

Abstract
Background and Purpose
Transient receptor potential vanilloid-4 (TRPV4) is a calcium-permeant ion channel that is known to affect vascular function. The ability of TRPV4 to cause a vasoconstriction in blood vessels has not yet been mechanistically examined. Further in neuronal cells, TRPV4 signalling can be potentiated by GPCR activation. Thus, we studied the mechanisms underlying the vascular contractile action of TRPV4 and the GPCR-mediated potentiation of such vasoconstriction, both of which are as yet unappreciated aspects of TRPV4 function.
Experimental Approach
The mechanisms of TRPV4-dependent regulation of vascular tone in isolated mouse aortae were studied using wire myography. TRPV4-dependent calcium signalling and prostanoid production was studied in cultured human umbilical vein endothelial cells (HUVECs).
Key Results
In addition to the well-documented vasorelaxation response triggered by TRPV4 activation, we report here a TRPV4-triggered vasoconstriction in the mouse aorta that involves a COX-generated Tx receptor (TP) agonist that acts in a MAPK and Src kinase signalling dependent manner. This constriction is potentiated by activation of the GPCRs for angiotensin (AT1 receptors) or proteinases (PAR1 and PAR2) via transactivation of the EGF receptor and a process involving PKC. TRPV4-dependent vascular contraction can be blocked by COX inhibitors or with TP antagonists. Further, TRPV4 activation in HUVECs stimulated Tx release as detected by an elisa.
Conclusion and Implications
We conclude that the GPCR potentiation of TRPV4 action and TRPV4-dependent Tx receptor activation are important regulators of vascular function and could be therapeutically targeted in vascular diseases.
Tables of Links
| TARGETS | |
|---|---|
| Ion channelsa | Enzymesd |
| TRPV4 channels | COX-1 |
| GPCRsb | COX-2 |
| AT1 receptors | cPLA2 |
| EP1 receptors | CYP450-4A |
| EP3 receptors | DAG lipase |
| TP receptors | MAPK |
| PAR1, proteinase-activated receptor1 | PKC |
| PAR2, proteinase-activated receptor2 | PLC |
| Catalytic receptorsc | Src-selective TK |
| EGFR, EGF receptors | TxA2 synthase (CYP5A1) |
| LIGANDS | |
|---|---|
| AG1478 | L-NAME |
| Angiotensin II | NO |
| GF109203X | PGE2 |
| Go6983 | Phenylephrine |
| GSK1016790A | PP1 |
| GSK2193874 | SC-51322 |
| HC067047 | TFLLR-NH2 |
| ICI 192605 | TxA2 |
| Indomethacin | U0126 |
| L-798106 | U46619 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a,b,c,d,,,).
Introduction
The transient receptor potential (TRP) superfamily comprises six families of polymodally activated non-selective cation channels (Clapham, 2003). The vanilloid family member TRPV4 was identified originally as a channel activated by hypotonic cell swelling (Liedtke et al., 2000; Peng and Hediger, 2002). Further studies in knockout mice have established the TRPV4 channel as an osmotic sensor in the CNS (Liedtke and Friedman, 2003). However, subsequent studies have also identified remarkable gating promiscuity with other TRPV4 channel activators including vascular shear stress (Gao et al., 2003; Wu et al., 2007), the phorbol-ester, 4-phorbol 12,13-didecanoate (4α-PDD; Watanabe et al., 2002a), temperature (Guler et al., 2002; Watanabe et al., 2002b), pressure (Suzuki et al., 2003) and low pH (Suzuki et al., 2003). TRPV4 channels are also triggered by anandamide and arachidonic acid subsequent to their P450-epoxygenase-dependent metabolism to the agonist, 5′,6′-epoxyeicosatrienoic acid (Watanabe et al., 2003), which can have antihypertensive activity. Pharmacologically, TRPV4 channels can be activated selectively with the synthetic agonist GSK1016790A (GSK101; Thorneloe et al., 2008; Willette et al., 2008). TRPV4 knockout mice have no overt malformations but have impaired hearing (Tabuchi et al., 2005) and osmoregulation (Liedtke and Friedman, 2003), abnormal thermosensation (Todaka et al., 2004), diminished pain sensitivity (Suzuki et al., 2003; Alessandri-Haber et al., 2006; Grant et al., 2007) and increased bone mass (Masuyama et al., 2008).
There is compelling evidence for the involvement of several TRP channels in the regulation of vascular tone (Watanabe et al., 2008; Earley and Brayden, 2010; Baylie and Brayden, 2011). TRPV4 channels are expressed principally in the endothelial cells (Mendoza et al., 2009; Bubolz et al., 2012; Sonkusare et al., 2012) but expression is also reported in the smooth muscle cells of selected vessels (Jia et al., 2004; Ducret et al., 2008). Activation of TRPV4 channels regulates endothelial calcium influx and triggers a NO-dependent vasodilation (Earley et al., 2009; Mendoza et al., 2009; Bubolz et al., 2012). In resistance arteries, Ca2+ influx through single TRPV4 channels and the activation of as few as three channels per cell causes maximum vessel dilatation through activation of endothelial cell intermediate conductance (IK) and small conductance (SK) potassium channels (Sonkusare et al., 2012). The majority of the TRPV4 channels are believed to be silent even during maximal stimulation (Sullivan et al., 2012). Interestingly, TRPV4 knockout mice are normotensive (Willette et al., 2008), although they show significantly reduced endothelium-dependent vascular dilation in response to blood flow (Hartmannsgruber et al., 2007). Although vasorelaxation is a widely recognized response to endothelial TRPV4 channel activation, contractile responses have not yet been evaluated. One of our aims was thus to study the vasoconstrictor actions mediated by TRPV4 channels.
Apart from the vasculature, TRPV4 channels are known to play an important role in other targets like neuronal cells. In such systems, signalling by GPCRs converge on TRP channels to modulate their activity. 5-HT (Ducret et al., 2008) and mGluR (Ene et al., 2007) receptors activate mammalian TRP channels (including TRPV4-like currents). Histamine and 5-HTare also reported to potentiate TRPV4 channel function in neuronal cells (Cenac et al., 2010). Indeed, molecular coupling between proteinase-activated receptor 2 (PAR2) and angiotensin AT1 receptors with TRPV4 and other TRP channels has also been documented in cell culture systems (Amadesi et al., 2004; Grant et al., 2007), but not in intact blood vessels. Further, although a number of GPCRs are also known to regulate vascular function (Tilley, 2011), the ability of GPCR signalling to interact with TRPV4 channel function in the vasculature has not yet been studied.
In order to understand the mechanisms underlying synergy between TRPV4 channels and GPCRs, we studied distinct prototype GPCRs known to be particularly important vascular regulators, PARs (PAR1 and PAR2) and the AT1 receptor. Unlike most GPCRs that are regulated by circulating hormones (e.g. angiotensin II, adrenaline), PARs are locally activated at sites of injury, inflammation or cell stress by proteolysis, which unmasks a tethered ligand (TL) activating sequence (Coughlin, 2000; Adams et al., 2011; Ramachandran et al., 2012). In vivo, enzymes of the coagulation cascade (thrombin, Factor VIIa/Xa, activated protein C) (Adams et al., 2011; Lee and Hamilton, 2012; Ramachandran et al., 2012) and other endogenous serine proteinases (trypsins; tissue kallikreins) trigger PAR function as do synthetic peptides derived from the TL sequence. The angiotensin II-activated AT1 receptor is a GPCR that typically activates PLC through Gαq coupling (de Gasparo et al., 2000). The octapeptide, angiotensin II, is the active component of the renin-angiotensin system that can be generated either systemically or locally in vessels via proteolytic processing of angiotensinogen to form the decapeptide, angiotensin I, which is converted by the dipeptidyl carboxypeptidase, ACE to active angiotensin II (Touyz and Schiffrin, 2000) that in turn stimulates the AT1 receptor. Both PARs and the AT1 receptor couple to several distinct G-proteins (Gαq, Gαi, Gα12/13) to trigger intracellular signalling. Depending on the agonist triggering receptor activation, these receptors can also exhibit biased signalling (Ramachandran et al., 2009; Zimmerman et al., 2012), where they recruit selective subsets of their entire signalling repertoire. Thus, in addition to regulating vascular tension via Gαq-triggered elevations of intracellular calcium, both PARs and AT1 receptors can activate vascular anabolic and inflammatory pathways involving β-arrestins, MAPK, Src, EGF receptor (EGFR) transactivation, PKC, and NF-κB activation.
We thus hypothesized that GPCR signalling represents a major mechanism for amplifying vascular TRPV4 channel function and that GPCR-TRPV4 channel interactions comprise a key signalling pathway for the endothelium-dependent regulation of vascular tone. We thus employed wire myography with murine isolated aorta tissue and cultured human umbilical vein endothelial cells to obtain new data showing that TRPV4 channels can regulate vasoconstriction via an endothelium-dependent release of a COX-generated TP receptor agonist (most likely TxA2 itself). Further, both the PARs and the AT1 receptor can modulate vascular endothelial function by potentiating TRPV4 channel activity via a mechanism that involves transactivation of the EGFR.
Methods
Animals, peptides and other reagents
Animal care and experimental procedures were approved by the Animal Resources Committee at the University of Calgary and were in accordance with the guidelines of the Canadian Council for Animal Care in Research. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; (McGrath et al., 2010). A total of 64 animals were used to perform the experiments described in the paper.
Six- to eight-week-old C57bl6 mice (Charles River Laboratories, Montreal, QC, Canada) or PAR knockout mice bred in house were used for all experiments. PAR1 and PAR2 knockout mice were a generous gift from Dr. Patricia Andrade-Gordon (Johnson & Johnson Pharmaceutical Research & Development) (Damiano et al., 1999).
Wire myograph studies
Isometric tension studies using wire myography were performed as described previously (McGuire et al., 2004; El-Daly et al., 2014). In brief, mice were killed either by injecting i.p. with 0.1 mL euthanyl (pentobarbital 240 mg·mL−1) and 0.5 mL heparin (1 mg·mL−1) or by cervical dislocation 10 min after i.p. administration of 0.5 mL of heparin (1 mg·mL−1). Blood vessels were flushed with 0.5 mL of 1 mg·mL−1 heparin transcardially and aortae were dissected free of perivascular adipose and connective tissue into ice-cold Krebs solution (115 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 1.2 mM NaH2PO4, 10.0 mM dextrose and 2.5 mM CaCl2), pH 7.4, aerated with 95% O2 and 5% CO2. Aorta rings were mounted in a Mulvany–Halpern myograph organ bath (610 multi-myograph system coupled to Chart5 system software, AD instruments, Colorado Springs, CO, USA). All assays were performed in Krebs buffer and resting tension (4.5 mN) was fixed for 1 h prior to all experiments. Tissue viability was assessed by contracting the tissue with 80 mM KCl. Endothelial integrity was assessed by contracting the tissue with 2.5 μM phenylephrine and monitoring ACh (0.2 or 1 μM)-dependent relaxation. In some assays, the endothelium was denuded by rubbing the intimal layer of the aorta gently with horsehair. In these vessels, the lack of endothelium was confirmed by a lack of relaxant response to Ach (10 μM) and the integrity of the smooth muscle layer was confirmed by monitoring isoproterenol (1 μM)-dependent relaxation in phenylephrine-contracted tissue. All studies examining contractile responses were performed in the presence of 100 μM L-NG-nitroarginine methyl ester (L-NAME). Contractile responses to GSK101 (in the presence and absence of signalling pathway inhibitors) was routinely monitored in the linear range of the concentration-effect curve (25 or 50 nM: Figure 1C).
Figure 1.
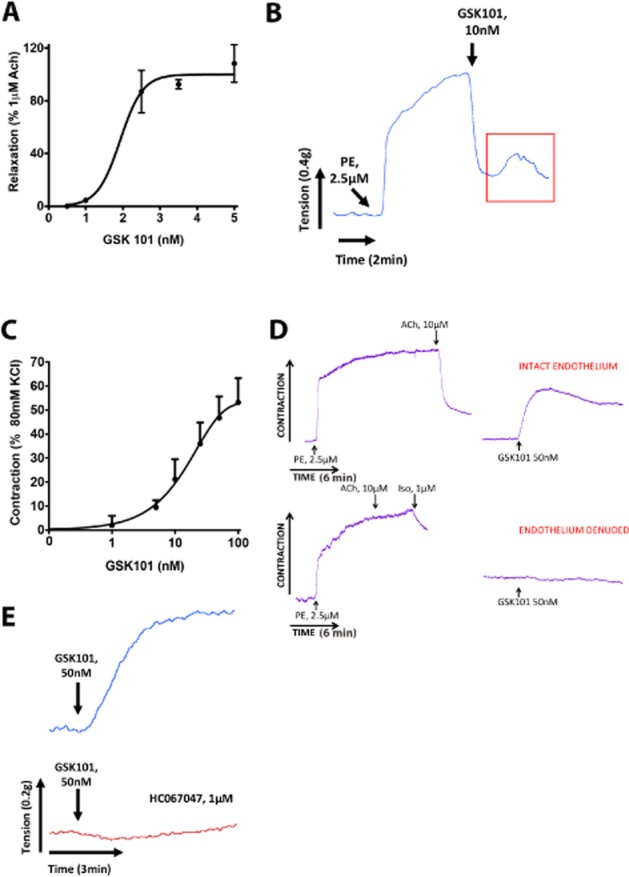
TRPV4 channel activation causes both relaxation and contraction responses in the mouse aorta. (A) Concentration effect curve for TRPV4-dependent relaxation responses to GSK101 in the mouse aorta. (B) In a phenylephrine (PE; 2.5 μM)-constricted mouse aorta, GSK101 (10 nM)-triggered relaxation is consistently followed by a contraction (box). (C) Concentration effect curve for TRPV4-dependent contraction in the mouse aorta. (D) TRPV4 channel activation triggers contraction in mouse aorta with intact endothelium (upper panel) but not in endothelium denuded mouse aorta (lower panel). Iso, isoprenaline. (E) GSK101 (50 nM)-triggered contraction is inhibited by the TRPV4 channel antagonist HC067047 (1 μM). Data are expressed as the mean ± SEM for A and C. n = 6.
Cell culture and Tx elisa
HUVECs were obtained from Invitrogen/Life Technologies, Burlington, ON, Canada, and cultured in basal media (Medium 200) supplemented with growth factor mixture (Large Vessel Endothelial supplement Cat # A1460801 containing FBS, hydrocortisone, human EGF, basic fibroblast growth factor, heparin and ascorbic acid; Invitrogen). HUVECs were expanded fourfold at each passage and cells stocks at passage three were then stored in liquid nitrogen for subsequent use in experiments up to passage 8. In order to assess endothelial cell TxA2 production, we used an enzyme immunoassay (Cayman Chemicals, Ann Arbor, MI, USA) to detect levels of the TxB2, a stable metabolite formed by rapid non-enzymatic hydrolysis of TxA2.
Calcium signalling
Calcium signalling was performed in HUVECs loaded with fluo-4-AM (Invitrogen) as described previously (Ramachandran et al., 2011). The concentration of GSK101 used to stimulate calcium signalling (0.5 nM) was at the low end of its concentration-effect curve to optimize detection of synergy between TRPV4 channel and GPCR signalling.
Data analysis
Results are expressed as the mean ± SEM, with n indicating the number of animals used for a particular set of experiments. Vascular tension data are expressed either as a % of the contractile response to 80 mM KCl or as a normalized contraction (% control) response, where the control group response was designated as 100% and all the experimental data sets (e.g. effect of inhibitors) are expressed as a percentage of that response. Vascular relaxation is expressed as a % of the relaxant response to 1 μM ACh. Statistical analysis was performed with GraphPad Prism 6 software (La Jolla, CA, USA). Data were analysed using unpaired Student's t-tests or one-way anova followed by Dunnett's post hoc test as appropriate. P < 0.05 values were considered to show significant differences between means.
Materials
Receptor-selective PAR1 (TFLLR-NH2) and PAR2 (2f-LIGRLO-NH2) activating peptides were synthesized as carboxy terminal amides (<95% purity by HPLC and mass spectrometry) by the peptide synthesis facility at the University of Calgary (peplab@ucalgary.ca). Porcine pancreatic trypsin (16 000 BAEE U·mg−1) was from Sigma (St. Louis, MO, USA; Cat. No. T0303). Human plasma thrombin was from Calbiochem (Etobicoke, ON, Canada; 3045 NIH U·mg−1, Cat. No. 605195). Angiotensin II was from Sigma (Cat. No. A9525). The TRPV4 antagonists, HC067047 (Everaerts et al., 2010) and GSK2193874 (Thorneloe et al., 2012), COX-1 inhibitor SC-560, COX-2 inhibitor NS-398, EP1 receptor antagonist SC-51322, EP3 receptor antagonist L-798,106, Src kinase inhibitor PP1, MAPK inhibitor U0126, PKC inhibitors Go6983 and GF109203X were from Tocris Bioscience (Bristol, UK). TRPV4 channel agonist GSK1016790A (GSK101; Thorneloe et al., 2008; Willette et al., 2008), acetylcholine, phenylephrine and all other chemicals were from Sigma (St. Louis, MO). Agonist and antagonist concentrations employed were chosen based on studies in the literature and previously published studies from our own laboratories (Laniyonu et al., 1994; Poole et al., 2013; El-Daly et al., 2014) and fully took into account recommended guidelines for judicious use of pharmacological inhibitors of signalling protein function (Bain et al., 2003; 2007; Cohen, 2010).
Results
Evaluation of TRPV4 channel-dependent contractile and relaxant response in the mouse aorta
As reported by previous studies (Sonkusare et al., 2012), we observed a vasorelaxation response to TRPV4 channel activation with GSK101 (0.1–5 nM) in the phenylephrine-constricted mouse aorta (Figure 1A, B). This response was blocked by pretreating the tissue with L-NAME, indicative of a NO-dependent relaxation of the blood vessels (not shown). Interestingly, we also observed that the TRPV4 channel-dependent relaxation response was consistently followed by a contractile response (Figure 1B). We therefore evaluated the ability of TRPV4 channels to trigger contractile responses in the mouse aorta. Indeed, in L-NAME-treated blood vessels at resting tension, we observed a concentration-dependent contractile response to GSK101 (Figure 1C and Supporting Information Figure S1A). This TRPV4-dependent contractile response was endothelium dependent (Figure 1D) and was abolished in blood vessels that had been mechanically stripped of endothelial cells. The integrity of the endothelium-denuded tissue was confirmed by monitoring isoprenaline-dependent vasorelaxation. The contractile response to TRPV4 channel activation was also abolished in the presence of the TRPV4 channel antagonists HC067047 (1 μM; Figure 1E) or GSK2193874 (Supporting Information Figure S1B).
Evaluation of the effect of kinase inhibitors on TRPV4-dependent contractions
Since PK inhibitors block responses to a number of contractile agents in the aorta, we tested the action of inhibitors targeting PKC [GF109203X (GFX) 0.3 μM and Go6983 0.3 μM], EGFR kinase (AG1478, 0.3 μM), p42/44 MAPK (U0126, 1 μM) and Src-selective TK (PP1, 1 μM) in regulating TRPV4-dependent contraction. TRPV4 channel-triggered contraction was modestly inhibited in vessels exposed to the PKC inhibitor Go6983 (0.3 μM) by 38.8 ± 14% and with GFX (0.3 μM) by 34.8 ± 18% but this did not reach statistical significance (Figure 2A and Supporting Information Figure S2A). Similarly, in vessels treated with the EGF kinase inhibitor, AG1478 (0.3 μM), TRPV4-dependent contractile responses were not statistically different from control GSK101-triggered contractions (Figure 2B). In contrast, treatment of blood vessels with the Src-selective TK inhibitor PP1 led to a substantial inhibition of 62 ± 18% of the GSK101 (50 nM) response. Similarly, treating vessels with the MAPK inhibitor (U0126, 1 μM) led to a 72 ± 2.5% inhibition of the GSK101 (50 nM) response (Figure 2C and Supporting Information Figure S2B). Significantly, none of these inhibitors affected the phenylephrine or PGE2-stimulated contractile responses (not shown).
Figure 2.
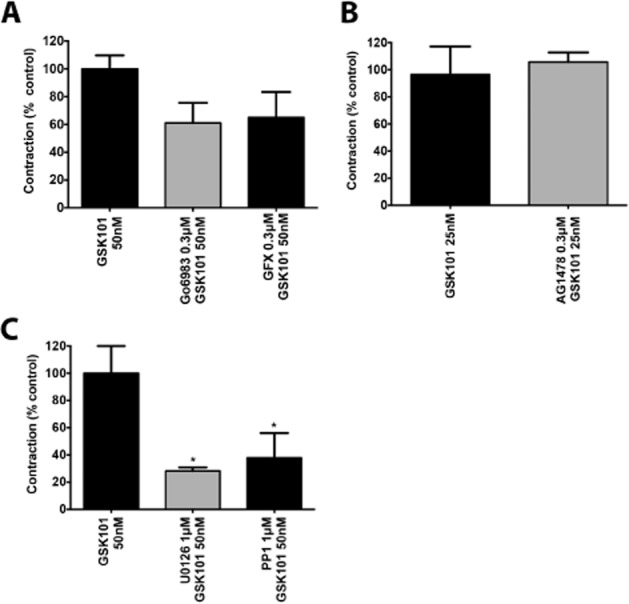
TRPV4 channel-mediated-contraction is MAPK and Src kinase dependent. (A) TRPV4-dependent contraction in the mouse aorta (GSK101, 50 nM) is not significantly inhibited by PKC inhibitors Go6983 (0.3 μM) or GFX (0.3 μM). (B) TRPV4-dependent contraction in the mouse aorta (GSK101, 25 nM) is not inhibited by EGFR kinase inhibition (AG1478, 0.3 μM). (C) TRPV4-dependent contraction in the mouse aorta (GSK101, 50 nM) is inhibited by the MAPK inhibitor U0126 (1 μM) and Src kinase inhibitor PP1 (1 μM). Data are expressed as the mean ± SEM. n = 4–6. * P < 0.05, significantly different from response triggered by 50 nM GSK101.
Evaluation of the role for COX pathways in TRPV4-dependent contraction
As we had found that the contractile response from TRPV4 channel activation was endothelium dependent, we hypothesized the involvement of COX-1 and COX-2-derived prostanoid products. By exposing the aorta to the non-selective COX inhibitor indomethacin (1 μM) prior to addition of GSK101, we were able to abolish the contractile response substantially (Figure 3A). Using the more selective inhibitors of COX-1 (SC560, 1 μM) and of COX-2 (NS-398, 2 μM), we observed 100% and 80 ± 10% inhibition of the TRPV4-dependent contraction, respectively (Figure 3A and Supporting Information Figure S2C). Of note, this inhibition was observed for a contractile concentration of GSK101 that was towards the top of its concentration-effect curve (Figure 1C).
Figure 3.
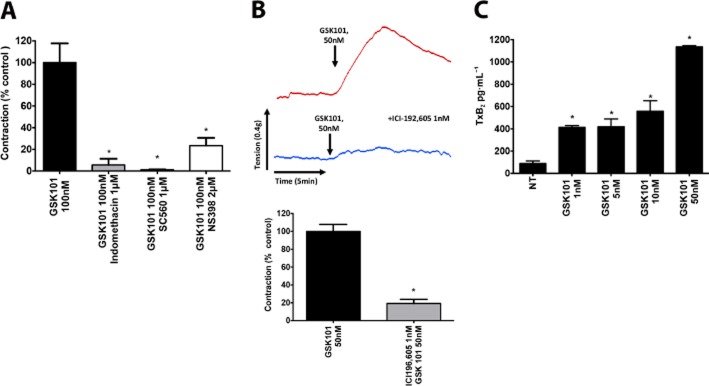
TRPV4 channel-mediated-contraction is Tx dependent. (A) TRPV4-dependent contraction in the mouse aorta (GSK101, 100 nM) is significantly inhibited by the non-selective COX inhibitor indomethacin (1 μM), COX-1 inhibitor SC-560 (1 μM) or the COX-2 inhibitor NS-398 (1 μM) and (B) the TP antagonist ICI192,605 (1 nM). (C) elisa detection of TxB2 (the stable metabolite of TxA2) in GSK101 (1–50 nM) treated HUVEC cells. Data are expressed as the mean ± SEM. n = 4–6. *P < 0.05, significantly different from control GSK101 response for Figures A and B. In (C), *P < 0.05 significantly different from levels in untreated HUVEC cells.
Having established that TRPV4 channel activation was leading to the generation of a contractile COX product, we sought to identify the receptor target for the contractile prostanoid being generated. Although PGE2 is a common ligand for four different receptors, EP1–4, activation of only EP1 and EP3 receptors leads to a contractile response (Woodward et al., 2011). We therefore used selective EP1 and EP3 receptor antagonists as well as and antagonist for the Tx receptor (TP), activation of which can also trigger vascular contractile responses (Woodward et al., 2011). Inhibition of the TRPV4-dependent contractile response was seen with the EP1 receptor antagonist (SC51322, 100 nM; 54 ± 7%) and the EP3 antagonist (L798,106, 100 nM; 17 ± 4%; not shown). Strikingly, pretreatment of the tissue with the TP receptor antagonist ICI192,605 (1 nM) led to a near-complete inhibition of GSK101-triggered vascular contraction (Figure 3B).
We wished to verify the specificity of the receptor antagonists used and therefore exposed the mouse aorta to the TxA2 mimetic U46619 (1 nM) in the presence of the EP1 and EP3 receptor antagonists (100 nM) and conversely exposed the tissue to PGE2 (0.25 μM) in the presence of the TP receptor antagonist ICI192,605 (1 nM). The response to U46619 was inhibited by the EP3 antagonist (L798,106, 100 nM) but not the EP1 receptor antagonist (SC51322, 100 nM) to the same extent as the GSK101 response. The contractile response generated by PGE2 was not inhibited by the TP receptor antagonist (ICI192,605, 1 nM; data not shown). We therefore conclude that the inhibition seen with the EP3 receptor antagonist was most likely due to its action on the TP receptor. In order to examine the involvement of the PGF2α receptors (PG F2-alpha receptor), which can also contract the mouse aorta, we exposed the tissue to PGF2α (0.3 μM) in the presence of the TP antagonist ICI192,605 (1 nM) and observe a 64% inhibition of the PGF2α-triggered contractile response. Thus, the identity of the contractile prostanoid being generated is most likely TxA2; however, a role for PGF2α or PGH2 acting on the TP receptor to cause contraction, cannot be ruled out.
To assess the biochemical pathway responsible for generating the contractile COX agonist, we used inhibitors of cytosolic PLA2 (cPLA2), DAG lipase, cytochrome p450A (CP450-4A) and Tx synthase to target different steps in the prostanoid biosynthesis pathway. Treating the tissue with the cPLA2 inhibitor (10 μM) modestly attenuated the contractile response to GSK101 (50 nM), while the DAG lipase inhibitor (RHC80267, 10 μM), the CYP450-4A enzyme suicide substrate inhibitor, 17-octadecanoic acid (ODYA, 20 μM) and the TxA2 synthase inhibitor ozagrel hydrochloride (50 nM) were without effect (Supporting Information Figure S3A–D).
Additionally, in order to further support our notion that the TRPV4 channel-generated contractile prostanoid was TxA2, we used HUVEC cultures to assess GSK101-stimulated TxA2 production using an elisa assay. We found that TRPV4 channel activation with GSK101 resulted in a concentration-dependent production of TxB2, the stable metabolite of TxA2 (Figure 3C).
As discussed above, because we found that the TRPV4 channel-triggered contraction, occurring through the release/action of a prostanoid that we believe to be TxA2, was blocked by p42/44 MAPK and Src kinase inhibition, we investigated the effect of these inhibitors on the action of the TxA2 analogue, U46619 (1 nM). In the mouse aorta, the TP receptor agonist U46619-mediated contractions were not blocked by the Src kinase inhibitor (1 μM), but were abolished by the MAPK inhibitor U01266 (1 μM; not shown). Our data thus indicated that the contractile action of receptor activation by the Tx analogue was dependent on MAPK activity, whereas the Src kinase signalling is involved in generating the contractile prostanoid.
Regulation of TRPV4 channel function by GPCR activation
Previous work on TRPV4 and other TRP channels has established a paradigm whereby GPCR activation can potentiate the channel function. Since GPCRs are key regulators of vascular function and, as we identified TRPV4 channels are endothelial ion channels capable of regulating vascular relaxation and contraction, we hypothesized that GPCR activation, as represented by the receptors for angiotensin (AT1) and serine proteinases (PARs 1 and 2), might modulate TRPV4 channel function in the blood vessels. We focused on PAR1 and PAR2, two members of the PAR family of GPCRs that are known to important regulators of cardiovascular function and are activated by coagulation proteinases (e.g. thrombin; factor VIIa/Xa), and can be triggered in the setting of stroke, turbulent flow, ischaemia reperfusion and vascular inflammation, for example, in atheromas (Ramachandran and Hollenberg, 2008; Ramachandran et al., 2012). AT1 receptors are known to regulate a range of cardiovascular responses (de Gasparo et al., 2000) and have been reported to colocalize with TRPV4 channels in blood vessels (Mercado et al., 2014). Together, these two receptor systems represent a link between traditional vascular GPCRs like the AT1 receptor and prototype pattern recognition GPCRs like PARs, which regulate the inflammatory innate immune response to change vascular function.
We used angiotensin II (10 nM) to activate the AT1 receptor, thrombin (5 U·mL−1) and the PAR1-selective receptor-activating peptide, TFLLR-NH2 (5 μM) to activate PAR1, and trypsin (10 U·mL−1) or the potent PAR2-selective agonist, 2-furoyl-LIGRLO-NH2 (25 μM) to activate PAR2 (Figure 4). Prior activation of PAR1, PAR2 or AT1 receptors caused an amplification of the TRPV4 channel-stimulated vascular contractile response but was without effect on the TRPV4-dependent vasorelaxation (Figure 4A). The magnitude of the TRPV4 channel-dependent contractile response seen in the presence of any of the three GPCR agonists was close to double than seen with GSK101-dependent TRPV4 channel activation. This amplification of the TRPV4-dependent contractile response caused by PAR1 and PAR2 agonists was not seen in aorta derived from PAR1 and PAR2 knockout mice respectively (data not shown). Similarly, in HUVECs, GSK101 (0.5 nM)-triggered calcium responses were enhanced by concentrations of agonist for AT1 receptors (angiotensin II, 50 nM) or PAR2 (2fLI, 10 nM) that did not on their own cause a significant acute increase in intracellular calcium (Figure 4B).
Figure 4.
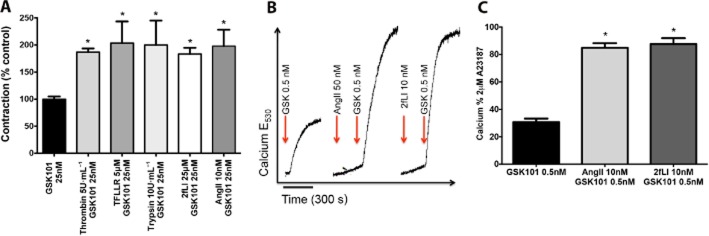
PAR1, PAR2 and AT1 receptor activation potentiates TRPV4-dependent contractile responses. (A) TRPV4-dependent contraction in the mouse aorta (GSK101, 25 nM) is potentiated by prior activation of PAR1 with thrombin (5 U·mL−1) or TFLLR-NH2 (5 μM), PAR2 activation with trypsin 10 U·mL−1 or 2FLIGRLO-NH2 (2FL; 25 μM) or AT1 receptor activation with angiotensin II (AngII; 10 nM). (B) Angiotensin II (50 nM) and 2fLIGRLO-NH2 (10 nM) activation of AT1 receptors and PAR2 potentiate TRPV4 channel calcium signalling in HUVECs (GSK101, 0.5 nM). Figure A data are expressed as the mean ± SEM. n = 6–8. *P < 0.05, significantly different from 25 nM GSK101 response. In C, *P < 0.05, significantly different from 0.5 nM GSK101-triggered response.
Role for EGFR transactivation
Our recent work found that transactivation of EGFR plays a role in the contractile actions of PARs 1 and 2 in the porcine coronary artery and others have found a role for EGFR transactivation in the stimulation of MAPK by angiotensin II. (Eguchi et al., 1998; El-Daly et al., 2014). We therefore investigated a role for such transactivation in regulating our observed GPCR-mediated potentiation of TRPV4 channel activation by GSK101. Indeed, when mouse aortae were treated with the EGFR kinase inhibitor, AG1478 (0.3 μM), the PAR2-mediated potentiation of TRPV4 channel-triggered contraction was significantly attenuated (Figure 5A). This inhibition of PAR2-dependent potentiation of TRPV4 channel action was not seen with the inactive tyrphostin analogue, AG(1)9. The potentiation of the TRPV4-dependent contractile response could also be reproduced by treating the mouse aorta with EGF itself (50 ng·mL−1) (Figure 5B). This action of EGF was also blocked by AG1478 (0.3 μM) but not by the inactive analog AG(1)9 (0.3 μM; Figure 5C).
Figure 5.
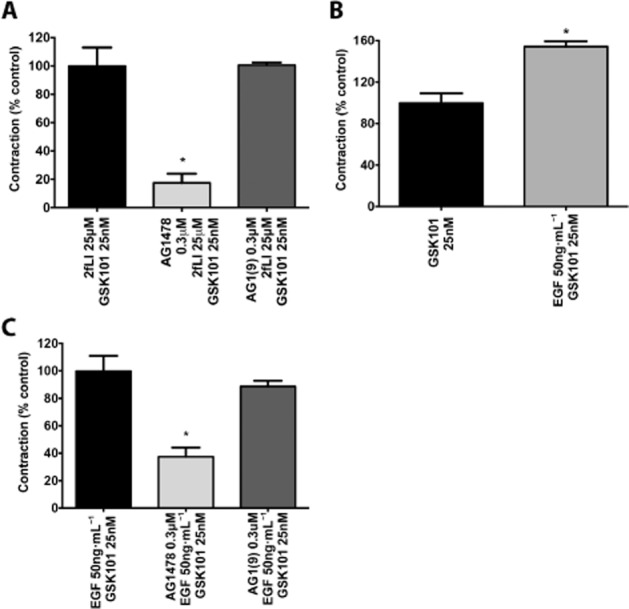
EGFR receptor transactivation underlies GPCR potentiation of TRPV4 channels. (A) PAR2 potentiation of TRPV4-dependent contraction (GSK101, 25 nM) in the mouse aorta is inhibited by EGFR kinase inhibitor AG1478 (0.3 μM) but not the inactive analogue AG1(9) (0.3 μM) *P < 0.05, significantly different from 25 μM 2fLI and 25 nM GSK101-triggered response (B) EGF (50 ng·mL−1) treatment can also potentiate TRPV4-dependent contraction (GSK101, 25 nM) in the mouse aorta. *P < 0.05, significantly different from 25 nM GSK101-triggered response (C) EGF (50 ng·mL−1) potentiation of TRPV4 contraction is inhibited by AG1478 (0.3 μM) but not the inactive analogue AG1(9) (0.3 μM). **P < 0.05, significantly different from 50 ng·mL−1 EGF and 25 nM GSK 101 triggered response. Data are expressed as the mean ± SEM. n = 3.
The two proposed mechanisms involved in GPCR-mediated transactivation of EGFR involve either a MMP-dependent release of heparin-binding EGF (HB-EGF), that in turn activates the EGFR or alternatively via an intracellular Src-mediated phosphorylation and activation of the EGFR in the absence of ligand binding (Daub et al., 1996; Prenzel et al., 1999; Gschwind et al., 2001). We attempted to inhibit the GPCR-dependent transactivation of EGFR and its enhancement of TRPV4 channel-mediated contractile responses using a number of MMP inhibitors (doxycycline, marimastat, BB94, GM6001). However, significant inhibition of the potentiation was not observed with any of the inhibitors (not shown). Since the Src kinase inhibitor blocked the TRPV4 contractile response itself, further studies to evaluate the role of Src kinase in the EGFR transactivation mechanism were not possible in this system.
In keeping with previous published work pointing to a role for PKC in regulating TRPV4 function, we hypothesized that GPCR activation could enhance TRPV4 function through a PKC-dependent phosphorylation of the channel (Peng et al., 2010). Indeed, pretreating the tissue with PKC inhibitors (GFX, 0.3 μM) or Go6983 (0.3 μM) we were able to substantially diminish the PAR2-dependent (Figure 6A) and EGF-dependent (Figure 6B) potentiation of TRPV4 channel-mediated contraction of the mouse aorta.
Figure 6.

GPCR and EGFR potentiation of TRPV4 channels is PKC dependent. (A) Trypsin (25 U·mL−1) or 2fLIGRLO-NH2 (25 μM) dependent PAR2 activation and potentiation of TRPV4 channel-mediated-contraction (GSK101, 25 nM) in the mouse aorta is inhibited by PKC inhibitors Go6983 (0.3 μM) (B) EGF (50 ng·mL−1) potentiation of TRPV4-dependent contraction (GSK101, 25 nM) in the mouse aorta is inhibited by PKC inhibitors Go6983 (0.3 μM) or GFX (0.3 μM). Data are expressed as the mean ± SEM. n = 3. *P < 0.05, significantly different from control group.
Discussion and conclusions
The main finding of our work is that TRPV4 channels, in addition to causing vasorelaxation, also trigger an endothelium-dependent TP receptor-mediated contractile response that involves Src and MAPK signalling pathways (Figure 7). Further, we show that the activation of PAR1, PAR2 and the AT1 receptors can markedly potentiate the TRPV4-mediated contractile response, involving a transactivation of the EGFR and PKC. Our data bear directly on the vascular role of TRPV4 channels, which has emerged recently as a key regulator of vascular relaxation responses (Hartmannsgruber et al., 2007; Earley and Brayden, 2010; Sonkusare et al., 2012).
Figure 7.
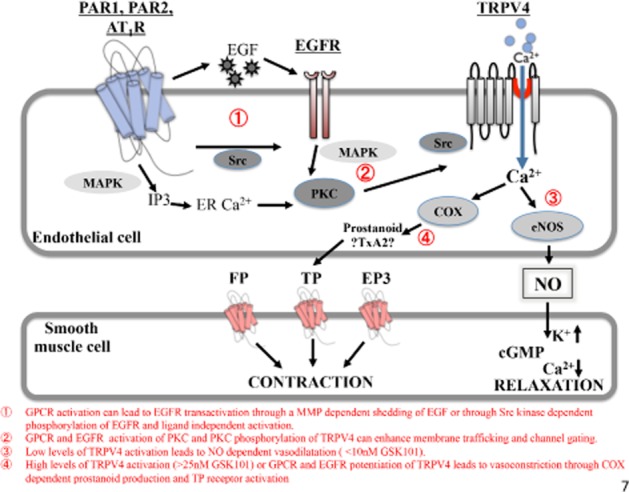
Overview of TRPV4 channel signalling and its modulation by GPCR-dependent EGFR transactivation in the vasculature. In the aorta bioassay, vasodilation is due to the release of the vasorelaxant, NO; vasoconstriction is caused by a prostanoid that acts on the TP receptor. The key findings are numbered in the figure and described below the diagram.
Our evidence that activation of TRPV4 channels triggers a vascular contraction via generation of a prostanoid targeting the TP receptor stems from the use of TP receptor antagonists and an analysis of Tx production by TRPV4 channel-activated HUVECs. In the bioassay, the TP antagonist, ICI192,605, blocked both the GSK101-induced contractions as well as the contractile action of both EP and TP agonists. Further, in cultured endothelial cells, using an elisa assay, we detected generation of Tx in response to TRPV4 channel activation, suggesting further that the TRPV4-triggered vascular contraction is a TxA2-mediated response. While the antagonist data clearly implicate the TP receptor in causing the contraction, the nature of the agonist (very likely Tx) remains to be identified unequivocally. Of note, we found that unlike the COX1 and COX2 inhibitors, the Tx synthase inhibitor did not inhibit the contractile response triggered by TRPV4 channel activation. Thus, it is possible that the contractile response was derived from PGH2 activation of the TP receptor as has been reported (Tesfamariam et al., 1989). Further work will thus be necessary to identify the chemical nature of the contractile agonist.
Two previous studies have noted a modest contractile response dependent on activation of TRPV4 channels, in the mouse mesenteric artery (Mendoza et al., 2009) and the rat pulmonary artery (Sukumaran et al., 2013). The responses in the mouse mesenteric artery, as in our study, are dependent on the presence of an intact endothelium. The TRPV4 channel-dependent contractile responses in the rat pulmonary artery were however observed in endothelium-denuded vessels suggesting TRPV4 channel expression and function in the smooth muscle of that vascular bed. The expression of TRPV4 channels in vascular smooth muscle cells has also been reported in other pulmonary and cerebral vessels (Earley et al., 2005). Thus, we suggest that the endothelium-dependent TRPV4-dependent contractile mechanism we describe here for a conductance vessel (aorta) may apply to some, but by no means all vascular beds. Further investigation of the TRPV4 channel-mediated regulation of other vascular beds is therefore indicated in future work.
While the platelets were originally established as the major source of Tx production (Hamberg et al., 1975), the ability of the endothelial cells also to produce Tx was established soon afterwards (Ingerman-Wojenski et al., 1981; Neri Serneri et al., 1983). Subsequent studies have identified diverse cellular response to TxA2 including up-regulation of adhesion proteins (Ishizuka et al., 1998), impaired insulin signalling (Song et al., 2009) and atherogenesis and importantly a documented shift towards Tx compared with prostacyclin synthesis in diseases such as diabetes and atherosclerosis (Udvardy et al., 1987). Since endothelial TRPV4 channel activation can occur in settings such as atherosclerosis where narrowing of the vessels would increase shear stress or result in turbulent flow, a role for endothelial TRPV4 channel activation as an important source for TxA2 generation can be postulated. Indeed, in hypertensive rats, shear stress, a proposed mechanism for activation of TRPV4 channels, can trigger the release of PGH2 and TxA2 leading to vasoconstriction (Huang et al., 2000). Thus, the endothelial cell TRPV4-stimulated production of TxA2 (or its equivalent) and its activation of TP receptors is likely to have effects on the cardiovascular system far beyond simply increasing vessel tension.
Our second main finding related to the enhancement of the endothelial TRPV4-induced contractile response by prior activation of AT1 receptors, PAR1 and PAR2 receptors. Interactions between GPCRs and TRP channels have been reported in a number of other settings. Histamine, 5-HTand PAR2 activating proteinases are reported to potentiate TRPV4 channel function in neuronal cells (Cenac et al., 2010; Poole et al., 2013) and other studies have documented the ability of phorbol-ester and bradykinin to increase phosphorylation of TRPV4 channels in a G-protein and AKAP79-dependent manner (Fan et al., 2009). In epithelial cells that co-express both PAR2 and TRPV4 channels, PAR2 potentiates TRPV4-dependent calcium signalling (Grant et al., 2007). In the vasculature, blocking TRPV4 channels can attenuate ACh-dependent vasodilation (Sonkusare et al., 2012) as well as 5-HT-dependent vasoconstriction (Xia et al., 2013) responses. A prominent role for PKC activation in regulating TRPV4 channel function has also been reported in a number of these studies as was originally reported some time back in an in vitro HEK cell transfection system (Xu et al., 2003). Our findings add to this literature, and show that TRPV4-dependent vasoconstriction can be augmented by local activation of a GPCR such as PAR1, PAR2 or AT1 receptors, and can occur through transactivation of the EGFR via a mechanism involving PKC. Of note, the non-selective MMP inhibitors we used in this study failed to reverse the transactivation of the EGFR caused by stimulation of the GPCRs as reported by Ullrich and colleagues (Daub et al., 1996). Recent studies have identified TNF-α converting enzyme (TACE, also known as ADAM-17) to be the MMP responsible for releasing EGF (Sahin et al., 2004; Blobel, 2005; Ohtsu et al., 2006). Further work with more selective inhibitors might shed light on this transactivation mechanism in endothelial cells. Since the EGF kinase inhibitor, AG1478 blocked GPCR-induced potentiation of TRPV4 channel action and since EGF itself also amplified TRPV4-induced contraction, we suggest that the mechanism for the MMP-independent transactivation of the EGF receptor is itself an important area for further focus. Unfortunately, we are not able to validate a role for Src kinase in this amplification process as Src-related signalling was involved in the TRPV4-dependent contractile response itself.
Given that activation of the GPCRs amplified the contractile responses, we expected that the relaxant response to GSK101 would also be enhanced. Unfortunately, the very steep, almost all-or-none concentration-effect curve for the relaxant action of GSK101 (Figure 1A) did not allow us to evaluate with any confidence, a possible GPCR-mediated amplification of the relaxant response. It has been reported that TRPV4-mediated maximal relaxation may require the opening of as few as three TRPV4 channels per endothelial cell, thus accounting for the steep concentration-effect curve (Sonkusare et al., 2012). Thus, the very tight coupling of TRPV4-mediated relaxant signalling response may mask any potentiation of TRPV4 channel activation by prior GPCR activation, in contrast with the contractile response.
The cooperative interactions between GPCRs, the EGFR and TRPV4 channels raises important questions regarding possible mechanisms that brings together the key players involved in mediating the vasoconstrictive response. Interestingly, in endothelial cells, TRPV4 channel expression has been localized to plasma membrane caveolar compartments (Saliez et al., 2008). Similar expression of the AT1 receptor is reported in caveolae (Takaguri et al., 2011) and caveolin-1 disruption can negatively regulate EGFR transactivation by angiotensin II. Both PAR1 (Russo et al., 2009) and PAR2 (Awasthi et al., 2007) expression has also been localized to caveolar microdomains. Thus, local signalling interaction between all of the key regulators of TRPV4-dependent vasoconstriction might be coordinated by caveolins. Indeed, monitoring expression of the TRPV4 channel and its interacting partners in different vascular beds or under different disease states could serve as a strategy for predicting its signalling response in the vasculature and could form the basis of interesting future studies.
In summary, we have identified an as yet unappreciated ability of TRPV4 channels to trigger blood vessel contraction and mechanisms involving GPCR activation that enhance this response. Our work adds a new dimension to hormonal regulation of vascular function and has direct relevance to therapeutically targeting this ion channel in disease states.
Acknowledgments
M. E.-D. was supported by a doctoral scholarship from the Egyptian Ministry of Higher Education. This work was funded by Operating Grants from the Canadian Institutes of Health Research.
Glossary
Abbreviations
- EGFR
EGF receptor
- GSK101
GSK1016790A
- PAR
proteinase-activated receptor
- TRPV4 channels
transient receptor potential vanilloid-4 channels
Author contributions
M. S., N. W. B., P. M., C. A., M. D. H. and R. R. conceived and designed research; M. S., M. E.-D., K. M. and R. R. performed research; M. S., K. M. and R. R. analysed data; C. A., M. D. H. and R. R. wrote the paper.
Conflict of interest
All authors assert that they have no conflict of interest related to the research described in this manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 (A) Representative trace showing the magnitude of TRPV4-dependent contraction relative to phenylephrine (PE; 2.5 μM) and KCl (80 mM) triggered vascular contractions. (B) Representative trace showing the effect of the TRPV4 inhibitor GSK2193874 on TRPV4-dependent vascular contractions.
Figure S2 (A) Representative trace showing the effect of Go6983 on TRPV4-dependent vascular contractions. (B) Representative trace showing the effect of the ERK MAP kinase inhibitor U0126 on TRPV4-dependent vascular contractions. (C) Representative trace showing the effect of Cox-1 and Cox-2 inhibitors on TRPV4-dependent vascular contractions.
Figure S3 Inhibitors of (A) cPLA2 (AACOCF3, 20 μM), (B) DAG lipase (RHC80267, 10 μM), (C) CP450-4A (ODYA-20 μM) and (D) thromboxane synthase (Ozagrel, 50 nM) do not inhibit TRPV4-dependent contraction.
References
- Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130:248–282. doi: 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Joseph EK, Reichling D, Levine JD. A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci. 2006;26:3864–3874. doi: 10.1523/JNEUROSCI.5385-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The concise guide to PHARMACOLOGY 2013/14: ion channels. Br J Pharmacol. 2013a;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol. 2013c;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013d;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadesi S, Nie J, Vergnolle N, Cottrell GS, Grady EF, Trevisani M, et al. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci. 2004;24:4300–4312. doi: 10.1523/JNEUROSCI.5679-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi V, Mandal SK, Papanna V, Rao LV, Pendurthi UR. Modulation of tissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler Thromb Vasc Biol. 2007;27:1447–1455. doi: 10.1161/ATVBAHA.107.143438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371(Pt 1):199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylie RL, Brayden JE. TRPV channels and vascular function. Acta Physiol (Oxf) 2011;203:99–116. doi: 10.1111/j.1748-1716.2010.02217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- Bubolz AH, Mendoza SA, Zheng X, Zinkevich NS, Li R, Gutterman DD, et al. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2 + entry and mitochondrial ROS signaling. Am J Physiol Heart Circ Physiol. 2012;302:H634–H642. doi: 10.1152/ajpheart.00717.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenac N, Altier C, Motta JP, d'Aldebert E, Galeano S, Zamponi GW, et al. Potentiation of TRPV4 signalling by histamine and serotonin: an important mechanism for visceral hypersensitivity. Gut. 2010;59:481–488. doi: 10.1136/gut.2009.192567. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Cohen P. Guidelines for the effective use of chemical inhibitors of protein function to understand their roles in cell regulation. Biochem J. 2010;425:53–54. doi: 10.1042/BJ20091428. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- Damiano BP, Cheung WM, Santulli RJ, Fung-Leung WP, Ngo K, Ye RD, et al. Cardiovascular responses mediated by protease-activated receptor-2 (PAR-2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. J Pharmacol Exp Ther. 1999;288:671–678. [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- Ducret T, Guibert C, Marthan R, Savineau JP. Serotonin-induced activation of TRPV4-like current in rat intrapulmonary arterial smooth muscle cells. Cell Calcium. 2008;43:315–323. doi: 10.1016/j.ceca.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Earley S, Brayden JE. Transient receptor potential channels and vascular function. Clin Sci (Lond) 2010;119:19–36. doi: 10.1042/CS20090641. [DOI] [PubMed] [Google Scholar]
- Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2 + signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol. 2009;297:H1096–H1102. doi: 10.1152/ajpheart.00241.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, Utsunomiya H, et al. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J Biol Chem. 1998;273:8890–8896. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- El-Daly M, Saifeddine M, Mihara K, Ramachandran R, Triggle CR, Hollenberg MD. Proteinase-activated receptors 1 and 2 and the regulation of porcine coronary artery contractility: a role for distinct tyrosine kinase pathways. Br J Pharmacol. 2014;171:2413–2425. doi: 10.1111/bph.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene FA, Kalmbach A, Kandler K. Metabotropic glutamate receptors in the lateral superior olive activate TRP-like channels: age- and experience-dependent regulation. J Neurophysiol. 2007;97:3365–3375. doi: 10.1152/jn.00686.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci U S A. 2010;107:19084–19089. doi: 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HC, Zhang X, McNaughton PA. Activation of the TRPV4 ion channel is enhanced by phosphorylation. J Biol Chem. 2009;284:27884–27891. doi: 10.1074/jbc.M109.028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Wu L, O'Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem. 2003;278:27129–27137. doi: 10.1074/jbc.M302517200. [DOI] [PubMed] [Google Scholar]
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol. 2007;578(Pt 3):715–733. doi: 10.1113/jphysiol.2006.121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20:1594–1600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408–6414. doi: 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci U S A. 1975;72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, et al. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS ONE. 2007;2:e827. doi: 10.1371/journal.pone.0000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Sun D, Koller A. Shear stress-induced release of prostaglandin H(2) in arterioles of hypertensive rats. Hypertension. 2000;35:925–930. doi: 10.1161/01.hyp.35.4.925. [DOI] [PubMed] [Google Scholar]
- Ingerman-Wojenski C, Silver MJ, Smith JB, Macarak E. Bovine endothelial cells in culture produce thromboxane as well as prostacyclin. J Clin Invest. 1981;67:1292–1296. doi: 10.1172/JCI110157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Kawakami M, Hidaka T, Matsuki Y, Takamizawa M, Suzuki K, et al. Stimulation with thromboxane A2 (TXA2) receptor agonist enhances ICAM-1, VCAM-1 or ELAM-1 expression by human vascular endothelial cells. Clin Exp Immunol. 1998;112:464–470. doi: 10.1046/j.1365-2249.1998.00614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Wang X, Varty L, Rizzo CA, Yang R, Correll CC, et al. Functional TRPV4 channels are expressed in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L272–L278. doi: 10.1152/ajplung.00393.2003. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laniyonu AA, Saifeddine M, Yang SG, Hollenberg MD. Tyrosine kinase inhibitors and the contractile action of G-protein-linked vascular agonists. Can J Physiol Pharmacol. 1994;72:1075–1085. doi: 10.1139/y94-150. [DOI] [PubMed] [Google Scholar]
- Lee H, Hamilton JR. Physiology, pharmacology, and therapeutic potential of protease-activated receptors in vascular disease. Pharmacol Ther. 2012;134:246–259. doi: 10.1016/j.pharmthera.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci U S A. 2003;100:13698–13703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525–535. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8:257–265. doi: 10.1016/j.cmet.2008.08.002. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JJ, Hollenberg MD, Bennett BM, Triggle CR. Hyperpolarization of murine small caliber mesenteric arteries by activation of endothelial proteinase-activated receptor 2. Can J Physiol Pharmacol. 2004;82:1103–1112. doi: 10.1139/y04-121. [DOI] [PubMed] [Google Scholar]
- Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, et al. TRPV4-mediated endothelial Ca2 + influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol. 2009;298:H466–H476. doi: 10.1152/ajpheart.00854.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, Nelson MT, et al. Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. J Gen Physiol. 2014;143:559–575. doi: 10.1085/jgp.201311050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri Serneri GG, Abbate R, Gensini GF, Panetta A, Casolo GC, Carini M. TxA2 production by human arteries and veins. Prostaglandins. 1983;25:753–766. doi: 10.1016/0090-6980(83)90001-1. [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, Suzuki H, et al. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2006;26:e133–e137. doi: 10.1161/01.ATV.0000236203.90331.d0. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Lewandrowski U, Muller B, Sickmann A, Walz G, Wegierski T. Identification of a protein kinase C-dependent phosphorylation site involved in sensitization of TRPV4 channel. Biochem Biophys Res Commun. 2010;391:1721–1725. doi: 10.1016/j.bbrc.2009.12.140. [DOI] [PubMed] [Google Scholar]
- Peng JB, Hediger MA. A family of calcium-permeable channels in the kidney: distinct roles in renal calcium handling. Curr Opin Nephrol Hypertens. 2002;11:555–561. doi: 10.1097/00041552-200209000-00012. [DOI] [PubMed] [Google Scholar]
- Poole DP, Amadesi S, Veldhuis NA, Abogadie FC, Lieu T, Darby W, et al. Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem. 2013;288:5790–5802. doi: 10.1074/jbc.M112.438184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153(Suppl. 1):S263–S282. doi: 10.1038/sj.bjp.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, et al. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol. 2009;76:791–801. doi: 10.1124/mol.109.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2) J Biol Chem. 2011;286:24638–24648. doi: 10.1074/jbc.M110.201988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov. 2012;11:69–86. doi: 10.1038/nrd3615. [DOI] [PubMed] [Google Scholar]
- Russo A, Soh UJ, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci U S A. 2009;106:6393–6397. doi: 10.1073/pnas.0810687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, et al. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. 2008;117:1065–1074. doi: 10.1161/CIRCULATIONAHA.107.731679. [DOI] [PubMed] [Google Scholar]
- Song P, Zhang M, Wang S, Xu J, Choi HC, Zou MH. Thromboxane A2 receptor activates a Rho-associated kinase/LKB1/PTEN pathway to attenuate endothelium insulin signaling. J Biol Chem. 2009;284:17120–17128. doi: 10.1074/jbc.M109.012583. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran SV, Singh TU, Parida S, Narasimha Reddy CE, Thangamalai R, Kandasamy K, et al. TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacol Res. 2013;78:18–27. doi: 10.1016/j.phrs.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Sullivan MN, Francis M, Pitts NL, Taylor MS, Earley S. Optical recording reveals novel properties of GSK1016790A-induced vanilloid transient receptor potential channel TRPV4 activity in primary human endothelial cells. Mol Pharmacol. 2012;82:464–472. doi: 10.1124/mol.112.078584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278:22664–22668. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Suzuki M, Mizuno A, Hara A. Hearing impairment in TRPV4 knockout mice. Neurosci Lett. 2005;382:304–308. doi: 10.1016/j.neulet.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Takaguri A, Shirai H, Kimura K, Hinoki A, Eguchi K, Carlile-Klusacek M, et al. Caveolin-1 negatively regulates a metalloprotease-dependent epidermal growth factor receptor transactivation by angiotensin II. J Mol Cell Cardiol. 2011;50:545–551. doi: 10.1016/j.yjmcc.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfamariam B, Jakubowski JA, Cohen RA. Contraction of diabetic rabbit aorta caused by endothelium-derived PGH2-TxA2. Am J Physiol. 1989;257(5 Pt 2):H1327–H1333. doi: 10.1152/ajpheart.1989.257.5.H1327. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, et al. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1 -piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J Pharmacol Exp Ther. 2008;326:432–442. doi: 10.1124/jpet.108.139295. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Science translational medicine. 2012;4:159ra148. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- Tilley DG. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res. 2011;109:217–230. doi: 10.1161/CIRCRESAHA.110.231225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaka H, Taniguchi J, Satoh J, Mizuno A, Suzuki M. Warm temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an essential role in thermal hyperalgesia. J Biol Chem. 2004;279:35133–35138. doi: 10.1074/jbc.M406260200. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- Udvardy M, Torok I, Rak K. Plasma thromboxane and prostacyclin metabolite ratio in atherosclerosis and diabetes mellitus. Thromb Res. 1987;47:479–484. doi: 10.1016/0049-3848(87)90463-4. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002a;277:13569–13577. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002b;277:47044–47051. doi: 10.1074/jbc.M208277200. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Murakami M, Ohba T, Takahashi Y, Ito H. TRP channel and cardiovascular disease. Pharmacol Ther. 2008;118:337–351. doi: 10.1016/j.pharmthera.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Willette RN, Bao W, Nerurkar S, Yue TL, Doe CP, Stankus G, et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: part 2. J Pharmacol Exp Ther. 2008;326:443–452. doi: 10.1124/jpet.107.134551. [DOI] [PubMed] [Google Scholar]
- Woodward DF, Jones RL, Narumiya S. International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63:471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- Wu L, Gao X, Brown RC, Heller S, O'Neil RG. Dual role of the TRPV4 channel as a sensor of flow and osmolality in renal epithelial cells. Am J Physiol Renal Physiol. 2007;293:F1699–F1713. doi: 10.1152/ajprenal.00462.2006. [DOI] [PubMed] [Google Scholar]
- Xia Y, Fu Z, Hu J, Huang C, Paudel O, Cai S, et al. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am J Physiol Cell Physiol. 2013;305:C704–C715. doi: 10.1152/ajpcell.00099.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Satoh E, Iijima T. Protein kinase C-mediated Ca2 + entry in HEK 293 cells transiently expressing human TRPV4. Br J Pharmacol. 2003;140:413–421. doi: 10.1038/sj.bjp.0705443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman B, Beautrait A, Aguila B, Charles R, Escher E, Claing A, et al. Differential beta-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci Signal. 2012;5:ra33. doi: 10.1126/scisignal.2002522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) Representative trace showing the magnitude of TRPV4-dependent contraction relative to phenylephrine (PE; 2.5 μM) and KCl (80 mM) triggered vascular contractions. (B) Representative trace showing the effect of the TRPV4 inhibitor GSK2193874 on TRPV4-dependent vascular contractions.
Figure S2 (A) Representative trace showing the effect of Go6983 on TRPV4-dependent vascular contractions. (B) Representative trace showing the effect of the ERK MAP kinase inhibitor U0126 on TRPV4-dependent vascular contractions. (C) Representative trace showing the effect of Cox-1 and Cox-2 inhibitors on TRPV4-dependent vascular contractions.
Figure S3 Inhibitors of (A) cPLA2 (AACOCF3, 20 μM), (B) DAG lipase (RHC80267, 10 μM), (C) CP450-4A (ODYA-20 μM) and (D) thromboxane synthase (Ozagrel, 50 nM) do not inhibit TRPV4-dependent contraction.