

Abstract
Background and Purpose
NaV1.8 ion channels have been highlighted as important molecular targets for the design of low MW blockers for the treatment of chronic pain. Here, we describe the effects of PF-01247324, a new generation, selective, orally bioavailable Nav1.8 channel blocker of novel chemotype.
Experimental Approach
The inhibition of Nav1.8 channels by PF-01247324 was studied using in vitro patch-clamp electrophysiology and the oral bioavailability and antinociceptive effects demonstrated using in vivo rodent models of inflammatory and neuropathic pain.
Key Results
PF-01247324 inhibited native tetrodotoxin-resistant (TTX-R) currents in human dorsal root ganglion (DRG) neurons (IC50: 331 nM) and in recombinantly expressed h Nav1.8 channels (IC50: 196 nM), with 50-fold selectivity over recombinantly expressed TTX-R hNav1.5 channels (IC50: ∼10 μM) and 65–100-fold selectivity over TTX-sensitive (TTX-S) channels (IC50: ∼10–18 μM). Native TTX-R currents in small-diameter rodent DRG neurons were inhibited with an IC50 448 nM, and the block of both human recombinant Nav1.8 channels and TTX-R from rat DRG neurons was both frequency and state dependent. In vitro current clamp showed that PF-01247324 reduced excitability in both rat and human DRG neurons and also altered the waveform of the action potential. In vivo experiments n rodents demonstrated efficacy in both inflammatory and neuropathic pain models.
Conclusions and Implications
Using PF-01247324, we have confirmed a role for Nav1.8 channels in both inflammatory and neuropathic pain. We have also demonstrated a key role for Nav1.8 channels in action potential upstroke and repetitive firing of rat and human DRG neurons.
Tables of Links
| TARGETS |
|---|
| Ion channels |
| Nav1.2 channels |
| Nav1.5 channels |
| Nav1.7 channels |
| Nav1.8 channels |
| Nav1.9 channels |
| LIGANDS |
|---|
| A-803467 |
| Gabapentin |
| PGE2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guideto PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Voltage-gated sodium channels (VGSCs) play an important role in action potential generation and propagation and mediate the rapid influx of sodium ions that underlies the rising phase of the action potential in sensory nociceptive neurons (Blair and Bean, 2002). The Na+ channels, Nav1.3 and Nav1.7, together with Nav1.8 and Nav1.9, have been highlighted as significant molecular targets for the design of low MW blockers for the treatment of chronic pain (see Dib-Hajj et al., 2009; 2010). It is well understood that non-selective inhibition of sodium channels contributes to the therapeutic efficacy of local anaesthetics (LA), for example lidocaine, and specific anticonvulsants used for treating chronic neuropathic pain, for example carbamazepine and mexiletine (Sindrup and Jensen, 2007; Gerner and Strichartz, 2008). However, such drugs often have a narrow therapeutic window, mainly because of the inhibition of sodium channels in the CNS and heart. Therefore, much effort has been focused on trying to identify subtype-selective inhibitors for the treatment of pain, with the hope of minimizing the risk of serious side effects. The Nav1.8 channel is highly expressed by small primary sensory neurons (Akopian et al., 1999; Coward et al., 2000; Djouhri et al., 2003), and a number of preclinical studies have implicated Nav1.8 channels in nociceptive processing (reviewed by Dib-Hajj et al., 2010). Genetic ablation of Nav1.8 channels in rodents results in deficits in nociception following inflammation, but not neuropathic pain (Akopian et al., 1999; Kerr et al., 2001; Nassar et al., 2005; Minett et al., 2014), while recent human genetic evidence suggest that gain of function mutations in Nav1.8 channels contribute to painful peripheral neuropathy (Faber et al., 2012; Huang et al., 2013; Han et al., 2014). Therefore, selective blockers of Nav1.8 channels could potentially treat a broad range of chronic pain states such as lower back pain, cancer pain and neuropathic pain (e.g. painful diabetic neuropathy). Hurdles in identifying Nav subtype-selective compounds are not surprising given the high degree of sequence homology, particularly around the LA binding site to which the majority of low MW compounds bind (see England and de Groot, 2009). A-803467 and A-887826 are examples of compounds in the public domain belonging to a broadly similar chemotype containing three aromatic rings that inhibit Nav1.8 channels with lack of frequency dependence and demonstrate preclinical efficacy in pain models (Jarvis et al., 2007; Zhang et al., 2010). A-803467 demonstrates high selectivity across all Nav subtypes tested, while A-887826 shows lower levels of selectivity with only threefold selectivity over hNav1.2 channels. However, A-803467 has poor solubility and low oral bioavailability in rats (F = 13%) (Jarvis et al., 2007). In vivo efficacy studies using the selective compound A-803467 has yielded results that contradict data from Nav1.8 channel knock-out mice in that pharmacological effects were observed in several rat models of neuropathic pain (Jarvis et al., 2007). In agreement with studies using knockdown or genetic ablation of Nav1.8 channels in rodents (Khasar et al., 1998; Akopian et al., 1999; Villarreal et al., 2005), A-803467 also showed efficacy in multiple models of inflammatory pain (Jarvis et al., 2007). Although specific compounds within the existing class of Nav1.8 channel blockers demonstrate selectivity over other Nav subtypes, their off-target binding to other proteins (and associated effects on pain processing) are difficult to characterize. Therefore, in order to fully interpret the role of Nav1.8 channels in different pain phenotypes and understand the discrepancies sometimes seen between pharmacological and genetic modulation of Nav1.8 channels, a new generation of selective compounds is required (associated with a different off-target profile).
In the present study, we report the discovery of PF-01247324, which represents a structurally novel highly selective, frequency-dependent Nav1.8 channel blocker that possesses high oral bioavailability in rat (F = 91%). We describe the biophysics of compound/channel interaction and exploit this tool to describe the role of Nav1.8 channels in both rat and human dorsal root ganglion (DRG) neuron excitability. We provide further evidence with a structurally novel compound that Nav1.8 channels have a role in both neuropathic and inflammatory pain by demonstrating analgesic efficacy in rodent behavioural models of pain.
Methods
Cell culture of sodium channel stable cell lines
HEK293 cells expressing human Nav1.8/β1 (Merck Millipore, Billerica, MA, USA) were grown in DMEM/F12 (with L-glutamine), 10% FBS, 1% non-essential amino acids, G418 400 μg·mL−1, puromycin 0.625 μg·mL−1, hygromycin 100 μg·mL−1. HEK293 cells expressing recombinant sodium channels other than hNav1.8, were grown in DMEM/high-glucose Dulbecco's modified, 10% FBS, 2 mM sodium pyruvate, and G418. All cell culture reagents were from Invitrogen (Life Technologies, Gent, Belgium).
Isolation of human DRG neurons for voltage-clamp recordings
Human, non-diseased, low post mortem injury L4 and L5 DRG were obtained from informed consent donors through the National Disease Research Interchange (Philadelphia, PA, USA), as approved by the institutional review board at the University of Pennsylvania. None of the donors had diabetes nor were not taking medication for chronic pain management. Tissue was shipped immediately in cell culture media on ice.
Ganglia were immediately dissected from nerve roots and minced. The DRG were enzymically digested at 37°C for 90–120 min with collagenase Type II (Worthington Biochemical Corporation, Lakewood, NJ, USA; 12 mg·mL−1) and dispase II (Roche, Burgess Hill, UK); 20 mg·mL−1) in a calcium-free solution consisting of (in mM): 132 NaCl, 5.4 KCl, 0.8 MgCl2, 10 HEPES, and 5 glucose, pH adjusted to 7.4 with NaOH. Digested DRGs were gently triturated, filtered through a 200 μM nylon mesh and centrifuged (400× g for 5 min). The pellet was resuspended in medium (DMEM/10% FBS + 50 mg·mL−1 gentamycin + 2 mM sodium pyruvate, ∼2.4 mL per ganglion), plated onto poly-D-Lysine-coated glass coverslips (BD Biosciences, San Jose, CA, USA) and incubated at 37°C for 1–48 h.
Isolation of human DRG neurons for current clamp recordings
Human, non-diseased, cervical (C4–C7) DRG were removed from organ donors with short post-mortem delay via dorsal laminectomy, following UK National Health Service (NHS) Blood and Transplant informed consent, NHS Trust site and Imperial Tissue Bank Research Ethics Committee approvals. None of the donors had diabetes nor were not taking medication for chronic pain management. The DRG were prepared as described previously (Anand et al., 2006). Briefly, ganglia were rinsed in Ham's F12 medium containing antibiotics (penicillin/streptomycin 100 μg·mL−1 each), minced and enzyme digested in Ham's F12 containing collagenase (0.2%), dispase (0.5%) and antibiotics for 3 h at 37°C, followed by papain (0.1%) digestion for 30 min at 37°C. Enzymes were aspirated and tissues triturated in Ham's F12 containing soybean trypsin inhibitor (1 mg·mL−1), to obtain a neuronal suspension, that was plated in Ham's F12 medium containing 10% FCS, 100 ng·mL−1 nerve growth factor (NGF), 50 ng·mL−1 glial cell-derived neurotrophic factor (GDNF) and neurotrophin-3 (NT3) (50 ng·mL−1), kanamycin (100 μg·mL−1), penicillin and streptomycin, in T25 flasks maintained at 37°C prior to transportation in a warm flask. Neuronal cultures were detached using TrypLE (Life Technologies) for 3–4 min, centrifuged, resuspended and replated on collagen-coated coverslips in Ham's F12 containing 10% FCS, NGF, GDNF and NT3, for electrophysiological recordings.
Isolation of rat DRG neurons for current clamp recordings
Rat DRG neuronal cultures were prepared as described by Passmore (2005). Notable differences were the absence of NGF in the growth medium and cells were plated on pre-coated coverslips (poly-D-lysine/laminin; BD Biosciences, San Jose, CA, USA). Healthy DRG neurons had a visible nucleolus 2–3 h after isolation and were patched within 12–24 h of isolation.
Manual patch electrophysiology
Coverslips containing either HEK293 cells expressing sodium channels or DRG neurons from rats or humans were placed in a recording chamber and perfused (approximately 1 mL·min−1) with an extracellular solution (ECS) containing (in mM): 132 NaCl, 5.4 KCl, 10 HEPES, 5 glucose, 1.8 CaCl2 and 0.8 MgCl2, pH 7.4 with NaOH for hNav1.1, hNav1.2, hNav1.4, hNav1.6, hNav1.7 and hNav1.8 channels. For hNav1.5 channels, the final sodium concentration was reduced by substituting 102 mM choline Cl. For whole-cell voltage-clamp recordings of tetrodotoxin-resistant (TTX-R) currents from human DRG neurons, ECS contained (in mM): 20 NaCl, 112 choline Cl, 5.4 KCl, 10 HEPES, 5 glucose, 1.8 CaCl2, 0.8 MgCl2, 0.05 CdCl2 and 0.0002 TTX, pH 7.4 with NaOH (300 mOsmol·L−1). For current clamp recordings from DRG neurons, ECS contained (in mM): 135 NaCl, 4.7 KCl, 1 MgCl2, 1 CaCl2, 10 glucose, 10 HEPES, pH 7.4 with NaOH (300 mOsmol·L−1). For voltage-clamp recordings, recording patch pipettes were filled with an intracellular solution containing (in mM): 110 CsF, 35 CsCl, 5 NaCl, 10 HEPES and 10 EGTA, pH 7.3 with NaOH (290 mOsmol·L−1), and had a resistance of 1–2 MΩ. For current clamp recordings the pipette solution contained (in mM): 140 KCl, 0.5 EGTA, 3 Mg-ATP, 5 HEPES, pH7.3 with KOH (290 mOsmol·L−1). All recordings were made at room temperature (22–24°C) using Axopatch 200B or Multiclamp 700B amplifiers and PCLAMP software (Molecular Devices, Sunnyvale, CA, USA). For current clamp recordings, resting membrane potential (RMP) and seal stability for each neuron was evaluated and neurons with RMP that was more positive than −45 mV were excluded. All compounds were dissolved in DMSO. The final maximal concentration of DMSO used (<0.3%) was found to have no significant effect on sodium currents. Solutions containing PF-01247324 or DMSO control were applied using a perfusion system (MSC-200, Bio-Logic SAS, Claix, France).
Analysis of electrophysiology data
Quantification of the number of action potentials per sweep was performed using Spike 2 software (CED, Cambridge, UK) whereby transient membrane depolarizations with an overshoot beyond 0 mV were accepted as action potentials. The number of action potentials for the last three (rat) or five (human) sweeps was summed in order to obtain a measure in control and in the presence of PF-01247324. The peak amplitude, 20–80% slope of the rising phase, voltage threshold and width at 50% peak, were measured for the second action potential of each sweep using a bespoke script in Spike 2 (CED). The last three (rat) or five (human) values in each condition were averaged in order to obtain a measure for each parameter in control and in the presence of PF-01247324. The data were analysed using a repeated measures mixed model with a compound symmetric covariance structure followed by t-tests using a pooled estimate of variance. Each dose of PF-01247324 was compared with its nearest preceding control value with statistical significance declared if P < 0.05. Concentration–response data were analysed using nonlinear least squares fit of the Logistic Equation (GraphPad Prism 5, San Diego, CA, USA) to provide IC50 values.
PatchXpress electrophysiology
ECS contained (in mM): 135 NaCl, 2 CaCl2, 5.4 KCl, 1 MgCl2, 10 glucose, and mM HEPES, pH 7.4, with NaOH. Intracellular solution contained (in mM): 135 CsF, 5 NaCl, 2 MgCl2,10 EGTA, 10 HEPES, pH 7.4 with NaOH. Na+ channel-expressing cells were harvested by trypsinization. Test compound effects were evaluated with voltage protocols identical to those used for h Nav1.8 channels in conventional patch-clamp and a steady-state inactivation curve was run for each cell before each compound addition and the V1/2 of inactivation calculated automatically. A detailed description of the PatchXpress protocol has been described (Castle et al., 2009).
In vivo methodology
All animal care and experimental procedures complied with guidelines and were approved by the Neusentis US Institutional Animal Use and Care Committee and were deemed to be as humane as possible. Animals were not used for multiple tests. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 68 mice and 188 rats were used.
Male CD-1 mice (23–30 g) and male Sprague Dawley rats (170–300 g) (Charles River, Raleigh, NC, USA) had free access to food and water and were maintained on a 12:12 h light/dark schedule. For rat PF-01247324 was formulated as solutions of 0, 10, 30, 100 mg·kg−1 in 0.5%MC/0.1%Tween 80 vehicle and dosed via oral gavage prior to behavioural testing. For mice, the dose range was 0, 30, 100, 350 and 1000 mg·kg−1. All studies were done in a blinded randomized manner.
Rat formalin model of persistent pain
Animals were tested for paw movement responses to an injection of a 2.5% formalin solution (50 μL in saline) using the Automated Nociception Analyzer (Yaksh et al., 2001). Animals were given PF-01247324, 4 h before formalin injection (n = 12 per group). The instrument recorded rapid foot movements counted in one minute bins continuously for 60 min. Treatment groups were assigned to balance treatments across days and across test chambers. The total number of flinches was counted over particular time intervals or phases. The phases summarized in this study were Phase 1: 0–9 min and Phase 2: 10–60 min.
Rat carrageenan thermal hyperalgesia
To induce a local inflammation, 50 μL of a 1% solution of λ-carrageenan in 0.9% saline was injected s.c. into the plantar surface of the right hind paw of the rat. PF-01247324 at various doses or vehicle was administered orally 1 h after carrageenan injection and behavioural testing was conducted 2 h following compound administration (n = 8 per group). To assess the thermally evoked paw withdrawal response, a commercially available Hargreaves Box was used (UCSD Department of Anesthesiology, San Diego, CA, USA; stimulus intensity 5.5 A). The time to withdrawal response was measured in seconds and assigned as the response latency. The thermal stimulus was automatically cut off after 20 s. The inflamed paw was tested in each animal and the data reported as the mean of three measurements taken within a 5 min time period.
Rat complete Freund's adjuvant (CFA) tactile allodynia
To induce a local inflammation, 150 μL of a saline/CFA emulsion was injected into the right hind paw of the rat 48 h prior to behavioural testing. PF-01247324 at various doses or vehicle was administered orally and behavioural testing was conducted 4 h following compound administration (n = 12 per group). Tactile testing was conducted as discussed by Chaplan et al., 1994. An upper threshold was set at 15 g.
Rat hypersensitivity following spinal nerve ligation SNL
A neuropathic pain model was induced, in rats, with nerve injury, using a modification of the method described by Kim and Chung (1992) in which only the L5 spinal nerve was ligated. Prior to testing the effect of PF-01247324, baseline measurements of allodynia were performed immediately prior to subject selection and compound administration. Only animals with baseline scores ≤6.6 were considered allodynic and utilized in further testing. Following baseline measurements, PF-01247324 10 or 30 mg·kg−1, vehicle or gabapentin 100 mg·kg−1 were administered 1–2 h prior to testing, (n = 6–8 per group). Test animals were placed in a box separated by walls with a wire mesh floor allowing access to the plantar surface of the paw. Tactile testing was conducted as described by Chaplan et al., 1994, A maximum threshold was set at 15 g.
Collection of plasma samples and measurement of concentrations of PF-02147324
Terminal blood samples were taken under deep CO2 anaesthesia by cardiac puncture. The approximate blood collection times following administration of PF-01247324 were, for rat formalin test, 5.25 h, for mouse rotarod, 1 h and for rat carrageenan test, 2.33 h and for rat CFA 4.25 h. No terminal blood samples were taken following rat SNL hypersensitivity testing, where exposures were determined in satellite animals as part of the Functional Observation Battery (FOB). Plasma was prepared by centrifugation within 1 h of blood collection and stored at −20°C until analysis. In the mouse experiments, whole brain was also taken for analysis and stored at −20°C.
Plasma proteins were precipitated by adding 200 μL of methanol to 25 μL of plasma in a 96-well plate. After capping and vortex mixing, samples were centrifuged for 20 min at 4000 rpm at 4 °C and then 10 μL of the supernatant were injected onto an LC-MS-MS system consisting of an HP Zorbax SB C8 75 × 4.6 mm 3.5 μm analytical column with detection on a SCIEX API 4000 Turbo Ion Spray MS system in positive ion mode (Perkin Elmer, Waltham, MA, USA).
Measurement of brain concentrations of PF-01247324
For the determination of brain tissue concentrations in mice and rats, brain tissue weights were recorded and then treated with 3× the brain weight of 80:20 acetonitrile : water with 2.5% formic acid containing 75 ng·mL−1 of the internal standard, PF-06542801. Brain tissue was homogenized for 2.5 min using the high-energy cell disrupter Mini-Beadbeater-96 (BioSpec Products, Bartlesville, OK, USA). The samples were centrifuged at 1800× g at 4°C for 20 min and analysed using LC-MS/MS in the same manner as the diluted plasma samples. The unbound fraction of PF-01247324 in rat brain homogenate was determined by equilibrium dialysis.
Data analysis
Statistical analyses were conducted using GraphPad Prism 5 (GraphPad). Data were analysed using a one-way anova (vehicle vs. PF-01247324 treatment) for formalin and thermal hyperalgesia and two-way anova for spinal nerve injury (SNL). Post hoc analysis was performed using either Dunnett's or Bonferroni tests.
Materials
PF-01247324 [6-amino-5-(2, 3, 5-trichloro-phenyl)-pyridine-2-carboxylic acid methylamide] was synthesized at Pfizer Laboratories (Sandwich, UK) as described in WO2006011050 (Lane et al., 2006). Gabapentin was supplied internally by Pfizer Laboratories and PGE2 was supplied by Sigma Aldrich (Gillingham, UK).
Results
The ability of PF-01247324 to block both native human TTX-R channels and human recombinant Nav1.8 channels was evaluated using whole-cell patch-clamp technique with a voltage protocol that set the holding potential to the half-inactivation voltage for each cell (Figure 1). PF-01247324 blocked both native and recombinant h Nav1.8 channels (IC50 at human TTX-R = 311 nM; IC50 at h Nav1.8 channels stably expressed in HEK293 = 196 nM). PF-01247324 was determined to be more than 50-fold selective over Nav1.5 channels (IC50 ∼10 μM) and exhibited a range of selectivity over TTX-sensitive (TTX-S) channels (∼65-fold for Nav1.2 to ∼100-fold for Nav1.7 channels). See Table 1 for summarized data. The IC50 at rat and mouse TTX-R channels was also determined to be 0.448 μM and 0.53 μM, respectively (see Supporting Information Table S1 SI), and the selectivity across rat TTX-S isoforms is shown in Supporting Information Table S2 in supplementary information. In addition PF-01247324 was evaluated in a broad screening panel of receptors, ion channels and enzymes (CEREP, Poitiers, France) and showed no or weak activity at these targets.
Figure 1.
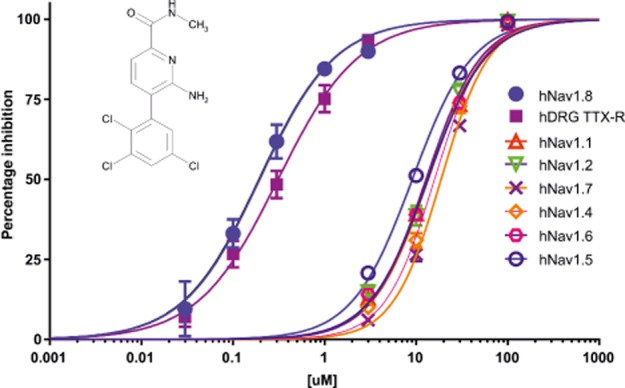
PF-01247324 is a selective Nav1.8 channel subtype-selective inhibitor. Selectivity was assessed across the family of human VGSC channels using manual patch-clamp electrophysiology. Testing was conducted at the specific half-inactivation voltage for each channel to more appropriately assess pharmacological selectivity (n = 5 for the human recombinantly expressed isoforms of the NaV1.8 channel and n = 4 for the TTX-R current in human DRG neurons). Cells were depolarized from a holding potential of −120 mV (or −150 mV for Nav1.5 channels) to a membrane potential that inactivated half of the available channels for 8 s followed by a 2 or 20 ms recovery at −120 mV and a 20 ms test pulse to 0 mV. Inset is the structure of PF-01247324.
Table 1.
PF-01247324 selectively blocks hNav1.8 channels over other human sodium channel isoforms recombinantly expressed in HEK293 cells as well as demonstrating comparable pharmacology at the native human channel
| Human Nav isoform/TTX-R | IC50 (μM) (95% CI) | Hill slope | n |
|---|---|---|---|
| 1.1 | 13.4 (12.4–14.78) | 1.4 | 5 |
| 1.2 | 12.8 (11.42–14.48) | 1.4 | 5 |
| 1.4 | 15.9 (14.54–17.42) | 1.6 | 5 |
| 1.5 | 9.0 (8.47–9.77) | 1.3 | 5 |
| 1.6 | 13.3 (12.40–14.47) | 1.2 | 5 |
| 1.7 | 18.7 (17.54–20.07) | 1.6 | 5 |
| 1.8 | 0.195 (0.156–0.246) | 1.05 | 5 |
| hTTX-R | 0.311 (0.254–0.384) | 0.995 | 4 |
Selectivity was assessed across the human VGSC isoforms using manual patch-clamp electrophysiology. Testing was conducted at the specific half-inactivation voltage for each channel (protocol details in Figure 1).
To determine the channel state or conformation with which PF-01247324 may interact, steady-state inactivation curves were generated using both recombinantly expressed hNav1.8 channels and TTX-R channels in rat DRG neurons at two different concentrations (1 and 3 μM) of PF-01247324 (Figure 2A–D). There was a concentration-dependent shift in the V1/2 value for steady-state inactivation for both recombinantly expressed hNav1.8 and native TTX-R channels in rat DRG neurons. The effect of PF-01247324 was reversible, indicating that rundown or a time-dependent shift in voltage-dependent properties was not responsible for the observed shift in V1/2. These data demonstrate that PF-01247324 preferentially inhibited inactivated channels in both recombinant and native systems. Additionally, PF-01247324 showed frequency-dependent block of both recombinant and native channels (Figure 2E and F).
Figure 2.
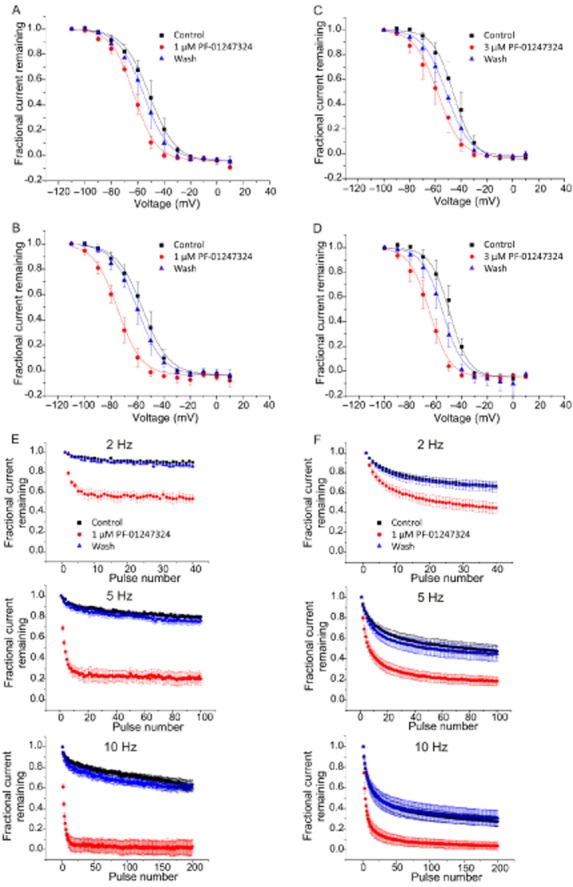
Inhibition by PF-01247324 is both use and state dependent at hNav1.8 channels and TTX-R in rat DRG neurons. Block of recombinant hNav1.8 and native TTX-R channels by PF-01247324 was assessed using voltage protocols designed to vary the amount of steady-state inactivation. Holding potential was −100 mV, and 8 second prepulses were given to various potentials, followed by a 20 ms step to 0 mV. Normalised fractional current vs. prepulse potential from hNav1.8 channels (A and B) and TTX-R in rat (C and D) illustrate shifts in V1/2 values observed in the presence of PF-01247324. Filled black squares are control, filled red circles are following application of PF-01247324 and filled blue triangles are wash. (A) and (C), show data following application of 1 μM PF-01247324 and (B) and (D), in the presence of 3 μM PF-01247324. Data represent means (± SEM) n = 6 per concentration for hNav1.8 channels, and n = 6 at 1 μM and n = 7 at 3 μM PF-0127324 for TTX-R in rat DRG neurons. Curve fits were generated by fitting Boltzmann equations to the data to obtain V1/2 and slope factors (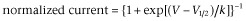 ) where V is the prepulse potential, V1/2 is the holding potential that gave half maximal activation, and k is the slope factor). Delta V1/2 = −7.67 ± 2.23 mV for hNav1.8 channels with 1 μM PF-01247324 compared with −16.62 ± 1.18 mV in the presence of 3 μM PF-01247324 and δV1/2 = −6.67 ± 0.5 mV for TTX-R channels in the presence of 1 μM PF-01247324 compared with −9.92 ± 0.89 mV in the presence of 3 μM PF-01247324. (E) Currents from hNav1.8 channels were repetitively evoked at a test pulse of 0 mV from a holding potential of −100 mV at 2, 5 and 10 Hz. 1 μM PF-01247324 (filled red circles) showed use-dependent block of the current at all frequencies tested. Filled black squares are control, and filled blue triangles are wash. Data represent means (± SEM), n = 9. (F) Same as in (E), but now, TTX-R currents were recorded from rat DRG neurons; 1 μM PF-01247324 (filled red circles) showed use-dependent block of the current at all frequencies tested. Filled black squares are control, filled blue triangles are wash. Data represent means (± SEM), n = 6.
) where V is the prepulse potential, V1/2 is the holding potential that gave half maximal activation, and k is the slope factor). Delta V1/2 = −7.67 ± 2.23 mV for hNav1.8 channels with 1 μM PF-01247324 compared with −16.62 ± 1.18 mV in the presence of 3 μM PF-01247324 and δV1/2 = −6.67 ± 0.5 mV for TTX-R channels in the presence of 1 μM PF-01247324 compared with −9.92 ± 0.89 mV in the presence of 3 μM PF-01247324. (E) Currents from hNav1.8 channels were repetitively evoked at a test pulse of 0 mV from a holding potential of −100 mV at 2, 5 and 10 Hz. 1 μM PF-01247324 (filled red circles) showed use-dependent block of the current at all frequencies tested. Filled black squares are control, and filled blue triangles are wash. Data represent means (± SEM), n = 9. (F) Same as in (E), but now, TTX-R currents were recorded from rat DRG neurons; 1 μM PF-01247324 (filled red circles) showed use-dependent block of the current at all frequencies tested. Filled black squares are control, filled blue triangles are wash. Data represent means (± SEM), n = 6.
To examine the basis of the interaction of PF-01247324 with hNav1.8 channels, site-directed mutagenesis was used to change two key residues that occur in the LA binding site (F1710A and Y1717A) in the S6 α-helical transmembrane segment of domain IV. We determined the potency of PF-01247324 at both the wild-type (WT) and double-mutant hNav1.8 channels using PatchXpress automated patch-clamp electrophysiology. There was a 65-fold difference in the IC50 value generated for the mutant versus WT hNav1.8 channels for PF-01247324 and a 135-fold shift for tetracaine (Figure 3A and B, Table 2), suggesting that PF-01247324 and other non-selective sodium channel inhibitors have overlapping binding sites.
Figure 3.
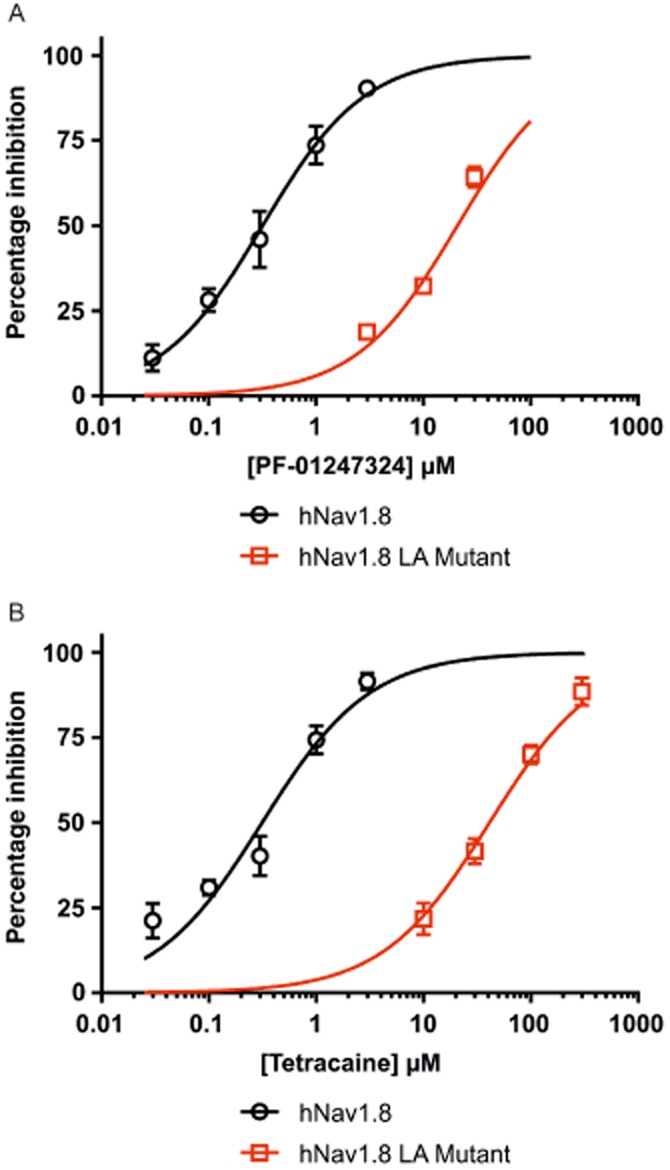
Potency and selectivity for Nav1.8 channels can be achieved at the LA binding site. Site-directed mutagenesis shows PF-01247324 and other non-selective sodium channel inhibitors have overlapping LA binding sites. All measurements were made from hNav1.8 channels stably expressed in HEK293 cells using PatchXpress automated patch-clamp system. The local anaesthetic mutant comprised the double mutation F1710A and Y1717A. Concentration– response curves for PF-01247324 (A) and tetracaine (B) illustrate a shift in the potency observed for the LA mutant versus WT hNav1.8 channels. Data represent means (± SEM) n = 3–13 per concentration.
Table 2.
Mutation of key residues for local anaesthetic binding alters the potency of block by PF-01247324
| WT hNav1.8 IC50 (μM) | LA mutant IC50 (μM) | |
|---|---|---|
| Tetracaine | 0.31 (0.24–0.39) | 41.22 (29.58–57.45) |
| PF-01247324 | 0.31 (0.25–0.40) | 20.86 (16.17–26.92) |
Table shows the IC50 values (95% CI) for each compound at both the WT and LA double-mutant (F1710A/Y1717A) hNaV1.8 channel as measured using the PatchXpress automated patch-clamp system. n = 3–13 per concentration tested.
Selective blockers of Nav1.8 channels are known to inhibit action potential firing in those DRG neurons where TTX-R currents play a prominent role (Jarvis et al., 2007; Zhang et al., 2010). Therefore, we evaluated the effects of PF-01247324 on action potential firing in both rat and human DRG neurons. 1 μM PF-01247324 significantly reduced evoked action potential firing in both rat and human DRG neurons from a holding potential of −60 mV (Figure 4A and B, respectively) in all cells tested and in the case of the latter, the trains of action potentials were not affected by the application of 500 nM TTX (P < 0.001 in rat, n = 10; P < 0.05 in human, n = 7). Currents mediated by TTX-R channels and, by inference, Nav1.8 channels have been suggested to carry the majority of the inward positive charge during the upstroke of the action potential (Blair and Bean, 2002). With respect to this we measured the properties of rat and human DRG neuron action potential waveform (second action potential in each sweep) including, peak amplitude, voltage threshold, the depolarizing slope between 20–80% of the peak amplitude (20–80% slope) and duration at 50% peak, before and after application of PF-01247324 in order to investigate the role of Nav1.8 channels in action potential generation. We observed a significant decrease in both peak amplitude and 20–80% slope of the rising phase of the action potential following application of 0.3 and 1.0 μM PF-01247324 to rat DRG neurons and 1 μM to human DRG neurons (Figure 5A and B). The percentage of decrease in the measured parameters of peak amplitude and slope were similar in both rat and human at 1 μM (Table 3). There was also a significant decrease in the voltage threshold of the action potential following application of 1 μM, but not 0.3 μM PF-01247324 to rat DRG neurons (P < 0.05), although this effect was not observed in the human DRG neurons at 1 μM (data not shown). The action potentials shown in Figure 5C illustrate the changes seen in the action potential waveform, and when these data are transformed into a phase plot, features of the action potential can be seen in more detail (Figure 5D). Boxed regions indicate the onset and offset of the shoulder, shown by the slowing rate of membrane voltage change. This is unaltered in both examples from rat or human following drug application. These data confirm a critical role for Nav1.8 channels in the upstroke of the action potential waveform found in both rat and human sensory neurons.
Figure 4.
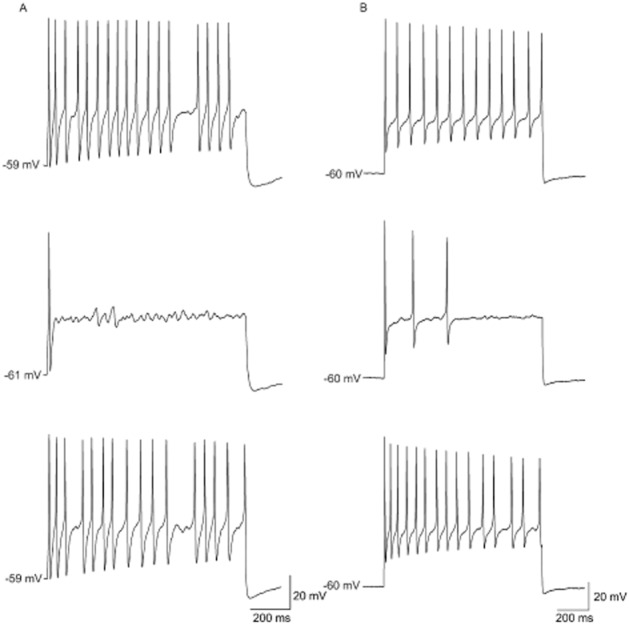
PF-01247324 attenuates neuronal firing in rat and human DRG neurons. The effect of 1 μM PF-01247324 on the excitability of a representative DRG neuron was measured using a 1 s step current injection from a holding potential of −60 mV. IC50 for PF-01247324 at TTX-R in rodent DRG neurons is 0.170 μM. (A) Representative recordings showing action potentials in a rat DRG neuron were inhibited by 1 μM PF-01247324 which reversed upon wash. Top trace is control, middle trace is with compound and bottom trace is wash (n = 12). (B) Representative recordings showing action potentials in a human DRG neuron were also inhibited by 1 μM PF-01247324 which reversed upon wash. (Top trace is control, middle trace is with compound and bottom trace is wash, n = 7). Current threshold for multiple action potential generation was first measured in order to consistently and routinely evoke action potential trains in DRG neurons with a range of excitability properties. This was performed by injecting a series of 600 ms depolarizing currents in either 50 pA (rat) or 200 pA (human) increments. Stimulated firing was examined using a series of 1 s long current steps from between 25 and 500 pA (rat) or 500 and 3000 pA (human) at 0.1 Hz. Action potential trains were reproducibly obtained by injecting 1.5–2.0× current threshold for evoking multiple action potentials.
Figure 5.
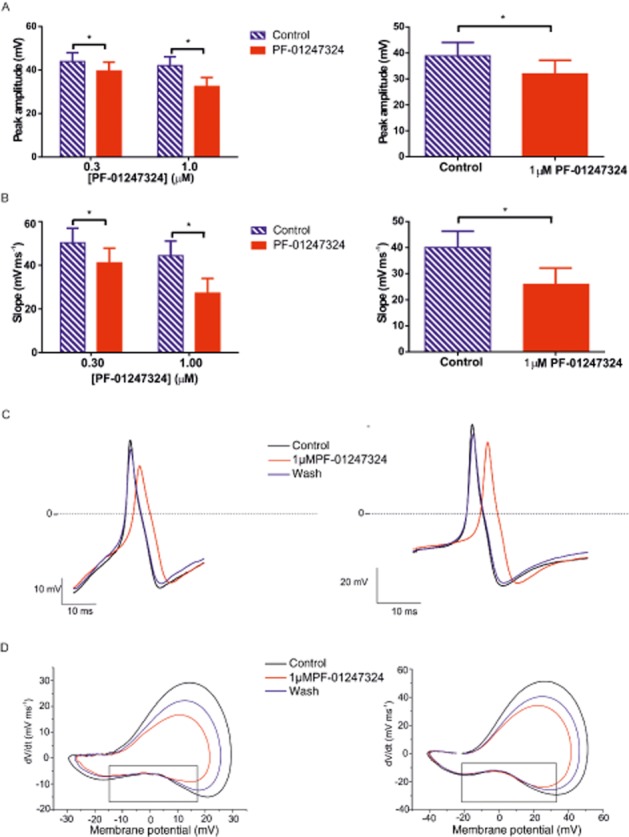
PF-01247324 altered the action potential waveform in rat and human DRG. The peak amplitude (mV) and slope (depolarising mV·ms−1) were measured for the second action potential in each sweep for the complete recording. The last three (rat) or five (human) values for peak and slope were averaged during control and application PF-01247324. (A) Bar charts (mean ± SEM) showing the change in the peak amplitude following application of either 0.3 and 1 μM PF-01247324 to action potentials recorded from rat DRG (left) and human DRG (right). (B) Bar charts (mean ± SEM ; n = 10 for rat DRG, n = 7 for human DRG) showing the change in the slope following application of either 0.3 and 1.0 μM PF-01247324 to action potentials recorded from rat DRG (left) and human DRG (right). * P < 0.05, ** P < 0.001; significantly different as indicated. (C) Single action potential waveform (second action potential in the train) showing the effect of drug treatment on the peak and slope. Action potentials from rat DRG neuron shown on the left and from human DRG neuron on the right. (D) Phase plots (dV·dt−1) from a representative recording from rat DRG neuron (bottom left) and human DRG neuron (bottom right) showing the effect of 1 μM PF-01247324 on the current potential. Boxed regions indicate the onset and offset of the shoulder of the action potential.
Table 3.
Change in action potential waveform parameters following application of PF-01247324
| [PF-01247324] (μM) | Species | n | Percentage decrease in peak amplitude (SEM) | Percentage decrease in slope (SEM) |
|---|---|---|---|---|
| 0.3 | Rat | 12 | 11.68 (2.91) | 23.98 (4.97) |
| 1.0 | Rat | 12 | 25.65 (4.70) | 47.05 (5.82) |
| 1.0 | Human | 7 | 18.55 (3.14) | 33.1 (5.61) |
Application of PF-01247324 effectively altered both the peak amplitude and depolarising slope of the action potential in both rat and human DRG. The peak amplitude and 20–80% slope of the rising phase were measured for the second action potential of each sweep. The last three (rat) or five (human) values in each condition were averaged in order to obtain a measure for each parameter in control and in the presence of PF-01247324. The percentage change in each parameter following application of either 0.3 or 1 μM PF-01247324 was calculated for each cell and is summarised with SEM.
Previous data have shown that acute application of PGE2 to rodent DRG neurons increases excitability (England et al., 1996). Therefore, given that the utility of a selective Nav1.8 channel blocker would be to attenuate pain during or following inflammation, we acutely applied 1 μM PGE2 to DRG neurons prior to evaluating the change in excitability following application of PF-01247324. The data in Figure 6A show that there was a significant reduction in the percentage inhibition of action potential number following application of 0.3 and 1.0 μM PF-01247324 in the presence of 1 μM PGE2. A representative time course shows clearly the enhanced excitability following acute application of 1 μM PGE2, followed by inhibition with 0.3 and 1.0 μM PF-01247324 (Figure 6B). Figure 6C shows examples of the raw traces corresponding to the numbered location on the time course shown in part B of the same figure.
Figure 6.
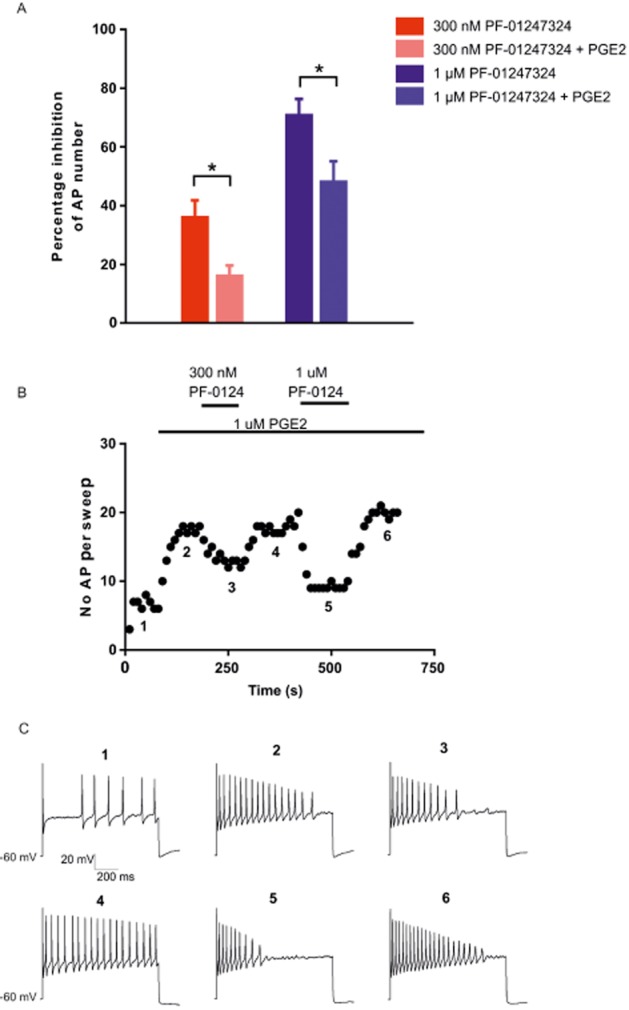
Increased concentrations of PF-01247324 are required to reduce excitability following exposure to 1 μM PGE2. 1 μM PGE2 was applied acutely to DRG neurons for ∼3 min prior to application of PGE2. (A) 1 μM PGE2 treatment caused a significant change in the percentage inhibition of action potential number following application of 0.3 and 1.0 μM PF-01247324. *P < 0.05, significantly different as indicated. n = 7–9 for 0.3 μM, n = 7–11 for 1 μM PF-01247324. (B) Representative time course from a DRG neuron. (C) Examples of the raw traces corresponding to the numbered location on the time course shown in (B).
In order to determine the oral bioavailability of PF-01247324 in vivo, rats (n = 3) were given doses of 2 mg·kg−1 (i.v.) and 5 mg·kg−1 (p.o.) and plasma measured at time intervals over 24 h. The results (Table 4) showed that systemic blood clearance (CL) of PF-01247324 was low in relation to hepatic blood flow (CL = 10 mL·min−1·kg−1 vs. hepatic blood flow 70 mL·min−1·kg−1). Comparison of dose-normalized exposure in animal dosed via i.v. and p.o. routes showed that oral bioavailability was 88%. Taking into account hepatic extraction (assumed equal to systemic CL), these results indicated that oral absorption of PF-01247324 was complete.
Table 4.
Pharmacokinetic parameters following i.v. and p.o. administration of PF-01247324 to male SD rats
| Parameter | Unit | Mean | SD | CV (%) |
|---|---|---|---|---|
| I.v. dose | ||||
| Dose | mg·kg−1 | 2 | – | – |
| AUC0−t | ng·h·mL−1 | 2895 | 1021 | 35 |
| AUC0−∞ | ng·h·mL−1 | 3066 | 911 | 33 |
| CL | ml·min−1·kg−1 | 11.7 | 3.8 | 33 |
| Vss | L kg−1 | 3.0 | 0.23 | 8 |
| P.o. dose | ||||
| Dose | mg·kg−1 | 5 | – | – |
| Cmax | ng·mL−1 | 474 | 39 | 8 |
| nM (unbound) | 50 | 4 | 8 | |
| AUC0-t | ng·h·mL−1 | 6716 | 1284 | 19 |
| AUC0-∞ | ng·h·mL−1 | 6955 | 1489 | 21 |
| F | % | 88 | 17 | 19 |
| Fa | fraction | 1.0 | 0.19 | 19 |
Male Sprague-Dawley rats (n = 3) were given either a bolus i.v. dose of 2 mg·kg−1 or a p.o. dose of 5 mg·kg−1 in 5% Tween 80 in glucose solution. Blood samples were taken from a cannula implanted in the superior vena cava at 5 min, 0.5, 1, 2, 4, 6 and 24 h post-dosing for i.v. or 0.25, 0.5, 1, 3, 6 and 24 h post-dose for p.o. Oral bioavailability (F) was calculated on the basis of comparison of dose-normalized AUC0-∞. The fraction of oral dose absorbed (Fa) was calculated from bioavailability assuming hepatic extraction was equal to systemic (i.v.) clearance (CL). Other abbreviations used in the table are: AUC0−t, area under the plasma concentration versus time curve from time 0 to last measured time-point; AUC0−∞, area under the plasma concentration versus time curve from time 0 to infinity; Vss, volume of distribution at steady state; Cmax, maximum measured concentration.
PF-01247324 was assessed in neuropathic, inflammatory and persistent pain models following oral administration. Data from the rat formalin assay are presented in Figure 7A and B. PF-01247324 reduced phase 2 flinching by 37% at 100 mg·kg−1, where, at the end of the experiment, the mean unbound plasma concentration was 0.89 μM (∼2× rat TTX-R IC50). There was a significant effect of 30 mg·kg−1 of PF-01247324 in the rat model carrageenan-induced thermal hyperalgesia and in CFA-induced mechanical hyperalgesia at exposures of 0.218 and 0.126 μM respectively (Figure 7C and D respectively). Anti-allodynic effects of PF-01247324 were also observed in the rat model of SNL at 1 and 2 h post treatment at doses of 10 mg·kg−1 (1 and 2 h) and 30 mg·kg−1 (2 h) (latter equivalent to 0.246 μM free plasma concentration); gabapentin (100 mg·kg−1) also effectively reduced allodynia (Figure 8). Sampling of brain tissue at the end of the formalin challenge experiment in rat indicated that PF-01247324 is able to penetrate into the CNS, whereby the mean unbound brain : plasma concentration ratio was 1.5. We therefore evaluated the effect of PF-01247324 in an FOB, which is a series of neurobehavioural tests (Moser, 1992). There were no differences between treatment groups following oral dosing in general overt behaviour, involuntary motor movements, gait, ease of removal from the cage and handling, eye observations, salivation, piloerection, stimulus reactivity, grip strength, righting reflex, diarrhoeal, respiration or core temperature up to a dose of 600 mg·kg−1 (1.01 μM unbound plasma concentration; data not shown). In a locomotor assay no adverse effects were seen up to and including 30 mg·kg−1 (0.258 μM unbound plasma concentration) (Supporting Information Tables S3–S5 in Supporting Information). We also evaluated the ability of PF-01247324 to alter motor coordination and balance as assessed by the rotarod test (Supporting Information Fig. S1). Doses of up to 1000 mg·kg−1 (1.22 μM unbound plasma concentration) of PF-01247324 did not impair the ability of mice to remain on the rotarod. No other phenotypes were observed in rats after drug intake. The Nav1.8 channels have recently been implicated in cardiac function (Chambers et al., 2010; van den Boogaard et al., 2014). However, an assessment of cardiovascular effects following systemic administration of PF-01247324 was not carried out in rats.
Figure 7.
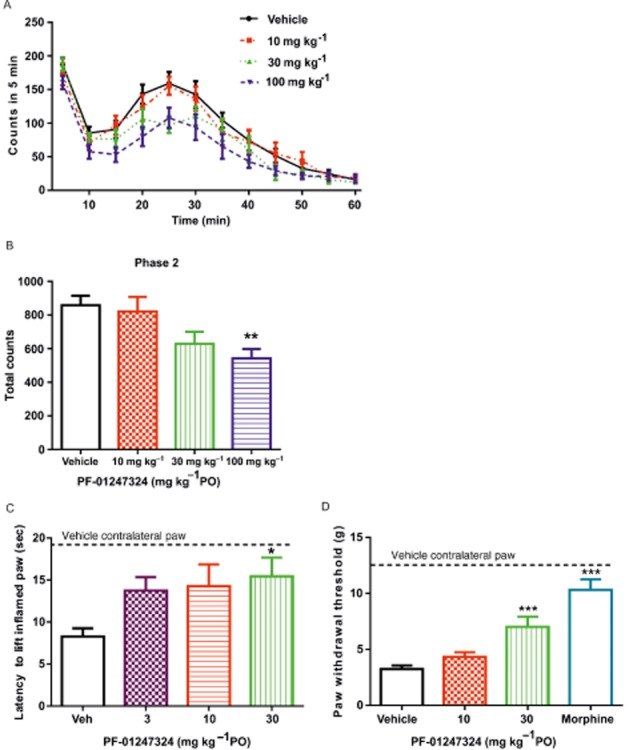
Antinociceptive effects of PF-1247324 in models of persistent and inflammatory pain. (A and B) PF-01247324 significantly reduced phase 2 flinching in a model of persistent pain, the rat formalin model, by 37% at 100 mg·kg−1, but not at the lower doses of 10 and 30 mg·kg−1. Data show mean ± SEM; n = 12. **P < 0.01, significantly different from vehicle-treated animals). PF-01247324 was administered orally 4 h prior to formalin injection. (C) PF-01247324 demonstrated a significant effect in the rat model of carrageenan-induced thermal hyperalgesia following oral administration at a dose of 30 mg·kg−1. Data show mean ± SEM; n = 8. *P < 0.05, significantly different from vehicle-treated animals). Also shown is the withdrawal latency for the non-inflamed paw from vehicle-treated animals (19.3 s). (D) PF-01247324 demonstrated a significant effect in the rat model of CFA-induced mechanical hyperalgesia following oral administration at a dose of 30 mg·kg−1 although not at the lower dose of 10 mg·kg−1. Data are mean ± SEM, n = 12. ***P < 0.0001, significantly different from vehicle-treated animals. Also shown is the withdrawal threshold for the non-inflamed paw from vehicle-treated animals (12.33 g).
Figure 8.
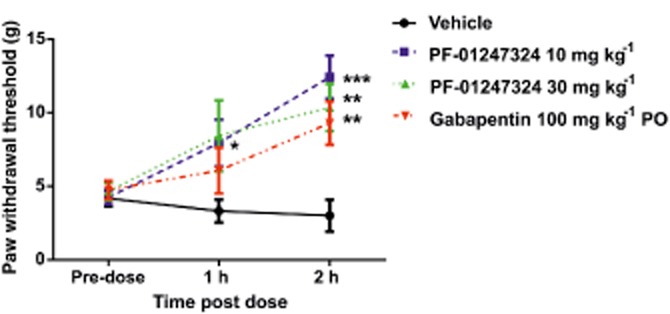
Antiallodynic effects of PF-01247324 in neuropathic pain. PF-01247324 reduced tactile allodynia in a spinal nerve ligation model (L5) of nerve injury. Doses of 10 and 30 mg·kg−1 PF-01247324 demonstrated significant antiallodynic effects 1 and 2 h after oral dosing. Data show mean ± SEM; n = 6–8. *P < 0.05, **P < 0.01, ***P < 0.0001, significantly different from pre-dosed values. Gabapentin was also tested for comparison as a positive control.
Discussion
PF-01247324 is a novel, highly selective blocker of Nav1.8 channels and binds to the inactivated state of the channel. Unlike previously described Nav1.8 channel blockers, this compound demonstrated frequency dependence. Although pharmacological selectivity is shown over the other human Nav subtypes, it is possible that off-target frequency dependence at other sodium channel subtypes could reduce the selectivity window. The majority of low MW Na+ channel blockers interact at the LA binding site, which because of a high level of sequence homology across Nav subtypes, seems an unlikely site for interaction of selective blockers (England and de Groot, 2009). However, a previous study has demonstrated overlap between the binding sites recognized by tetracaine and A-803467 using site-directed mutagenesis, suggesting that isoform selectivity can be achieved at sites close to that for LA binding (Browne et al., 2009). Using site-directed mutagenesis, we have demonstrated that PF-01247324 also interacts with key aromatic residues within the LA binding site. However, further studies are required to define residues underlying subtype selectivity.
Nav1.8 channels have been shown to contribute to the upstroke of the action potential and support high repetitive firing because of a depolarized voltage-dependence of activation/inactivation and rapid recovery from inactivation (Renganathan et al., 2001; Cummins et al., 2007; Rush and Cummins, 2007). Application of PF-01247324 was able to attenuate repetitive firing in both rodent and human DRG neurons, the latter case being the first demonstration of a selective inhibitor of Nav1.8 channels reducing excitability in human sensory neurons. Using the action potential as a command voltage and by implementing an ionic substitution method Blair and Bean (2002) have suggested a prominent role for Nav1.8 channels in the depolarizing phase and also in the shoulder of the nociceptive sensory neuron action potential. Application of PF-01247324 reversibly attenuated both the peak amplitude and the slope of the action potentials evoked in rat and human DRG (findings which to our knowledge represent the first direct pharmacological evidence confirming a specific role for Nav1.8 channels in generating the action potential upstroke in the cell body of a sensory neuron). In contrast, the shoulder of the action potential was unchanged following application of PF-01247324 in both rat and human DRG neurons suggesting that, contrary to Blair and Bean's suggestion, Nav1.8 channels do not have such a prominent role in the shoulder of the action potential. Following sensitization of DRG neurons with the inflammatory mediator PGE2, PF-01247324 was still able to attenuate excitability in vitro, although higher concentrations were needed to achieve the same reduction of firing observed in the absence of PGE2 (likely because of increased availability of Nav1.8 channels following phosphorylation).
Nav1.8 channels are highly (but not exclusively) expressed in nociceptors (Amaya et al., 2000; Shields et al., 2012), and their expression and function is modulated by agents that cause pain (England et al., 1996; Fjell et al., 1999; Kerr et al., 2001). Furthermore, it is up-regulated following inflammation and chronic nerve injury in human tissue (Coward et al., 2000; Coggeshall et al., 2004; Renton et al., 2005; Black et al., 2008), and gain of function mutations of Nav1.8 channels in humans contribute to painful peripheral neuropathy (Faber et al., 2012; Huang et al., 2013; Han et al., 2014). Genetic knockout and antisense knockdown studies have suggested a role for Nav1.8 channels in inflammatory pain (Khasar et al., 1998; Akopian et al., 1999; Villarreal et al., 2005), and prior to the disclosure of A-803467, data supporting a role for Nav1.8 channels in neuropathic pain was equivocal (Kerr et al., 2001; Roza et al., 2003; Nassar et al., 2005; Thakor et al., 2009). Knockdown studies have supported a role for Nav1.8 channels in neuropathic pain (Khasar et al., 1998; Dong et al., 2007; Ruangsri et al., 2011) although non-specific effects on other proteins should be considered (see Jackson and Linsley, 2010), while neuropathic pain develops normally (with the exception of cold allodynia in chronic constriction injury model) in Nav1.8 channel knockout mice (Kerr et al., 2001; Nassar et al., 2005). In addition, destruction of Nav1.8 channel-expressing neurons (using floxed diphtheria toxin) had no effect on neuropathic pain behavioural measures (Abrahamsen et al., 2008). Moreover, a comparative study of Nav1.3, Nav1.7, Nav1.8 and Nav1.9 channel knockout mice using different neuropathic pain models has been interpreted as demonstrating a specific role for Nav1.7 channels in mediating neuropathic pain (including sympathetic sprouting) (Minett et al., 2014). A-803467 attenuated pain sensitivity in models of both nerve injury and inflammation induced pain (Jarvis et al., 2007), which provided the first pharmacological evidence supporting a role for Nav1.8 channels in inflammatory and neuropathic pain. PF-01247324 is between 50- and 100-fold selective over other human sodium channel subtypes and is orally bioavailable achieving unbound plasma exposures equivalent to between 1× and 3× IC50. In agreement with studies using A-803467, PF-01247324 also showed efficacy in a rodent models of neuropathic pain, providing additional pharmacological evidence using a different chemotype, that the Nav1.8 channel has a role in neuropathic pain.
A role for Nav1.8 channels in nociceptive or inflammatory pain is less equivocal and in agreement with data from studies using A-803467, PF-01247324 showed analgesic activity in the rat model of carrageenan-induced thermal hyperalgesia and CFA-induced mechanical hyperalgesia at 30 mg·kg−1 (unbound plasma exposure of 0.218 and 0.126 μM, respectively), which was also devoid of any effect in a rat locomotor assay. Unlike A-803467 and studies showing a lack of change in the pain behaviour in the formalin model following genetic ablation of Nav1.8 channels (Nassar et al., 2005), PF-01247324 was effective at reducing nociception in the persistent phase of the formalin test at 100 mg·kg−1. However, although there is some suggestion of CNS effects at this dose (equivalent to 0.89 μM free plasma exposure) in a rat locomotor assay, there were no differences between treatment groups for other rat neurobehavioural endpoints measured at the equivalent dose. In addition, doses up to 1000 mg·kg−1 (equivalent to 1.22 μM free plasma exposure) were shown to have no effect on motor coordination in the mouse rotarod assay.
In conclusion, we here describe the actions of PF-01247324, which represents a second-generation highly selective Nav1.8 channel blocker that has high oral bioavailability in rats (F = 91%). We have shown that, unlike previously described selective Nav1.8 channel blockers, the inhibition of both hNav1.8 and native TTX-R channels in rat DRG neurons by PF-01247324 is both state- and frequency-dependent. In addition, the compound interacts with key residues within the local anaesthetic binding site. Application of PF-01247324 to rodent and human DRG neurons attenuated excitability in vitro, this being the first reported data showing the effect of a selective Nav1.8 channel blocker on excitability in human DRG. PF-01247324 produced antinociceptive effects in both inflammatory and neuropathic pain models in rat, providing pharmacological evidence from a novel chemotype that suggests Nav1.8 channels have a role in both inflammatory and neuropathic pain. The oral bioavailability and subtype selectivity of PF-01247324 provide an ideal tool compound to further investigate the role of Nav1.8 channels in pain processing.
Acknowledgments
The authors would like to acknowledge Phil Stanley who provided the statistical analysis for the in vitro current clamp data and Dr Gareth Waldron who helped with the interpretation of the CNS safety studies. We thank Dr Marco Sinisi for helping with organ donor DRG retrieval, and NC3R for funding UK human DRG collection.
Glossary
Abbreviations
- CFA
complete Freund's adjuvant
- DRG
dorsal root ganglion
- LA
local anaesthetic
- SNL
spinal nerve ligation
- TTR-R
tetrodotoxin-resistant
- TTX
tetrodotoxin
- TTX-S
tetrodotoxin-sensitive
- VGSC
voltage-gated sodium channel
Author contributions
C. E. P., A. R. B., J. W. T., M. D. F., J. H.M., A. J. A., A. J. C. L., B. M. A., D. M. P., A. C. G., R. L. P., G. S., A. J. K., M. K. and S. B. performed the research. C. E. P., M. K., S. B., A. W. B., S. E., M. L. C., R. R., P. B., R. P. B. and E. B. S. designed the research study. U. A. and P. A. contributed essential reagents. C. E. P., A. R. B., J. W. T., M. D. F., J. H. M., A. J. A., A. J. C. L., B. M. A., D. M. P., A. C. G., R. L. P., G. S., A. J. K., M. K. and S. B. analysed the data. C. E. P. and E. B. S. wrote the paper.
Conflicts of interest
All authors with the exception of G. S., M. K., S. E., A. B., U. A. and P. A. are employees of Pfizer, Ltd. S. E. and A. B. are current employees of AbbVie. M. K. is currently an employee of Mission Therapeutics. U. A. and Professor P. A. are employed by the Faculty of Medicine at Imperial College London.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 PF-1247324 did not impair motor function. PF-01247324 was dosed 1 h prior to rotarod testing, n = 9–12 per group. The positive control diazepam was formulated in 10%EtOH/90% (40% PEG 200/60% sterile water) and dosed s.c. at 3 mg·kg−1 30 min prior to rotarod testing. Rotarod speed was set at 10 r.p.m. Each mouse was placed on the rotating rod and given three opportunities to remain for 10 min. The longest lapsed time among the three trials was recorded to the nearest second. PF-01247324 did not significantly alter motor coordination and balance as assessed by the rotarod assay at doses up to 1000 mg·kg−1 when compared with vehicle-treated animals. Data show mean (± SEM). ***P < 0.0001.
Table S1 Potency of PF-01247324 was assessed at native TTX-R currents from DRG neurons from common preclinical species. IC50 values for TTX-R currents recorded from rat, mouse, cynomolgus monkey and pig DRG neurons. Also shown for comparison is the IC50 value at human native TTX-R currents. n = 2–6 observations per concentration. 95% confidence intervals are stated in parentheses.
Table S2 Selectivity for PF-01247324 across rat TTX-S isoforms. The IC50 for PF-01247324 at recombinantly expressed rat TTX-S isoforms was determined using manual patch electrophysiology. Cells were depolarized from a holding potential of −120 mV to a membrane potential that inactivated half of the available channels for 8 s followed by a 20 ms recovery at −120 mV and a 20 ms test pulse to 0 mV. n ≥ 4 per recording. Also shown for comparison is the IC50 for PF-01247324 at TTX-R in rat DRG.
Table S3 Locomotor activity: inner (central) zone as assessed as part of a FOB. Oral doses of 30, 100, 300 and 600 mg·kg−1 were assessed in a FOB between 3 and 6 h post dose. The FOB is a series of non-invasive observational measures and assessments of behavioural signs, which included recording of the animals' movements in an open-field arena over a 5 min period (using a Noldus Ethovision Software), in order to assess autonomic nervous system, neuromuscular and sensorimotor function (Moser, 1992). Clinical signs (home cage observations) were also evaluated. Numerical data were summarized as group means, and treatment groups were compared with vehicle-treated animals using Student's t-test for unpaired data (Microsoft Excel). *P = <0.05, **P = <0.01, ***P = <0.001 n = 6 animals per group.
Table S4 Locomotor activity: outer zone as assessed as part of a FOB. Oral doses of 30, 100, 300 and 600 mg·kg−1 were assessed in an FOB between 3 and 6 h post dose. The FOB is a series of non-invasive observational measures and assessments of behavioural signs, which included recording of the animals' movements in an open-field arena over a 5 min period (using a Noldus Ethovision Software), autonomic nervous system, neuromuscular and sensorimotor function (Moser, 1992). Clinical signs (home cage observations) were also evaluated. Numerical data were summarized as group means, and treatment groups were compared with vehicle-treated animals using Student's t-test for unpaired data (Microsoft Excel). *P = <0.05, **P = <0.01, ***P = <0.001 n = 6 animals per group.
Table S5 Locomotor activity: total distance moved both zones as assessed as part of a FOB. Oral doses of 30, 100, 300 and 600 mg·kg−1 were assessed in a FOB between 3 and 6 h post dose. The FOB is a series of non-invasive observational measures and assessments of behavioural signs, which included recording of the animals' movements in an open-field arena over a 5 min period (using a Noldus Ethovision Software), autonomic nervous system, neuromuscular and sensorimotor function (Moser, 1992). Clinical signs (home cage observations) were also evaluated. Numerical data was summarized as group means, and treatment groups were compared with vehicle-treated animals using Student's t-test for unpaired data (Microsoft Excel). *P = <0.05, **P = <0.01, ***P = <0.001 n = 6 animals per group. The change at 600 mg·kg−1 dose was not significant because of greater variation in the group.
References
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Decosterd I, Samad T, Plumpton C, Tate S, et al. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- Anand U, Otto WR, Casula MA, Day NC, Davis JB, Bountra C, et al. The effect of neurotrophic factors on morphology, TRPV1 expression and capsaicin responses of cultured human DRG sensory neurons. Neurosci Lett. 2006;399:51–56. doi: 10.1016/j.neulet.2006.01.046. [DOI] [PubMed] [Google Scholar]
- Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol. 2008;64:644–653. doi: 10.1002/ana.21527. [DOI] [PubMed] [Google Scholar]
- Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22:10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boogaard M, Smemo S, Burnicka-Turek O, Arnolds TE, van de Werken HJ, Klaus P, et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest. 2014;124:1844–1852. doi: 10.1172/JCI73140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne LE, Blaney FE, Yusaf SP, Clare JJ, Wray D. Structural determinants of drugs acting on the Nav1.8 channel. J Biol Chem. 2009;284:10523–10536. doi: 10.1074/jbc.M807569200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle N, Printzenhoff D, Zellmer S, Antonio B, Wickenden A, Silvia C. Sodium channel inhibitor drug discovery using automated high throughput electrophysiology platforms. Comb Chem High Throughput Screen. 2009;12:107–122. doi: 10.2174/138620709787047993. [DOI] [PubMed] [Google Scholar]
- Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152. doi: 10.1038/ng.516. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Tate S, Carlton SM. Differential expression of tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 in normal and inflamed rats. Neurosci Lett. 2004;355:45–48. doi: 10.1016/j.neulet.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Coward K, Plumpton C, Facer P, Birch R, Carlstedt T, Tate S, et al. Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain. 2000;85:41–50. doi: 10.1016/s0304-3959(99)00251-1. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: implications for mechanisms of pain. Pain. 2007;131:243–257. doi: 10.1016/j.pain.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Black JA, Waxman SG. Voltage gated sodium channels: therapeutic targets for pain. Pain Med. 2009;10:1260–1269. doi: 10.1111/j.1526-4637.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- Djouhri L, Fang X, Okuse K, Wood JN, Berry CM, Lawson SN. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol. 2003;550:739–752. doi: 10.1113/jphysiol.2003.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XW, Goregoaker S, Engler H, Zhou X, Mark L, Crona J, et al. Small interfering RNA-mediated selective knockdown of NaV1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience. 2007;146:812–821. doi: 10.1016/j.neuroscience.2007.01.054. [DOI] [PubMed] [Google Scholar]
- England S, de Groot MJ. Subtype selective targeting of voltage-gated sodium channels. Br J Pharmacol. 2009;158:1413–1425. doi: 10.1111/j.1476-5381.2009.00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber CG, Lauria G, Merkies IS, Cheng X, Han C, Ahn HS, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A. 2012;109:19444–19449. doi: 10.1073/pnas.1216080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell J, Cummins TR, Dib-Hajj SD, Fried K, Black JA, Waxman SG. Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Brain Res Mol Brain Res. 1999;67:267–282. doi: 10.1016/s0169-328x(99)00070-4. [DOI] [PubMed] [Google Scholar]
- Gerner P, Strichartz GR. Sensory and motor complications of local anaesthetics. Muscle Nerve. 2008;37:421–425. doi: 10.1002/mus.20967. [DOI] [PubMed] [Google Scholar]
- Han C, Vasylyev D, Macala LJ, Gerrits MM, Hoeijmakers JG, Bekelaar KJ, et al. The G1662S NaV1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J Neurol Neurosurg Psychiatry. 2014;85:499–505. doi: 10.1136/jnnp-2013-306095. [DOI] [PubMed] [Google Scholar]
- Huang J, Yang Y, Zhao P, Gerrits MM, Hoeijmakers JG, Bekelaar K, et al. Small-fiber neuropathy NaV1.8 mutation shifts activation to hyperpolarised potentials and increases excitability of dorsal root ganglion neurons. J Neurosci. 2013;33:14087–14097. doi: 10.1523/JNEUROSCI.2710-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci USA. 2007;104:8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BJ, Souslova V, McMahon SB, Wood JN. A role for the TTX-resistant sodium channel Nav1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport. 2001;12:3077–3080. doi: 10.1097/00001756-200110080-00019. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium channel mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/s0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG Group NCRRGW. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Lane CAL, Maw GN, Rawson DJ, Thompson LR. 2006. Pyridine derivatives. PCT Int. Appl. WO 2006011050.
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett MS, Falk S, Santana-Varela S, Bogdanov YD, Nassar MA, Heegaard AM, et al. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 2014;6:301–312. doi: 10.1016/j.celrep.2013.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser VC. Applications of a neurobehavioural screening battery. J Am Coll Toxicol. 1992;10:661–669. [Google Scholar]
- Nassar MA, Levato A, Stirling LC, Wood JN. Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Mol Pain. 2005;22:1–24. doi: 10.1186/1744-8069-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore GM. Dorsal root ganglion neurones in culture: a model for identifying novel analgesic targets. J Pharmacol Toxicol Methods. 2005;51:201–208. doi: 10.1016/j.vascn.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG. Contribution of NaV1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- Renton T, Yiangou Y, Plumpton C, Tate S, Bountra C, Anand P. Sodium channel Nav1.8 immunoreactivity in painful human dental pulp. BMC Oral Health. 2005;5:5. doi: 10.1186/1472-6831-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roza C, Laird JM, Souslova V, Wood JN, Cervero F. The tetrodotoxin-resistant sodium channel NaV1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J Physiol. 2003;550:921–926. doi: 10.1113/jphysiol.2003.046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruangsri S, Lin A, Mulpuri Y, Lee K, Spigelman I, Nishimura I. Relationship of axonal voltage-gated sodium channel 1.8 (NaV1.8) mRNA accumulation to sciatic nerve injury-induced painful neuropathy in rats. J Biol Chem. 2011;286:39836–39847. doi: 10.1074/jbc.M111.261701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Cummins TR. Painful research: identification of a small molecule inhibitor that selectively targets NaV1.8 sodium channels. Mol Interv. 2007;7:192–195. doi: 10.1124/mi.7.4.4. [DOI] [PubMed] [Google Scholar]
- Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, et al. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153:2017–2030. doi: 10.1016/j.pain.2012.04.022. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Jensen TS. Are sodium channel blockers useless in peripheral neuropathic pain? Pain. 2007;128:6–7. doi: 10.1016/j.pain.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Thakor DK, Lin A, Matsuka Y, Meyer EM, Ruangsri S, Nishimura I, et al. Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol Pain. 2009;5:14. doi: 10.1186/1744-8069-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal CF, Sachs D, Cunha FQ, Parada CA, Ferreira SH. The role of NaV1.8 sodium channel in the maintenance of chronic inflammatory hypernociception. Neurosci Lett. 2005;386:72–77. doi: 10.1016/j.neulet.2005.04.060. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Ozaki G, McCumber D, Rathbun M, Svensson C, Malkmus S, et al. An automated flinch detecting system for use in the formalin nociceptive bioassay. J Appl Physiol. 2001;90:2386–2402. doi: 10.1152/jappl.2001.90.6.2386. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Shieh CC, Chapman ML, Matulenko MA, Hakeem AH, Atkinson RN, et al. A-887826 is a structurally novel, potent and voltage-dependent NaV1.8 sodium channel blocker that attenuates neuropathic tactile allodynia in rats. Neuropharmacology. 2010;59:201–207. doi: 10.1016/j.neuropharm.2010.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 PF-1247324 did not impair motor function. PF-01247324 was dosed 1 h prior to rotarod testing, n = 9–12 per group. The positive control diazepam was formulated in 10%EtOH/90% (40% PEG 200/60% sterile water) and dosed s.c. at 3 mg·kg−1 30 min prior to rotarod testing. Rotarod speed was set at 10 r.p.m. Each mouse was placed on the rotating rod and given three opportunities to remain for 10 min. The longest lapsed time among the three trials was recorded to the nearest second. PF-01247324 did not significantly alter motor coordination and balance as assessed by the rotarod assay at doses up to 1000 mg·kg−1 when compared with vehicle-treated animals. Data show mean (± SEM). ***P < 0.0001.
Table S1 Potency of PF-01247324 was assessed at native TTX-R currents from DRG neurons from common preclinical species. IC50 values for TTX-R currents recorded from rat, mouse, cynomolgus monkey and pig DRG neurons. Also shown for comparison is the IC50 value at human native TTX-R currents. n = 2–6 observations per concentration. 95% confidence intervals are stated in parentheses.
Table S2 Selectivity for PF-01247324 across rat TTX-S isoforms. The IC50 for PF-01247324 at recombinantly expressed rat TTX-S isoforms was determined using manual patch electrophysiology. Cells were depolarized from a holding potential of −120 mV to a membrane potential that inactivated half of the available channels for 8 s followed by a 20 ms recovery at −120 mV and a 20 ms test pulse to 0 mV. n ≥ 4 per recording. Also shown for comparison is the IC50 for PF-01247324 at TTX-R in rat DRG.
Table S3 Locomotor activity: inner (central) zone as assessed as part of a FOB. Oral doses of 30, 100, 300 and 600 mg·kg−1 were assessed in a FOB between 3 and 6 h post dose. The FOB is a series of non-invasive observational measures and assessments of behavioural signs, which included recording of the animals' movements in an open-field arena over a 5 min period (using a Noldus Ethovision Software), in order to assess autonomic nervous system, neuromuscular and sensorimotor function (Moser, 1992). Clinical signs (home cage observations) were also evaluated. Numerical data were summarized as group means, and treatment groups were compared with vehicle-treated animals using Student's t-test for unpaired data (Microsoft Excel). *P = <0.05, **P = <0.01, ***P = <0.001 n = 6 animals per group.
Table S4 Locomotor activity: outer zone as assessed as part of a FOB. Oral doses of 30, 100, 300 and 600 mg·kg−1 were assessed in an FOB between 3 and 6 h post dose. The FOB is a series of non-invasive observational measures and assessments of behavioural signs, which included recording of the animals' movements in an open-field arena over a 5 min period (using a Noldus Ethovision Software), autonomic nervous system, neuromuscular and sensorimotor function (Moser, 1992). Clinical signs (home cage observations) were also evaluated. Numerical data were summarized as group means, and treatment groups were compared with vehicle-treated animals using Student's t-test for unpaired data (Microsoft Excel). *P = <0.05, **P = <0.01, ***P = <0.001 n = 6 animals per group.
Table S5 Locomotor activity: total distance moved both zones as assessed as part of a FOB. Oral doses of 30, 100, 300 and 600 mg·kg−1 were assessed in a FOB between 3 and 6 h post dose. The FOB is a series of non-invasive observational measures and assessments of behavioural signs, which included recording of the animals' movements in an open-field arena over a 5 min period (using a Noldus Ethovision Software), autonomic nervous system, neuromuscular and sensorimotor function (Moser, 1992). Clinical signs (home cage observations) were also evaluated. Numerical data was summarized as group means, and treatment groups were compared with vehicle-treated animals using Student's t-test for unpaired data (Microsoft Excel). *P = <0.05, **P = <0.01, ***P = <0.001 n = 6 animals per group. The change at 600 mg·kg−1 dose was not significant because of greater variation in the group.