

Abstract
Background and Purpose
Cognitive deficits in patients with Alzheimer's disease, Parkinson's disease, traumatic brain injury and stroke often involve alterations in cholinergic signalling. Currently available therapeutic drugs provide only symptomatic relief. Therefore, novel therapeutic strategies are needed to retard and/or arrest the progressive loss of memory.
Experimental Approach
Scopolamine-induced memory impairment provides a rapid and reversible phenotypic screening paradigm for cognition enhancement drug discovery. Male C57BL/6J mice given scopolamine (1 mg·kg−1) were used to evaluate the ability of LS-1–137, a novel sigma (σ1) receptor-selective agonist, to improve the cognitive deficits associated with muscarinic antagonist administration.
Key Results
LS-1–137 is a high-affinity (Ki = 3.2 nM) σ1 receptor agonist that is 80-fold selective for σ1, compared with σ2 receptors. LS-1–137 binds with low affinity at D2-like (D2, D3 and D4) dopamine and muscarinic receptors. LS-1–137 was found to partially reverse the learning deficits associated with scopolamine administration using a water maze test and an active avoidance task. LS-1–137 treatment was also found to trigger the release of brain-derived neurotrophic factor from rat astrocytes.
Conclusions and Implications
The σ1 receptor-selective compound LS-1–137 may represent a novel candidate cognitive enhancer for the treatment of muscarinic receptor-dependent cognitive deficits.
Tables of Links
| LIGANDS |
|---|
| Atropine |
| BDNF |
| Dopamine |
| Haloperidol |
| NE-100 |
| PRE-084 |
| Quinuclidinyl benzilate |
| Scopolamine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Alexander et al., 2013a,b,c,d,,,).
Introduction
Impairment in learning and memory is seen in (i) aged populations, (ii) patients with neurodegenerative diseases including Alzheimer's disease (AD) and Parkinson's disease, (iii) stroke patients and (iv) patients with traumatic brain injury (Scarpini et al., 2003; Tarawneh and Galvin, 2010; Ruscher et al., 2011). Although the causes of cognitive impairments vary, previous studies have suggested that alteration in cholinergic neurotransmission may play an important role in the disruption of learning and memory (Francis et al., 1999; Craig et al., 2011; Dumas and Newhouse, 2011). According to the cholinergic hypothesis, age-dependent cognitive decline is primarily related to impairment in cholinergic neurotransmission (Bartus et al., 1982; Coyle et al., 1983; Kirk et al., 1994).
Administration of the competitive cholinergic muscarinic receptor antagonist scopolamine causes cognitive dysfunctions similar to those observed in normal aging and AD (Ebert and Kirch, 1998; Bejar et al., 1999; Klinkenberg and Blokland, 2010). A scopolamine-dependent model of cognitive deficits has been used to screen for potential cognition-enhancing drugs (Flood and Cherkin, 1986; Ennaceur and Meliani, 1992; Antonini et al., 2009; Klinkenberg and Blokland, 2010). Since the sigma σ1 receptor is known to potently modulate cholinergic neurotransmission at different levels (van Waarde et al., 2011), we investigated the effect of our novel σ1 receptor-selective ligand, LS-1–137, on scopolamine-dependent cognitive deficits.
The σ1 receptors are expressed in the hippocampus, frontal cortex and olfactory bulb. In the periphery, σ1 receptors are expressed in the heart, lungs, kidneys, liver and gonads (Vilner et al., 1995; Kitaichi et al., 2000; Hayashi and Su, 2005). This receptor has been identified as an endoplasmic reticulum (ER) resident protein located on the interface of the ER and mitochondria (Hayashi and Su, 2007).
The σ1 receptor is a 25 kDa protein that shares no amino acid sequence homology with any of the known mammalian proteins (Seth et al., 1998; Hayashi and Su, 2005; Kourrich et al., 2012). The σ1 receptor possesses two transmembrane spanning regions. The carboxy terminus of the σ1 receptor is involved in chaperone activity and ligand binding (Hayashi and Su, 2007; Kourrich et al., 2012). Recently, Fontanilla and co-workers suggested that the hallucinogen N,N-dimethyltryptamine may be the endogenous ligand (Fontanilla et al., 2009). In addition, the neurosteroids progesterone, pregnonolone sulfate and dehydroepiandrosterone sulfate have appreciable affinity for the σ1 receptor (Maurice et al., 1998; Kourrich et al., 2012).
Rather than being a classical receptor signalling unit, σ1 receptors probably act as intracellular modulators of a variety of signal transduction systems. The σ1 receptor has also been postulated to act as a molecular chaperone that regulates cellular survival and synaptogenesis (Hayashi and Su, 2007; Tsai et al., 2009; Fujimoto et al., 2012). Ligands selective for σ1 receptors have been reported to exert antidepressant-like, anxiolytic and analgesic actions in preclinical studies (Hayashi et al., 2011). In addition, σ1 receptor occupation has been linked to neurorestorative effects and has been suggested as a novel therapeutic target for the treatment of cognition deficit (Maurice et al., 1994; 1996; Urani et al., 1998; Maurice, 2004b; Espallergues et al., 2007; Antonini et al., 2011; van Waarde et al., 2011; Kourrich et al., 2012).
Our laboratory previously reported on the neuroprotective properties of the σ1 receptor ligand, LS-1–137, which is a novel N-(benzylpiperidin)phenylacetamide that binds to the σ1 receptor with high affinity (Ki value = 3.2 nM) and exhibits 80-fold binding selectivity for the σ1, over the σ2 receptor (Huang et al., 1998; 2001). LS-1–137 was found to protect HT-22 neuronal cells from glutamate-induced cell death and LS-1–137 was found in vivo to significantly reduce the ischaemic lesion volume in rats using a transient middle cerebral artery occlusion model of stroke (Luedtke et al., 2012). In this communication, we continue to characterize LS-1–137 by examining its (i) intrinsic efficacy in vitro, (ii) ability to moderate scopolamine-dependent cognitive deficits in C57BL/6J mice and (iii) potency to release brain-derived neurotrophic factor (BDNF) from rat astrocytes.
Methods
Animals
All animal care and experimental procedures complied with the ‘Guide for the Care and Use of Laboratory Animals’ (Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, 1996) and were approved by the Institutional Animal Care and Use Committee at University of North Texas Health Science Center. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 66 animals were used in the experiments described here.
Male C57BL/6J (6–8 weeks) mice were obtained from Jackson Laboratories. Mice were habituated for 1 week in the animal facility before behavioural studies were initiated. Mice were housed under 12 h light/12 h dark conditions with free access to food and water. The animals were randomly assigned into eight different groups.
Preparation of drug
The σ1-selective test drug, LS-1–137, was synthesized by the NIMH Chemical Synthesis and Drug Supply Program as described previously (Huang et al., 1998; 2001; Luedtke et al., 2012). Scopolamine hydrobromide was purchased from Sigma-Aldrich (St. Louis, MO, USA). The σ1 receptor agonist (+/−)-PPCC (methyl(1R,2S/1S,2R)-2-[4-hydroxy-4-phenylpiperidin-1-yl)methyl]-1-(4-methylphenyl)cyclopro-panecarboxylate) oxalate (Cat# 3870), the σ1 receptor agonist PRE-084 (Cat# 0589) and the σ1 receptor antagonist NE-100 (Cat# 3133) was purchased from TOCRIS/B&D Systems (Bristol, UK). For the animal behavioural studies, scopolamine (1 mg·kg−1) and test drugs were prepared on the day of the experiment. Scopolamine was dissolved in sterile water and test drugs in 5% dimethyl sulfoxide. Drugs were administered via intraperitoneal injection. Test drug was injected 15 min prior to scopolamine hydrobromide administration. The relevant behavioural testing was started 30 min after the scopolamine injection. For the co-immunoprecipitation studies, test drugs were dissolved in culture medium.
σ1 receptor–binding immunoglobulin protein (BiP) association assay
CHO cells were maintained in MEM/alpha culture medium supplemented with 10% heat inactivated FBS and 2 mM of Glutamax (Invitrogen, Carlsbad, CA, USA). CHO cells were plated in 6-well plates and treated with test compounds dissolved in culture medium for 30 min at 37°C. The reaction was terminated by removing the culture medium containing the test drug and replacing it with 3 mL of PBS. CHO cells were harvested and suspended in 50 mM HEPES (pH 7.4) followed by cross-linking with 50 μg·mL−1 of dithiobis succinimidyl propionate (Thermo Scientific, Watham, MA, USA). The reaction was stopped by adding Tris/HCl 50 mM (pH 8.8). After a 15 min incubation on ice, cells were lysed with 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X/100, 0.3% sodium deoxycholate, 0.1% SDS buffer containing a protease inhibitor cocktail (Roche Complete; Roche Applied Science, Mannheim, Germany). After centrifugation at 12 000× g for 1 min, the supernatant was incubated with σ1 receptor antibody (Abcam, Cambridge, UK, #ab53852) overnight at 4°C. The cell lysate was then incubated with Sepharose Protein-A (Invitrogen) for 90 min. After centrifugation at 12 000× g for 1 min, the supernatant was discarded and the pellet was suspended in 0.5 mL lysis buffer. The pellet suspension was centrifuged at 12 000× g for 20 min, supernatant was discarded and the pellet was suspended in 0.5 mL 2× sample buffer/bMCE buffer. After centrifugation at 12 000 g for 1 min, supernatant was analysed by elisa assay as described in the manufacturer's protocol (USCNK Life Sciences, Hubei, China, SEC343Mu).
Active avoidance test
The active avoidance test is an associative learning test (Olton and Isaacson, 1968; Shetty et al., 2013) where mice were placed in an acrylic T-shaped apparatus with black coloured walls and a clear ceiling. The T-shaped apparatus was divided into three compartments, a stem and two goal arms. Each compartment was separated with a removable door.
The apparatus rested on a grid floor made of stainless steel rods that were 3 mm thick and 7 mm apart. A 0.69 mA scrambled shock was delivered to the grid floor by pressing a foot pedal connected to a programmable animal shocker (San Diego Instruments, San Diego, CA, USA).
In this test, a mouse was placed in the start box and the door was immediately removed to signal the start of the trial. On the first trial, the mouse was shocked in the first arm entered and was allowed to escape to the opposite arm, designated as the correct arm for the remainder of the session. On subsequent trials, a shock was initiated if the mouse did not enter the correct arm in less than 5 s and the shock continued until the mouse entered the correct arm. Upon entering the correct arm, the mouse was allowed to rest for 10 s and was then returned to a holding cage.
In these experiments, there was an interval of 1 min between trials. The session ended when the mouse met the following three criteria: (i) the mouse chose the correct arm in the last four out of the five trials, (ii) the mouse avoided the shock by entering the correct arm in less than 5 s and (iii) the mouse chose the correct arm and avoided the shock in the last two consecutive trials. The ability to learn the avoidance task was considered inversely proportional to the number of trials a mouse takes to reach criterion.
Spatial learning and memory/water maze
Spatial learning was evaluated using the Morris water maze (Morris, 1984; D'Hooge and De Deyn, 2001), slightly modified from the method previously reported (Shetty et al., 2013). The apparatus consisted of a white polyethylene circular pool, 120 cm in diameter and 50 cm deep. The pool was filled with water to a height of 34 cm (15 cm below the top edge of the tank). The water temperature was 24 ± 1°C. The water was made cloudy by the addition of white, non-toxic paint. Four equally spaced points served as the start locations, dividing the tank into four equally spaced quadrants. A platform (10 × 10 cm), located in the northwest quadrant, was submerged 1 cm below the water so the mice could climb onto the platform. A video camera, mounted above the centre of the tank, was connected to a computer running an ANY-maze video tracking system software package (Stoelting, Wood Dale, IL, USA) to record the behaviour of the mice. The tank was located in a portion of the room with external cues on the wall, which could be used by the animals for orientation of the platform. The testing was conducted in two phases: (i) a straight alley pre-training phase and (ii) a spatial discrimination acquisition phase.
Straight alley (pre-training phase)
The straight alley pre-training phase is essential because this is when the mice first learn the motor components of swimming and climbing onto a platform. They also learn that there will be a platform hidden below the surface of the water and that it is available for them to escape from the water.
For the pre-training phase, the mouse was placed at end of a 10 × 60 cm acrylic alley and allowed to swim to the other end, where the 10 × 10 cm platform was located. The time for the mouse to swim to and climb onto the platform was recorded. Visual cues were removed by placing a black curtain around the tank. For this study, mice received one session with five trials per day. The pre-training phase was conducted for two days (Friday and Monday). There was an inter-trial interval of 5 min between each of the five trials.
Spatial discrimination acquisition phase
In the acquisition phase, the mouse learns and remembers the location of the platform. This phase was started immediately following the last day of the pre-training phase. In the acquisition phase, the black curtain was removed and mice were allowed to use visual cues in the room to locate the hidden platform. The mouse was placed in a circular pool with its back end first and its head facing the side of the pool to avoid any distress and bias. The tracking system was started immediately after the mouse was placed in the pool. The trial ended either when the animal climbed onto the platform or after 90 s elapsed. Each testing day consisted of five trials, with each trial starting from one of the four possible starting locations, from where the mouse swam until it located the platform. Once the mouse had reached the platform, it was allowed to rest in the holding cage for a 90 s inter-trial interval. The path length (distance to the platform), latency (time taken to reach the platform) and swim speed were recorded by a tracking system (ANY-maze). For this study, each mouse received a single session for three subsequent days (Tuesday–Thursday). Drugs were injected every day prior to testing.
Radioligand binding studies
The binding properties of membrane-associated receptors were characterized using a radioligand filtration binding assay. The muscarinic receptor-binding protocol was identical to that described previously (Luthin and Wolfe, 1984). Briefly, frozen male Sprague Dawley rat brains were obtained from BioChemed (Winchester, VA, USA). Rat cortex was dissected on a chilled glass plate. The homogenized cortical tissue was stored at −80°C in binding buffer (50 mM Tris-HCl, 150 mM NaCl, 10 μM EDTA at pH 7.4). At the time of the experiment, rat cortical tissue was thawed on ice, re-homogenized, diluted in binding buffer and incubated with [3H]-quinuclidinyl benzilate ([3H]-QNB; PerkinElmer, Waltham, MA, USA) and drugs at 37°C for 60 min. Atropine (1 μM) was used to define the non-specific binding. The final assay volume was 4.0 mL. Binding was terminated by the addition of the cold wash buffer (10 mM Tris–HCl, 150 mM NaCl, pH = 7.5) and filtration over a glass-fibre filter (Pall A/B filters, #66198, Fisher Scientific, Pittsburgh, PA, USA). For these competitive radioligand binding studies, the IC50 values were determined using a one site fit analysis. The IC50 values were converted to equilibrium dissociation constants (Ki values) using the Cheng and Prusoff (1973) correction assuming a Kd value for the binding of [3H]-QNB of 0.04 nM. Mean Ki values ± SEM are reported for at least three independent experiments (Cheng and Prusoff, 1973).
BDNF release assay
Primary astrocytes were derived from the cerebral cortex of 3 day post-natal Sprague Dawley rats. Briefly, after removal of the brain from the cranial cavity, the meninges were removed and the cerebral cortex was dissected and placed into a sterile tube containing 2 mL of the dissociation solution [1.9 mL PBS, pH 7.2 with 0.25% trypsin (final concentration)]. After the tube was incubated at 37°C for 10 min, it was centrifuged at 400× g for 5 min. The resulting pellet was resuspended in 2 mL of plating media (DMEM with sodium pyruvate/10% FBS + 1% penicillin/streptomycin) and dissociated further with the aid of a fire-polished Pasteur pipet. The cells were then centrifuged and resuspended in 2 mL of plating media. After these resuspension and centrifugation steps were repeated twice, the cell suspension was filtered through a 70 μm cell strainer into a 50 mL conical tube. The filtrate was diluted by adding 5 mL of plating media (DMEM F-12 media supplemented with 2 mM glutamine, non-essential amino acids, 10% fetal calf serum, and 1% Pen-Strep). The cells were counted and plated into antibody coated 96-well plates (37°C with 5% CO2). The media were changed every 48 h.
LS-1–137-mediated release of BDNF was determined using a method previously described (Su et al., 2012). Briefly, a 96-well Nunc MaxiSorp polystyrene plate (Thermo Fisher Scientific, Rochester, NY, USA) was pre-coated with an anti-BDNF monoclonal antibody (Promega, Madison, WI, USA, G700B). After rinsing off the unbound antibody and blocking the plate to minimize non-specific binding, the culture medium was added for 2 h to equilibrate the cell growth environment. Rat astrocytes were then plated at a density of 10 000 cells·per well). After the cells attached to the surface, cells were treated with LS-1–137. BDNF standards, ranging in concentration from 0 to 500 pg·mL−1, were added in parallel wells. The plate was then incubated with the polyclonal anti-human BDNF antibody (Promega G164C). Anti-IgY-HRP tertiary antibody (Promega G767A; final concentration = 0.5 μg·mL−1) was used to detect specifically bound polyclonal antibody. The BDNF release was quantified by measuring the absorbance at 450 nm using a Viktor3 ELISA plate reader (PerkinElmer).
NMDA analysis
Recombinant receptors
cDNAs encoding rat GluN1 and GluN2A were generous gifts from Dr. David Lynch (University of Pennsylvania). HEK 293 cells were transiently transfected with recombinant NMDA receptor subunits using PolyJet™ DNA in vitro tranfection reagent (SignaGen Laboratories, Rockville, MD, USA). HEK 293 cells were washed and placed in fresh DMEM containing 10% FBS and antibiotic (penicillin 100 U·mL−1). GluN1 along with GluN2A cDNA (0.5:0.5 μg) was added to cells growing exponentially on one poly-L-lysine-coated coverslip placed in a 35 mm culture dish. Transfected cells were used for electrophysiological analysis 24 to 48 h after the transfection.
Electrophysiology
Whole-cell patch recordings were made at room temperature (22–25°C) at a holding potential of −60 mV. Patch pipettes of borosilicate glass (M1B150F, World Precision Instruments, Inc., Sarasota, FL, USA) were pulled (Flaming/Brown, P-87/PC, Sutter Instrument Co., Novato, CA, USA) to a tip resistance of 7–8 MΩ. The pipette solution contained (in mM): 140 KCl, 2 MgCl2, 0.2 EGTA, 10 HEPES, 2 Na2-ATP, 0.2 Na3-GTP at pH 7.2. A coverslip containing transfected cells was placed in a small chamber (∼1.5 mL) on the stage of an inverted light microscope (Olympus IMT-2) and superfused continuously (5–8 mL·min−1) with the following external solution containing (in mM): 125 NaCl, 5.5 KCl, 5.0 CaCl2, 20 HEPES, 10 D-glucose, 10 μM glycine; pH 7.3. NMDA was prepared in extracellular solution and was applied (10 s) to cells via gravity flow using a Y-shaped tube positioned near the target cell. With this system, the 10 to 90% rise time of the junction potential at the open tip was 60–120 ms. NMDA-evoked currents from the whole-cell configuration were obtained using a patch clamp amplifier (Axopatch 200A, Axon Instruments, Foster City, CA, USA) equipped with a CV201A headstage. The currents were low-pass filtered at 5 kHz, monitored on an oscilloscope and a chart recorder (Gould TA240) and stored on a computer (pClamp 6.05, Axon Instruments) for subsequent analysis. To monitor the possibility that access resistance changed over time or during different experimental conditions, at the initiation of each recording, the current response was measured and stored on a digital oscilloscope to a 5 mV voltage pulse. This stored trace was continually referenced throughout the recording. If a change in access resistance was observed throughout the recording period, the patch was aborted and that data were not included in the analysis.
Data analysis
The effects of treatment on the active avoidance and BDNF release were analysed via one-way anova. The effects of treatment on Morris water maze data were analysed using two-way anova with treatment and test day as between-group factors. Planned individual comparisons between different treatment groups were performed using a single degree-of-freedom F-test involving the error term from the overall anova. The effect of LS-1–137 on recombinant NMDA receptors was analysed using a paired t-test. Significance for all statistical comparisons was set at P ≤ 0.05. The data were analysed using Prism 6.0a; GraphPad Software (San Diego, CA, USA).
Results
Binding properties
The phenylacetamide LS-1–137 binds with high affinity at σ1 receptors (Ki value = 3.2 nM), 80-fold σ1 versus σ2 receptor-binding selectivity and low affinity at the D2-like (D2, D3 and D4) dopamine receptor subtypes (Ki values >500 nM; Luedtke et al., 2012).
σ1 receptor–BiP association assay for intrinsic efficacy
The intrinsic efficacy of LS-1–137 was evaluated by determining its ability to modulate σ1 receptor-BiP association. As shown in Figure 1, LS-1–137 (10 μM) caused the dissociation of σ1 receptor/BiP complex and the LS-1–137-dependent dissociation of σ1 receptors from BiP was blocked by NE-100. Based upon these results, LS-1–137 is acting as an agonist at σ1 receptors. One-way anova indicated that the effect of treatment was significant [F(4,33) = 55.7; P < 0.0001].
Figure 1.
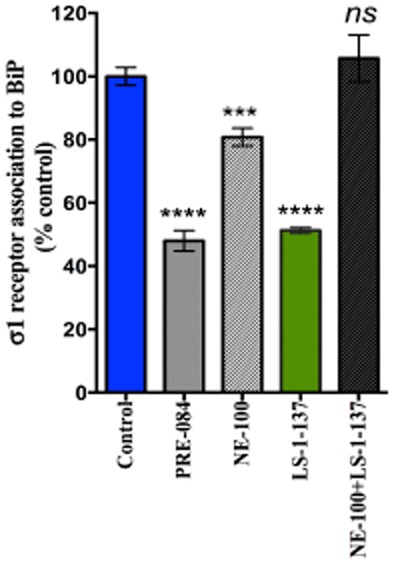
LS-1–137 regulation of σ1 receptor-BiP complex formation. The efficacy of LS-1–137 was compared with the known σ1 receptor agonist PRE-084 and antagonist NE-100. Vehicle (control) or test drugs were applied into culture medium for 30 min at 37°C. Cell lysates were collected and used for the detection of σ1 receptors coupled to BiP. Test drugs were applied at 1 or 10 μM final concentration (n ≥ 3). Data is presented as the percent (%) of untreated control cells ± SEM. ***P = 0.004 versus control; ****P < 0.001 versus control; ns P > 0.05 not significant.
Active avoidance task
The ability of LS-1–137 to attenuate the disruptive effects of scopolamine (1 mg·kg−1) in male C57BL/6J mice was evaluated using an active avoidance task. LS-1–137 was tested at a dose of 3 mg·kg−1 and reference compounds PRE-084, PPCC and NE-100 were tested at 5 mg·kg−1. All drugs were administered via i.p. injection 15 min prior to the administration of scopolamine and the active avoidance task was started 30 min following the administration of scopolamine.
Vehicle-treated mice met the criterion in an average of 14.6 ± 1.1 trials (Figure 2A). Scopolamine-treated animals met the criterion after 40.2 ± 2.3 trials. For mice treated with LS-1–137 (3 mg·kg−1) plus scopolamine, the animals met the criteria in 29.0 ± 1.9 trials. One-way anova indicated a significant effect of LS-1–137 treatment compared with vehicle controls [F (3, 24) = 45.8; P < 0.0001]. Also, the administration of LS-1–137 in the absence of scopolamine did not affect the performance of the mice in the active avoidance task compared with the vehicle treated group.
Figure 2.
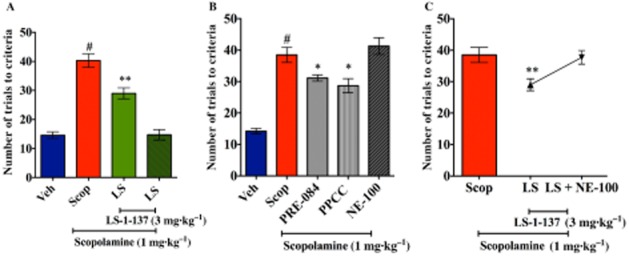
Modulation of scopolamine effects on learning by LS-1–137 using the active avoidance task. An active avoidance task was used to assess the effects of scopolamine on short-term learning. All test drugs were administered prior to starting the test. Male C57BL/6J mice were pretreated with LS-1–137 (LS) or vehicle (Veh) at 15 min prior to scopolamine (Scop) administration. The test was started 30 min after the scopolamine injection. The data presented in the figure panels represents the number of trials for an animal to meet the criteria. Data are reported as the means ± SEM. for n ≥ 6 animals per group. *P < 0.05 vs. scopolamine and #P < 0.001 vs. vehicle group). Panel A (left): comparison of the effects of vehicle versus scopolamine (1 mg·kg−1) versus scopolamine plus LS-1–137 (3 mg·kg−1) versus vehicle plus LS-1–137. Panel B (middle): comparison of the effects of vehicle versus scopolamine (1 mg·kg−1) versus scopolamine in the presence of PRE-084, PPCC or NE-100. Panel C (right): ability of NE-100 to attenuate the inhibition of the effect of scopolamine by LS-1–137. *P < 0.05, **P < 0.008.
We then tested the reference σ1 receptor agonists PRE-084 and PPCC, as well as the antagonist NE-100, in this assay (Figure 2B). Both PRE-084 and PPCC significantly attenuated the effect of scopolamine (PRE-084, 31.1 ± 1.0 trials; PPCC, 28.6 ± 2.2 trials; scopolamine, 38.5 ± 2.4 trials). One-way anova indicated a significant effect of treatment [F (4, 36) = 27.52; P < 0.0001]. However, NE-100 was not able to ameliorate the effect of scopolamine.
To further validate the σ1 receptor-mediated activity of LS-1–137 in this assay, we co-administered LS-1–137 (3 mg·kg−1) and NE-100 (5 mg·kg−1). Fifteen minutes after the injection of LS-1–137 and NE-100, scopolamine was administered to the mice. NE-100 blocked the effect of LS-1–137 (37.7 ± 2.1 trials; Figure 2C). One-way anova indicated that the effect of the antagonist NE-100 on LS-1–137 treatment was significant [F (2, 23) = 4.57; P = 0.02].
Spatial learning and memory studies
The efficiency of the mice to locate the hidden platform over three consecutive days using the Morris water maze task is shown in Figure 3. The testing was started 30 min after the i.p. injection of scopolamine. Latencies to reach the platform (Figure 3, left panel) decreased for all the groups over the 3 day testing period. Overall, the scopolamine and the scopolamine plus LS-1–137-treated mice had higher latencies than the vehicle and the LS-1–137-treated mice. On days 2 and 3, the scopolamine plus LS-1–137-treated mice had lower latencies than the scopolamine-treated mice. A two-way anova revealed significant main effects of treatment [F (3,33) = 31.54; P < 0.001] and days of testing [F (2,66) = 15.44; P < 0.001], but showed no interaction of treatment and days [F (6,66) = 0.804; P = 0.57].
Figure 3.
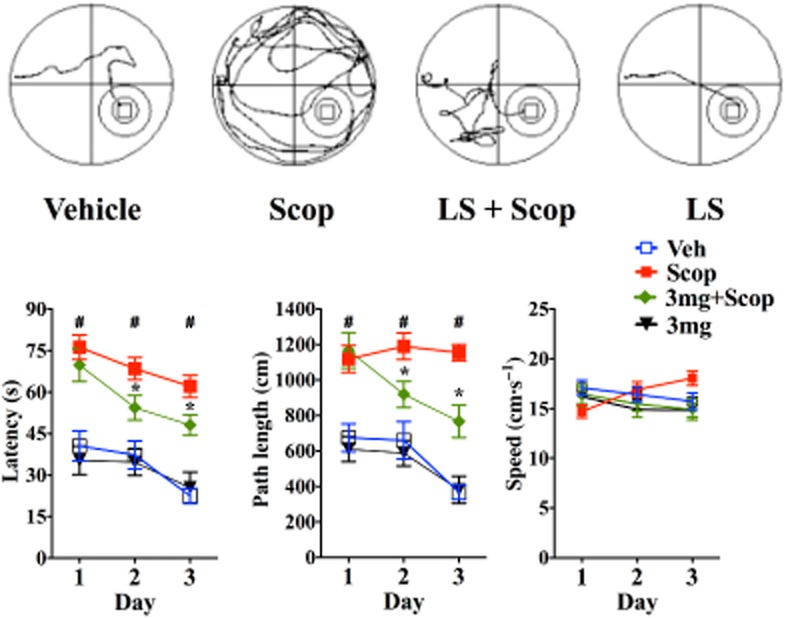
Effect of scopolamine (Scop) and LS-1–137 on latency, path length and speed in Morris water maze. Male C57BL/6J mice were pretreated with LS-1–137 (LS; 3 mg·kg−1) 15 min prior to Scop administration (1 mg·kg−1). Testing was started 30 min after Scop administration. Top panel: representative swim paths of animals in different groups (left to right: vehicle control vs. Scop vs. Scop plus LS-1–137 (LS) vs. LS-1–137) is shown. Bottom panel: data are presented as the mean ± SEM for n ≥ 8 animals per group for the daily effects of scopolamine, vehicle control, LS-1–137 in the presence of scopolamine or LS-1–137. Bottom left panel: time taken to reach the platform (latency in seconds). Bottom middle panel: average distance travelled to the platform (path length in cm). Bottom right panel: swim speed in cm·s−1. *Compared with Scop-treated group, #compared with vehicle control group. P ≤ 0.05 is considered significant.
Path lengths to reach the platform were also quantified. Path lengths decreased for all of the groups over the 3 day testing period (Figure 3, middle panel). The scopolamine and scopolamine plus LS-1–137-treated mice had a longer path length than the other mice to reach the platform. On days 2 and 3, the scopolamine plus LS-1–137-treated mice performed better (shorter path length) than the scopolamine-treated ones. A two-way anova revealed significant main effects of treatment [F (3,33) = 34.01; P < 0.001] days of testing [F (2,66) = 11.05; P < 0.001] and a significant interaction of treatment and days [F (6,66) = 2.26; P = 0.048].
While the swim speed of most groups remained constant, the swim speed of the scopolamine-treated mice increased slightly over the 3 day testing period (Figure 3, right panel). A two-way anova yielded a significant interaction of days of testing and treatment [F (6, 66) = 4.711, P < 0.0005] and no main effect of treatment [F (3,33) = 0.8094; P = 0.4978] or days [F (2,66) = 0.1858; P = 0.8308].
Muscarinic binding
Both the active avoidance task and the water maze studies indicated that LS-1–137 was capable of partially attenuating the cognitive deficits caused by the administration of the muscarinic antagonist scopolamine. One possible mechanism of action might be that LS-1–137 was competitively binding to the muscarinic binding sites and blocking scopolamine binding in vivo. To test this possibility, we determined the affinity of LS-1–137 for muscarinic receptors using the radioligand 3H-QNB, which is a high-affinity (Kd = 0.04 nM) muscarinic receptor-selective radioligand. Competitive radioligand binding studies were conducted using rat brain cortex tissue and 3H-QNB to compare the ability of scopolamine and LS-1–137 to competitively inhibit radioligand binding (Hulme et al., 1978). There was a >1000-fold difference in affinity of LS-1–137 (Ki = 1447 ± 894 nM, n = 3) for 3H-QNB binding sites compared with scopolamine (Ki = 0.61 ± 0.16 nM, n = 4). A composite competitive binding curve for scopolamine and LS-1–137 is shown in Figure 4.
Figure 4.
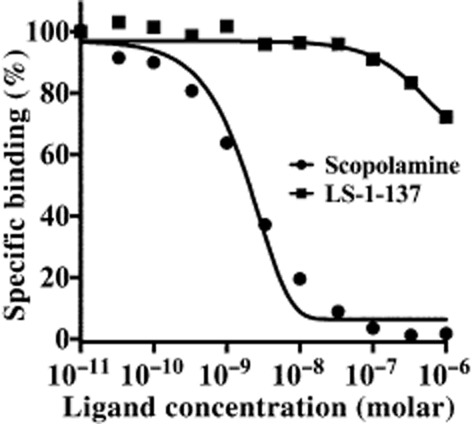
Comparison of the binding of scopolamine and LS-1–137 to muscarinic receptors using 3H-QNB and rat cortical brain tissue. Competitive radioligand binding studies were conducted using murine brain cortex tissue and 3H-QNB to label muscarinic receptors. Non-specific binding was defined using 1 μM atropine. This figure shows a composite competition curve for the specific binding of 3H-QNB as a function of inhibitor concentration, where each point is the mean inhibition from n = 4 for scopolamine and n = 3 for LS-1–137. IC50 values for each independent experiment were converted to Ki values using the Cheng and Prusoff equation (1973). The mean Ki values ± SEM for 3H-QNB binding sites were found to be 1447 ± 894 nM (n = 3) for LS-1–137 and 0.61 ± 0.16 nM (n = 4) for scopolamine.
Effect of LS-1–137 on recombinant NMDA receptors
NMDA receptors have been reported to play a key role in synaptic plasticity and memory (Huang and Dillon, 1999; Hunt and Castillo, 2012). It has been reported that σ receptor ligands may have bidirectional modulation on NMDA receptor function, depending on ligand structure and the ligand concentrations that are used. This modulation of NMDA receptor function might occur directly or via an interaction with σ1 receptors (Nishikawa et al., 2000; Kume et al., 2002; Martina et al., 2007). To examine whether an indirect or a direct modulation of NMDA receptors might contribute to our observed in vivo effects of LS-1–137 on cognitive function presented here or the neuroprotective effects we reported previously (Luedtke et al., 2012), we assessed the effect of LS-1–137 on NMDA responses recorded from HEK 293 cells transiently expressing rat recombinant GluN1NR2A receptors. GluN1NR2A receptors were selected for these studies because they are predominantly expressed at synaptic sites in mature neurons and are involved in long-term potentiation (Tovar and Westbrook, 2002; Massey et al., 2004) and ischaemic brain damage (Liu et al., 2007).
First, to examine whether LS-1–137 can directly modulate NMDA receptors, LS-1–137 (1 and 10 μM) was co-applied with 20 μM NMDA (∼EC30; Huang et al., 2010). As shown in Figure 5A, application of LS-1–137 had no effect on NMDA-activated currents. The relative currents are 99 ± 3.1% (n = 8) and 101 ± 2.6% (n = 11; for 1 μM and 10 μM of LS-1–137, respectively) of control values (P > 0.05, paired t-test).
Figure 5.
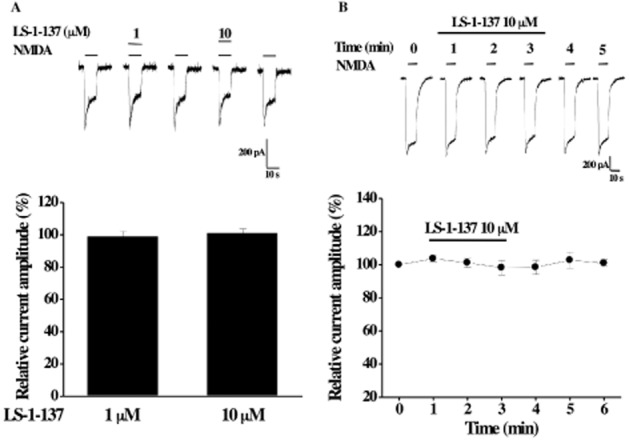
Effect of LS-1–137 on rat recombinant GluN1GluN2A NMDA receptors transiently expressed in HEK 293 cells. (A, top) Representative traces of whole-cell NMDA (20 μM)-activated currents recorded from same cell is shown. LS-1–137 (1 or 10 μM) was co-applied with NMDA to the cell for 10 s. (Bottom) Summary data of the effect of LS-1–137 on NMDA response are shown. (B, top) Representative recording of NMDA-activated currents recorded before, during the 3 min perfusion with 10 μM LS-1–137 and the after washout is shown. (Bottom) Summary data of effect of LS-1–137 treatment on NMDA response are shown. All current amplitudes are normalized to NMDA response in the absence of LS-1–137 (as 100%). Each data point represents mean ± SEM from at ≥4 cells.
Second, since σ1 receptors are endogenously expressed in HEK 293 cells and able to modulate ion channel activities (Fontanilla et al., 2009), we examined whether LS-1–137 might modulate NMDA receptor activity via activation of σ1 receptors. LS-1–137 (10 μM) was added to the external solution and continuously perfused through the bath for 3 min. Our previous studies have shown that the onset for the effect of σ1 receptor agonist on voltage-gated calcium channels occurs at approximately 3 min (Tchedre et al., 2008). NMDA-activated currents were recorded before (control), during LS-1–137 perfusion and after washout. As shown in Figure 5B, LS-1–137 treatment did not have a significant effect on NMDA response (P > 0.05, paired t-test). The relative currents were 98 ± 4.3% of control at 3 min of LS-1–137 perfusion. Taken together, our data suggest that LS-1–137 does not have a direct effect on GluN1NR2A NMDA receptors.
BDNF release assay
There have been several recent reports that σ1 receptor agonists can increase secretion of BDNF in both cultured cell lines and rat hippocampus tissue (Fujimoto et al., 2012; Hashimoto, 2013; Ring and Regan, 2013). We found that LS-1–137 could elicit the release of BDNF from primary rat astrocytes in a dose-dependent manner (Figure 6).
Figure 6.
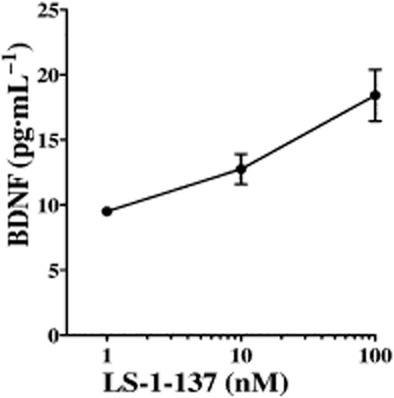
LS-1–137 induced BDNF release from rat astrocytes. LS-1–137 induced BDNF release in a concentration-dependent manner from rat primary cortical astrocytes. Cells were treated with LS-1–137 at the indicated concentrations for 18 h. BDNF release into the media was measured by elisa. The data are representative of three independent experiments and expressed as the mean ± SEM BDNF (pg·mL−1). The mean baseline for this assay (vehicle control) was 1.51 ± 0.18 pg·mL−1.
Discussion
We have reported that the phenylacetamide LS-1–137 was a σ1 receptor versus σ2 receptor-selective compound, devoid of D2-like dopamine receptor-binding activity (i) capable of inhibiting glutamate-dependent cell death using the HT-22 murine hippocampal cell line and (ii) exhibiting neuroprotective properties in a rat transient middle cerebral artery occlusion stroke model (Huang et al., 1998; Luedtke et al., 2012). In the present work, we have extended those findings by showing that LS-1–137 (i) acted as a potent agonist at σ1 receptors using both in vitro (Hayashi and Su, 2007) and in vivo assays, (ii) exhibited cognition-enhancing properties in vivo, (iii) exhibited low affinity binding at muscarinic and NMDA receptors and (iv) modulated BDNF release. The results of this study provided additional evidence suggesting that the use of σ1 receptor agonists, in general, represent a potential therapeutic strategy for the treatment of cognitive decline associated with decreased cholinergic tone and that LS-1–137 may represent a novel σ1 receptor-selective agonist for the treatment of cognitive dysfunction.
The results of our active avoidance studies indicated that LS-1–137 was able to attenuate scopolamine-induced cognitive deficits in C57BL/6J mice without interfering with the normal learning process in the active avoidance task or the Morris water maze (D'Hooge and De Deyn, 2001). The commercially available σ1 agonists, PRE-084 and PPCC, were also found to be effective in attenuating scopolamine-induced learning impairment, while the σ1 antagonist, NE-100, was not able to reverse the scopolamine-induced cognitive deficit. These observations are consistent with previous reports that σ1 receptor agonists exhibit anti-amnesic properties (Matsuno et al., 1994; Senda et al., 1997; Urani et al., 1998; Maurice, 2004b; Espallergues et al., 2007; Maurice and Su, 2009; van Waarde et al., 2011; Niitsu et al., 2012; Ito et al., 2013).
To further validate that LS-1–137 is mediating its action through the stimulation of σ1 receptors, we co-administered LS-1–137 and NE-100 and found that NE-100 blocked the effect of LS-1–137. This attenuation of the effects of LS-1–137 by NE-100 is consistent with the BiP co-immunoprecipitation assay result indicating that LS-1–137 acts as an agonist at σ1 receptors both in vitro and in vivo.
As previously mentioned, in the absence of scopolamine LS-1–137 did not enhance or attenuate performance in the active avoidance task or in the Morris water maze, suggesting that σ1 receptor compounds may mediate their action only in the presence of atypical or pathological conditions (Urani et al., 1998; Espallergues et al., 2007). This observation may derive from the fact that, rather than being a classical receptor signalling unit, the σ1 receptor acts as an intracellular signal transduction modulator of multiple signalling pathways.
The σ1 receptors localized at the mitochondria-associated endoplasmic reticulum (ER) membrane (MAM) regulate a variety of cellular functions including (i) Ca2+ signalling between ER and mitochondria, (ii) neuronal differentiation, (iii) ion channel activation and (iv) cellular survival. Under resting conditions, σ1 receptors are bound to the ER chaperone protein BiP. Ligand stimulation or ER Ca2+ depletion causes a dissociation of σ1 receptors and BiP. The unbound σ1 receptor chaperones the unstable inositol 1–4-5-triphosphate (IP3) receptor and prevents IP3 receptor degradation. Stabilization of the IP3 receptors increases Ca2+ flow into the mitochondria. This increase of Ca2+ in the mitochondria results in the activation of enzymes that trigger ATP production via the Krebs cycle. Thus, σ1 receptors are postulated to drive the bioenergetics within the cell (Hayashi and Su, 2007; Ishikawa and Hashimoto, 2010; Ortega-Roldan et al., 2013).
The σ1 receptor has also been reported to regulate metabotropic receptor signalling including amplification of NMDA-sensitive glutamatergic receptor, dopaminergic and IP3-related metabotropic receptor activity. σ1 receptor activation also modulates Ca2+ signalling at the ER, regulates IP3 receptor activity and modulates neurotransmitter release (Su and Hayashi, 2003; Hayashi and Su, 2005). The σ1 receptor has also been postulated to regulate ER stress, cellular redox, cellular survival and synaptogenesis (Hayashi and Su, 2007; Fujimoto et al., 2012).
In addition to our studies demonstrating LS-1–137 neuroprotective properties in a rat transient middle cerebral artery occlusion stroke model (Luedtke et al., 2012), there have been a number of studies suggesting that σ1 receptor activation can lead to neuroprotection. Antonini et al. (2009; 2011,) demonstrated that occupation of the rat σ1 receptor by PPCC and (-)-MR22 can prevent cognitive impairment caused by the combined administration of the muscarinic receptor antagonist atropine and immunotoxin 192 IgG-saporin, which leads to a loss of cholinergic neurons in the basal forebrain. Those authors suggest that the observed PPCC and (-)-MR22 mediated neuroprotection might be due to stabilization of IP3 receptors that prolong the flow of calcium into the mitochondria. Other studies suggest that σ1 receptor stimulation can lead to the activation of PLC/PKC pathways, which promotes neurogenesis (Maurice et al., 2001; Villard et al., 2011).
The neuroprotective properties of σ1 agonists have also been studied in the context of neurodegenerative disorders. The cognitive impairments and toxicity induced in mice after intracerebral injection of amyloid peptides was prevented by the administration of the σ1 receptor-selective agonist PRE-084 (Meunier et al., 2006; Villard et al., 2011).
In the last several years, there have been reports that agonist-mediated σ1 receptor chaperone activity may be linked to the secretion and the regulation of BDNF (Fujimoto et al., 2012; Hashimoto, 2013; Ring and Regan, 2013). BDNF plays a crucial role in regulating neuron growth, repair and synaptic plasticity. Increased BDNF levels in vivo have been shown to correlate with increased long-term potentiation, neurogenesis, learning and improved memory (Yamada et al., 2002; Yamada and Nabeshima, 2004), whereas reduced levels of BDNF have been implicated in the pathogenesis of neurodegenerative disorders (Mattson et al., 2004a,b). It is postulated that the chaperone activity of σ1 receptor leads to increased release of BDNF, which, in turn, modulates signalling pathways that promote neuroprotection (Yagasaki et al., 2006). Fujimoto et al. (2012) proposed that the antiamnesic properties of the σ1 receptor agonist SA-4503 might be due, at least in part, to its ability to enhance release of the BDNF in the hippocampus (Fujimoto et al., 2012).
In this study, we demonstrated that LS-1–137 can dose dependently cause the release of BDNF from neonatal rat astrocytes. Scopolamine is known to modulate the BDNF expression (Lindefors et al., 1992). Therefore, it is possible that the diminution of the in vivo effects of scopolamine that we observed following LS-1–137 administration might be due, in part, to increased release of BDNF.
Additional studies will be required to further define the mechanism of action of LS-1–137 on BDNF release. In a previous study it was reported that SA-4503 increased BDNF release from the rat neuroblastoma cell line B104 in a dose-dependent manner by potentiating the post-translational processing of BDNF, without affecting BDNF mRNA expression levels. SA-4503 was reported to decrease intracellular levels of both pro-BDNF and mature BDNF, whereas extracellular levels of mature BDNF increased (Fujimoto et al., 2012). The chaperone activity of σ1 receptor has also been proposed to promote the secretion of mature BDNF from its precursor pro-BDNF (Hashimoto, 2013). Although the mechanism of action of LS-1–137 in currently undefined, the observed dose-dependent effect of LS-1–137 on BDNF release is consistent with a number of recent studies suggesting that σ1 receptor agonism potentiates BDNF release (Fujimoto et al., 2012; Hashimoto, 2013; Ring and Regan, 2013; Francardo et al., 2014; Peviani et al., 2014).
While the neuroprotective properties of LS-1–137 may play a role in the attenuation of the behavioural effects of scopolamine, it seems more likely that the ability of σ1 receptor agonists to modulate cholinergic transmission may be more germane to the in vivo effects of LS-1–137 reported in this communication. Several studies have shown σ1 receptor activation increases cholinergic neurotransmission in (i) rat hippocampal slices and (ii) the rat hippocampus and cortex. These effects were blocked by the σ1 receptor antagonist haloperidol (Junien et al., 1991; Matsuno et al., 1992; 1993; 1994; 1995,,,). Other studies demonstrated that the cognitive effects of σ1 receptor ligands can be prevented by σ1 receptor antisense oligonucleotides but not by the administration of a mismatch oligodeoxynucleotide (Matsuno et al., 1994; Senda et al., 1997; Maurice et al., 2001).
One caveat to this proposal is the possibility that the observed LS-1–137-dependent attenuation of scopolamine-induced cognitive deficits might be the result of LS-1–137 binding off-target at muscarinic or NMDA receptors. However, we evaluated the binding properties of LS-1–137 at these receptors and found that LS-1–137 is essentially devoid of muscarinic and NMDA receptor-binding activity.
In conclusion, based upon the results presented in this communication and our previous studies using a rodent model of stroke, the phenylacetamide LS-1–137 is a novel σ1 versus σ2 receptor-selective agonist that is devoid of D2-like dopaminergic and muscarinic receptor-binding activity. Furthermore, LS-1–137 exhibits neuroprotective and cognitive-enhancing properties in vivo and has neurotrophic factor regulatory properties.
Acknowledgments
The authors would like to thank Jessica Wong for technical assistance in the behavioural testing. We would also like to thank Michelle Taylor for editorial assistance in the preparation of this manuscript. LS-1–137 was generously synthesized for us by Chunyang Jin, PhD, and Kenneth Rehder, PhD, of the NIMH Chemical Synthesis and Drug Supply Program.
Glossary
Abbreviations
- AD
Alzheimer's disease
- BDNF
brain-derived neurotrophic factor
- BiP
binding immunoglobulin protein
- ER
endoplasmic reticulum
- PPCC
methyl(1R,2S/1S,2R)-2-[4-hydroxy-4-phenylpiperidin-1-yl)methyl]-1-(4-methylphenyl)cyclopro-panecarboxylate
- QNB
quinuclidinyl benzylate
Author contributions
Maninder Malik: Involved in the design and performance of the behavioural pharmacology aspects of the experiments. Involved in the interpretation of experimental data. Also involved in manuscript writing, graphic presentation and editing. Claudia Rangel-Barajas: Involved in the design and interpretation of behavioural experimental data. Involved in statistical analysis of experimental data. Also involved in manuscript writing and editing. Nathalie Sumien: Involved in the design and interpretation of behavioural experimental data. Involved in statistical evaluation of experimental data. Also involved in manuscript writing and editing. Chang Su: Involved in the design, performance and interpretation of studies designed to evaluate the effect of novel σ1 receptor-selective compounds on BDNF release from rat astrocytes. Meharvan Singh: Involved in the design and interpretation of studies designed to evaluate the effect of novel σ1 receptor-selective compounds on BDNF release from rat astrocytes. Also involved in manuscript writing and editing. Zhenglan Chen: Involved in the design and performance of the NMDA electrophysiology studies designed to investigate the possible off-site binding of our σ1 selective ligand LS-1–137. Ren-Qi Huang: Involved in the design, interpretation of the NMDA electrophysiology studies designed to investigate the possible off-site binding of our σ1 selective ligand LS-1–137. Also involved in the manuscript writing and editing for the sections related to the electrophysiology studies. Johann Meunier: Involved in the design and interpretation of studies designed to evaluate the intrinsic efficacy of novel σ1 receptor-selective compounds. Tangui Maurice: Involved in the design and interpretation of studies designed to evaluate the intrinsic efficacy of novel σ1 receptor-selective compounds. Also involved in manuscript writing and editing. Robert H. Mach: Involved in the design and interpretation of all medicinal chemical and molecular pharmacological aspects of the experiments. Involved in the interpretation of experimental data. Also involved in manuscript writing and editing. Robert R. Luedtke: Involved in the design and interpretation of all aspects of the experiments. Also involved in manuscript writing, graphic presentation and editing.
Conflict of interest
Johann Meunier is an employee of Amylgen. Tangui Maurice is the Scientific Director of Amylgen and a scientific advisory board member of Anavex Life Sciences. The BiP dissociation experiments were performed at Amylgen. Anavex has no role in the present research.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013a;170:1449–1458. [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-Gated Ion Channels. Br J Pharmacol. 2013c;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013d;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini V, Prezzavento O, Coradazzi M, Marrazzo A, Ronsisvalle S, Arena E, et al. Anti-amnesic properties of (+/−)-PPCC, a novel sigma receptor ligand, on cognitive dysfunction induced by selective cholinergic lesion in rats. J Neurochem. 2009;109:744–754. doi: 10.1111/j.1471-4159.2009.06000.x. [DOI] [PubMed] [Google Scholar]
- Antonini V, Marrazzo A, Kleiner G, Coradazzi M, Ronsisvalle S, Prezzavento O, et al. Anti-amnesic and neuroprotective actions of the sigma-1 receptor agonist (-)-MR22 in rats with selective cholinergic lesion and amyloid infusion. J Alzheimers Dis. 2011;24:569–586. doi: 10.3233/JAD-2011-101794. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Dean RL, III, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- Bejar C, Wang RH, Weinstock M. Effect of rivastigmine on scopolamine-induced memory impairment in rats. Eur J Pharmacol. 1999;383:231–240. doi: 10.1016/s0014-2999(99)00643-3. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science. 1983;219:1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- Craig LA, Hong NS, McDonald RJ. Revisiting the cholinergic hypothesis in the development of Alzheimer's disease. Neurosci Biobehav Rev. 2011;35:1397–1409. doi: 10.1016/j.neubiorev.2011.03.001. [DOI] [PubMed] [Google Scholar]
- D'Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev. 2001;36:60–90. doi: 10.1016/s0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- Dumas JA, Newhouse PA. The cholinergic hypothesis of cognitive aging revisited again: cholinergic functional compensation. Pharmacol Biochem Behav. 2011;99:254–261. doi: 10.1016/j.pbb.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert U, Kirch W. Scopolamine model of dementia: electroencephalogram findings and cognitive performance. Eur J Clin Invest. 1998;28:944–949. doi: 10.1046/j.1365-2362.1998.00393.x. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, Meliani K. Effects of physostigmine and scopolamine on rats’ performances in object-recognition and radial-maze tests. Psychopharmacology (Berl) 1992;109:321–330. doi: 10.1007/BF02245880. [DOI] [PubMed] [Google Scholar]
- Espallergues J, Lapalud P, Christopoulos A, Avlani VA, Sexton PM, Vamvakides A, et al. Involvement of the sigma1 (sigma1) receptor in the anti-amnesic, but not antidepressant-like, effects of the aminotetrahydrofuran derivative ANAVEX1-41. Br J Pharmacol. 2007;152:267–279. doi: 10.1038/sj.bjp.0707386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood JF, Cherkin A. Scopolamine effects on memory retention in mice: a model of dementia? Behav Neural Biol. 1986;45:169–184. doi: 10.1016/s0163-1047(86)90750-8. [DOI] [PubMed] [Google Scholar]
- Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science. 2009;323:934–937. doi: 10.1126/science.1166127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francardo V, Bez F, Wieloch T, Nissbrandt H, Ruscher K, Cenci MA. Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain. 2014;137:1998–2014. doi: 10.1093/brain/awu107. [DOI] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Hayashi T, Urfer R, Mita S, Su TP. Sigma-1 receptor chaperones regulate the secretion of brain-derived neurotrophic factor. Synapse. 2012;66:630–639. doi: 10.1002/syn.21549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K. Sigma-1 receptor chaperone and brain-derived neurotrophic factor: emerging links between cardiovascular disease and depression. Prog Neurobiol. 2013;100:15–29. doi: 10.1016/j.pneurobio.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su T. The sigma receptor: evolution of the concept in neuropsychopharmacology. Curr Neuropharmacol. 2005;3:267–280. doi: 10.2174/157015905774322516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Tsai SY, Mori T, Fujimoto M, Su TP. Targeting ligand-operated chaperone sigma-1 receptors in the treatment of neuropsychiatric disorders. Expert Opin Ther Targets. 2011;15:557–577. doi: 10.1517/14728222.2011.560837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RQ, Dillon GH. Effect of extracellular pH on GABA-activated current in rat recombinant receptors and thin hypothalamic slices. J Neurophysiol. 1999;82:1233–1243. doi: 10.1152/jn.1999.82.3.1233. [DOI] [PubMed] [Google Scholar]
- Huang RQ, Singh M, Dillon GH. Genistein directly inhibits native and recombinant NMDA receptors. Neuropharmacology. 2010;58:1246–1251. doi: 10.1016/j.neuropharm.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Hammond PS, Whirrett BR, Kuhner RJ, Wu L, Childers SR, et al. Synthesis and quantitative structure-activity relationships of N-(1-benzylpiperidin-4-yl)phenylacetamides and related analogues as potent and selective sigma1 receptor ligands. J Med Chem. 1998;41:2361–2370. doi: 10.1021/jm980032l. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hammond PS, Wu L, Mach RH. Synthesis and structure-activity relationships of N-(1-benzylpiperidin-4-yl)arylacetamide analogues as potent sigma1 receptor ligands. J Med Chem. 2001;44:4404–4415. doi: 10.1021/jm010384j. [DOI] [PubMed] [Google Scholar]
- Hulme EC, Birdsall NJ, Burgen AS, Mehta P. The binding of antagonists to brain muscarinic receptors. Mol Pharmacol. 1978;14:737–750. [PubMed] [Google Scholar]
- Hunt DL, Castillo PE. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol. 2012;22:496–508. doi: 10.1016/j.conb.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council. Guide for the Care and Use of Laboratory Animals. Washington, D.C: National Academy Press; 1996. [Google Scholar]
- Ishikawa M, Hashimoto K. The role of sigma-1 receptors in the pathophysiology of neuropsychiatric diseases. J Recept Ligand Channel Res. 2010;3:25–36. [Google Scholar]
- Ito K, Hirooka Y, Sunagawa K. Brain sigma 1 receptor stimulation improves mental disorder and cardiac function in mice with myocardial infarction. J Cardiovasc Pharmacol. 2013;62:222–228. doi: 10.1097/FJC.0b013e3182970b15. [DOI] [PubMed] [Google Scholar]
- Junien JL, Roman FJ, Brunelle G, Pascaud X. JO1784, a novel sigma ligand, potentiates [3H]acetylcholine release from rat hippocampal slices. Eur J Pharmacol. 1991;200:343–345. doi: 10.1016/0014-2999(91)90593-f. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk CJ, Reddy NL, Fischer JB, Wolcott TC, Knapp AG, McBurney RN. In vitro neuroprotection by substituted guanidines with varying affinities for the N-methyl-D-aspartate receptor ionophore and for sigma sites. J Pharmacol Exp Ther. 1994;271:1080–1085. [PubMed] [Google Scholar]
- Kitaichi K, Chabot JG, Moebius FF, Flandorfer A, Glossmann H, Quirion R. Expression of the purported sigma(1) (sigma(1)) receptor in the mammalian brain and its possible relevance in deficits induced by antagonism of the NMDA receptor complex as revealed using an antisense strategy. J Chem Neuroanat. 2000;20:375–387. doi: 10.1016/s0891-0618(00)00106-x. [DOI] [PubMed] [Google Scholar]
- Klinkenberg I, Blokland A. The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci Biobehav Rev. 2010;34:1307–1350. doi: 10.1016/j.neubiorev.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Su TP, Fujimoto M, Bonci A. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012;35:762–771. doi: 10.1016/j.tins.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume T, Nishikawa H, Taguchi R, Hashino A, Katsuki H, Kaneko S, et al. Antagonism of NMDA receptors by sigma receptor ligands attenuates chemical ischemia-induced neuronal death in vitro. Eur J Pharmacol. 2002;455:91–100. doi: 10.1016/s0014-2999(02)02582-7. [DOI] [PubMed] [Google Scholar]
- Lindefors N, Ernfors T, Falkenberg T, Hefti F. Septal cholinergic afferents regulate expression of brain-derived neutrotrophic factor and beta-nerve growth factor mRNA in rat hippocampus. Exp Brain Res. 1992;88:771–779. doi: 10.1007/BF02259130. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedtke RR, Perez E, Yang SH, Liu R, Vangveravong S, Tu Z, et al. Neuroprotective effects of high affinity sigma 1 receptor selective compounds. Brain Res. 2012;1441:17–26. doi: 10.1016/j.brainres.2011.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthin GR, Wolfe BB. Comparison of [3H]pirenzepine and [3H]quinuclidinylbenzilate binding to muscarinic cholinergic receptors in rat brain. J Pharmacol Exp Ther. 1984;228:648–655. [PubMed] [Google Scholar]
- Martina M, Turcotte ME, Halman S, Bergeron R. The sigma-1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels in rat hippocampus. J Physiol. 2007;578:143–157. doi: 10.1113/jphysiol.2006.116178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K, Matsunaga K, Mita S. Increase of extracellular acetylcholine level in rat frontal cortex induced by (+)N-allylnormetazocine as measured by brain microdialysis. Brain Res. 1992;575:315–319. doi: 10.1016/0006-8993(92)90096-r. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Matsunaga K, Senda T, Mita S. Increase in extracellular acetylcholine level by sigma ligands in rat frontal cortex. J Pharmacol Exp Ther. 1993;265:851–859. [PubMed] [Google Scholar]
- Matsuno K, Senda T, Matsunaga K, Mita S. Ameliorating effects of sigma receptor ligands on the impairment of passive avoidance tasks in mice: involvement in the central acetylcholinergic system. Eur J Pharmacol. 1994;261:43–51. doi: 10.1016/0014-2999(94)90298-4. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Senda T, Kobayashi T, Mita S. Involvement of sigma 1 receptor in (+)-N-allylnormetazocine-stimulated hippocampal cholinergic functions in rats. Brain Res. 1995;690:200–206. doi: 10.1016/0006-8993(95)00618-z. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004a;27:589–594. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Wan R, Guo Z. Prophylactic activation of neuroprotective stress response pathways by dietary and behavioral manipulations. NeuroRx. 2004b;1:111–1116. doi: 10.1602/neurorx.1.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T. Improving Alzheimer's disease-related cognitive deficits with sigma1 (σ1) receptor agonists. Drug News Perspect. 2002;15:617–625. doi: 10.1358/dnp.2002.15.10.740241. [DOI] [PubMed] [Google Scholar]
- Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T, Su TP, Parish DW, Nabeshima T, Privat A. PRE-084, a sigma selective PCP derivative, attenuates MK-801-induced impairment of learning in mice. Pharmacol Biochem Behav. 1994;49:859–869. doi: 10.1016/0091-3057(94)90235-6. [DOI] [PubMed] [Google Scholar]
- Maurice T, Roman FJ, Su TP, Privat A. Beneficial effects of sigma agonists on the age-related learning impairment in the senescence-accelerated mouse (SAM) Brain Res. 1996;733:219–230. doi: 10.1016/0006-8993(96)00565-3. [DOI] [PubMed] [Google Scholar]
- Maurice T, Su TP, Privat A. Sigma1 (sigma 1) receptor agonists and neurosteroids attenuate B25-35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience. 1998;83:413–428. doi: 10.1016/s0306-4522(97)00405-3. [DOI] [PubMed] [Google Scholar]
- Maurice T, Phan VL, Privat A. The anti-amnesic effects of sigma1 (sigma1) receptor agonists confirmed by in vivo antisense strategy in the mouse. Brain Res. 2001;898:113–121. doi: 10.1016/s0006-8993(01)02152-7. [DOI] [PubMed] [Google Scholar]
- Maurice T, Meunier J, Feng B, Ieni J, Monaghan DT. Interaction with sigma(1) protein, but not N-methyl-D-aspartate receptor, is involved in the pharmacological activity of donepezil. J Pharmacol Exp Ther. 2006;317:606–614. doi: 10.1124/jpet.105.097394. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Ieni J, Maurice T. The anti-amnesic and neuroprotective effects of donepezil against amyloid b24-35 peptide-induced toxicity in mice involved an interaction with the sigma1 receptor. Br J Pharmacol. 2006;149:998–1012. doi: 10.1038/sj.bjp.0706927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neuroscie Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Niitsu T, Iyo M, Hashimoto K. Sigma-1 receptor agonists as therapeutic drugs for cognitive impairment in neuropsychiatric diseases. Curr Pharm Des. 2012;18:875–883. doi: 10.2174/138161212799436476. [DOI] [PubMed] [Google Scholar]
- Nishikawa H, Hashino A, Kume T, Katsuki H, Kaneko S, Akaike A. Involvement of direct inhibition of NMDA receptors in the effects of sigma-receptor ligands on glutamate neurotoxicity in vitro. Eur J Pharmacol. 2000;404:41–48. doi: 10.1016/s0014-2999(00)00595-1. [DOI] [PubMed] [Google Scholar]
- Olton DS, Isaacson RL. Hippocampal lesions and active avoidance. Physiol Behav. 1968;3:719–724. [Google Scholar]
- Ortega-Roldan JL, Ossa F, Schnell JR. Characterization of the human sigma-1 receptor chaperone domain structure and binding immunoglobulin protein (BiP) interactions. J Biol Chem. 2013;288:21448–21457. doi: 10.1074/jbc.M113.450379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peviani M, Salvaneschi E, Bontempi L, Petese A, Manzo A, Rossi D, et al. Neuroprotective effects of the sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol Dis. 2014;62:218–232. doi: 10.1016/j.nbd.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Ring RM, Regan CM. Captodiamine, a putative antidepressant, enhances hypothalamic BDNF expression in vivo by synergistic 5-HT2c receptor antagonism and sigma-1 receptor agonism. J Psychopharmacol. 2013;27:930–939. doi: 10.1177/0269881113497614. [DOI] [PubMed] [Google Scholar]
- Ruscher K, Shamloo M, Rickhag M, Ladunga I, Soriano L, Gisselsson L, et al. The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke. Brain. 2011;134:732–746. doi: 10.1093/brain/awq367. [DOI] [PubMed] [Google Scholar]
- Scarpini E, Scheltens P, Feldman H. Treatment of Alzheimer's disease: current status and new perspectives. Lancet Neurol. 2003;2:539–547. doi: 10.1016/s1474-4422(03)00502-7. [DOI] [PubMed] [Google Scholar]
- Senda T, Matsuno K, Kobayashi T, Mita S. Reduction of the scopolamine-induced impairment of passive-avoidance performance by sigma receptor agonist in mice. Physiol Behav. 1997;61:257–264. doi: 10.1016/s0031-9384(96)00447-7. [DOI] [PubMed] [Google Scholar]
- Seth P, Fei YJ, Li HW, Huang W, Leibach FH, Ganapathy V. Cloning and functional characterization of a sigma receptor from rat brain. J Neurochem. 1998;70:922–931. doi: 10.1046/j.1471-4159.1998.70030922.x. [DOI] [PubMed] [Google Scholar]
- Shetty RA, Forster MJ, Sumien N. Coenzyme Q(10) supplementation reverses age-related impairments in spatial learning and lowers protein oxidation. Age (Dordr) 2013;35:1821–1834. doi: 10.1007/s11357-012-9484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, Cunningham RL, Rybalchenko N, Singh M. Progesterone increases the release of brain-derived neurotrophic factor from glia via progesterone receptor membrane component 1 (Pgrmc1)-dependent ERK5 signaling. Endocrinology. 2012;153:4389–4400. doi: 10.1210/en.2011-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TP, Hayashi T. Understanding the molecular mechanism of sigma-1 receptors: towards a hypothesis that sigma-1 receptors are intracellular amplifiers for signal transduction. Curr Med Chem. 2003;10:2073–2080. doi: 10.2174/0929867033456783. [DOI] [PubMed] [Google Scholar]
- Tarawneh R, Galvin JE. Potential future neuroprotective therapies for neurodegenerative disorders and stroke. Clin Geriatr Med. 2010;26:125–147. doi: 10.1016/j.cger.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchedre KT, Huang RQ, Dibas A, Krishnamoorthy RR, Dillon GH, Yorio T. Sigma-1 receptor regulation of voltage-gated calcium channels involves a direct interaction. Invest Ophthalmol Vis Sci. 2008;49:4993–5002. doi: 10.1167/iovs.08-1867. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Tsai S-Y, Hayashi T, Mori T, Su T-P. Sigma-1 receptor chaperones and diseases. Cent Nerv Syst Agents Med Chem. 2009;9:184–189. doi: 10.2174/1871524910909030184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urani A, Privat A, Maurice T. The modulation by neurosteroids of the scopolamine-induced learning impairment in mice involves an interaction with sigma1 (σ1) receptors. Brain Res. 1998;799:64–77. doi: 10.1016/s0006-8993(98)00469-7. [DOI] [PubMed] [Google Scholar]
- Villard V, Espallergues J, Keller E, Vamvakides A, Maurice T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (sigma1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J Psychopharmacol. 2011;25:1101–1117. doi: 10.1177/0269881110379286. [DOI] [PubMed] [Google Scholar]
- Vilner BJ, John CS, Bowen WD. Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 1995;55:408–413. [PubMed] [Google Scholar]
- van Waarde A, Ramakrishnan NK, Rybczynska AA, Elsinga PH, Ishiwata K, Nijholt IM, et al. The cholinergic system, sigma-1 receptors and cognition. Behav Brain Res. 2011;221:543–554. doi: 10.1016/j.bbr.2009.12.043. [DOI] [PubMed] [Google Scholar]
- Yagasaki Y, Numakawa T, Kumamaru E, Hayashi T, Su TP, Kunugi H. Chronic antidepressants potentiate via sigma-1 receptors the brain-derived neurotrophic factor-induced signaling for glutamate release. J Biol Chem. 2006;281:12941–12949. doi: 10.1074/jbc.M508157200. [DOI] [PubMed] [Google Scholar]
- Yamada K, Nabeshima T. Interaction of BDNF/TrkB signaling with NMDA receptor in learning and memory. Drug News Perspect. 2004;17:435–438. doi: 10.1358/dnp.2004.17.7.863702. [DOI] [PubMed] [Google Scholar]
- Yamada K, Mizuno M, Nabeshima T. Role for brain-derived neurotrophic factor in learning and memory. Life Sci. 2002;70:735–774. doi: 10.1016/s0024-3205(01)01461-8. [DOI] [PubMed] [Google Scholar]