

Abstract
Most hepatocellular carcinomas (HCCs) develop in the context of chronic liver inflammation. Oxidative stress is thought to play a major role in the pathogenesis of HCC development. In this study, we examined whether cold-inducible RNA-binding protein (Cirp) controls reactive oxygen species (ROS) accumulation and development of HCC by using murine models of hepatocarcinogenesis and human liver samples. Cirp expression, ROS accumulation, and CD133 expression were increased in the liver of tumor-harboring mice. Cirp deficiency reduced production of interleukin-1β and interleukin-6 in Kupffer cells, ROS accumulation, and CD133 expression, leading to attenuated hepatocarcinogenesis. Thioacetamide treatment enhanced hepatic expression of CD133 and phosphorylated signal transducer and activator of transcription 3 (STAT3), which was prevented by treatment with the antioxidant butylated hydroxyanisole. Intriguingly, the risk of human HCC recurrence is positively correlated with Cirp expression in liver. Cirp appears to play a critical carcinogenic function and its expression might be a useful biomarker for HCC risk prediction.
Keywords: CD133, HCC, recurrence, ROS, stem cell
Hepatocellular carcinoma is the most common form of liver cancer and the third leading cause of cancer deaths worldwide, and is usually associated with a very poor prognosis.1 In addition to chronic exposure to toxic chemicals, chronic infections with hepatitis B virus or hepatitis C virus as well as non-alcoholic steatohepatitis are the major risk factors for HCC.2,3 Oxidative stress is thought to play a major role in the pathogenesis of cancer development.4,5 The great majority of HCC cases arise in the setting of chronic liver disease and usually present when that disease process is at a cirrhotic stage, a final irreversible stage of chronic inflammation.6,7 Progression of chronic liver disease is associated with hepatocyte death and a subsequent inflammatory response, both of which involve ROS accumulation in the liver.8 Thus, the risk of hepatocarcinogenesis depends on background liver factors. However, the underlying molecular mechanisms linking ROS and cancer remain to be fully explored.
The concept that tumors are maintained by dedicated stem cells, the so-called cancer stem cell hypothesis, has attracted much interest. According to this hypothesis, cancer cannot be viewed as simple monoclonal expansions of functionally equal tumor cells. Instead, only a small minority of tumor cells, the cancer stem cells or tumor-initiating cells, have the ability to maintain the malignant population.9,10 CD133, a transmembrane glycoprotein, is a valuable marker of cancer stem cells.11 Sox2, a member of the Sox HMG box family of transcription factors, has been shown to participate in reprogramming of adult somatic cells to a pluripotent stem cell state and has been implicated in tumorigenesis in various organs.12,13 In the malignant transformation of foregut basal progenitor cells, Sox2 cooperates with inflammation-mediated STAT3 activation.14
The chemical thioacetamide (TAA) can induce liver cirrhosis and cancer of the bile ducts when given to rats over a period of several months.15 Previously, we found that mice given TAA for 10 months develop HCC rather than cholangiocellular carcinoma subsequent to appearance of liver fibrosis, thus providing a model that closely mimics the natural history of human chronic liver disease.16 In addition, the histology of the TAA-exposed rat liver was reported to resemble human liver cirrhosis.17 Thus, the mouse TAA model may be suitable for studying the relationship between inflammation and hepatocarcinogenesis and allows studies to be carried out that are of relevance to human chronic liver disease such as hepatitis C virus-related liver disease and non-alcoholic steatohepatitis.
Cold-inducible RNA-binding protein (Cirp, also called Cirbp and hnRNP A18), originally identified as the first mammalian cold shock protein,18 is induced by cellular stresses such as UV irradiation and hypoxia.19–21 and is suggested to mediate the preservation of neural stem cells.22 In response to the stress, Cirp, which migrates from the nucleus to the cytoplasm, affects post-transcription expression of its target mRNAs23–25 and functions as a damage-associated molecular pattern molecule that promotes inflammatory responses when expressed extracellularly.26 Cirp also affects cell growth and cell death induced by TNF-α or genotoxic stress.27,28 Recently, Cirp was found to link inflammation and tumorigenesis in colitis-associated cancer.29 However, the involvement of Cirp in HCC development is not well understood.
Here we examined whether Cirp plays a role in tumorigenesis in the liver by using Cirp-deficient (Cir−/−) mice and found that Cirp increases ROS accumulation and CD133 expression, leading to enhanced liver tumorigenesis. This represents the first report of the functional link between Cirp and hepatocarcinogenesis.
Materials and Methods
Mice and treatment
The generation of Cirp−/− mice has been described previously.30 The animals showed neither gross abnormality nor hepatic inflammation. Sex- and age-matched C57BL/6 WT and Cirp−/− mice (8–12 weeks old) were given 0.03% TAA (Sigma, St. Louis, MO, USA) in drinking water. After 2 and 10 months on normal chow, mice were killed and analyzed for presence of fibrosis and HCCs. Wild-type mice were fed either butylated hydroxyanisole-containing (0.7%) or regular chow and treated with TAA for 8 weeks. Two-week old male WT and Cirp−/− mice were injected i.p. with 25 mg/kg DEN (Sigma). After 8 months on normal chow, mice were killed and their livers were removed and analyzed for the presence of HCCs. Kupffer cells were isolated as described before.31
Biochemical and immunochemical analyses
Real-time qPCR, immunoblotting, and immunohistochemistry were previously described.18,29,32 Reverse transcription–PCR was carried out with primers for mouse Cirp, 5′-gaggactcagcttcgacacc-3′ and 5′-ctccctgtcctttaccacca-3′ and mouse CD133, 5′-tcaaagggacccagaaactg-3′ and 5′-gccttgttcttggtgttggt-3′. Antibodies used were: anti-actin (Sigma); anti-Sox2, anti-STAT3, anti-phospho-STAT3 (Cell Signaling Technology, Danvers, MA, USA); anti-PRMO1 (CD133; Abnova, Newmarket, UK); and anti-8-OHdG (Japan Institute for the Control of Aging, Shizuoka, Japan). Anti-Cirp antibody was described before.30 Protein oxidation was assessed by the OxyBlot Protein Oxidation Detection Kit (Merck Millipore, Billerica, MA, USA). Immunohistochemistry was carried out using ImmPRESS reagents (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer's recommendations. Hydroxyproline content was assessed using Hydroxyproline Assay Kit (BioVision, Milpitas, CA, USA).
Human tissue samples
Hepatocellular carcinoma tissues and non-cancerous liver tissues were obtained from 12 patients who had undergone curative hepatectomy for HCC at the Kinki University Hospital (Osaka-Sayama, Japan). The specimens used were routinely processed, formalin-fixed, and paraffin-embedded. After H&E staining, all samples were diagnosed as HCC. The HCC and non-cancerous specimens were frozen and stocked at −80°C. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the institutional review boards. Written informed consent was obtained from all patients for subsequent use of their resected tissues.
Statistical analysis
Data are presented as means ± SEM. Differences were analyzed by Student's t-test. P values <0.05 were considered significant.
Results
Attenuated hepatocarcinogenesis in Cirp−/− mice
Hepatocellular carcinoma is a paradigm for inflammation-induced cancer, as it most frequently develops on grounds of chronic hepatitis, consecutive cellular damage, and compensatory regeneration.6 Wild-type and Cirp−/− mice were given 0.03% TAA in drinking water. After 8 weeks of TAA treatment, we observed inflammation and formation of fibrotic septa.18 Wild-type and Cirp−/− mice given TAA for 10 months developed severe inflammation, fibrosis, and well-differentiated HCCs (Figs1a,S1a). Tumor sizes and numbers were considerably smaller in Cirp−/− mice relative to similarly treated WT mice (Fig.1b). However, no significant difference in liver fibrosis was found between WT and Cirp−/− mice (Fig. S1b).
Fig 1.
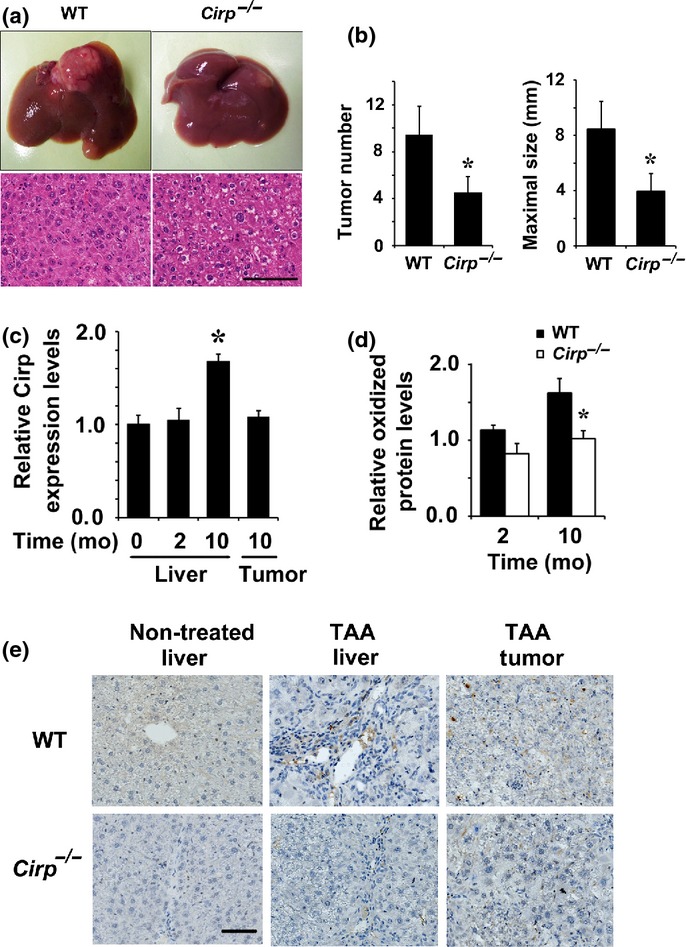
Attenuated hepatocarcinogenesis in Cirp−/− mice. (a) Wild-type and Cirp−/− mice were treated with thioacetamide (TAA) for 10 months. Typical examples of macroscopic tumorigenesis in the TAA model are shown. In the lower panels, liver tumor sections were examined using H&E staining. Scale bar = 200 μm. (b) Numbers and maximum sizes of tumors in mice were determined (WT mice, n = 8; Cirp−/− mice, n = 8). Results are means ± SEM. *P < 0.05 versus WT mice. (c) WT mice were treated with TAA for 2 or 10 months (mo) and liver RNA was extracted from non-cancerous tissues (liver) and tumors. Relative mRNA amounts of Cirp were determined by real-time quantitative PCR and normalized to the amount of actin mRNA. The amount of mRNA in untreated liver was given an arbitrary value of 1.0. Results are means ± SEM (n = 5). *P < 0.05 versus non-treated liver. (d) Lysates of non-tumor liver tissue from TAA-treated mice were extracted at the indicated time points and assessed by immunoblotting and quantified using image analysis software. The amount of protein oxidation in untreated liver was given an arbitrary value of 1.0. Results are means ± SEM (n = 4). *P < 0.05 versus WT mice. (e) Representative immunostaining images of non-tumor and tumor tissues with anti-8-hydroxy-2′-deoxyguanosine antibody.
Cirp deficiency attenuated ROS accumulation in TAA-treated liver
Cirp expression was increased in the liver of tumor-harboring mice given TAA for 10 months (Fig.1c). A causal link between oxidative stress and cancer was proposed.4,16,33 When mice were given TAA for 10 months, WT livers had significantly higher levels of oxidized protein than Cirp−/− livers (Fig.1d). Immunohistochemical analysis showed that 8-OHdG-specific signals were observed in periportal cells (Fig.1e), including but not limited to Kupffer cells (Fig. S2), and suggested that ROS accumulation was attenuated in Cirp−/− mice (Fig.1e).
Decreased expression of phosphorylated STAT3, Sox2, and CD133 in Cirp−/− mice
Accumulation of ROS, which was enhanced by Cirp (Fig.1d,e), activates the transcription factor STAT3 through src homology-containing phosphatase 1/2 inactivation in the absence of IKKβ.34 Deletion of Cirp decreased expression of phosphorylated STAT3 in non-treated and TAA-treated livers (Fig.2). Activation of STAT3 is required for liver cancer stem cell expansion and HCC formation.34,35 Correspondingly, Cirp−/− mice were found to have lower levels of Sox2 and CD133 protein than WT mice (Fig.2a,b). Immunohistochemical analysis was carried out to identify the cells expressing Sox2 in the liver. In the normal liver, neither hepatocytes nor inflammatory cells expressed Sox2 protein, whereas in TAA-treated livers, Sox2 was expressed in periportal parenchymal cells. Deletion of Cirp decreased the number of Sox2-positive cells in TAA-treated livers (Fig.2c). Cirp deficiency was associated with decreased CD133 mRNA levels both in livers and tumors (Fig.2e). However, the direct causal link between STAT3 and cancer stem cells has not been established in this study. The reduced stem cell marker expression seen in Cirp−/− mice would be due to the secondary effects associated with reduced STAT3 activation and inflammation. Thus, Cirp-mediated STAT3 activation in inflammatory cells might regulate cancer stem cell expansion through augmented inflammatory response in the liver.
Fig 2.
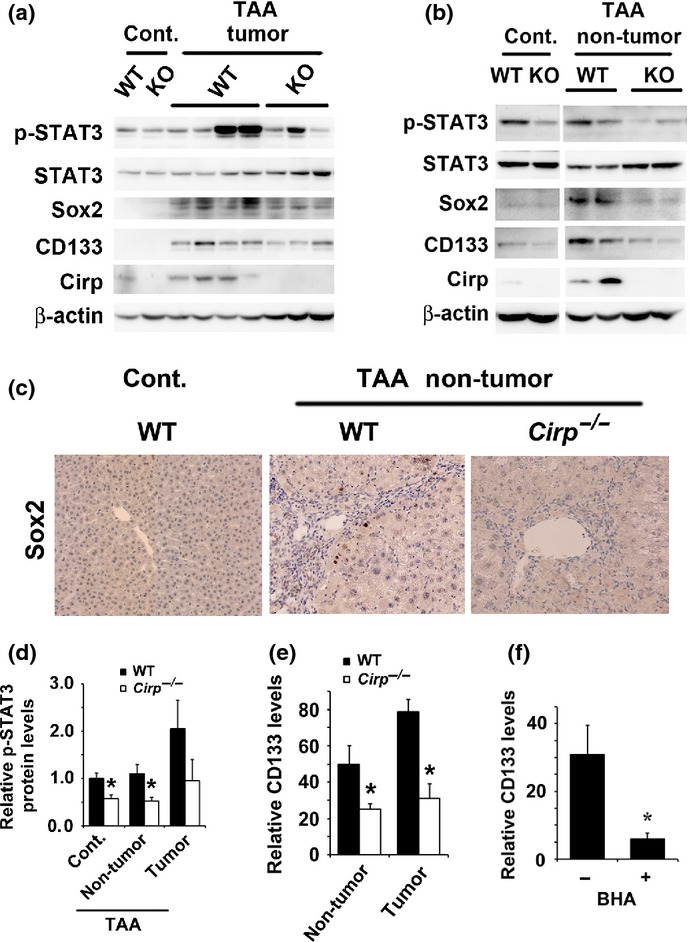
Decreased expression of phosphorylated (p-)STAT3, Sox2, and CD133 in Cirp−/− mice. Wild-type and Cirp−/− mice were treated with thioacetamide (TAA) for 10 months. (a, b) Homogenates of tumors (a) and non-tumor liver tissues (b) were gel-separated and immunoblotted with the indicated antibodies. Representative data are shown. Cont., non-treated liver. (c) Representative immunostaining images of non-tumor tissues with anti-Sox2 antibody. Cont., non-treated liver. (d) The p-STAT3/actin ratio was measured. Quantification of Western blot bands was carried out using densitometry. *P < 0.05 compared with WT mice. (e) RNA was extracted from tumors and non-tumor tissues. Relative mRNA amounts of CD133 were determined by real-time quantitative PCR and normalized to the amount of actin mRNA. The amount of mRNA in untreated liver was given an arbitrary value of 1.0. Results are means ± SEM (n = 6 per group). *P < 0.05 compared with WT mice. (f) WT mice were fed either butylated hydroxyanisole (BHA)-containing (0.7%) or regular chow and treated with TAA for 8 weeks. Relative amounts of mRNA were determined by real-time quantitative PCR and normalized to the amount of actin mRNA. The amount of CD133 mRNA in untreated liver was given an arbitrary value of 1.0. Results are means ± SEM (n = 4). *P < 0.05 compared with mice fed regular chow.
To evaluate the contribution of oxidative stress to CD133 upregulation in the liver, we placed a group of mice on chow diet supplemented with the antioxidant butylated hydroxyanisole. Mice kept on this diet showed a significant reduction in expression of CD133 and phosphorylated STAT3 (Figs2f,S3). Thus, CD133 expression is upregulated at least partially through mechanisms that may depend on ROS accumulation.
Cirp deficiency attenuated proinflammatory cytokine production in Kupffer cells
We confirmed that Cirp was mainly expressed in the resident liver macrophages called Kupffer cells when mice were given TAA for 10 months (Fig.3a). Cirp disruption significantly inhibited production of IL-1β and IL-6 in Kupffer cells (Fig.3b) and attenuated inflammatory response and hepatocyte death (Fig.3c–e). To examine the effect of Cirp on acute liver injury, serum ALT was measured in mice given TAA for 2 months. No significant difference was found between WT and Cirp−/− mice (Fig.3c). Chronic inflammation results in accumulation of ROS.16 Cirp would induce liver inflammation by enhancing cytokine production in Kupffer cells, leading to ROS accumulation.
Fig 3.
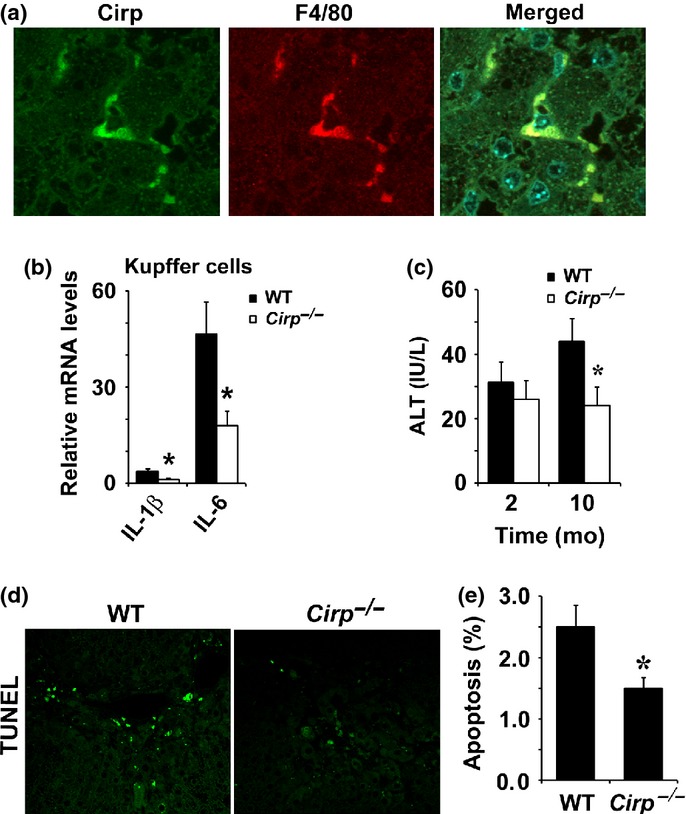
Cold-inducible RNA-binding protein (Cirp) deficiency attenuated proinflammatory cytokine production in Kupffer cells. (a) Immunohistochemistry was carried out on liver sections of thioacetamide (TAA)-treated WT mice. Cells stained with indicated antibodies were identified by confocal microscopy. (b) Production of interleukin (IL)-1β and IL-6 mRNA was measured by real-time quantitative PCR in Kupffer cells from WT and Cirp−/− mice. The amount of mRNA in hepatocytes was given an arbitrary value of 1.0. Results are means ± SEM (n = 4). *P < 0.05 compared with WT mice. (c) ALT levels in serum were determined after 2 and 10 months (mo) of TAA treatment. Results are means ± SEM (n = 5). *P < 0.05 compared with WT mice. (d, e) Extent of hepatocyte apoptosis was determined by TUNEL staining. Results are means ± SEM (n = 6). *P < 0.05 compared with WT mice.
Cirp deficiency inhibited DEN-induced hepatocarcinogenesis
After DEN injection on postnatal day 14, WT mice develop well-differentiated HCCs but have neither chronic inflammation nor fibrosis in the liver.32,33,36 Tumor numbers and sizes were smaller in Cirp−/− mice relative to similarly treated WT mice, but the difference in tumor size was not significant between WT and Cirp−/− mice (Fig.4a,b). Cirp expression was increased in the liver of tumor-harboring mice, but not in tumors (Fig.4c). Wild-type mice had significantly higher levels of oxidized protein than Cirp−/− mice (Fig.4d). Cirp−/− tumors were found to have lower levels of CD133 protein than WT tumors (Fig.4e). There was no significant difference in Sox2 expression between WT and Cirp−/− tumors (data not shown).
Fig 4.
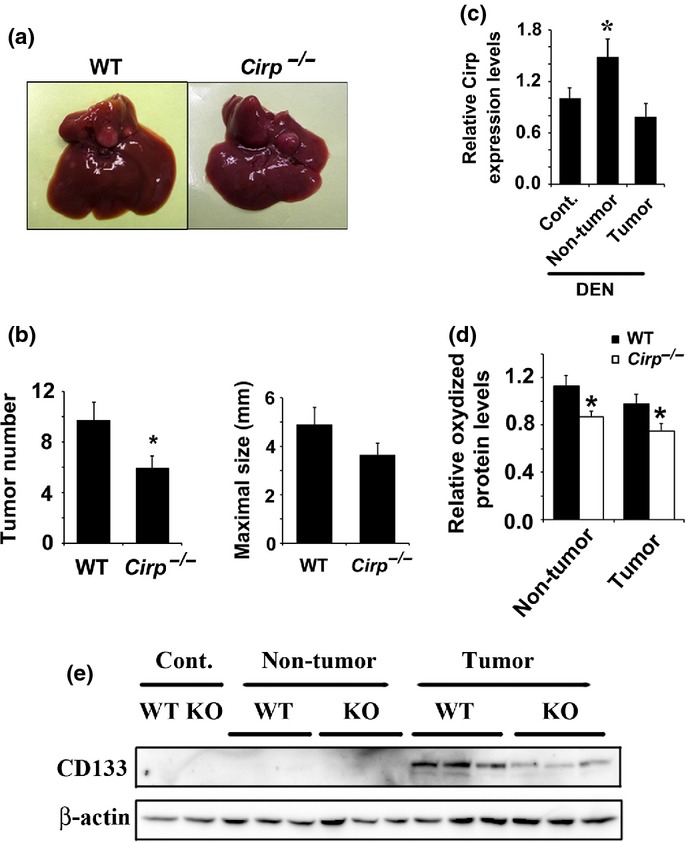
Cold-inducible RNA-binding protein (Cirp) deficiency inhibited DEN-induced hepatocarcinogenesis. WT and Cirp−/− mice were injected with diethylnitrosamine (DEN) injection (25 mg/kg) and killed 8 months later. (a) Representative livers of WT and Cirp−/− mice. (b) Tumor number (>0.5 mm) and tumor size in livers of WT (n = 8) and Cirp−/− mice (n = 8). Data are means ± SEM. *P < 0.05 compared with WT mice. (c) Liver RNA was extracted from non-treated livers (Cont.), non-tumor tissues, and tumors. Relative mRNA amounts of Cirp were determined by real-time quantitative PCR and normalized to the amount of actin mRNA. The amount of mRNA in untreated liver was given an arbitrary value of 1.0. Results are means ± SEM (n = 5). *P < 0.05 compared with non-treated liver (Cont.). (d) Lysates of non-tumor liver tissue from DEN-injected mice were extracted, assessed by immunoblotting, and quantified using image analysis software. The amount of protein oxidation in untreated liver was given an arbitrary value of 1.0. Results are means ± SEM (n = 4). *P < 0.05 versus WT mice. (e) Lysates of non-treated liver (Cont.), tumor, and non-cancerous liver tissue were assessed by immunoblotting with the indicated antibodies.
Association between risk of HCC recurrence and Cirp expression in human liver
Intrahepatic HCC development after hepatectomy is caused by de novo HCC development and/or metastasis from the resected HCCs. The risk of the former depends on background liver factors, such as liver fibrosis, whereas the risk of the latter mainly depends on the characteristics of the resected HCCs.37 Enhanced ROS accumulation in non-cancerous liver was associated with increased risk for HCC recurrence after hepatectomy.16 In mouse models, Cirp is involved in ROS accumulation and enhances de novo HCC development. We examined whether this hypothesis is applicable to humans. Immunohistochemical analysis showed that Cirp was mainly expressed in the periportal areas (Fig.5a). In patients with HCC recurrence after hepatectomy, Cirp expression levels in the non-tumor tissues were significantly higher than in those without HCC recurrence (Figs5b,c,S4). These data, coupled with the contribution of Cirp to tumorigenesis in mice, suggest that elevated Cirp expression in inflammatory cells is associated with increased risk of human HCC development or recurrence.
Fig 5.
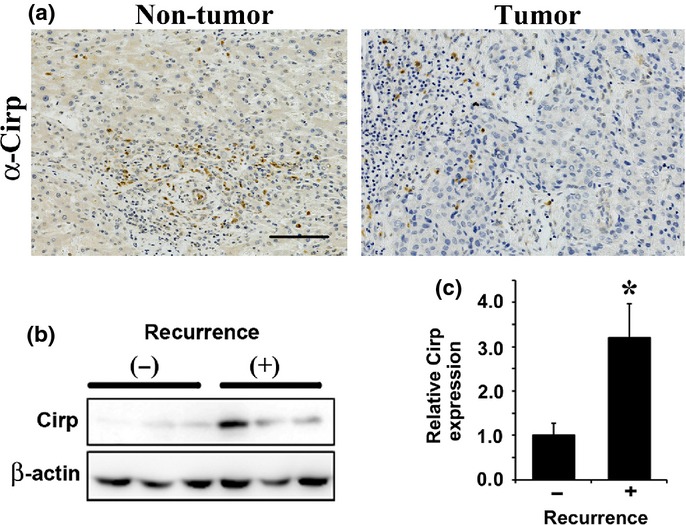
Association between risk of hepatocellular carcinoma (HCC) recurrence and cold-inducible RNA-binding protein (Cirp) expression in human liver. (a) Representative immunostaining images of non-tumor and HCC tissues with anti-Cirp antibody. Scale bar = 100 μm. (b) Lysates of non-cancerous liver tissue from patients who underwent partial hepatectomy for HCC with (+) or without (−) HCC recurrence in the future were extracted and assessed by immunoblotting with the indicated antibodies. Representative data are shown. (c) Cirp protein levels in the liver were quantified using image analysis software. Results are means ± SEM (n = 6 per group). *P < 0.05 versus patients without HCC recurrence.
Discussion
Hepatic progenitor cells are bipotential cells residing in normal liver. Their proliferation is observed in reactive conditions of the liver and in primary liver cancers. Hepatic progenitor cells, found in the periportal areas, are believed to represent liver stem cells. After genetic deregulation of the self-renewal pathway, liver stem cells may transform into cancer stem cells.10,38 Here we found that Cirp disruption decreased expression of cancer stem cell marker CD133 in the non-tumorous liver and tumor of tumor-harboring mice and inhibited tumorigenesis. Cirp might promote hepatocarcinogenesis through cancer stem/progenitor cell expansion.
Reactive oxygen species and cellular oxidant stress are associated with cancer, and elevated ROS level is a hallmark of highly invasive cancer and high risk for HCC recurrence.18,39,40 By contrast, there is a hypothesis that keeping ROS levels low within cancer stem cells or tumor-initiating cells is an important feature of stemness and offers protection against the cell toxicities of ROS.41,42 However, in proliferative neural stem cells, high levels of ROS regulate self-renewal by driving PI3K/AKT signaling.43 After Apc loss, Lgr5+ intestinal stem cell/progenitor cell expansion is a critical process during colorectal tumor initiation and is dependent on RAC1-driven ROS production.44 In the absence of p38MAPK, upregulation of Sox2 expression is dependent on ROS accumulation.16 Thus, the role of ROS in stem cell maintenance, cellular transformation, and cancer stem cell survival appears to be contextually and tissue-specific. Our data suggest that Cirp disruption downregulated hepatic CD133 expression partially through reduced ROS accumulation.
Cold-inducible RNA-binding protein released into the circulation stimulates the release of TNF-α from macrophages through Toll-like receptor 4 and nuclear factor-κB activation, and triggers an inflammatory response to hemorrhagic shock and sepsis.26 Cirp promotes the development of intestinal inflammation and colorectal tumors through regulating apoptosis and production of TNF-α and IL-23.29 In bone marrow-derived macrophages, expression of IL-1β was decreased in the absence of Cirp.29 Here we have shown that Cirp upregulated expression of IL-1β and IL-6 in Kupffer cells, resident liver macrophages. Given that chronic liver inflammation results in ROS accumulation in the liver,16 the production of proinflammatory cytokine would be one of mechanisms by which Cirp regulates ROS accumulation. It should be noted, however, that the direct causal link between Cirp and ROS production has not been established in this study. In this regard, the reduced ROS accumulation seen in the absence of Cirp might be due to the secondary effects associated with reduced inflammation. Consistently, 8-OHdG-positive cells were mainly found in the periportal areas where many inflammatory cells are located.
In inflammatory bowel disease, chronic inflammation upregulates Cirp expression and a major site of Cirp action is an inflammatory cell.29 The relative proportion of inflammatory cells is lower in HCC tissues than in the surrounding liver tissues. Consistently, Cirp expression was increased in the liver of tumor-harboring mice given TAA for 10 months, but not in tumors. Deletion of Cirp decreased expression of phosphorylated STAT3 in TAA-treated livers but not in tumors, which is also caused by the decreased proportion of inflammatory cells in tumors. Additionally, in tumor cells, the main cellular component of HCC tissues, STAT3 activation could be regulated independently of Cirp expression. STAT3 is required for liver cancer stem cell expansion.34,35 Cirp-mediated STAT3 activation in inflammatory cells might regulate cancer stem cell expansion through augmented inflammatory response in the liver.
Hepatocellular carcinoma is an appropriate target for surveillance programs for early cancer detection. Currently, liver ultrasonography combined with serum α-fetoprotein measurement is the standard method of HCC surveillance. However, such a surveillance program has several limitations, including low sensitivity, high cost, and suboptimal adherence to the surveillance.45 The ability to reliably predict an individual's risk of HCC, such that surveillance strategies could be appropriately personalized, would represent a substantial advance in surveillance programs. A number of molecular markers predictive for HCC have been reported,45,46 but are not yet feasible for practical management. Here we showed that Cirp expression was significantly increased in non-cancerous liver samples from patients with post-operative recurrence in the future. Furthermore, in two murine HCC models, Cirp expression was found to increase in livers of tumor-harboring mice. Cirp expression could be a marker for predicting the risk of HCC development. Hypoxia that is enhanced in chronic inflammatory diseases upregulates Cirp expression by a mechanism that involves neither hypoxia-inducible factor-1 nor mitochondria.21 This may be one explanation for Cirp induction in the liver. However, the exact mechanisms by which Cirp expression is upregulated in the liver remain to be elucidated. Recently, Cirp was reported to be released into the circulation.26 The analysis of the Cirp level in the serum may increase the identification rate of patients with a higher risk of HCC development. A future large-scale study of Cirp in patients with chronic hepatitis at different stages will be crucial to determine whether the Cirp expression in the serum is able to predict the risk of cancer and prognosis of patients with chronic liver disease.
In summary, Cirp could enhance tumorigenesis by controlling ROS accumulation and might be associated with stem cell maintenance. Modulation and measurement of Cirp expression might hold promise for advanced treatment and personalized management in patients with chronic liver disease.
Acknowledgments
This research was supported by grants from Yasuda Medical Foundation, Takeda Science Foundation, Japan Foundation for Research and Promotion of Endoscopy and Japan Society for the Promotion of Science.
Glossary
Abbreviations
- Cirp
Cold-inducible RNA-binding protein
- DEN
Diethylnitrosamine
- HCC
Hepatocellular carcinoma
- IL
Interleukin
- 8-OHdG
8-hydroxy-2′-deoxyguanosine
- qPCR
Quantitative PCR
- ROS
Reactive oxygen species
- STAT3
Signal transducer and activator of transcription 3
- Sox2
Sex determining region Y-box 2
- TAA
Thioacetamide
- TNF
Tumor necrosis factor
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. (a) Thioacetamide treatment induced liver fibrosis in mice. (b) Mice were treated with thioacetamide (TAA) for 10 months. No significant difference in hydroxyproline contents was found between WT and Cirp−/− mice.
Fig. S2. Immunohistochemistry was carried out on liver sections of thioacetamide (TAA)-treated WT mice.
Fig. S3. Treatment with the antioxidant decreased protein level of phosphorylated STAT3.
Fig. S4. Representative immunostaining images of non-tumor livers of patients with (a–c) and without (d–f) hepatocellular carcinoma recurrence.
References
- Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–46. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- Moudgil V, Redhu D, Dhanda S, Singh J. A review of molecular mechanisms in the development of hepatocellular carcinoma by aflatoxin and hepatitis B and C viruses. J Environ Pathol Toxicol Oncol. 2013;32:165–75. doi: 10.1615/jenvironpatholtoxicoloncol.2013007166. [DOI] [PubMed] [Google Scholar]
- White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol. 2012;10:1342–59. doi: 10.1016/j.cgh.2012.10.001. .e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames BN. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science. 1983;221:1256–64. doi: 10.1126/science.6351251. [DOI] [PubMed] [Google Scholar]
- Woo RA, Poon RY. Activated oncogenes promote and cooperate with chromosomal instability for neoplastic transformation. Genes Dev. 2004;18:1317–30. doi: 10.1101/gad.1165204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaou K, Sarris M, Talianidis I. Molecular pathways: the complex roles of inflammation pathways in the development and treatment of liver cancer. Clin Cancer Res. 2013;19:2810–6. doi: 10.1158/1078-0432.CCR-12-1961. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Kudo M. Molecular link between liver fibrosis and hepatocellular carcinoma. Liver Cancer. 2013;2:365–6. doi: 10.1159/000343851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Oishi N, Yamashita T, Kaneko S. Molecular biology of liver cancer stem cells. Liver Cancer. 2014;3:71–84. doi: 10.1159/000343863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Chan KW, Hu L, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–56. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Boumahdi S, Driessens G, Lapouge G, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511:246–50. doi: 10.1038/nature13305. [DOI] [PubMed] [Google Scholar]
- Lengerke C, Fehm T, Kurth R, et al. Expression of the embryonic stem cell marker SOX2 in early-stage breast carcinoma. BMC Cancer. 2011;11:42. doi: 10.1186/1471-2407-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Jiang M, Lu Y, et al. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell. 2013;12:304–15. doi: 10.1016/j.stem.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta DN. Production of cancer of the bile ducts with thioacetamide. Nature. 1955;175:257. doi: 10.1038/175257a0. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Kudo M, Umemura A, et al. p38α inhibits liver fibrogenesis and consequent hepatocarcinogenesis by curtailing accumulation of reactive oxygen species. Cancer Res. 2013;73:215–24. doi: 10.1158/0008-5472.CAN-12-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann T, Müller A, Machnik G, et al. Biochemical and morphological studies on production and regression of experimental liver cirrhosis induced by thioacetamide in Uje: WIST rats. Z Versuchstierkd. 1987;30:165–80. [PubMed] [Google Scholar]
- Nishiyama H, Itoh K, Kaneko Y, et al. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J Cell Biol. 1997;137:899–908. doi: 10.1083/jcb.137.4.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita J. Cold shock response in mammalian cells. J Mol Microbiol Biotechnol. 1999;1:243–55. [PubMed] [Google Scholar]
- Wellmann S, Bührer C, Moderegger E, et al. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci. 2004;117:1785–94. doi: 10.1242/jcs.01026. [DOI] [PubMed] [Google Scholar]
- Yang C, Carrier F. The UV-inducible RNA-binding protein A18 (A18 hnRNP) plays a protective role in the genotoxic stress response. J Biol Chem. 2001;276:47277–84. doi: 10.1074/jbc.M105396200. [DOI] [PubMed] [Google Scholar]
- Saito K, Fukuda N, Matsumoto T, et al. Moderate low temperature preserves the stemness of neural stem cells and suppresses apoptosis of the cells via activation of the cold-inducible RNA binding protein. Brain Res. 2010;1358:20–9. doi: 10.1016/j.brainres.2010.08.048. [DOI] [PubMed] [Google Scholar]
- De Leeuw F, Zhang T, Wauquier C, et al. The cold-inducible RNA-binding protein migrates from the nucleus to cytoplasmic stress granules by a methylation-dependent mechanism and acts as a translational repressor. Exp Cell Res. 2007;313:4130–44. doi: 10.1016/j.yexcr.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Yang R, Weber DJ, Carrier F. Post-transcriptional regulation of thioredoxin by the stress inducible heterogenous ribonucleoprotein A18. Nucleic Acids Res. 2006;34:1224–36. doi: 10.1093/nar/gkj519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morf J, Rey G, Schneider K, et al. Cold-inducible RNA-binding protein modulates circadian gene expression posttranscriptionally. Science. 2012;338:379–83. doi: 10.1126/science.1217726. [DOI] [PubMed] [Google Scholar]
- Qiang X, Yang WL, Wu R, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19:1489–95. doi: 10.1038/nm.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artero-Castro A, Callejas FB, Castellvi J, et al. Cold-inducible RNA-binding protein bypasses replicative senescence in primary cells through extracellular signal-regulated kinase 1 and 2 activation. Mol Cell Biol. 2009;29:1855–68. doi: 10.1128/MCB.01386-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Itoh K, Higashitsuji H, et al. Cirp protects against tumor necrosis factor-alpha-induced apoptosis via activation of extracellular signal-regulated kinase. Biochim Biophys Acta. 2006;1763:290–5. doi: 10.1016/j.bbamcr.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Kashida H, Watanabe T, et al. Stress response protein Cirp links inflammation and tumorigenesis in colitis-associated cancer. Cancer Res. 2014;74:6119–28. doi: 10.1158/0008-5472.CAN-14-0471. [DOI] [PubMed] [Google Scholar]
- Masuda T, Itoh K, Higashitsuji H, et al. Cold-inducible RNA-binding protein (Cirp) interacts with Dyrk1b/Mirk and promotes proliferation of immature male germ cells in mice. Proc Natl Acad Sci U S A. 2012;109:10885–90. doi: 10.1073/pnas.1121524109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- Sakurai T, He G, Matsuzawa A, et al. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–65. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- He G, Yu GY, Temkin V, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–97. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung SJ, Yoon JH, Yu SJ. STAT3&Cytochrome P450 2C9: a novel signaling pathway in liver cancer stem cells. Biomed Pharmacother. 2012;66:612–6. doi: 10.1016/j.biopha.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A. 2006;103:10544–51. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Matsuyama Y, Tanaka E, et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J Hepatol. 2003;38:200–7. doi: 10.1016/s0168-8278(02)00360-4. [DOI] [PubMed] [Google Scholar]
- Lo RC, Ng IO. Hepatic progenitor cells: their role and functional significance in the new classification of primary liver cancers. Liver Cancer. 2013;2:84–92. doi: 10.1159/000343844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8. [PubMed] [Google Scholar]
- Smith J, Ladi E, Mayer-Proschel M, Noble M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc Natl Acad Sci U S A. 2000;97:10032–7. doi: 10.1073/pnas.170209797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grek CL, Tew KD. Redox metabolism and malignancy. Curr Opin Pharmacol. 2010;10:362–8. doi: 10.1016/j.coph.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Belle JE, Orozco NM, Paucar AA, et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. 2011;8:59–71. doi: 10.1016/j.stem.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myant KB, Cammareri P, McGhee EJ, et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12:761–73. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Han KH. Epidemiology and surveillance of hepatocellular carcinoma. Liver Cancer. 2012;1:2–14. doi: 10.1159/000339016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi P, Cheng SQ, Wang H, Li N, Chen YF, Gao CF. Serum microRNAs as biomarkers for hepatocellular carcinoma in Chinese patients with chronic hepatitis B virus infection. PLoS One. 2011;6:e28486. doi: 10.1371/journal.pone.0028486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (a) Thioacetamide treatment induced liver fibrosis in mice. (b) Mice were treated with thioacetamide (TAA) for 10 months. No significant difference in hydroxyproline contents was found between WT and Cirp−/− mice.
Fig. S2. Immunohistochemistry was carried out on liver sections of thioacetamide (TAA)-treated WT mice.
Fig. S3. Treatment with the antioxidant decreased protein level of phosphorylated STAT3.
Fig. S4. Representative immunostaining images of non-tumor livers of patients with (a–c) and without (d–f) hepatocellular carcinoma recurrence.