renal Physiol. 2011;301:F943–52. doi: 10.1152/ajprenal.00170.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palmer SC, Prickett TC, Espiner EA, Yandle TG, Richards AM. Regional release and clearance of C-type natriuretic peptides in the human circulation and relation to cardiac function. Hypertension. 2009;54:612–8. doi: 10.1161/HYPERTENSIONAHA.109.135608. [DOI] [PubMed] [Google Scholar]
- 46.Lok DJ, Klip IT, Voors AA, Lok SI, Bruggink-Andre de la Porte PW, Hillege HL, et al. Prognostic value of N-terminal pro C-type natriuretic peptide in heart failure patients with preserved and reduced ejection fraction. European journal of heart failure. 2014;16:958–66. doi: 10.1002/ejhf.140. [DOI] [PubMed] [Google Scholar]
- 47.Gulberg V, Moller S, Henriksen JH, Gerbes AL. Increased renal production of C-type natriuretic peptide (CNP) in patients with cirrhosis and functional renal failure. Gut. 2000;47:852–7. doi: 10.1136/gut.47.6.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindeman RD, Tobin J, Shock NW. Longitudinal studies on the rate of decline in renal function with age. J Am Geriatr Soc. 1985;33:278–85. doi: 10.1111/j.1532-5415.1985.tb07117.x. [DOI] [PubMed] [Google Scholar]
- 49.Weinstein JR, Anderson S. The aging kidney: physiological changes. Adv Chronic Kidney Dis. 2010;17:302–7. doi: 10.1053/j.ackd.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Damman K, Ng Kam Chuen MJ, MacFadyen RJ, Lip GY, Gaze D, Collinson PO, et al. Volume status and diuretic therapy in systolic heart failure and the detection of early abnormalities in renal and tubular function. J Am Coll Cardiol. 2011;57:2233–41. doi: 10.1016/j.jacc.2010.10.065. [DOI] [PubMed] [Google Scholar]
- 51.Borgeson DD, Stevens TL, Heublein DM, Matsuda Y, Burnett JC. Activation of myocardial and renal natriuretic peptides during acute intravascular volume overload in dogs: functional cardiorenal responses to receptor antagonism. Clin Sci (Lond) 1998;95:195–202. [PubMed] [Google Scholar]
- 52.Ng LL, Geeranavar S, Jennings SC, Loke I, O'Brien RJ. Diagnosis of heart failure using urinary natriuretic peptides. Clin Sci (Lond) 2004;106:129–33. doi: 10.1042/CS20030234. [DOI] [PubMed] [Google Scholar]
- 53.Hu P, Wang J, Hu B, Lu L, Xuan Q, Qin YH. Increased urinary C-type natriuretic peptide excretion may be an early marker of renal tubulointerstitial fibrosis. Peptides. 2012;37:98–105. doi: 10.1016/j.peptides.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 54.Shin SJ, Wen JD, Lee YJ, Chen IH, Tsai JH. Increased C-type natriuretic peptide mRNA expression in the kidney of diabetic rats. J Endocrinol. 1998;158:35–42. doi: 10.1677/joe.0.1580035. [DOI] [PubMed] [Google Scholar]
- 55.Cataliotti A, Giordano M, De Pascale E, Giordano G, Castellino P, Jougasaki M, et al. CNP production in the kidney and effects of protein intake restriction in nephrotic syndrome. Am J Physiol Renal Physiol. 2002;283:F464–72. doi: 10.1152/ajprenal.00372.2001. [DOI] [PubMed] [Google Scholar]