

Abstract
The glutamate delta-1 (GluD1) receptor is highly expressed in the forebrain. We have previously shown that loss of GluD1 leads to social and cognitive deficits in mice, however, its role in synaptic development and neurotransmission remains poorly understood. Here we report that GluD1 is enriched in the medial prefrontal cortex (mPFC) and GluD1 knockout mice exhibit a higher dendritic spine number, greater excitatory neurotransmission as well as higher number of synapses in mPFC. In addition abnormalities in the LIMK1-cofilin signaling, which regulates spine dynamics, and a lower ratio of GluN2A/GluN2B expression was observed in the mPFC in GluD1 knockout mice. Analysis of the GluD1 knockout CA1 hippocampus similarly indicated the presence of higher spine number and synapses and altered LIMK1-cofilin signaling. We found that systemic administration of an N-methyl-d-aspartate (NMDA) receptor partial agonist d-cycloserine (DCS) at a high-dose, but not at a low-dose, and a GluN2B-selective inhibitor Ro-25-6981 partially normalized the abnormalities in LIMK1-cofilin signaling and reduced excess spine number in mPFC. The molecular effects of high-dose DCS and GluN2B inhibitor correlated with their ability to reduce the higher stereotyped behavior and depression-like behavior in GluD1 knockout mice. Together these findings demonstrate a critical requirement for GluD1 in normal spine development in the cortex and hippocampus. Moreover, these results identify inhibition of GluN2Bcontaining receptors as a mechanism for reducing excess dendritic spines and stereotyped behavior which may have therapeutic value in certain neurodevelopmental disorders.
Keywords: Glutamate, Dendritic spine, GRID1, GluD1, GluN2B
1. Introduction
The ionotropic glutamate receptor family of proteins is comprised of four subtypes NMDA, AMPA, kainate and delta receptors. Glutamate delta-1 (GluD1) and glutamate delta-2 (GluD2) form the delta family of iGluRs and are distinct from other iGluRs in that they do not exhibit typical agonist-induced ion channel currents (Traynelis et al., 2010; Naur et al., 2007; Yadav et al., 2011; Ady et al., 2014). Instead recent studies have demonstrated a crucial role of the delta receptors in synapse formation by interacting with presynaptic proteins such as Neurexin1 (Uemura et al., 2010; Matsuda et al., 2010; Ryu et al., 2012; Yasumura et al., 2012). Although the synaptic function of GluD2 expressed in Purkinje cells has been extensively studied, the function of GluD1 in native system remains poorly understood. We have previously shown that deletion of GluD1 leads to social deficits and emergence of depression-like and aggressive behavior (Yadav et al., 2012). Furthermore, loss of GluD1 leads to abnormal cognitive functions including, enhanced working memory and deficits in both contextual and cued fear conditioning as well as reversal learning of spatial memory (Yadav et al., 2013). We have also found that social deficits in the GluD1 knockout (KO) mice were amenable to modulation of NMDA receptors by agonist d-cycloserine (Yadav et al., 2012), however, the precise molecular mechanism of this effect is not clear. In agreement with our behavioral studies which suggest that loss of GluD1 leads to emergence of behavioral phenotypes relevant to neuropsychiatric disorders, single nucleotide polymorphism and copy-number variation studies have demonstrated an association of GRID1 gene, that codes for the GluD1 receptor, with autism (Griswold et al., 2012; Glessner et al., 2009; Nord et al., 2011; Smith et al., 2009), as well as schizophrenia and bipolar disorder (Fallin et al., 2005; Edwards et al., 2012; Guo et al., 2007; Greenwood et al., 2011).
GluD1 is highly expressed in the forebrain including the cortex and hippocampus (Lomeli et al., 1993; Gao et al., 2007; Yadav et al., 2012) and recent studies also indicate expression in cerebellar interneurons (Konno et al., 2014). In the cortex and hippocampus high level of GluD1 mRNA and protein appears in pyramidal neurons (Gao et al., 1993; Lein et al., 2007; Konno et al., 2014; Hepp et al., 2014). GluD1 appears to be particularly localized to spinelike structures in the hippocampus that represent excitatory synapses (Hepp et al., 2014). In this study we have found that pyramidal neurons in adult GluD1 KO medial prefrontal cortex (mPFC) and hippocampus have higher dendritic spine number that may occur due to impaired spine pruning or excessive spine generation. We also observed abnormalities in LIMK1-cofilin signaling which is involved in regulating spine dynamics and a lower NMDA receptor GluN2A/GluN2B subunit expression ratio suggesting a potential impairment in the GluN2B to GluN2A developmental switch (Williams et al., 1993). Moreover, inhibition of GluN2B-containing receptors was found to reverse signaling abnormalities and spine density as well as stereotyped behavior and depression-like behavior in GluD1 KO mice. These results for the first time demonstrate a critical role of GluD1 in maintaining spine dynamics and ability of GluN2B inhibition in reversing the higher spine density which may have implications for disorders such as autism where higher number of spines is a commonly observed neuropathology (Penzes et al., 2011).
2. Materials and methods
2.1. Animal housing
GluD1 KO mice were generously provided by Dr. Jian Zuo, St. Jude’s Children’s Hospital (Gao et al., 2007). GluD1 KO mice were generated and genotyped as previously described (Yadav et al., 2012). Mice were group housed in the animal house facility at a constant temperature (22 ± 1 °C) and a 12-h light–dark cycle with free access to food and water. Behavioral testing was performed between 9:00 a.m and 4:00 p.m in animals between 6 and 8 weeks of age. Female mice were not used for the experiments to avoid the confounding effects of the estrus cycle on behavioral and neurochemical measures. All experimental mice were placed in the experimental room at least 60 min before beginning of any experimental protocol. All behavioral procedures were video-recorded and scored by a scorer blind to the genotype of the animal via a random coding system of the video files. In this study strict measures were taken to minimize pain and suffering to animals in accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the National Institutes of Health. All experimental protocols were approved by the Creighton University Institutional Animal Care and Use Committee Policies and Procedures.
2.2. Materials
Tetrodotoxin was purchased from Alomone Labs (Jerusalem, Israel), bicuculline methiodide and picrotoxin were purchased from Tocris (Minneapolis, MN, USA), d-cycloserine (DCS), escitalopram and desipramine were purchased from Sigma-–Aldrich (St. Louis, MO, USA). Ro-25-6981 was purchased from Abcam chemicals (Cambridge, MA, USA).
2.3. Drug treatment
DCS was dissolved in 0.9% saline and was made fresh 1 h before the experiment. Stock solution of Ro-25-6981 was made in distilled water and stored at −20 °C. Ro-25-6981 was diluted with 0.9% saline to the working concentration 1 h before the experiment. Desipramine and escitalopram were diluted in 0.9% saline fresh before each experiment.
2.4. Golgi staining
Golgi staining was performed with FD Rapid GolgiStain Kit as per the protocol of the supplier (FD NeuroTechnologies, Inc., Columbia, MD, USA). Brains were collected from wildtype and GluD1 KO mice at postnatal days 14, 30, 60 and 180 and kept in mixture of solution A and B (mix in 1:1 ratio) for 14 days followed by solution C for 72 h and frozen at −70 °C in methyl butane and dry ice. 100 μm thick sections were cut using cryostat and collected on gelatin coated glass slides. Sections were stained, dehydrated and treated with xylene subsequently. Images were taken using Axioscope Z 1051-078 microscope (Zeiss, Chester, VA, USA) with Q-Capture PRO7 software. 100× oil objective was used to take the images of spines, quantification was performed on dendrites beginning at 75 μm distal to the soma. Areas of interest were cropped and spine numbers were counted manually by an observer blind to the genotype. Dendritic spines of the IInd and IIIrd branch point of first order apical dendrites of CA1 pyramidal neurons were used for analysis. The dendritic spines of the Ist branch point of first order dendrites were used for the analysis in infralimbic cortex and layer II–III of prelimbic cortex. A total of 3–4 mice were used for each group and 10–15 neurons from each animal were analyzed and averaged.
2.5. Diolistic labeling and Imaris analysis
Diolistic labeling was performed as previously described (Shen et al., 2008) with minor modifications. The animals were anesthetized and perfused with 0.1 M phosphate buffer (PB) followed by 1.8% paraformaldehyde (PFA). The brain was removed and postfixed for 1 h, sectioned coronally (150 μm thickness) at room temperature, and the sections were stored in PB until diolistic labeling. Tungsten particles (1.3 μm diameter) (Bio-Rad Laboratories, Hercules, California, USA) coated with the lipophilic carbocyanine dye DiI (Molecular Probes, Life Technologies, Grand Island, NY, USA) were delivered diolistically into the section at 100 psi using a Helios Gene Gun system (Bio-Rad Laboratories, Hercules, California, USA) with modified chamber (O’Brien and Lummis, 2006). A polycarbonate filter with 3.0 μm pore size was capped on top of the gun barrel to filter large clusters of particles.
DiI was allowed to diffuse along neuron dendrites and axons in PB containing 0.01% (w/v) thimerosal (Sigma–Aldrich, St. Louis, MO, USA) for 24–48 h at 4 °C, and then labeled sections were fixed again in 4% PFA for 1 h. After brief washing in PB, sections were mounted onto glass slides in Gelvatol mounting medium. Imaging of labeled sections were performed using LSM 510 confocal microscope (Zeiss, Chester, VA, USA) at Integrated Biomedical Imaging Facility of Creighton University. DiI was excited using the Helium/Neon at 543 nm laser line. Image of each DiI- positive neurons were acquired using a 63× oil immersion objective, using a frame size of 512 × 512 pixels which generated an image with field size 146.25 × 146.25 μm and pixel scale 0.29 × 0.29 μm. The neurons were scanned at 1 μm intervals along the z-axis (depending on the depth of whole neuron), in order to acquire sufficient resolution to conduct spine counting and measurement, the frame size was increased to 2048 × 2048 pixels which generated a pixel size 0.07 × 0.07 μm. The dendrite that was clearly connected to the soma and fully separable from crossing dendrites was cropped. The cropped dendrite was scanned at 0.1 μm intervals along the z-axis (depending on the depth of the dendrite) using the same objective. The filament module of Imaris software 7.6.3, a 3-D imaging software (Bitplane, South Windsor, CT, USA) was used to quantify the spine density, and morphology. The minimum end segment diameter (spine head) was set at ≥0.20 μm. Automatic quantification of dendritic protrusions and spine classification into mushroom (mature), stubby, thin and long or filopodialike was performed using Imaris XT. For quantitative analysis, image was rendered by the surpass module of Imaris software. Spine quantification commenced on dendrites beginning at 75 μm distal to the soma, and after the 2nd and 3rd branch points and the minimum length of dendrite quantified was 25 μm. Counts were made by rotating the segment in 3-D to be able to quantify spines in the z-axis. For each neuron, 2–4 dendrites were analyzed. For each animal at least 15–20 neurons were analyzed and averaged and a total of 3–5 mice were used for each group. The dendrites for analysis were chosen using the same rule as for Golgi staining. The rules of morphological classification of dendritic spines of Imaris XT were adopted from (McKinney et al., 1999; Thomas et al., 2009). Briefly, rules for spines classification were as follows; stubby: length (spine) < 1 μm; mushroom: lengths (spine) < 3 μm and maximum width (head) > mean width (neck) × 2; long and thin: length (spine) > 1 μm and mean width (head) ≥ mean width (neck). Remaining spines were categorized as filopodia-like were >3 μm in length, lacking distinct head. Since Imaris XT software follows a sequential process of classification from stubby to mushroom to thin and long and finally to filopodia-like, there is no overlap in the spine categorization due to the above mentioned spine rules.
2.6. Whole-cell electrophysiology
Whole-cell electrophysiology was performed as previously described (Hillman et al., 2011) with minor modifications. Briefly, acute prefrontal cortex slices were obtained from mice at postnatal days (P18–25) in accordance with the approved protocols of Creighton University Institutional Animal Care and Use Committee. After isoflurane anesthesia mice were decapitated and brains were removed rapidly and placed in ice-cold artificial cerebrospinal fluid (ACSF) of the following composition (in mM): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 3 MgCl2 and 10 glucose saturated with 95% O2/5% CO2. 300–350 μm thick coronal sections were prepared using vibrating microtome (Leica VT1200, Buffalo Grove, IL, USA). Whole-cell patch recordings were obtained from layer II–III prelimbic pyramidal neurons in voltage-clamp configuration with an Axopatch 200B (Molecular Devices, Sunnyvale, CA, USA). Glass pipette with a resistance of 5–8 mOhm were filled with an internal solution consisting of (in mM) 110 cesium gluconate, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA,2 Na2ATP, and 0.3 Na2GTP (pH 7.35). QX314 was added to block voltage-gated sodium channels. The recording ACSF contained (in mM) 1.5 CaCl2 and 1.5 MgCl2. mEPSCs were recorded in the presence of 1 μM tetrodotoxin and 100 μM picrotoxin or 20 μM bicuculline. Whole-cell recordings with a pipette access resistance less than 20 mOhm and that changed less than 20% during the duration of recording were included. Signal was filtered at 2 kHz and digitized at10 kHz using an Axon Digidata 1440A analog-to-digital board (Molecular Devices, CA). Recordings were analyzed using Minianalysis (Synaposoft, Atlanta, GA, USA) with an amplitude threshold set at 6 pA and frequency, amplitude and decay of mEPSC currents were determined. The root mean square (RMS) noise (pA) was measured for each recording. No difference was observed between wildtype and GluD1 KO recordings (wildtype = 2.105 ± 0.255, GluD1 KO = 2.170 ± 0.097).
2.7. Immunohistochemistry
Brains were fixed by perfusion of the animals under deep anesthesia with 4% (w/v) paraformaldehyde in 0.1 M sodium phosphate buffer (PBS, pH 7.2); and thereafter removed and immersed overnight at 4 °C in the same solution. Brains were then cryoprotected by soaking in 20% (w/v) sucrose in PBS (12 h, 4 °C) and 30% (w/v) sucrose in PBS (8 h, 4 °C) and frozen in dry ice-cooled isopenthane at −70 °C. Thereafter, 30 μm thick coronal sections were cut and post fixed with the fixative solution for 2 h at 37 °C, before the immunohistochemical staining. After a 15 min treatment with 0.3% (v/v) Triton X-100 (in PBS), blocking was performed by treating the sections with goat and donkey serum diluted in PBS (10%, v/v) for 2 h at room temperature. Afterward, sections were incubated with primary antibodies, diluted with PBS containing 3% goat and donkey serum (anti-PDS-95 mouse monoclonal, 1:300, Thermo scientific, Rockford, IL and anti-synaptophysin rabbit polyclonal, 1:50, Invitrogen, Camarillo, CA) for 24–36 h at 4 °C. Sections were washed with PBS (10 min × 3), and incubated with secondary antibodies (goat anti mouse Alexa Fluor 595 and donkey anti rabbit Alexa Fluor 488, 1:250, Life Technologies, Grand Island, NY) diluted with PBS containing 3% goat and donkey serum for 2 h at room temperature. After washing (PBS,10 min × 4), the wet sections were placed on slides and coverslipped with anti-fluorescence fading fluid (Fluoromount-G, SouthernBiotech, Birmingham, Alabama). Control sections in which primary antibodies were omitted showed no labeled cells. Images of equivalent regions, 1024 × 1024 pixels, were captured using a Leica TCS SP8 MP confocal microscope using a 63×, 1.4 numerical aperture oil-immersion objective at a 4× zoom. An optical section (0.8-mm thick) was taken from each tissue section, and at least five sections per region of interest were analyzed for each animal. All microscope settings were unchanged from section to section. We manually quantified the synaptic puncta to obtain a more accurate assessment of PSD-95/synaptophysin contacts. Images were exported to Imaris software and PSD-95 and synaptophysin paired confocal images were adjusted for contrast/brightness to optimize detection of the faintly stained PSD-95 immunostained puncta. A grid of 1 μm2 was chosen to count the total distribution of PSD-95 puncta/μm2. The corresponding synaptophysin-immunostained image was then superimposed under the PSD-95, all PSD-95/synaptophysin contacts were then quantified. Quantification of synaptic punctawas performed by a person blind to the genotype. Only direct PSD-95/synaptophysin contacts were counted, i.e., PSD-95 puncta in apposition to synaptophysin immunoreactive puncta, were then assessed with Image J for their corresponding staining intensities by placement of a circle 32 pixels2 over each of the thresholded puncta. It is important to note that in Image J, thresholding an image does not change the value of the puncta intensity from the non-thresholded image (Cao et al., 2013). Groups of animals were compared using unpaired t-test.
2.8. Synaptoneurosome preparation and western blot analysis
Synaptoneurosome preparation and western blotting analysis were performed as previously described (Yadav et al., 2012). For synaptoneurosomal preparations, 28–35 days old wildtype and GluD1 KO mice were anesthetized using isoflurane anesthesia, mice were then decapitated and thereafter all experimental procedures were conducted on ice. The hippocampus and mPFC was crudely dissected out and put into synaptoneurosomal buffer at 4 °C. The freshly isolated tissue was homogenized in synaptoneurosome buffer (10 mM HEPES, 1 mM EDTA, 2 mM EGTA, 0.5 mM DTT, 10 μg/ml leupeptin, and 50 μg/ml soybean trypsin inhibitor, pH 7.0) as previously described (Villasana et al., 2006), additionally containing 5 mg/ml pepstatin, 50 mg/ml Aprotonin and 0.5 mM phenylmethane sulfonylfluoride (PMSF). The homogenate was diluted further with the same volume of synaptoneurosome buffer and briefly and gently sonicated. The sample was loaded into a 1.0 ml Luerlock syringe (BD syringes) and filtered twice through three layers of a pre-wetted 100 μm pore nylon filter CMN-0105-D (Small Parts Inc., Logansport, IN, USA) held in a 13 mm diameter filter holder XX3001200 (Milipore, MA, USA). The resulting filtrate was loaded into a 1 ml Luer-lock syringe and filtered through a pre-wetted 5 μm pore hydrophilic filter CMN-0005-D (Small Parts Inc., Logansport, IN, USA) held in a 13 mm diameter filter holder. The resulting filtrate was centrifuged at 1000 × g for 10 min. The pellet obtained corresponded to the synaptoneurosome fraction. Isolated synaptoneurosomes were resuspended in 75 μl of buffer solution containing 0.32 M sucrose, and 1 mM NaHCO3 (pH 7.0).
For western blotting, protein estimation was determined using the Bradford assay, and protein was prepared and loaded on 10% or 12% sodium dodecyl sulfate gel in equal amount (15 μg/well). Gels were transferred to nitrocellulose membrane (GE Healthcare, Piscataway, NJ, USA) using Biorad mini protean tetra cell (Bio-Rad Laboratories Inc., Hercules, California, USA). Transfer was followed by blocking with 5% bovine serum albumin (BSA) in Tris-buffered saline with 1% Tween 20 (TBST) for 1 h at room temperature. The primary antibodies; p-Cofilin, Cofilin, LIMK1 and PAK1 (Cell Signaling Technology, MA, USA), 1:1000; p-LIMK1 (Thr 508) (Novus Biologicals, Littleton, CO, USA), 1:500; p-PAK1 (Thr212) (Santa Cruz Biotechnology Inc, Dallas, Texas, USA), 1:1000; GluN2A and GluN2B (Millipore, Temecula, California, USA), 1:1000; GluD1 (Almone lab, Har Hotzvim Hi-Tech Park, Jerusalem, Israel), 1:1000 were used. Membranes were incubated overnight at 4 °C followed by washing, and were then incubated with horse-radish peroxidase conjugated anti-rabbit or antimouse secondary antibody 1:5000; (Cell Signaling Technology, Danvers, MA, USA) for 1 h at room temperature followed by washing with TBST. Blots were developed using Pierce® ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL, USA) and images were taken using Precision Illuminator Model B95 (Imaging Research Inc., Germany) with a MTI CCD 72S camera and analyzed using MCID Basic software version 7.0 (Imaging Research, St. Catharines, ON, Canada). The X-ray film processor used was model- BMI No 122106 (Brown’s Medical imaging, Omaha, NE, USA). For analysis of protein expression, the optical density of each phosphorylated protein was normalized to corresponding total protein and other (non-phospho) proteins were normalized to actin.
2.9. Hole-board testing
Hole-board test was performed as previously described (Irie et al., 2012). Briefly the hole-board apparatus consisted of a chamber with transparent Plexiglas walls and a white square base (40 × 40 cm) which had 16 equally spaced holes (3 cm diameter) in a 4 × 4 configuration. The base was raised 5 cm from the bottom of the chamber. Mice were placed in center of the chamber and activity was video recorded for a test period of 10 min. Head-dipping behavior was scored. An “exploratory dip” was defined as a dip in a hole different from a previous one while “stereotyped dip” was defined as a dip in the same hole as previous one. The number of stereotyped dips and total dips were recorded for the entire session.
2.10. Forced swim test
The forced swim test was performed as previously described (Yadav et al., 2012) with minor modifications. Wildtype and GluD1 KO mice were intraperitoneally administered with saline, desipramine (30 mg/kg), escitalopram (30 mg/kg), DCS (30 or 320 mg/kg) or Ro-25-6981 (10 mg/kg), 30 min prior to performing the forced swim test. All final solutions were prepared in saline. Videotapes were scored by an observer blind to the genotype of the mice. Mice were judged immobile when they remained floating passively in the water, with minor movements to keep their heads above the water. The videos were scored for the total number of immobile events during the 6 min duration of the test. Each event was of a 5 s duration and was counted as immobile if the mouse spent 3 s or more being immobile.
2.11. Locomotor activity
A custom-made circular open-field chamber (27.9 cm diameter × 35.6 cm wall height) was used to assess the locomotor activity; the chamber was bisected by two photobeams (Suryavanshi et al., 2014). Locomotion was counted via an automated photobeam break counter, indicating spatial movement when each photobeam was interrupted (Med Associates, Inc., St. Albans, VT, USA). Animals were acclimatized for 15 min, animals were briefly removed from the chamber and injected (i.p.) with saline, d-cycloserine (DCS) (30 or 320 mg/kg) or Ro-258691 (10 mg/kg) and then placed back into the chamber and locomotor behavior was recorded for 60 min. The total beam breaks after the administration of saline, DCS or Ro-258691 was measured. At the end of the open field session, animals were returned to their home cages for 15 min. Afterward animals were placed in a square open field 25 × 25 cm arena with grid marking (6.25 × 6.25 cm) on the bottom and spontaneous activity was recorded for 15 min. The total time that each mouse spent in the central four squares was tallied (Yadav et al., 2012). Scoring was done by an experimenter blind to the genotype of the mice.
2.12. Statistical analysis
Data were analyzed using Student’s unpaired t-test with Welch’s correction, one-way ANOVA (analysis of variance) or two-way ANOVA with post-hoc multiplecomparisons test. Differences were considered significant if P ≤ 0.05. Prism 4 (GraphPad Software Inc., San Diego, CA, USA) was used for analysis and representation.
3. Results
3.1. Higher dendritic spine density and excitatory neurotransmission in medial prefrontal cortex of GluD1 knockout mice
High GluD1 mRNA expression is observed in the cortex in rodents (Lomeli et al., 1993; Yadav et al., 2012; Hepp et al., 2014) and appears to be localized to pyramidal neurons (Lomeli et al., 1993; Lein et al., 2007; Konno et al., 2014; Hepp et al., 2014). Recent studies also found high protein expression of GluD1 in the cortex (Konno et al., 2014; Hepp et al., 2014). We further tested the protein expression in synaptoneurosomal preparation using a new commercially available antibody by western blotting. GluD1 was found to be highly expressed in the mPFC in wildtype mice but no expression was observed in GluD1 KO indicating the specificity of this antibody (Fig. 1A). We examined whether loss of GluD1 has an impact on the dendritic spines which mediate excitatory neurotransmission in pyramidal neurons in mPFC. Analysis of Golgi-impregnated tissue at postnatal day 14 (P14) and P30 was performed. In wildtype mice a reduction in spine density was observed in the pyramidal neurons of prelimbic cortex (layer II–III) from P14 to P30 (P = 0.035, unpaired t-test; Fig. 1B). This finding is consistent with activity-dependent pruning, synapse elimination and stabilization of synapses during adolescence (Rakic et al., 1986). Interestingly no reduction in the spine density from P14 to P30 was observed in GluD1 KO mice (P = 0.9271) suggesting a deficit in pruning of spines or an excessive generation of new spines or a combination thereof (Fig. 1B). Higher density of spines was also observed at P30 in GluD1 KO mice in the infralimbic cortex (P = 0.0066, unpaired ttest; Fig. 1B). We found a linear increase in spine density from P14 to P30 in GluD1 KO in the infralimbic cortex but not in prelimbic cortex. These differences in the two cortical regions are likely due to differences in the contribution of impaired spine pruning and new spine formation which regulates spine number. In order to further evaluate morphological features of dendritic spines in GluD1 KO mice we performed diolistic labeling of fixed vibratome slices and 3-D reconstruction analysis using Imaris software. Mice at P30 were used for this study. Consistent with the Golgi analysis, diolistic labeling revealed higher number of spines in the prelimbic cortex of GluD1 KO mice (P = 0.0002; Fig. 1C). Further analysis revealed that both mushroom and filopodia-like spine number was significantly higher in GluD1 KO mice (P = 0.0001 and P = 0.0453 respectively; Fig. 1C). Previous observation indicates that the proportion of stubby spines is drastically reduced while those of thin spines increases from P15 to adulthood (Harris et al., 1992). Since we did not observe a significant difference in the proportion of either stubby or thin and long spines in GluD1 KO adult compared to wildtype adult, it suggest that the spine composition in GluD1 KO represents a more adult rather than younger spine profile but with more spine (filopodia) formation and possibly less spine (mushroom) reduction (Harris and Kater., 1994; Dickstein et al., 2012). We observed a noticeable difference in the spine density in Golgi and DiI stained neurons, the DiI stained neurons showed higher spine density compared to Golgi stained neuron. This variability may be due to the different analysis parameters applied in the two methods; we performed 2-D manual analysis for Golgi staining while 3-D automated analysis using Imaris software for DiI staining. However, these differences may also arise due to the ability of DiI staining to better resolve spines as previously reported by Shen et al. (2009). DiI is uniformly incorporated into the cellular membrane through lateral diffusion and more efficiently labels the dendritic spines. Another advantage of fluorescent labeling is to produce spine image of better resolution and larger sample size (Gan et al., 2000; Wallace and Bear, 2004). Using whole-cell recordings we further tested whether the abnormalities in spine density have an impact on synaptic function. mEPSCs were recorded from prelimbic cortex neurons. Since recordings were performed at −70 mV in the presence of extracellular Mg2+, NMDA receptors will be blocked and therefore the mEPSCs predominantly represent AMPA receptor activity. We found higher frequency of mEPSCs in pyramidal neurons from prelimbic cortex (P = 0.0489; Fig. 1D) consistent with the higher number of mushroom-like spines. No difference in the amplitude or decay of mEPSCs was observed suggesting that mature synapses in GluD1 KO are more but are not dissimilar in AMPA receptor function.
Fig. 1.
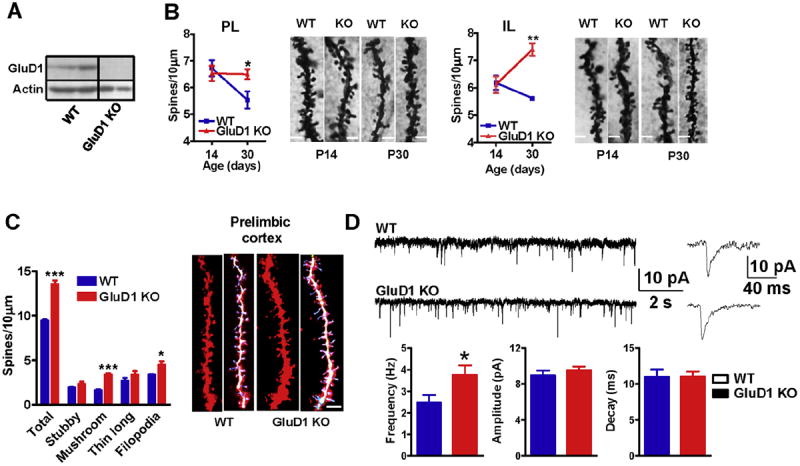
GluD1 KO mice exhibit impaired spine development and higher excitatory neurotransmission in the medial prefrontal cortex. A. Expression of GluD1 in synaptoneurosome from mPFC using western blotting. Lack of immunoreactivity demonstrates specificity of the antibody for GluD1. B. Golgi impregnation and spine analysis was performed from wildtype and GluD1 KO mice (n = 3–4 animals/age group and 15–20 neurons/animal). Impaired reduction in spine number over development was observed in prelimibic and infralimbic cortex in GluD1 KO mice with no difference in spine number between day 14 and day 30 old animals (P > 0.05, unpaired t-test). Significantly higher spine density was observed in GluD1 KO mice compared to wildtype (*P < 0.05, **P < 0.01, unpaired t-test). The bar in the image represents 2 μm. C. Diolistic labeling and spine analysis was performed from wildtype and GluD1 KO mice (4–5 animals/genotype and 15–20 neurons/animal). Significantly higher number of total spines, mushroom-like as well as filopodialike spines were found in the neurons in prelimbic cortex of mPFC of GluD1 KO mice (***P = 0.0002, ***P = 0.0001 and *P = 0.0453, unpaired t-test). The bar in the image represents 5 μm. D. Whole-cell voltage-clamp recordings from layer II–III pyramidal cells from prelimibic cortex were performed. A higher frequency of mEPSCs was observed in the pyramidal neurons in GluD1 KO mice (*P = 0.0489, unpaired t-test; N = 6 animals/group. Parameter from 2 to 3 recordings/individual animal were averaged and used for comparison.). No significant difference was observed in the amplitude or decay of mEPSCs. Recordings were performed at −70 mV in the presence of picrotoxin (100 μM) and tetrodotoxin (1 μM). Signal was filtered at 2 kHz and digitized at 10 kHz.
3.2. Abnormal signaling mechanisms regulating spine dynamics in medial prefrontal cortex of GluD1 knockout mice are corrected by modulating NMDA receptor function
We tested the possibility that abnormal mechanisms underlying spine dynamics may be responsible for the higher number of spines in mPFC neurons of GluD1 KO mice. Cofilin is an actinbinding protein whose activation depolymerizes actin filaments which can induce remodeling of dendritic spines (dos Remedios et al., 2003; Shi et al., 2009; Pontrello et al., 2012). Since, cofilin is inactivated by phosphorylation at Ser3 and reactivated by dephosphorylation (Morgan et al., 1993) we examined the cofilin phosphorylation at Ser3. We found a lower proportion of phosphorylated form of cofilin in synaptoenurosomes from mPFC in GluD1 KO mice (P = 0.0495, unpaired t-test; Fig. 2A). Cofilin phosphorylation is regulated by LIMK1 and phosphorylation of LIMK1 at Thr 508 by PAK1or ROCK is an important physiological mechanism of LIMK1 activation (Edwards et al., 1999; Maekawa et al., 1999). Thus, we tested the levels of phosphorylated and total LIMK1 and PAK1. We did not observe any change in the expression of p-PAK1 but the expression of p-LIMK1 was found to be lower in GluD1 KO mPFC (P = 0.001, unpaired t-test; Fig. 2A). These results suggest that abnormality in LIMK1-cofilin signaling which regulates spine turnover may underlie the higher spine number in the mPFC neurons in GluD1 KO mice.
Fig. 2.
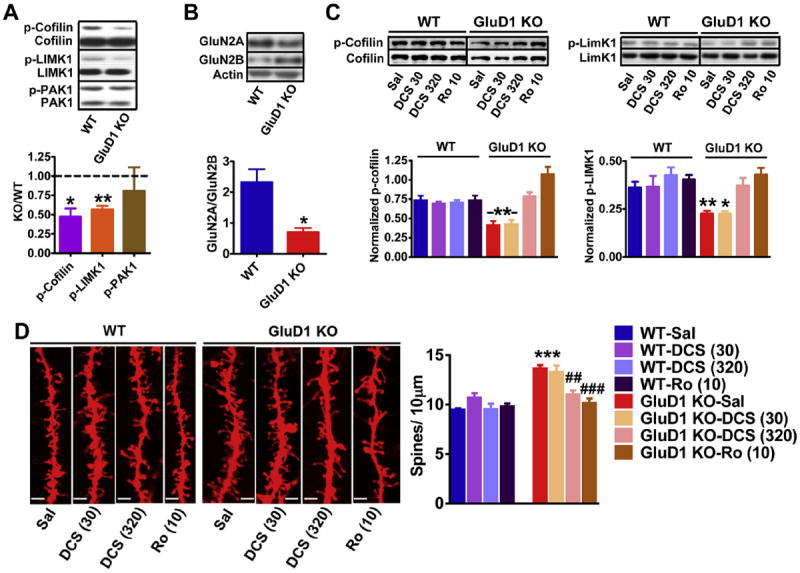
Inhibition of GluN2B-containing receptors reverses the downreguated p-cofilin-p-LIMK1 signaling and higher spine density in GluD1 KO mice. A. The levels of total and phosophorylated form of cofilin, LIMK1 and PAK1 were measured in synaptoneurosome preparation from mPFC of wildtype and GluD1 KO mice. Lower expression of p-cofilin (*P = 0.0495, unpaired t-test) and p-LIMK1 (**P = 0.001, unpaired t-test) was found in GluD1 KO mice. No difference in the expression of total proteins or p-PAK1 was observed. N = 5–8 animals/group. B. Significantly lower GluN2A/GluN2B expression ratio was observed in the synaptoneurosome from mPFC (*P = 0.0105, N = 4 animals per group). C. Mice were injected intraperitoneally with saline, DCS (30 or 320 mg/kg) or Ro-25-6981 (10 mg/kg). DCS (320 mg/kg) and Ro-25-6981 (10 mg/kg) normalized the lower p-cofilin and p- LIMK1 levels close to wildtype levels (*P < 0.05, **P < 0.01 compared to wildtype-saline, N = 4). D. Brain was collected after 30 min of systemic administration with saline, DCS or Ro-25-6981 and processed for diolistic labeling and spine analysis (4–5 animals/group and 15–20 neurons/animal). Representative images of dendrites from different treatment groups are shown. The bar in the image represents 5 μm. The total spine number was higher in GluD1 KO mice and was not significantly affected by DCS at 30 mg/kg (P > 0.05 compared to KO-saline, one-way ANOVA). Total spine density in GluD1 KO was however found to be reduced by DCS at 320 mg/kg and Ro-25-6981 (**P < 0.01 and ***P < 0.001 compared to KO-saline, one-way ANOVA).
Since, the activation of cofilin has been suggested to underlie activity-dependent and NMDA receptor mediated dendritic spine remodeling (Shi and Ethell, 2006; Pontrello et al., 2012), we tested whether there are abnormalities in expression of NMDA receptor subunits and whether modulation of NMDA receptor function can normalize phosphorylated cofilin and LIMK1 levels. We evaluated the expression of the predominant subtypes of NMDA receptors GluN2A and GluN2B subunits. Similar to our previous observation a trend for higher GluN2B expression was observed in mPFC (Yadav et al., 2012) while an opposite trend of lower GluN2A subunit expression was observed in GluD1 KO mice. An important characteristic in neuronal developmental is the switch in the NMDA receptor subunit composition from GluN2B to GluN2A subunit (Williams et al.,1993). Whenwe took the ratio of GluN2A to GluN2B expression it was significantly lower in GluD1 KO mice (unpaired t-test; P = 0.0105) suggesting impairment in the developmental switch (Fig. 2B). In an effort to normalize the GluN2A/GluN2B function we tested the effect of DCS which has approximately full efficacy at GluN2A-containing receptors but is a partial agonist for GluN2B-containing receptors (Dravid et al., 2010). Although low dose of DCS (30 mg/kg i.p.) was ineffective in normalizing the pcofilin and p-LIMK1 levels (wildtype-saline vs. GluD1 KO-saline and DCS 30; P < 0.01, one-way ANOVA, Dunnett’s multiple comparison test), higher dose of DCS (320 mg/kg i.p.) was able to normalize both the p-cofilin and p-LIMK1 levels in GluD1 KO (wild type saline vs. GluD1 KO DCS 320; P > 0.05; Fig. 2C). Since higher dose of DCS is likely to have a strong inhibitory effect on GluN2B-containing receptors we further tested whether inhibition of GluN2B-containing receptors alone is sufficient to normalize this signaling pathway. We found that systemic administration of high-affinity GluN2Bselective inhibitor Ro-25-6981 (10 mg/kg i.p.) partially normalized the p-cofilin and p-LIMK1 level in mPFC in GluD1 KO mice to wildtype-saline levels (P > 0.05, wildtype-saline versus GluD1 KORo- 25-6981, one-way ANOVA; Fig. 3C). We observed that Ro-25-6981 not only normalized p-cofilin levels but that it increased the p-cofilin levels beyond control wildtype. We further tested whether similar modulation of NMDA receptor is able to correct the spine abnormality in the mPFC of GluD1 KO mice. We found that the basal level of the total spine density was higher in GluD1 KO mice compared to wildtype in the vehicle groups (Fig. 2D; P < 0.001, oneway ANOVA with Bonferroni’s multiple comparison test). Treatment with low dose of DCS (30 mg/kg) did not show any effect on total spine density (P > 0.05), however, DCS at 320 mg/kg significantly reduced the total spine density in prelimbic pyramidal neurons (P < 0.01; Fig. 2D). Moreover, Ro-25-6981 (10 mg/kg i.p.) also normalized the total spine density in GluD1 KO mice (P < 0.001).
Fig. 3.
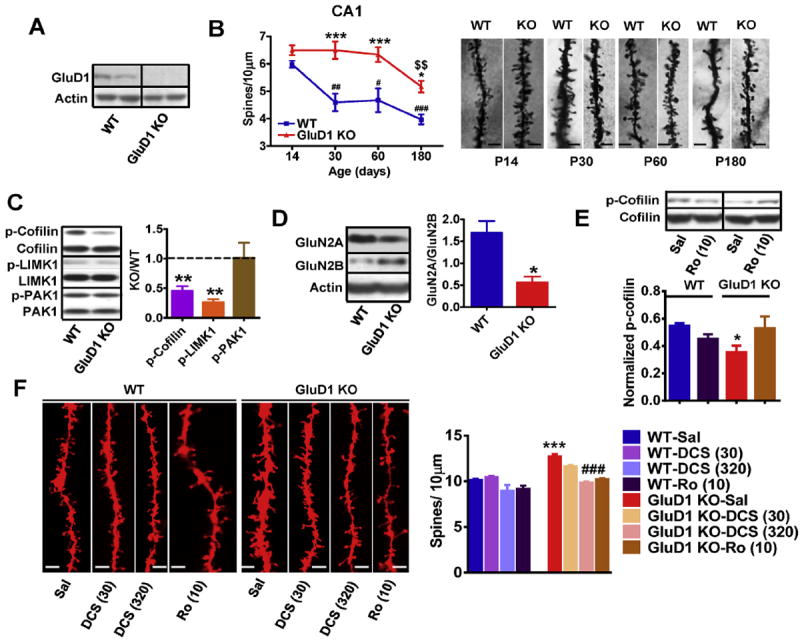
The abnormalities in spine development and signaling are conserved between the hippocampus and mPFC in the GluD1 KO mice. A. High expression of GluD1 was observed in synaptoneurosome from hippocampus using western blotting. B. Golgi impregnation and spine analysis was performed from wildtype and GluD1 KO mice (n = 3–4 animals/age group and 15–20 neurons/animal). Higher spine density was observed in the apical dendrites of CA1 neurons of GluD1 KO mice from postnatal days 30–60 (***P < 0.001, two-way ANOVA Bonferroni’s multiple comparisons test) and postnatal day 180 (P < 0.05). Reduction in spine number observed in wildtype mice (##P < 0.01, day 30; #P < 0.05, day 60 and ###P < 0.001, day 180) was absent in GluD1 KO mice from day 14 to day 60. Significant reduction in spine number from day 60 to day 180 age was observed in GluD1 KO mice ($$P < 0.01). C. The levels of total and phosophorylated form of cofilin, LIMK1 and PAK1 were measured in synaptoneurosome preparation from hippocampus. Lower expression of p-cofilin (**P = 0.0007, unpaired t-test) and p-LIMK1 (**P = 0.0002, unpaired t-test) were found in GluD1 KO mice. No difference in the expression of total proteins or p-PAK1 was observed. N = 5–8 animals/group. D. The ratio of GluN2A/GluN2B expression was significantly lower in GluD1 KO mice hippocampus (*P = 0.0196). E. Systemic administration of Ro-25-6981 (10 mg/kg) normalized the lower p-cofilin levels in hippocampus (N = 4 animals for each group). F. The total spine number was higher in GluD1 KO mice and was not significantly affected by DCS at 30 mg/kg (P > 0.05 compared to KO-saline, one-way ANOVA). Total spine density in GluD1 KO was however found to be reduced by DCS at 320 mg/kg and Ro-25-6981 (P < 0.001 compared to KO-saline, one-way ANOVA). N = 4–5 for each group.
3.3. Abnormality in spine pruning and spine dynamics signaling is conserved in the hippocampus of GluD1 knockout mice
GluD1 subunit is highly expressed in the rodent hippocampus during early postnatal days and in the adult (Lomeli et al., 1993; Gao et al., 2007; Konno et al., 2014; Hepp et al., 2014). We also found high expression of GluD1 in the hippocampus (Fig. 3A). We examined whether the loss of GluD1 also impacts spine density in apical dendrites of CA1 hippocampal neurons. Golgiimpregnation was performed and number of dendritic spines of first order dendrites at IInd and IIIrd dendritic branch points of CA1 pyramidal neurons were analyzed. We found that in wildtype animals the spine density reduced significantly over adulthood (P < 0.01, P14 compared to P30; P < 0.05, P14 compared to P60 and P < 0.001, P14 compared to P180, one-way ANOVA; Fig. 3B). In contrast, in GluD1 KO mice the spine density remained unchanged from P14 up to P60 (P > 0.05; Fig. 3B). A reduction in spine density was observed from P60 to P180 in GluD1 KO mice (P < 0.01). The spine density in GluD1 KO mice was significantly higher compared to wildtype (P < 0.01, P30-60 and P < 0.05, P180, two-way ANOVA with Bonferroni multiple comparison test). We also analyzed spine density in the dentate gyrus granule neurons and found higher density of spines in GluD1 KO mice at both P14 and P30 (P < 0.0001 and P = 0.0108; unpaired t-test1). We further tested whether the abnormality in spine dynamics signaling is also impaired in hippocampus similar to mPFC. We found a lower proportion of phosphorylated form of cofilin in synaptoenurosomes from hippocampus in GluD1 KO mice (P = 0.0007, unpaired t-test; Fig. 3C) as well as lower expression of p-LIMK1 (P = 0.0002 for hippocampus, unpaired t-test) but no change in p-PAK1. We also found that similar to the mPFC the ratio of expression of GluN2A/GluN2B subunit was significantly lower in GluD1 KO mice (P = 0.0196; Fig. 3D). We further found that treatment with Ro-25-6981 (10 mg/kg i.p.) although did not significantly increase the pcofilin level compared to saline but it normalized p-cofilin in GluD1 KO to wildtype-saline level (P > 0.05, wildtype-saline versus GluD1 KO-Ro-25-6981, one-way ANOVA with Dunnett’s multiple comparison test; Fig. 3E). Moreover, DCS (320 mg/kg i.p.) and Ro-25-6981 (10 mg/kg i.p.) also normalized the spine density in the hippocampus (Fig. 3F; P < 0.001, one-way ANOVA with Bonferroni’s multiple comparison test). Thus loss of GluD1 in hippocampus leads to similar changes as observed in mPFC. We also analyzed the effect of Ro-25-6981 on dendritic diameter but we did not see any significant effect (wildtype saline = 0.769 ± 0.032, wildtype-Ro = 0.741 ± 0.0370, Glud1 KO saline = 0.796 ± 0.0408, GluD1 KO-Ro = 0.770 ± 0.0593).
We also evaluated the synapse number using a complementary immunohistochemical method. We found that the colocalized PSD95-synaptophysin puncta, that represent excitatory synapses, were significantly higher in the mPFC of GluD1 KO at both P14 and P30 (P = 0.005 and P = 0.0029, Fig. 4A and B). We also tested whether higher excitatory synapse number is limited to mPFC or occurs in hippocampus region which is also enriched with GluD1. We found that in the stratum radiatum of hippocampus CA1 region the synaptic puncta were significantly higher at both P14 and P30 (P = 0.0452 and P = 0.0467, Fig. 4C and D). Together these results suggest that loss of GluD1 leads to hyperconnectivity in the cortex and hippocampus.
Fig. 4.
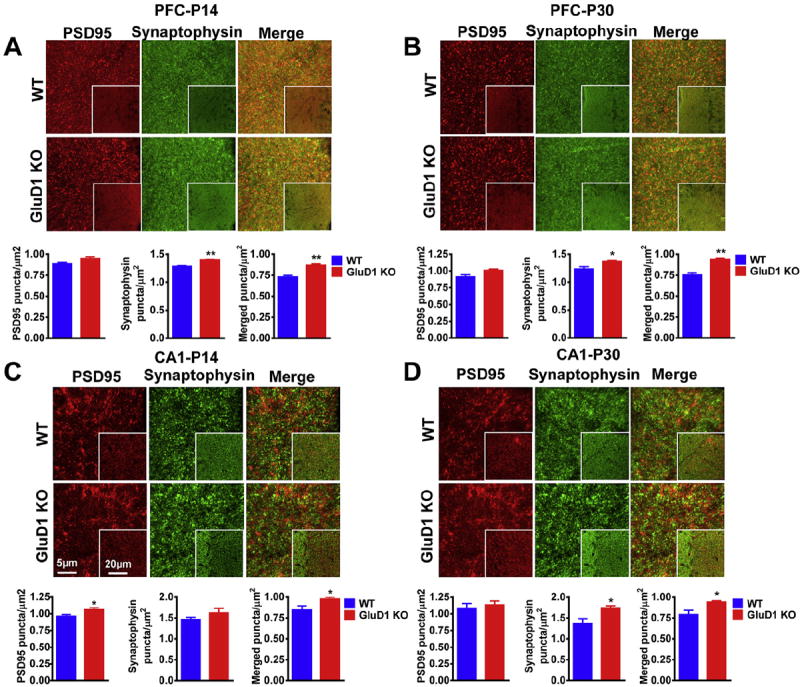
Higher synaptic punctas in the mPFC and CA1 hippocampal area of GluD1 KO mice. PSD-95 (red) and synaptophysin (green) antibodies were used to stain synapses (merged) in the mPFC and stratum radiatum of the hippocampal area CA1. A. GluD1 KO showed significantly higher synaptophysin puncta (P = 0.0014, unpaired t-test) and higher synaptic density (P = 0.005) in the mPFC at postnatal day 14. B. At postnatal day 30 GluD1 KO showed higher synaptophysin puncta as well as synaptic density (P = 0.0413 and P = 0.0029, respectively). C. GluD1 KO showed higher PSD95 puncta and synaptic density (P = 0.0343 and P = 0.0452, respectively) at postnatal day 14 in stratum radiatum of the hippocampal area CA1. D. A significantly higher synaptophysin puncta and synaptic density (P = 0.0429, P = 0.0467) was observed in the stratum radiatum of the hippocampal area CA1 of GluD1 KO mice at postnatal day 30. N = 4 animals per group. *P < 0.05, **P < 0.01. The scale bar is 5 μm and 20 μm for inset images. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4. Reduction of stereotyped behavior and depression-like behavior in GluD1 knockout mice by inhibition of GluN2B receptors
We further assessed the possible behavioral outcome of modulation of NMDA receptors which normalized the signaling and spine density in GluD1 KO mice. We used the hole-board test which measures repetitive or stereotyped behavior which is one of the core features of the neurodevelopmental disorders such as autism (Crawley, 2007). We evaluated the effect of DCS (30 and 320 mg/kg i.p.) and Ro-25-6981 (10 mg/kg i.p.) on the repetitive behavior. We found a significant genotype (P < 0.05) and treatment (P < 0.05) effect but no interaction effect (P > 0.05; twoway ANOVA; Fig. 5A). Post-hoc Bonferroni test indicate a significant difference in the saline-wildtype and saline-GluD1 KO groups (P < 0.01). We further compared within-genotype drug effect using one-way ANOVA. No significant treatment differences were observed in wildtype mice (P > 0.05, Dunnett’s test). Consistent with the ability of high-dose DCS and Ro-25-6981 in reversing the spine abnormalities these agents reversed the higher stereotyped head-dipping behavior in GluD1 KO mice (P < 0.05; Fig. 5A). In contrast, DCS at 30 mg/kg did not significantly affect the stereotyped behavior in GluD1 KO mice (P > 0.05). Two-way ANOVA analysis of the total head-dips was conducted. No genotype or interaction effect was observed, however, a significant treatment effect was observed (P < 0.05, two-way ANOVA; Fig. 5B). Further analysis of within-genotype treatment effect revealed significant effect of DCS 320 mg/kg on total head-dips in GluD1 KO mice (P < 0.05, one-way ANOVA), however, this increase cannot explain the lowering of the stereotyped head-dips since an increase in total head-dips should typically increase the incidence of stereotyped head-dips. We further studied the effect of DCS (30 and 320 mg/kg) and Ro-25-6981 (10 mg/kg) on locomotor activity and time-in-center in open field as a measure of anxiety-like behavior. In locomotor activity no significant genotype or interaction effect was observed, however, a significant treatment effect was observed (P < 0.05, two-way ANOVA; Supplementary Fig. 3A). Withingenotype analysis did not reveal any significant treatment effect in either genotype (P > 0.05, one-way ANOVA). In time-incenter no significant genotype, treatment or interaction effect was observed (P > 0.05, two-way ANOVA; Supplementary Fig. 3B). These data support the finding that the effect of treatment on stereotypy cannot be explained entirely due to changes in locomotion or anxiety-like behavior.
Fig. 5.
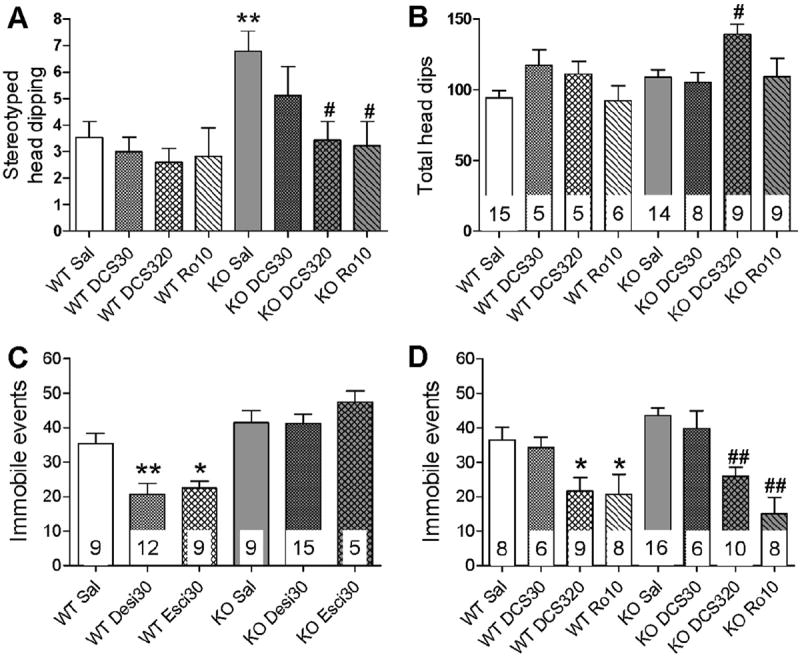
High-dose DCS and Ro-25-6981 reduce the stereotyped and depression-like behavior in GluD1 KO mice. A. The stereotyped head-dipping behavior was measured in the hole-board test (N = 5–15/group). A significant genotype (P < 0.05) and treatment (P < 0.05) effect was observed (two-way ANOVA) and GluD1 KO mice (saline) exhibited significantly higher stereotyped head-dips compared to wildtype mice (saline) (**P < 0.01; Bonferroni post-hoc comparison). DCS at 320 mg/kg and Ro-25-6981 (10 mg/kg) but not DCS at 30 mg/kg significantly reduced the stereotyped head-dips in GluD1 KO mice (#P < 0.05, within-genotype one-way ANOVA; Dunnett’s test compared to saline group). No effect of any of the treatments was observed in the wildtype mice (P > 0.05, one-way ANOVA). B. A significant treatment effect (P < 0.05) was observed in the analysis of total headdips (two-way ANOVA) with no genotype or interaction effect. Within-genotype analysis revealed significant increase in total head-dips by DCS (320 mg/kg) (#P < 0.05; one-way ANOVA). C. Animals were injected with saline, desipramine (30 mg/kg i.p.) or escitalopram (30 mg/kg i.p.), 30 min before the forced-swim test (N = 5–15/group). The number of immobile events was measured over a duration of 6 min. Two-way ANOVA with genotype and treatment as factors revealed a significant genotype (P < 0.001), treatment (P < 0.05) and interaction (P < 0.05) effect. A significant reduction in the number of immobile events was observed in wildtype mice after treatment with desipramine and escitalopram (*P < 0.05 and **P < 0.01, within-genotype one-way ANOVA compared to wildtype-saline) but not in GluD1 KO mice (P > 0.05). D. Animals were administered saline, DCS (30 or 320 mg/kg i.p.) or Ro-25-6981 (10 mg/kg i.p.), 30 min before the forced-swim test (N = 6–16/group). Two-way ANOVA revealed no significant genotype or interaction effect (P > 0.05), however, a significant treatment effect was observed (P < 0.001; two-way ANOVA). A significant reduction in the number of immobile events was produced by DCS (320 mg/kg i.p.) and Ro-25-6981 (10 mg/kg i.p.) both in wildtype mice (*P < 0.05 compared to wildtype-saline, within-genotype one-way ANOVA) and GluD1 KO mice (##P < 0.01 compared to KO-saline, within-genotype one-way ANOVA).
We have previously shown that GluD1 KO mice exhibit depression-like behavior (Yadav et al., 2012). Here we tested the sensitivity of depression-like behavior in wildtype and GluD1 KO mice to traditional antidepressants; tricyclic antidepressant (TCA) desipramine (30 mg/kg i.p.) and selective-serotonin reuptake inhibitor (SSRI) escitalopram (30 mg/kg i.p.) in the forced-swim test. Two-way ANOVAwith genotype and treatment as factors revealed a significant genotype (P < 0.001), treatment (P < 0.05) and interaction (P< 0.05) effect (Fig. 5C). Within-genotype testing revealed that in wildtype mice both agents led to a reduction in immobile events in forced-swim test (P < 0.01 for desipramine and P < 0.01 for escitalopram, one-way ANOVA, Dunnett’s test). Interestingly, these agents were not able to reduce immobility in the GluD1 KO mice (P > 0.05, one-way ANOVA). We further tested the effect of NMDA receptor agents on depression-like behavior. Two-way ANOVA revealed no significant genotype or interaction effect (P > 0.05), however, a significant treatment effect was observed (P < 0.001, two-way ANOVA; Fig. 5D). One-way ANOVA for within-genotype effects indicated that DCS (320 mg/kg) and Ro-25-6981 (10 mg/kg) reduced the immobility in wildtype mice (P < 0.05). More interestingly these agents also reduced immobility in GluD1 KO mice (P < 0.01, one-way ANOVA; Fig. 5D). Low-dose DCS (30 mg/kg) was ineffective in both wildtype and GluD1 KO mice (P > 0.05). Together these results demonstrate the selectivity of GluN2B inhibition to reduce depression-like behavior in GluD1 KOmice. In addition these results indicate that high-dose DCS and GluN2B inhibitor in addition to partially reversing the molecular and spine abnormalities also reverse behavioral abnormalities in GluD1 KO mice.
4. Discussion
Our studies demonstrate that GluD1 is necessary for normal spine development and the loss of GluD1 leads to higher spine density and impairs the normal reduction of spines in the mPFC and hippocampus. Moreover, the presence of higher proportion of active cofilin suggests that spines may be dynamic in GluD1 KO mice. We also found that the ratio of GluN2A/GluN2B expression was lower in GluD1 KO mice which together with spine reduction deficits suggest that loss of GluD1 leads to features relevant to abnormal development. We found that inhibition of GluN2B receptors normalized the signaling mechanism that contribute to spine dynamics and reduced the spine number and also reversed the stereotyped behavior and depression-like behavior in GluD1 KO mice.
4.1. Regulation of dendritic spine development in cortico-limbic circuit by GluD1
Despite the high expression of GluD1 in the forebrain its role in synaptic regulation remains unknown. We have found for the first time a role of GluD1 in vivo in regulating spine development. In agreement with the phenomenon of spine pruning during early development (Rakic et al., 1986) we observed a significant reduction in spine number in the mPFC and CA1 apical dendrites during the 3rd to 4th postnatal week in wildtype mice (Figs. 1 and 4). In contrast, in the GluD1 KO mice the spine density was constant from postnatal days 14–30, suggesting impaired pruning. Moreover, a reduction in spine density from postnatal days 60–180 was observed in both wildtype and GluD1 KO mice in CA1 neurons suggesting that although loss of GluD1 impairs the early developmental pruning it does not impact the reduction in spine density during normal aging (Dickstein et al., 2012). An alternative interpretation of these data is that loss of GluD1 increases the formation of new spines and thereby masking ongoing pruning. Additionally in the hippocampus spine pruning may be delayed and not completely impaired. Spine formation, retraction and stabilization is a dynamic process. It is thought that activity and experience-dependent processes that underlie spine pruning lead to spine stabilization and reduction in the overall turnover characterized by generation and retraction of spines (Holtmaat and Svoboda, 2009). The presence of higher filopodia-like structures and higher proportion of F-actin severing protein cofilin suggest that spinogenesis is more dynamic with higher cycling between spine formation and retraction in the neurons of GluD1 KO mice. It is possible that the presence of GluD1 is required for spine stabilization by interacting with the candidate trans-synaptic partner Neurexin1 (Uemura et al., 2010). However, this interpretation will require further live imaging studies where the generation and removal of spines can be evaluated in real time.
Cofilin activation and inactivation is regulated by LIMK1 which in turn is activated by either PAK1or ROCK (Edwards et al., 1999; Maekawa et al., 1999). Cofilin can also be independently regulated by calcineurin-dependent activation of phosphatase Slingshot-1L (SSH1L) (Zhao et al., 2012). Since we found no change in the basal p-PAK1 in GluD1 KO mice we predict that ROCK or calcineurin pathways may underlie the lower p-cofilin levels in GluD1 KO mice. Future studies will address the precise upstream molecules that lead to cofilin and spine abnormalities. Recent studies have demonstrated that the localization of cofilin in dendritic spines is regulated by β-arrestin-2 in response to NMDA receptor activation (Pontrello et al., 2012). NMDA receptor activates calcineurin which dephosphorylates cofilin (Wang et al., 2005; Carlisle et al., 2008; Meng et al., 2002) leading to elongation and higher turnover of spines (Pontrello et al., 2012). We found that GluN2A/GluN2B ratio was abnormal in GluD1 KO and inhibition of GluN2B-containing receptors in GluD1 KO mice partly corrected the LIMK1-cofilin signaling and reduced the spine number suggesting that a fine balance of NMDA receptor activity must be maintained to regulate spine dynamics. Furthermore, in the mPFC GluN2B inhibition increased p-cofilin beyond wildtype saline (Fig. 2). This higher level of p-cofilin may arise due to greater role of GluN2B in signaling in GluD1 KO due to its higher expression. Moreover, it is likely that GluN2B activity has both LIMK1-dependent and LIMK1- independent (potentially through calcineurin) effects on p-cofilin, since only higher p-cofilin but not p-LIMK1 levels were observed in response to Ro-25-6981.
4.2. Relevance of the neuropathology in GluD1 knockout mice to mental disorders
The occurrence of social interaction deficits, stereotyped behavior, and reversal learning deficits together with the typical spine pathology suggest that loss of GluD1 leads to phenotypes relevant to neuropsychiatric disorders particularly autism where higher spine density has been reported (Yadav et al., 2012, 2013, Figs.1 and 3; Penzes et al., 2011). We also observed a lower GluN2A/GluN2B expression ratio which suggests that the switch in GluN2B to GluN2A may be impaired in GluD1 KO and it will be interesting to address whether other features of developmental delay occur in GluD1 KO mice. We found a higher number of dendritic spines, and higher frequency of mEPSC in the mPFC of GluD1 KO mice, suggestive of hyperconnectivity in the mPFC (Fig. 1). Inhibition of GluN2B-containing receptors was found to reduce the number of spines in mPFC and hippocampus as well as occurrence of repetitive behavior and depression-like behavior in GluD1 KO mice. One of the limitations of this finding is that drug treatment may be correcting other synaptic deficits or leading to other peripheral effects, that are responsible for the efficacy of drugs for abnormal behaviors, which we have not presently explored. Nonetheless, it should be noted that dorsolateral PFC and hippocampus (Mineur et al., 2013) in humans plays a critical role in governing stereotyped behavior and depression-like behavior (Forbes and Grafman, 2010; Nelson and Guyer, 2011; Yizhar, 2012; Kadota et al., 2010) and the ability of GluN2B inhibition and high-dose of DCS, which is an FDA approved therapeutic agent, to reverse excess spine abnormality has implications for treatment of certain neurodevelopmental disorders such as autism where higher spine density is observed.
Supplementary Material
Acknowledgments
This work was supported by Health Future Foundation (SMD), LB692 Faculty Development Award (SMD), EPSCoR Award (SMD, BJM), National Institute of Health (NIH) #1R21MH098270 (SMD) and a grant from NE DHHS (Stem Cell 2014-08). The project was also supported by G20RR024001 from National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. We thank Dr. Suneet Mehrotra and Dr. Thomas F. Murray for providing technical support and resources for Gene Gun and Imaris analysis.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.neuropharm.2015.02.013.
References
- Ady V, Perroy J, Tricoire L, Piochon C, Dadak S, Chen X, Dusart I, Fagni L, Lambolez B, Levenes C. Type 1 metabotropic glutamate receptors (mGlu1) trigger the gating of GluD2 delta glutamate receptors. EMBO Rep. 2014;15:103–109. doi: 10.1002/embr.201337371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Manzerra P, Marcora E, Kennedy MB. SynGAP regulates steady-state and activity-dependent phosphorylation of cofilin. J Neurosci. 2008;28:13673–13683. doi: 10.1523/JNEUROSCI.4695-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Cong, Rioult-Pedotti Mengia S, Migani Paolo, Yu Crystal J, Tiwari Rakesh, Parang Keykavous, Spaller Mark R, Goebel Dennis J, Marshall John. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol. 2013;11(2):e1001478. doi: 10.1371/journal.pbio.1001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol (Zurich, Switz) 2007;17:448–459. doi: 10.1111/j.1750-3639.2007.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein DL, Weaver CM, Luebke JI, Hof PR. Dendritic spine changes associated with normal aging. Neuroscience. 2013;251:21–32. doi: 10.1016/j.neuroscience.2012.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83:433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- Dravid SM, Burger PB, Prakash A, Geballe MT, Yadav R, Le P, Vellano K, Snyder JP, Traynelis SF. Structural determinants of D-cycloserine efficacy at the NR1/NR2C NMDA receptors. J Neurosci. 2010;30:2741–2754. doi: 10.1523/JNEUROSCI.5390-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AC, Aliev F, Bierut LJ, et al. Genome-wide association study of comorbid depressive syndrome and alcohol dependence. Psychiatr Genet. 2012;22:31–41. doi: 10.1097/YPG.0b013e32834acd07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Fallin MD, Lasseter VK, Avramopoulos D, et al. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among ashkenazi jewish case-parent trios. Am J Hum Genet. 2005;77:918–936. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes CE, Grafman J. The role of the human prefrontal cortex in social cognition and moral judgment. Annu Rev Neurosci. 2010;33:299–324. doi: 10.1146/annurev-neuro-060909-153230. [DOI] [PubMed] [Google Scholar]
- Gao J, Maison SF, Wu X, et al. Orphan glutamate receptor delta1 subunit required for high-frequency hearing. Mol Cell Biol. 2007;27:4500–4512. doi: 10.1128/MCB.02051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Lazzeroni LC, Murray SS, et al. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the consortium on the genetics of schizophrenia. Am J Psychiatry. 2011;168:930–946. doi: 10.1176/appi.ajp.2011.10050723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold AJ, Ma D, Cukier HN, et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. 2012;21:3513–3523. doi: 10.1093/hmg/dds164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo SZ, Huang K, Shi YY, et al. A case-control association study between the GRID1 gene and schizophrenia in the Chinese Northern Han Population. Schizophr Res. 2007;93:385–390. doi: 10.1016/j.schres.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implication for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12(7):2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- Hepp R, Hay YA, Aguado C, Lujan R, Dauphinot L, Potier MC, Nomura S, Poirel O, Mestikawy SE, Lambolez B, Tricoire L. Glutamate receptors of the delta family are widely expressed in the adult brain. Brain Struct Funct. 2014 Jul 8; doi: 10.1007/s00429-014-0827-4. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Gelfin G, Bloch B, Levin R, Edelman S, Javitt DC, Kremer I. A randomized add-on trial of high-dose D-cycloserine for treatment-resistant depression. Int J Neuropsychopharmacol. 2013;16:501–506. doi: 10.1017/S1461145712000910. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC, Gelfin Y, Gorelik E, Bar M, Blanaru M, Kremer I. Controlled trial of D-cycloserine adjuvant therapy for treatment-resistant major depressive disorder. J Affect Disord. 2006;93:239–243. doi: 10.1016/j.jad.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Hillman BG, Gupta SC, Stairs DJ, Buonanno A, Dravid SM. Behavioral analysis of NR2C knockout mouse reveals deficit in acquisition of conditioned fear and working memory. Neurobiol Learn Mem. 2011;95:404–414. doi: 10.1016/j.nlm.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Irie F, Badie-Mahdavi H, Yamaguchi Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc Natl Acad Sci U S A. 2012;109:5052–5056. doi: 10.1073/pnas.1117881109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota H, Sekiguchi H, Takeuchi S, Miyazaki M, Kohno Y, Nakajima Y. The role of the dorsolateral prefrontal cortex in the inhibition of stereotyped responses. Exp Brain Res. 2010;203:593–600. doi: 10.1007/s00221-010-2269-4. [DOI] [PubMed] [Google Scholar]
- Konno K, Matsuda K, Nakamoto C, Uchigashima M, Miyazaki T, Yamasaki M, Sakimura K, Yuzaki M, Watanabe M. Enriched expression of GluD1 in higher brain regions and its involvement in parallel fiber-interneuron synapse formation in the cerebellum. J Neurosci. 2014;34:7412–7424. doi: 10.1523/JNEUROSCI.0628-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science (New York, N Y) 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli H, Sprengel R, Laurie DJ, Kohr G, Herb A, Seeburg PH, Wisden W. The rat delta-1 and delta-2 subunits extend the excitatory amino acid receptor family. FEBS Lett. 1993;315:318–322. doi: 10.1016/0014-5793(93)81186-4. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science (New York, N Y) 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Miura E, Miyazaki T, et al. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science (New York, N Y) 2010;328:363–368. doi: 10.1126/science.1185152. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Obayemi A, Wigestrand MB, Fote GM, Calarco CA, Li AM, Picciotto MR. Cholinergic signaling in the hippocampus regulates social stress resilience and anxiety- and depression-like behavior. Proc Natl Acad Sci U S A. 2013;26(110):3573–3578. doi: 10.1073/pnas.1219731110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TE, Lockerbie RO, Minamide LS, Browning MD, Bamburg JR. Isolation and characterization of a regulated form of actin depolymerizing factor. J Cell Biol. 1993;122:623–633. doi: 10.1083/jcb.122.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naur P, Hansen KB, Kristensen AS, et al. Ionotropic glutamate-like receptor delta2 binds D-serine and glycine. Proc Natl Acad Sci U S A. 2007;104:14116–14121. doi: 10.1073/pnas.0703718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson EE, Guyer AE. The development of the ventral prefrontal cortex and social flexibility. Dev Cogn Neurosci. 2011;1:233–245. doi: 10.1016/j.dcn.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Roeb W, Dickel DE, et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur J Hum Genet. 2011;19:727–731. doi: 10.1038/ejhg.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JA, Lummis SC. Diolistic labeling of neuronal cultures and intact tissue using a hand-held gene gun. Nat Protoc. 2006;1:1517–1521. doi: 10.1038/nprot.2006.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM. Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci U S A. 2012;109:E442–E451. doi: 10.1073/pnas.1118803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28:631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, Goldman-Rakic PS. Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science (New York, N Y) 1986;232:232–235. doi: 10.1126/science.3952506. [DOI] [PubMed] [Google Scholar]
- Ryu K, Yokoyama M, Yamashita M, Hirano T. Induction of excitatory and inhibitory presynaptic differentiation by GluD1. Biochem Biophys Res Commun. 2012;417:157–161. doi: 10.1016/j.bbrc.2011.11.075. [DOI] [PubMed] [Google Scholar]
- Shen H, Sesack SR, Toda S, Kalivas PW. Automated quantification of dendritic spine density and spine head diameter in medium spiny neurons of the nucleus accumbens. Brain Struct Funct. 2008;213:149–157. doi: 10.1007/s00429-008-0184-2. [DOI] [PubMed] [Google Scholar]
- Shen HW, Toda S, Moussawi K, Bouknight A, Zahm DS, Kalivas PW. Altered dendritic spine plasticity in cocaine-withdrawn rats. J Neurosci. 2009;29(9):2876–2884. doi: 10.1523/JNEUROSCI.5638-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci. 2009;29:8129–8142. doi: 10.1523/JNEUROSCI.4681-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M, Spence MA, Flodman P. Nuclear and mitochondrial genome defects in autisms. Ann N Y Acad Sci. 2009;1151:102–132. doi: 10.1111/j.1749-6632.2008.03571.x. [DOI] [PubMed] [Google Scholar]
- Suryavanshi PS, Ugale RR, Yilmazer-Hanke D, Stairs DJ, Dravid SM. GluN2C/GluN2D subunit-selective NMDA receptor potentiator CIQ reverses MK-801-induced impairment in prepulse inhibition and working memory in Y-maze test in mice. Br J Pharmacol. 2014;171:799–809. doi: 10.1111/bph.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Ritter B, Verbich D, Sanson C, Bourbonniere L, McKinney RA, McPherson PS. Intersectin regulates dendritic spine development and somatodendritic endocytosis but not synaptic vesicle recycling in hippocampal neurons. J Biol Chem. 2009;284:12410–12419. doi: 10.1074/jbc.M809746200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M. Trans-synaptic interaction of GluRdelta2 and neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- Villasana LE, Klann E, Tejada-Simon MV. Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods. 2006;158:30–36. doi: 10.1016/j.jneumeth.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace W, Bear MF. A morphological correlate of synaptic scaling in visual cortex. J Neurosci. 2004;24:6928–6938. doi: 10.1523/JNEUROSCI.1110-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by slingshot via calcineurin. J Biol Chem. 2005;280:12683–12689. doi: 10.1074/jbc.M411494200. [DOI] [PubMed] [Google Scholar]
- Williams K, Russell SL, Shen YM, Molinoff PB. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993;10:267–278. doi: 10.1016/0896-6273(93)90317-k. [DOI] [PubMed] [Google Scholar]
- Yadav R, Gupta SC, Hillman BG, Bhatt JM, Stairs DJ, Dravid SM. Deletion of glutamate delta-1 receptor in mouse leads to aberrant emotional and social behaviors. PLoS One. 2012;7:e32969. doi: 10.1371/journal.pone.0032969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R, Hillman BG, Gupta SC, Suryavanshi P, Bhatt JM, Pavuluri R, Stairs DJ, Dravid SM. Deletion of glutamate delta-1 receptor in mouse leads to enhanced working memory and deficit in fear conditioning. PLoS One. 2013;8:e60785. doi: 10.1371/journal.pone.0060785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R, Rimerman R, Scofield MA, Dravid SM. Mutations in the transmembrane domain M3 generate spontaneously open orphan glutamate delta1 receptor. Brain Res. 2011;1382:1–8. doi: 10.1016/j.brainres.2010.12.086. [DOI] [PubMed] [Google Scholar]
- Yasumura M, Yoshida T, Lee SJ, Uemura T, Joo JY, Mishina M. Glutamate receptor delta1 induces preferentially inhibitory presynaptic differentiation of cortical neurons by interacting with neurexins through cerebellin precursor protein subtypes. J Neurochem. 2012;121:705–716. doi: 10.1111/j.1471-4159.2011.07631.x. [DOI] [PubMed] [Google Scholar]
- Yizhar O. Optogenetic insights into social behavior function. Biol Psychiatry. 2012;71:1075–1080. doi: 10.1016/j.biopsych.2011.12.029. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zhao JW, Gao ZL, Ji QY, Wang H, Zhang HY, Yang YD, Xing FJ, Meng LJ, Wang Y. Regulation of cofilin activity by CaMKII and calcineurin. Am J Med Sci. 2012;344:462–472. doi: 10.1097/MAJ.0b013e318244745b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.