

Abstract
Polycations that are degradable by reduction of disulfide bonds are developed for applications in delivery of nucleic acids. This paper surveys methods of synthesis of bioreducible polycations and discusses current understanding of the mechanism of action of bioreducible polyplexes. Emphasis is placed on the relationship between the biological redox environment and toxicity, trafficking, transfection activity, and in vivo behavior of bioreducible polycations and polyplexes.
Keywords: polycations, disulfides, gene delivery, polyplexes, bioreducible
1. Introduction
Biodegradable polymers have a long tradition in biomedical applications. In particular, hydrolytically degradable aliphatic polyesters, polyorthoesters, and polyanhydrides have become part of biomaterials, devices, and drug delivery systems that are in widespread clinical use. Polymers that are degradable by reduction of disulfide bonds represent a growing class of biodegradable polymers that are based on alternative mechanism of degradation. Bioreducible polymers received only a limited attention in biomedical applications prior to early 2000's, when they began to be explored in nucleic acid delivery. In contrast to limited biomedical use, numerous disulfide-containing polymers, such as vulcanized rubber, have been used extensively in various industrial applications.[1] Likewise, disulfide linkers have been prominent in drug and protein bioconjugation approaches.[2] Due to the unique dynamic nature of the disulfide bond, which can be easily formed and cleaved, disulfide-containing polymers are finding applications in a broad range of sophisticated adaptable and self-repair materials and in material design based on dynamic combinatorial libraries.[3-5] To limit the scope of this article, we will define bioreducible polycations as those containing multiple disulfide bonds whose cleavage leads to generation of low-molecular-weight degradation products. Thus, for example, polymers that utilize disulfide bonds for conjugation of other molecules, such as drugs, or block copolymers in which the blocks are connected with a disulfide will not be discussed.
Bioreducible polycations are the most widely investigated group of bioreducible polymers in biomedical applications.[6] Bioreducible polycations form polyelectrolyte complexes with nucleic acids and these complexes, termed polyplexes, then protect the nucleic acids and facilitate their transport across cellular membranes (Scheme 1). A delivery vector should protect the nucleic acids in the extracellular space and during intracellular trafficking. Following successful transport to the intracellular site of action; typically cytoplasm or nucleus; the delivery vector should then selectively release the nucleic acids. The contradictory requirements for delivery systems to stably encapsulate nucleic acids in the extracellular space and in enzyme-rich intracellular compartments, while readily release them in other intracellular compartments led to the development of a variety of stimulus-responsive delivery systems. Using a simplified view of the nucleic acid delivery process and the known compartmentalization of disulfide reducing activity in the biological environment, it has been hypothesized that bioreducible polycations can provide such extracellular protection and selective intracellular release of nucleic acids. High disulfide reducing capacity in biological environment for the most part coincides with the locations in which the release of nucleic acids is desirable. Bioreducible polycations have thus been developed as stimulus-responsive polymers to improve efficiency of nucleic acid delivery based on the above understanding of the intracellular trafficking and distribution of reducing activity within the cell and organism.
Scheme 1.

Principle of nucleic acid delivery by bioreducible polycations.
2. Biological redox environment
Bioreducible polycations tap into a complex system of redox processes that are involved in a large number of vital biological pathways and mechanisms in living organism. Importantly for the use of bioreducible polycations in nucleic acid delivery, there is a redox potential gradient between extracellular environment and various subcellular organelles.
The cellular redox state exists in dynamic equilibrium and may change depending on a variety of factors including the stage of the cell cycle and biological status of the cell in general.[7] The redox state of a cell is a sum of multiple redox couples of which glutathione (GSH) is believed to be the most important for the behavior of bioreducible polycations. GSH is the most abundant intracellular thiol. GSH concentrations reach mM levels inside the cell but only μM levels in the blood plasma into which it is continuously secreted through the hepatic vein by the liver.[8, 9]
GSH participates in multiple critical cellular processes, including protein and DNA synthesis, amino acid transport, enzyme activity, metabolism and cell protection against oxidative stress.[10] The intracellular GSH exists either in its oxidized (GSSG) or the dominant reduced form (GSH). The ratio between the reduced and oxidized form of GSH is maintained and determined by the activity of GSH reductase, NADPH concentration, and transaldolase activity. The GSH/GSSG ratio and absolute concentrations of GSH and GSSG are indicators of the overall redox environment of the cell.[7] The GSH concentration and redox ratio (GSH:GSSG) vary among subcellular compartments as a result of the different roles of GSH in the different compartments.[11]
The majority of intracellular GSH is found in the cytosol (1–11 mM), which is also the principal place of GSH biosynthesis.[12-14] The most reducing environment in the cell is present in the nucleus, where it is required for DNA synthesis and repair and to maintain transcription factors in reduced state.[15-17] The nuclear GSH levels are typically greater than the cytosolic levels and can reach up to 20 mM.[11, 17, 18] Another major location of GSH in the cell is in mitochondria (~5 mM).[19, 20] Both mitochondrial and nuclear GSH pools are at least partially independent of the cytosolic pool. In contrast to the highly reducing environment found in the nucleus and mitochondria, the redox environment in endoplasmic reticulum (ER) is oxidizing.[21, 22] The GSH redox ratio in ER typically ranges from 1:1 to 3:1. These values are significantly lower than even within the extracellular space where the GSH redox ratio is ~7:1.[8] Despite some ability to reduce disulfide bonds,[23] the acidic environment of endosomes and lysosomes is generally oxidizing with the GSH redox ratio ranging from 1:1 to 3:1.[24-29] Limited extent of disulfide reduction in endosomes and lysosomes can proceed with the help of reducing enzyme gamma-interferon-inducible lysosomal thiol reductase in combination with cysteine.[29, 30]
Cellular turnover of GSH is associated with its transport out of cells. Transported GSH can participate in reductive reactions that may involve cell membrane and the immediate vicinity of the cell membrane. Furthermore, despite the oxidizing nature of the extracellular environment, the additional presence of redox-active thiols in various proteins and thiol-containing enzymes on the cellular plasma membrane mediate disulfide reduction at the cell surface.[31-33] The redox activity of the cell surface is closely correlated with the levels of oxidoreductase enzymes at the plasma membrane.[34-36] The total levels of redox-active thiols on the surface of cells are estimated to range from ~4 to ~30 nmol/106 cells.[37, 38]
3. Thiol-disulfide exchange reaction
Disulfide bonds and their reactions are important for various biological processes including redox homeostasis, correct assembly of proteins, and metabolic regulation of enzymatic activity.[39] Because of the biological importance of disulfides, cells evolved robust mechanism for the formation, cleavage, and reshuffling of disulfide bonds in proteins. These mechanisms mostly rely on thiol-disulfide exchange reactions. Thiol-disulfide exchange is the reaction of a thiol with a disulfide that involves formation of a new disulfide and a thiol (Equation 1). Thiol-disulfide exchange is the main mechanism of degradation of bioreducible polycations in the cells.
From a chemical perspective, thiol-disulfide exchange is a remarkable reaction. It involves cleavage and formation of a strong covalent bond, yet it proceeds reversibly at room temperature in water at neutral pH with a relatively fast rate. The half-life of the reaction is ~2 h at the thiol and disulfide concentrations in mM range.[40] Thiol-disulfide exchange involves multiple equilibria and all possible thiols, symmetrical disulfides (RSSR), as well as mixed disulfides (RSSR’) as products (Equation 1 and 2).
| (1) |
| (2) |
Thiolate (RS−) is the active nucleophile in the thiol-disulfide exchange and thus the reaction rate depends strongly on the solution pH. The thiol in GSH has a pKa of 9.65 and thus only about 0.1% of it is present as thiolate at neutral pH = 7. The thiol-disulfide exchange is effectively switched-off even under slightly acidic conditions.[41] In addition to pH and acidity of the participating thiols, the rate of thiol-disulfide exchange is greatly affected by steric effects. The steric effects are most pronounced when the thiols are fully substituted at the α carbon to sulfur. For example, the reaction of penicillamine (Pen) with mixed Pen-GSH disulfide is ~100,000-fold slower than the reaction of Pen with GSSG disulfide.[42] The rate of thiol-disulfide exchange also depends on the presence of charged substituents in the participating thiols and disulfides.[43, 44] For example, the rate of reaction of GSH with positively charged peptide disulfides was up to 2 orders of magnitude faster than the rate of the same reaction with negatively charged peptide disulfides.[44]
In cells, thiol-disulfide exchanges are catalyzed by a class of proteins named thiol-disulfide oxidoreductases (Figure 1). The prototypical enzyme of this group is protein disulfide isomerase (PDI). PDI can catalyze thiol-disulfide exchange reactions in a wide range of proteins, peptides and low molecular weight thiols and disulfides.[35] The activity of PDI and other oxidoreductases depends on a pair of cysteines that are usually arranged in a Cys-X-X-Cys motif (where X is any amino acid). This motif is found in a domain that shares structural homology with the small (10 kDa) redox protein thioredoxin.[39] PDI has two such thioredoxin domains (Cys-Gly-His-Cys) that are independent and functionally non-equivalent and they cycle between dithiol and disulfide states.[45] Depending on the redox environment and the nature of the substrate, PDI can catalyze the formation, reduction, or isomerization (reshuffling) of disulfide bonds. If the PDI active site is in the oxidized (disulfide) state, the enzyme oxidizes protein thiols. Conversely, when the active site is in a reduced (thiol) form, PDI can catalyze the reduction of disulfides. In a disulfide reduction (Figure 1), the PDI thiolate attacks the disulfide bond in the oxidized molecule and forms a transient mixed-disulfide bond. In a subsequent exchange, the remaining thiolate attacks the mixed disulfide bond and resolves it. The overall result of this reaction is the oxidation of PDI and the concomitant reduction of the initially oxidized molecule. The redox state of the active site in either product can be regenerated for another catalytic cycle with another redox molecule such as GSH.[39]
Figure 1.
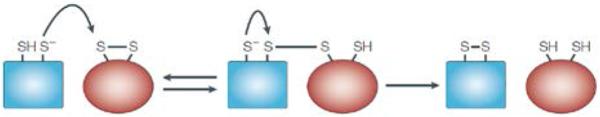
Mechanism of thiol-disulfide exchange by oxidoreductases. Reproduced with permission.[39] Copyright 2002, Nature Publishing Group.
In cells, disulfide bonds are formed predominantly in the lumen of the ER. The oxidation of thiols to form disulfides is catalyzed rapidly in the ER by several different oxidoreductases, including PDI. To a lesser extent, PDI and similar enzymes also participate in thiol-disulfide exchange reactions in other parts of the cell, including plasma membrane. Thiol-disulfide oxidoreductases, such as thioredoxin, are also present in the reducing environment of the cytoplasm where they usually catalyze reduction of disulfide bonds.
4. Synthesis of bioreducible polycations
Two main strategies have been used to prepare polyplexes containing bioreducible polycations. In the first strategy, polyplexes are prepared by directly mixing nucleic acids with the synthesized bioreducible polycation. In the second strategy, polyplexes are first prepared with non-bioreducible cationic molecules and disulfide bonds are introduced subsequently within the formed polyplexes. Although each strategy has its own set of advantages and disadvantages, current trends appear to favor the first method.
Bioreducible polycations can be synthesized by several methods either with or without involvement of sulfur chemistry (Figure 2). Michael polyaddition, radical polymerization, and ring-opening polymerization are all compatible with disulfide-containing monomers and allow direct preparation of bioreducible polycations. Only ring-opening polymerization and Michael polyaddition, however, allow easy direct synthesis of bioreducible polycations with disulfide bonds in the polymer backbone. Oxidation of thiolated oligo- and polyamines or their crosslinking using bioreducible low-molecular-weight crosslinkers are versatile alternatives to the above polymerization methods that can be performed with virtually any oligo- and polyamine. The following sections will discuss the main synthetic methods of bioreducible polycations. The first part focuses on methods that do not utilize sulfur chemistry (i.e., they use molecules that already contain disulfides that will become part of the polycations). The subsequent two parts focus on methods that rely on thiol-disulfide chemistry to prepare bioreducible polycations.
Figure 2.
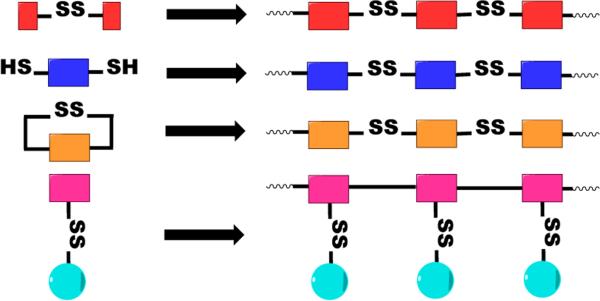
Synthetic approaches to bioreducible polycations.
4.1. Synthesis from disulfide-containing monomers and crosslinkers
4.1.1. Poly(amido amine)s
Poly(amido amine)s (PAA) transpired as one of the most successful and most widely investigated classes of bioreducible polycations. PAA are a family of water-soluble polymers discovered and developed by Paolo Ferruti in the 1960's.[46] PAA are synthesized by Michael polyaddition of primary or secondary aliphatic amines to the CH2=CH bond of bisacrylamides (Figure 3). The Michael polyaddition is typically performed under mild conditions in water or in a mixture of water with alcohols. Michael polyaddition is compatible with monomers that contain variety of functional groups, including disulfides, alcohols, amides, tertiary amines, and guanidinium moieties.[46-49] As a result, bioreducible PAA can be easily prepared using either disulfide-containing amines such as N,N’-dimethylcystamine[50] or bisacrylamides such as N,N’-cystaminebisacrylamide (CBA, 1).[51] Vast majority of the reported bioreducible PAA uses commercially available CBA as the disulfide-containing monomer. Indeed, synthesis of the very first bioreducible PAA was reported in 2004 using the reaction of 2-methylpiperazine with CBA.[52]
Figure 3.

Synthesis of bioreducible PAA by Michael polyaddition and examples of common amines used in the synthesis of linear and branched PAA.
PAA are polyelectrolytes that contain regularly spaced tertiary amines and amides along the polymer chain. Acid-base behavior of PAA depends on the structure of the amine monomer and the nature of the side substituents. Unlike most polycations used in nucleic acid delivery (i.e., PEI), the ionization behavior of individual amines in PAA is usually similar to the behavior of low-molecular-weight amines. In other words, the ionization of the individual amines in PAA is not affected by the ionization state of the entire macromolecule and is thus not significantly influenced by molecular weight of PAA.[53] This behavior is due to the relatively long distance between the ionizable groups in PAA.[46]
Primary amines and secondary diamines act as bifunctional monomers and their use leads to the synthesis of linear PAA. The use of primary diamines (8, 9) leads to either insoluble crosslinked PAA or soluble branched PAA if the reaction is performed at low temperatures or low monomer concentrations. In some cases, primary diamines such as 8 can be used in their mono-protonated form to prepare soluble PAA.[54] Linear bioreducible PAA can be prepared from primary diamines by protecting one of the amines with a protecting group removable in acidic conditions.[55] The use of mixed primary/secondary amines (10, 11) also leads to branched PAA but degree of branching can be controlled by stoichiometry and reaction temperature due to different reactivities of the primary and secondary amines.[56] In this type of Michael polyaddition, the amine reactivity typically decreases in the following order: secondary amine (original) > primary amine >> secondary amine (formed). As a result, reaction of equal molar ratio of a triamine monomer (11) and bisacrylamide monomers produces linear polymers.[57] The topology of these polymers can be also controlled by polymerization temperature because the reactivity of the formed secondary amines is significantly enhanced by elevating reaction temperature above ~50 °C, leading to the formation of hyperbranched polymers.[58] The versatility of Michael polyaddition was recently demonstrated even on monomers such as cyclam (12) and bicyclam CXCR4 antagonist Plerixafor (13) containing multiple secondary amines.[59, 60]
4.1.2. Disulfide crosslinkers
Controlled crosslinking of existing oligoamines and polyamines is among the simplest ways of preparing bioreducible polycations. This approach has been widely applied to the synthesis of bioreducible PEI by reacting oligoethylenimine with small-molecule crosslinkers containing disulfide bond.[61-63] Several commercially available crosslinkers are suitable for this purpose. Three different crosslinker chemistries are commonly used as shown in Figure 4. Bisacrylamide crosslinker (1) reacts with either primary or secondary amines by Michael addition, resulting in the formation of secondary or tertiary amines. Imidoester crosslinker (14) reacts only with primary amines and forms permanently charged amidinate salts. Reactive ester crosslinkers, such as N-hydroxysuccinimidyl ester (15), react with primary or secondary amines to form amides.[64] Thus, crosslinkers 1 and 14 preserve the overall charge of the crosslinked polycation, albeit with the basicity of the amine changed. In contrast, crosslinker 15 decreases the total number of protonizable amines in the polycations. The net positive charge of polycations prepared with 15 is further decreased due to the formation of carboxylic acid groups by partial hydrolysis of the active ester groups during the crosslinking reaction. Because of the multivalent nature of the polycations used and random nature of the crosslinking, the synthesized bioreducible polycations are usually branched or hyperbranched polymers. Although this decreases the reaction control and definition of the product, the simplicity of the crosslinking reaction offers the possibility to synthesize small polycation libraries and to rapidly screen numerous polycations.[62]
Figure 4.

Bioreducible crosslinkers used in the synthesis of bioreducible polycations.
Bioreducible crosslinkers are also applicable as a way of directly stabilizing the polyplexes. In such case, the polyplexes are formed with non-bioreducible oligo- or polycations and treated with a suitable low-molecular-weight crosslinker. The treatment reversibly stabilizes the polyplexes by creating crosslinked network of bioreducible polycations within the polyplex.[65-70] While the polyplex crosslinking is experimentally a simple method, the characterization of the properties of the bioreducible polycations within the polyplexes is challenging and thus rarely performed.
4.1.3. Miscellaneous methods
Alternative synthetic strategies that do not involve reactions on the sulfur include grafting of cationic oligomers to non-charged polymer backbones such as dextran or hyaluronic acid by a disulfide bond.[71-75] A representative example of such approach utilizes dextran modified with disulfide-containing functional groups that are capable of initiating controlled radical polymerization of N,N-dimethylaminoethylmethacrylate (DMAEMA).[73]
Although radical polymerization has been used to synthesize variety of polycations, there are no available reports of direct synthesis of bioreducible polycations. That is despite the fact that unlike thiols, which have high chain-transfer constants, the transfer constants of disulfides are significantly smaller and monomers with disulfides in the sidechain can be readily polymerized by free radical polymerization as well as controlled radical polymerizations such as RAFT and ATRP.
Interesting use of disulfide exchange for stabilization of polyplexes based on hyperbranched bioreducible PAA was reported by the You lab. The polyplexes were heated to 50 °C for 15 min to trigger disulfide-disulfide exchange that resulted in the internal crosslinked structure within the polyplexes.[76]
4.2. Synthesis by oxidation of thiols
4.2.1. Polypeptides
Pioneered by the Rice lab, bioreducible polypeptides represent one of the earliest examples of bioreducible polycations used in gene delivery.[77] Synthesis of cationic peptides that contain two or more cysteine residues and subsequent oxidation of the cysteine thiols leads to high molecular weight bioreducible polypeptides. The cysteine thiols can be oxidized using either oxygen or other mild oxidizing agents such as dimethylsulfoxide.[78-81] When higher stability of the disulfide bonds in reducing environment is required, penicillamine with sterically hindered thiol can be used in place of cysteine (Figure 5). Bioreducible polypeptides combine the exquisite control of amino acid sequence of peptides with the benefits of high molecular weight polycations in protecting nucleic acids. This approach represents a viable alternative of preparing high molecular weight repetitive polypeptides with a better control over the amino acid sequence and molecular weight than traditional chemical synthesis. This approach is also an easier alternative to polypeptide synthesis using genetic engineering methods, which provide the ultimate control over the properties of polypeptides.[82]
Figure 5.
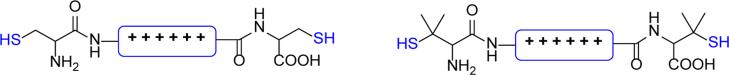
Cationic peptides used in the synthesis of bioreducible polypeptides based on cysteine (left) and penicillamine with sterically hindered thiol (right).
The polypeptides can be synthesized from multiple different peptide building blocks and thus present a straightforward way of preparing polymers that combine polyplex-forming capability with a biological function of the used peptides.[81, 83-86] Examples of such multifunctional bioreducible polypeptides include those that combine membrane-active peptides rich in arginine (e.g., Cys-(Arg)8-Cys) or endosomolytic peptides rich in histidine (e.g., Cys-Lys-(His)3-Lys-(His)3-Cys) with nuclear localizating peptides (e.g., Cys-Gly-Ala-Gly-Pro-(Lys)3-Arg-Lys-Val-Cys).[87, 88] This approach can be also used to incorporate non-peptide functionalities such as poly(ethylene glycol) (PEG) and carbohydrates into the bioreducible polypeptides.[77, 83, 89-91] A notable weakness of these multifunctional bioreducible polypeptides is the random distribution of the individual peptide blocks along the polymer chain. The Rice lab recently reported an improved synthesis, named iterative reducible ligation that now allows controlling the order of the individual peptide blocks in bioreducible polypeptides.[92, 93]
Similar to the crosslinking approaches discussed above, bioreducible polypeptides can be prepared in situ after formation of polyplexes using cysteine-flanked cationic peptides. When polyplexes are formed using this method, the local concentration of the thiol groups increases substantially, which leads to an easy aerial oxidation without the need for any additional oxidizing agents. One of the advantages of this approach, also called DNA template-assisted polymerization, is that using peptides with three and more cysteine residues results in the formation of polyplexes stabilized by disulfide crosslinks without the need for additional reagents or synthetic steps.[94]
4.2.2. Thiolated polycations
Similar to bioreducible polypeptides, introduction of thiol groups to the structure of polycations allows easy synthesis of bioreducible polycations by thiol oxidation. Oxidation of thiol-containing polycations offers tremendous versatility because virtually any polycation with primary, secondary or tertiary amines can be thiolated.[95] Various methods of thiolation have been reported (Figure 6). Among the most common are methods that use reaction of iminothiolane with primary amines of a polycation such as PEI and poly(L-lysine) (PLL).[96-98] Using iminothiolane conserves the number of positive charges of the polycation and is applicable to both conventional synthesis of polycations as well as in situ formation of bioreducible polycations after polyplex formation with thiolated polycations. For example, the Kataoka lab reported synthesis of thiolated block copolymer of PEG and PLL using iminothiolane. After formation of polyplexes with siRNA, oxidation of the thiols in the core of the polyplexes yielded bioreducible PLL.[99] Alternative thiolation chemistries that were used to synthesize bioreducible PEI include the use of thiirane and methylthiirane, which provides polycations with sterically hindered thiols (Figure 6).[100, 101]
Figure 6.

Common thiolation methods of polyamines.
Several attempts have been made to improve the above random thiolation strategies by precisely controlling the location and number of thiol groups in polycations. Synthesis of well-defined bioreducible linear PEI by oxidation of α,ω-bisthiol-oligoethylenimines has been described by Park and colleagues.[102] Similar approach that relies on oxidation of α,ω-bisthiol polymers has been applied to methacrylate-based polycations by our lab. Taking advantage of the ability of reversible addition-fragmentation chain transfer (RAFT) polymerization to prepare polymers with well-defined endgroups, we synthesized N,N-dimethylaminoethyl methacrylate (DMAEMA) copolymers with terminal thiol groups (Figure 7). Linear bioreducible poly(DMAEMA) polycations were then prepared by oxidation of the terminal thiols with dimethylsulfoxide.[103, 104]
Figure 7.
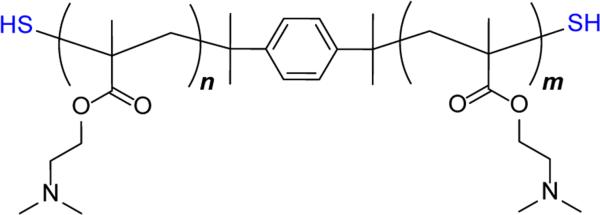
Structure of well-defined α,ω-bisthiol polycation based on N,N-dimethylaminoethyl methacrylate prepared by RAFT polymerization.[105]
4.3. Synthesis by thiol-disulfide exchange polymerization of cyclic disulfides
Polymerization of cyclic cationic disulfides by thiol-disulfide exchange is another method of synthesis of bioreducible polycations which includes reactions directly involving sulfur. A prototypical example of such reaction is the ring opening polymerization of lipoic acid or similar cyclic disulfides like asparagusic acid.[106] Historically, probably the first example of a bioreducible polycation prepared by this method has been described by Balakirev and colleagues in 2000.[107] The authors prepared a cationic derivative of lipoic acid which was polymerized by opening the 1,2-dithiolane ring using a thiol-containing peptide or a reducing agent dithiothreitol. With the exception of polymerized liposomes,[108] the use of this nearly forgotten polymerization has been recently promoted by the Matile lab who used it to synthesize guanidinium-rich cell-penetrating polydisulfides (Figure 8).[109] An alternative example of thiol-disulfide exchange polymerization not based on lipoic acid was reported by the Miller lab who used cyclic disulfides based on spermine.[110] Overall, this synthetic method has a number of attractive features that makes it a prime target for further exploration in nucleic acid delivery.
Figure 8.

Thiol-disulfide exchange polymerization of cationic lipoic acid derivative.[109]
5. Nucleic acid delivery properties of bioreducible polycations
5.1. Cellular uptake and trafficking
The main function of bioreducible polycations in nucleic acid delivery is to safely deliver the nucleic acids to their intracellular site of action. This requires that polyplexes are efficiently taken up by cells and routed through appropriate trafficking pathways to avoid premature release and degradation of the nucleic acids (Scheme 1). Current understanding of bioreducible polyplexes suggests that their uptake and intracellular trafficking follows similar principles as conventional non-bioreducible polyplexes.[111-113] Here, we will focus mostly on the unique features of bioreducible polyplexes.
Similar to conventional polyplexes, bioreducible polyplexes rely on a charge-mediated attachment to the heparan sulfate proteoglycans on the cell surface to facilitate internalization.[114] In addition, however, oxidoreductases (e.g., PDI) and redox-active thiols on the surface of plasma membrane may participate in thiol-disulfide exchanges with bioreducible polycations and affect cellular uptake. Evidence for the involvement of PDI in the uptake of disulfide-containing polycations has been reported as early as 1990.[32] The ability of the plasma membrane thiols to participate in thiol-disulfide exchange has been also confirmed in multiple studies with films based on bioreducible polycations.[56, 115-118] Involvement of the surface thiols in cell uptake of polyplexes based on bioreducible PAA was substantiated in our recent study.[119] Cell uptake of bioreducible polyplexes was significantly higher than uptake of non-bioreducible polyplexes based on chemically similar PAA. Blocking plasma membrane thiols with cell impermeable thiol blocker DTNB abrogated the enhanced uptake of the bioreducible polyplexes. Increased cell uptake of bioreducible polyplexes was also reported by the Bae lab in a study that compared bioreducible PEI with a control non-degradable PEI.[96] Recent work by the Leroux lab with PAMAM dendrimers implicated cell surface oxidoreductases in the process of disulfide reduction and suggested their involvement in enhanced cell uptake of bioreducible polyplexes.[120] Although direct comparison with chemically similar polycations was not reported, several studies by the S. W. Kim lab have shown greatly enhanced uptake of polyplexes based on a series of bioreducible PAA when compared with PEI polyplexes.[121, 122] Despite the growing supporting evidence, it is fair to mention that not all studies support the involvement of surface thiols in cell uptake of disulfide-containing polymers and polyplexes.[23, 123]
Multiple endocytic pathways may participate in the internalization of polyplexes but only some of them facilitate transfection. In one of rare mechanistic studies of intracellular trafficking of bioreducible polyplexes, Braeckmans and colleagues have shown flotillin-dependent endocytosis and a PAK1-dependent phagocytosis-like mechanism as the two major cellular internalization pathways.[124, 125] However, it was the flotillin-1-dependent endocytosis that was responsible for transfection of the bioreducible PAA polyplexes. Clathrin- and caveolae-mediated endocytotic pathways had only limited role in uptake and trafficking of the bioreducible polyplexes. These findings were similar to a previous report on PEI polyplexes, suggesting that the presence of disulfide bonds does not impart unique trafficking properties.[114] In contrast, results reported by Shen and colleagues point to caveolae- and PI3K-mediated endocytosis as those responsible for transfection of polyplexes based on bioreducible PAA.[126] Different chemical structures of the used polycations and different cell lines may explain the reported differences.
Following endocytic uptake, polyplexes have to escape from the endo/lysosomal pathway into cytoplasm in order to avoid routing into lysosomes. Once in the cytoplasm, the polyplexes are exposed to a highly reducing environment in which bioreducible polycations can be degraded. As a result, the nucleic acid can be efficiently released from the polyplexes. Multiple studies support rapid disulfide reduction and cytoplasmic release of nucleic acids from bioreducible polyplexes. Using confocal microscopy, Kim and colleagues showed more efficient cytoplasmic release of siRNA from polyplexes based on bioreducible PAA when compared with polyplexes based on PEI. The authors suggested that strong electrostatic interactions and lack of intracellular degradation of PEI was behind the observed differences.[127] Further evidence for efficient cytoplasmic siRNA and DNA release from bioreducible polyplexes was provided by a study with arginine-grafted bioreducible PAA (Figure 9).[122, 128] The study corroborated the relationship between cytoplasmic GSH and siRNA release by showing that cytoplasmic localization of siRNA was diminished after GSH depletion with buthionine sulphoximine (BSO). No such effect was observed for PEI polyplexes. Similar effect of bioreducible polycations on intracellular release of plasmid DNA was reported by the Vinogradov lab.[129] Confocal microscopy showed that more diffused signal of fluorescently labeled DNA was observed in the cytoplasm when the DNA was delivered by bioreducible PEI than when delivered by non-reducible PEI. The Leong lab developed FRET-based method to study release of DNA from polyplexes.[130] Recent publication indicates that the half-life of intracellular DNA release from bioreducible PAA polyplexes is between 5 and 17 hours, depending on the method of polyplex preparation.[131] Unfortunately, no direct comparison between bioreducible and non-bioreducible polyplexes was conducted to allow determination of the effect of intracellular degradation of bioreducible polycations on the rate of DNA release.
Figure 9.
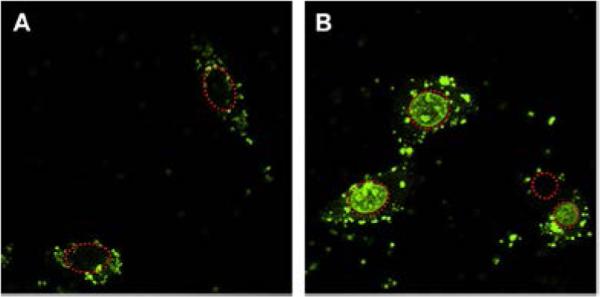
Improved cytoplasmic release of DNA from polyplexes prepared with bioreducible PAA. Intracellular distribution of fluorescently labeled DNA delivered with PEI (A) or bioreducible PAA (B). (red dashed circles indicate cell nuclei) Reproduced with permission.[122] Copyright 2009, Elsevier Ltd.
5.2. Transfection activity
The most often cited rationale for the use of bioreducible polycations is their potential to facilitate intracellular release of nucleic acids as a result of fast intracellular disulfide reduction. This in turn is expected to improve transfection due to enhanced availability of the nucleic acids. Bioreducible polycations have been successfully used to transfect a variety of nucleic acids (plasmid DNA, siRNA, mRNA, miRNA), often with superior transfection activity when compared with commercial transfection agents. Yet, despite intensive research spanning more than a decade, the evidence to support the main underlying assumption is weak and the available studies paint more complicated picture of the mechanism of action of bioreducible polycations than originally conceived.
We conducted a head-to-head comparison of transfection properties of chemically analogous bioreducible and non-bioreducible PAA with the goal of assessing the role of bioreducibility on polyplex transfection.[132] We have evaluated polyplexes of plasmid DNA, mRNA, siRNA, and antisense oligonucleotides (AON) in a panel of human pancreatic cell lines containing different levels of innate GSH (Table 1). Overall, no clear relationship was found between cellular GSH concentration and relative transfection of bioreducible polyplexes. Multiple previous studies attempted to demonstrate such association between intracellular GSH and activity of bioreducible polyplexes by biochemically altering GSH levels in the cells. The evidence overall suggests that biochemically changing cellular GSH levels leads to changes in transfection, albeit the differences are usually small. In general, artificially increasing the cellular GSH leads to an increase in transfection activity of bioreducible polyplexes.[51, 79, 107] Contradicting reports that show a transfection decrease, however, are also available.[80]
Table 1.
Relative transfection of bioreducible PAA in a panel of pancreatic cancer cell line with increasing cellular GSH content.a (Adapted from [132])
| Nucleic acid | Cell line (GSH concentration) |
|||
|---|---|---|---|---|
| Panc-1 (1.1 mM) | MiaPaCa (2.4 mM) | AsPC-1 (4.7 mM) | Panc-28 (7.5 mM) | |
| Plasmid DNA | 0.9 | 3.2 | 5.4 | 0.1 |
| mRNA | 5.6 | 8.8 | 2.0 | 54 |
| siRNA | 0.7 | 1.0 | 0.9 | 0.8 |
| AON | 0.7 | 1.2 | 1.6 | 1.3 |
Values in the table were calculated by dividing transfections of polyplexes made with bioreducible PAA with transfections of non-bioreducible PAA polyplexes.
When evaluating the effect of the type of delivered nucleic acid, we have found that only mRNA benefited consistently across the panel of cell lines from using bioreducible polycations (Table 1). The bioreducible PAA mediated mRNA transfection that was 2-54-fold higher than transfection mediated by the non-bioreducible PAA. Bioreducible PAA showed increased plasmid DNA transfection in only 2 out of 4 tested cell lines. Likewise, results from the Engbersen lab show that increasing disulfide content in bioreducible PAA does not result in straightforward effect on transfection.[133] In our studies, bioreducible PAA showed no benefit over non-bioreducible PAA in delivery of small nucleic acids (siRNA, AON), an observation confirmed in another recent study.[119] It is worth noting that mRNA polyplexes had by far the highest stability against polyelectrolyte exchange. Polyelectrolyte exchange reactions are widely believed to be the main mechanism of intracellular release of nucleic acids from polyplexes. Several other studies confirm that using bioreducible polyplexes benefits mRNA transfection when compared with non-degradable polycations.[79, 80]
The above discussion confirms high transfection ability of bioreducible polyplexes. There is one class of bioreducible polyplexes, however, in which the presence of disulfides results in markedly decreased transfection. Crosslinking polyplexes with bioreducible crosslinkers (Figure 4) often leads to several orders of magnitude loss of transfection activity. For example, the Kissel lab reported more than a 1,000-fold decrease in luciferase expression of PEI polyplexes crosslinked with (15).[70] Similar decrease in transfection was observed by the Seymour lab for PLL polyplexes crosslinked with (14).[66] An interesting aspect of these polyplexes is that the most likely reason for the decreased transfection was a compromised endosomal escape due to restricted polycation mobility caused by to the covalent crosslinking.[134, 135] Pre-treatment with a reducing agent or direct injection of the polyplexes into the cytoplasm or the nucleus of the cells restored transfection activity of the crosslinked polyplexes. From the perspective of the site of disulfide reduction, these findings suggest that extracellular or endosomal disulfide reduction were not major contributors to disulfide reduction in the crosslinked polyplexes.
5.3. Toxicity
Toxicity of polycations is a well-known occurrence and a major concern for clinical application of polycation-based nucleic acid delivery.[136-138] The half-maximal cytotoxic dose (IC50) of PEI in cell viability assays in vitro is typically in the range 5-20 μg/mL. The maximum tolerated dose (MTD) of linear PEI (22 kDa) in mice is reported to be around 2.4 mg/kg body weight.[139] For comparison, MTD in mice of a common anticancer agent doxorubicin is 8-10 mg/kg body weight and MTD of another widely used anticancer drug cisplatin is 4 mg/kg body weight.[140, 141]
A generally accepted 2-phase model of polycation cytotoxicity has been proposed by the Mikos lab[136] and corroborated by the work of the Moghimi lab.[138, 142] Phase 1 of polycation cytotoxicity occurs within the first hour of exposure and includes disruption of cellular membranes and induction of membrane leakage due to pore formation.[138, 143, 144] One of the characteristic features of this phase is the release of lactate dehydrogenase and redistribution of phosphatidylserine from the inner plasma membrane to the outer cell surface. Phase 2 cytotoxicity originates from intracellular effects of polycations on mitochondrial membrane, which lead to induction of apoptosis demonstrated by significant activation of the effector caspase 3.[145-147] Another likely contributing factor to the apoptotic effect of polycations is leakage of lysosomal proteases into cytoplasm as a result of lysosomal lysis by the polycations.[148]
Toxicity of polycations is strongly dependent on their molecular weight and charge density. Rapid intracellular degradation of bioreducible polycations should thus contribute to decreased cytotoxicity. The extent of cytotoxicity reduction, however, will strongly depend on the kinetics and location of the degradation of the bioreducible polycations. If degradation occurs at least partially in the extracellular space, as suggested by some,[120, 149] both phases of polycation cytotoxicity may be positively affected. If the degradation occurs in endo/lysosomes, then only phase 2 events may be positively affected, while phase 1 effects will be observed. If degradation occurs in the cytoplasm, then only phase 2 events associated with mitochondrial damage, but not those associated with lysosomal lysis, may be positively affected. Little can be gained if the degradation kinetics is much slower than the kinetics of toxicity. It is thus unlikely that phase 1 cytotoxicity, which occurs within the first hour of exposure, can be reduced to any significant extent by the use of bioreducible polycations unless they are rapidly degraded in the extracellular space.
Most published reports show that bioreducible polycations do indeed exhibit decreased in vitro cytotoxicity when compared with control non-bioreducible polycations.[51, 63, 102, 122, 133, 150] While IC50 values are not always reported, bioreducible polycations often exhibit 5-25-fold higher IC50 when compared with non-bioreducible polycations of similar chemical structure and molecular weight. For example, the cytotoxicity of bioreducible polycation prepared from thiolated PEI was 8–19 times lower than that of the control PEI.[96] In our recent study, bioreducible PAA showed IC50 that were, depending on the cell line, about 12- to 25-fold higher than IC50 of comparable non-bioreducible PAA.[151] The cytotoxicity was closely tied to disulfide content in bioreducible PAA and to total GSH levels in the used cells (Figure 10). Bioreducible PAA also exhibited greatly decreased activation of caspases and extent of apoptosis. In contrast to the beneficial effect on phase 2 cytotoxicity, bioreducibility had only negligible effect on phase 1 cytotoxicity, suggesting a primarily intracellular site of degradation of the bioreducible PAA. Surprisingly, plasma membrane thiols selectively increased cytotoxicity of bioreducible PAA, while having no effect on the cytotoxicity of non-reducible PAA. This was most likely due to increased cell uptake and membrane perturbation by bioreducible PAA due to the formation of mixed disulfides with cell surface thiols.
Figure 10.

Cytotoxicity of bioreducible polycations decreases with increasing disulfide content and cellular GSH levels. (Adapted from [151])
Decreased toxicity of bioreducible polycations has been well-documented in vitro but corresponding evidence that would demonstrate similar decrease in toxicity in vivo is lacking. Our own anecdotal unpublished data suggest that bioreducible polycations have comparable acute toxicity in vivo when compared with non-bioreducible control polycations with similar chemical structure.
5.4. In vivo properties
The above discussion demonstrated beneficial features of bioreducible polycations in delivery of nucleic acids in vitro. Majority of bioreducible polycations is intended for use in systemic delivery of nucleic acids. It is therefore important to establish if their advantageous features in vitro translate into improved properties in vivo. In particular, we need to address the following questions that address the validity of the main rationale behind the use of bioreducible polycations (i.e., extracellular stabilization combined with easy intracellular degradation). First, are disulfides in bioreducible polycations sufficiently stable during systemic circulation to protect the nucleic acids and facilitate delivery to target tissues? Second, does rapid intracellular degradation of bioreducible polycations result in enhanced transfection activity in vivo? Third, does the decrease in toxicity of bioreducible polycations observed in vitro translate into improved safety profile in vivo? These questions would be best answered in head-to-head comparative studies in which bioreducible polycations are assessed against non-bioreducible polycations with similar chemical structure and properties. Unfortunately, such studies are exceedingly rare and there is currently insufficient amount of evidence to provide definitive answer to the above questions.
Although the environment in blood plasma is mostly oxidizing, the presence of low concentrations of small molecule thiols (cysteine and GSH) means that degradation of bioreducible polycations cannot be completely ruled out. Furthermore, abundant protein thiols (e.g., serum albumin) can chemically react with bioreducible polycations by thiol-disulfide exchange and potentially modify the pattern with which proteins bind to polyplexes and thus alter their in vivo fate. Several past studies with bioreducible polycations support sufficient stability of the disulfides in plasma circulation.[66, 70, 152] Experience with drug-antibody conjugates such as Mylotarg in which the drug was conjugated to the antibody using a linker with sterically hindered disulfide further supports the compatibility of disulfide-containing delivery systems with systemic delivery.[2] Extended plasma circulation was successfully demonstrated using polyplexes based on bioreducible polypeptides prepared from Cys(Lys)10Cys and PLL polyplexes based on the use of disulfide crosslinkers.[66, 152, 153] For example, polyplexes based on disulfide crosslinker 14 exhibited plasma circulations times that increased with increasing content of the crosslinker. Almost 40% of the injected dose was found in circulation for highly crosslinked polyplexes, whereas only 5.8% of control non-crosslinked polyplexes remained in circulation 30 min post-injection.[66] These findings were also confirmed for disulfide-crosslinked PEI polyplexes.[70] Several recent biodistribution studies demonstrated the ability of bioreducible PAA to deliver nucleic acids to tumors.[154, 155] Improved transfection activity of bioreducible polyplexes was demonstrated by the Wagner lab. Head-to-head comparison of PEI polyplexes stabilized with either bioreducible crosslinker 15 or analogous non-reducible crosslinker showed not only increased transfection but also a longer duration of transgene expression after intratumoral injection. Targeted gene expression in tumor tissue was also achieved after systemic administration of the bioreducible polyplexes.[156]
5.5. In vivo delivery of therapeutic nucleic acids
Growing number of studies report successful application of bioreducible polycations to deliver therapeutic nucleic acids. Plasmid DNA and siRNA have been successfully delivered with bioreducible polycations and demonstrated promise in the treatment of various diseases in a range of animal models.
Notable progress in the application of bioreducible PAA has been made by the S. W. Kim lab in multiple therapeutic areas. Plasmid encoding hypoxia-inducible vascular endothelial growth factor (RTP-VEGF) was delivered by bioreducible PAA in a rabbit myocardial infarct model. The results showed a 4-fold increase in VEGF protein expression in the region of the infarct compared to control PEI polyplexes.[157] A significant decrease in blood glucose level in type II diabetic mice was observed after a single intravenous administration of polyplexes prepared with exendin-4 plasmid and bioreducible PAA grafted with arginine.[158] Another promising application of the arginine-grafted bioreducible PAA was reported for in vivo delivery of human erythropoietin plasmid DNA to produce long-term therapeutic erythropoiesis to treat anemia. Intravenous administration of a single dose of the polyplexes significantly increased hematocrit levels over a 60-day period in rats.[159] The same polyplexes were also applied to treat myocardial infarction in rats.[160]
Bioreducible polypeptide prepared from Cys-(D-Arg)9-Cys was used to deliver heme oxygenase-1 gene to treat hypoxic-ischemic brain injury in rats.[161] The same bioreducible polypeptide was also successfully applied to the delivery of suicide gene herpes simplex virus-thymidine kinase (HSV-tk) in combination with small molecule drug ganciclovir to treat spinal cord tumors in rats by intratumoral administration.[162]
In addition to plasmid DNA, bioreducible polycations have been successfully used to deliver therapeutic siRNA. Bioreducible PAA modified with cholesterol to increase stability of siRNA polyplexes was used for systemic VEGF siRNA delivery to successfully treat breast cancer in nude mice.[163] Bioreducible PEI was conjugated with N-acetylglucosamine and indocyanine-green to deliver TGFβ1 siRNA to image and treat liver fibrosis in mice.[164] The above described bioreducible polypeptide based on D-Arg could be also used for intratumoral administration of the polyplexes containing VEGF siRNA in a subcutaneous squamous cell carcinoma tumor model.[165] Wang and Lin et al. have applied bioreducible PEI in human telomerase reverse transcriptase (hTERT) siRNA delivery to treat subcutaneous HepG2 liver tumor in nude mice. The in vivo study showed significant inhibition of tumor growth after intratumoral administration of the bioreducible polyplexes compared with the control PEI.[63]
6. Conclusions
Bioreducible polycations continue to justify their rising importance in nucleic acid delivery. Growing body of literature demonstrate their safety and suitability to deliver a broad range of nucleic acids both in vitro and in vivo. Multiple therapeutic application areas of bioreducible polycations emerged ranging from cancer to cardiovascular diseases. Yet, despite the tremendous progress in the last decade, more than a few fundamental questions about the mechanism of action of bioreducible polyplexes remain. Future growth will, in great part, depend on deeper understanding of the relative contributions of the various cellular and body redox pools to the properties and behavior of bioreducible polyplexes. Increased focus on in vivo properties and applications in additional disease models will further underscore the future success of this class of nucleic acid delivery vectors.
Acknowledgements
This study was supported in part by the National Institutes of Health (CA 109711, EB 014570).
Biography
 David Oupický is a Parke-Davis Professor of Pharmaceutics and Co-Director of the Center for Drug Delivery and Nanomedicine at the University of Nebraska Medical Center (UNMC). He received his Ph.D. in Macromolecular Chemistry with Prof. Karel Ulbrich at the Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic. He was a postdoc at the University of Birmingham where he worked with Prof. Len Seymour on non-viral gene delivery. After 10 years as a faculty at Wayne State University in Detroit, he joined UNMC in 2013. His research interests include synthesis of novel polymers and development of drug and nucleic acid delivery systems.
David Oupický is a Parke-Davis Professor of Pharmaceutics and Co-Director of the Center for Drug Delivery and Nanomedicine at the University of Nebraska Medical Center (UNMC). He received his Ph.D. in Macromolecular Chemistry with Prof. Karel Ulbrich at the Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic. He was a postdoc at the University of Birmingham where he worked with Prof. Len Seymour on non-viral gene delivery. After 10 years as a faculty at Wayne State University in Detroit, he joined UNMC in 2013. His research interests include synthesis of novel polymers and development of drug and nucleic acid delivery systems.
 Jing Li studied Pharmaceutical Engineering at Southeast University in Nanjing and completed her Ph.D. in 2012 at Wayne State University in Detroit under the mentorship of Prof. Oupický. She is currently a Postdoctoral Fellow at the Center for Drug Delivery and Nanomedicine at UNMC. Her research interests are focused on the design of polymeric nucleic acid delivery vectors and development of combination therapeutic approaches that target the chemokine networks in cancer and inflammatory conditions.
Jing Li studied Pharmaceutical Engineering at Southeast University in Nanjing and completed her Ph.D. in 2012 at Wayne State University in Detroit under the mentorship of Prof. Oupický. She is currently a Postdoctoral Fellow at the Center for Drug Delivery and Nanomedicine at UNMC. Her research interests are focused on the design of polymeric nucleic acid delivery vectors and development of combination therapeutic approaches that target the chemokine networks in cancer and inflammatory conditions.
Footnotes
This paper provides an overview of the current status of research into the chemistry of bioreducible polycations and their use in nucleic acid delivery.
References
- 1.Bang EK, Lista M, Sforazzini G, Sakai N, Matile S. Chem. Sci. 2012;3:1752. [Google Scholar]
- 2.Wu AM, Senter PD. Nat. Biotechnol. 2005;23:1137. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 3.Corbett PT, Leclaire J, Vial L, West KR, Wietor J-L, Sanders JK, Otto S. Chem. Rev. 2006;106:3652. doi: 10.1021/cr020452p. [DOI] [PubMed] [Google Scholar]
- 4.Yoon JA, Kamada J, Koynov K, Mohin J, Nicolaÿ R, Zhang Y, Balazs AC, Kowalewski T, Matyjaszewski K. Macromolecules. 2011;45:142. [Google Scholar]
- 5.Otto S, Furlan RL, Sanders JK. Science. 2002;297:590. doi: 10.1126/science.1072361. [DOI] [PubMed] [Google Scholar]
- 6.Son S, Namgung R, Kim J, Singha K, Kim WJ. Acc. Chem. Res. 2012;45:1100. doi: 10.1021/ar200248u. [DOI] [PubMed] [Google Scholar]
- 7.Schafer FQ, Buettner GR. Free Radic. Biol. Med. 2001;30:1191. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 8.Jones DP, Carlson JL, Samiec PS, Sternberg P, Jr., Mody VC, Jr., Reed RL, Brown LA. Clin. Chim. Acta. 1998;275:175. doi: 10.1016/s0009-8981(98)00089-8. [DOI] [PubMed] [Google Scholar]
- 9.Filomeni G, Rotilio G, Ciriolo MR. Biochem. Pharmacol. 2002;64:1057. doi: 10.1016/s0006-2952(02)01176-0. [DOI] [PubMed] [Google Scholar]
- 10.Meister A, Anderson ME. Annu. Rev. Biochem. 1983;52:711. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 11.Smith CV, Jones DP, Guenthner TM, Lash LH, Lauterburg BH. Toxicol. Appl. Pharmacol. 1996;140:1. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 12.Gilbert HF. Adv. Enzymol. 1990;63:69. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 13.Kosower NS, Kosower EM. Int. Rev. Cytol. 1978;54:109. doi: 10.1016/s0074-7696(08)60166-7. [DOI] [PubMed] [Google Scholar]
- 14.Hwang C, Lodish HF, Sinskey AJ. Methods Enzymol. 1995;251:212. doi: 10.1016/0076-6879(95)51123-7. [DOI] [PubMed] [Google Scholar]
- 15.Arrigo AP. Free Radic. Biol. Med. 1999;27:936. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Bishopric NH, Discher DJ, Murphy BJ, Webster KA. Mol. Cell Biol. 1996;16:1035. doi: 10.1128/mcb.16.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellomo G, Vairetti M, Stivala L, Mirabelli F, Richelmi P, Orrenius S. Proc. Natl. Acad. Sci. U. S. A. 1992;89:4412. doi: 10.1073/pnas.89.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soboll S, Grundel S, Harris J, Kolb-Bachofen V, Ketterer B, Sies H. Biochem. J. 1995;311(Pt 3):889. doi: 10.1042/bj3110889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wahllander A, Soboll S, Sies H, Linke I, Muller M. FEBS Lett. 1979;97:138. doi: 10.1016/0014-5793(79)80069-1. [DOI] [PubMed] [Google Scholar]
- 20.Lash LH, Putt DA, Matherly LH. J. Am. Soc. Nephrol. 2002;13:292A. [Google Scholar]
- 21.Braakman I, Helenius J, Helenius A. EMBO J. 1992;11:1717. doi: 10.1002/j.1460-2075.1992.tb05223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang C, Sinskey AJ, Lodish HF. Science. 1992;257:1496. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Chen H, Vlahov IR, Cheng J-X, Low PS. Proc. Nat. Acad. Sci. U.S.A. 2006;103:13872. doi: 10.1073/pnas.0601455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen WC, Ryser HJ, LaManna L. J. Biol. Chem. 1985;260:10905. [PubMed] [Google Scholar]
- 25.Fivaz M, Vilbois F, Thurnheer S, Pasquali C, Abrami L, Bickel PE, Parton RG, van der Goot FG. EMBO J. 2002;21:3989. doi: 10.1093/emboj/cdf398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phan UT, Arunachalam B, Cresswell P. J. Biol. Chem. 2000;275:25907. doi: 10.1074/jbc.M003459200. [DOI] [PubMed] [Google Scholar]
- 27.Saito G, Amidon GL, Lee KD. Gene Ther. 2003;10:72. doi: 10.1038/sj.gt.3301859. [DOI] [PubMed] [Google Scholar]
- 28.Collins DS, Unanue ER, Harding CV, Immunol J. 1991;147:4054. [PubMed] [Google Scholar]
- 29.Go YM, Jones DP. Biochim. Biophys. Acta. 2008;1780:1273. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. J. Controlled Rel. 2011;152:2. doi: 10.1016/j.jconrel.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 31.Ryser HJ, Mandel R, Ghani F. J. Biol. Chem. 1991;266:18439. [PubMed] [Google Scholar]
- 32.Feener EP, Shen WC, Ryser HJP. J. Biol. Chem. 1990;265:18780. [PubMed] [Google Scholar]
- 33.Sahaf B, Heydari K, Herzenberg LA, Herzenberg LA. Arch. Biochem. Biophys. 2005;434:26. doi: 10.1016/j.abb.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 34.Donoghue N, Hogg PJ. Methods Enzymol. 2002;348:76. doi: 10.1016/s0076-6879(02)48628-4. [DOI] [PubMed] [Google Scholar]
- 35.Gilbert HF. J. Biol. Chem. 1997;272:29399. doi: 10.1074/jbc.272.47.29399. [DOI] [PubMed] [Google Scholar]
- 36.Donoghue N, Yam PTW, Jiang XM, Hogg PJ. Protein Sci. 2000;9:2436. doi: 10.1110/ps.9.12.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang XM, Fitzgerald M, Grant CM, Hogg PJ. J. Biol. Chem. 1999;274:2416. doi: 10.1074/jbc.274.4.2416. [DOI] [PubMed] [Google Scholar]
- 38.Laragione T, Bonetto V, Casoni F, Massignan T, Bianchi G, Gianazza E, Ghezzi P. Proc. Natl. Acad. Sci. U. S. A. 2003;100:14737. doi: 10.1073/pnas.2434516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sevier CS, Kaiser CA. Nat. Rev. Mol. Cell Biol. 2002;3:836. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- 40.Szajewski RP, Whitesides GM. J. Am. Chem. Soc. 1980;102:2011. [Google Scholar]
- 41.Otto S, Furlan RLE, Sanders JKM. Drug Discov. Today. 2002;7:117. doi: 10.1016/s1359-6446(01)02086-4. [DOI] [PubMed] [Google Scholar]
- 42.Rabenstein DL, Theriault Y. Can. J. Chem. 1984;62:1672. [Google Scholar]
- 43.Wu C, Wang S, Brülisauer L, Leroux J-C, Gauthier MA. Biomacromolecules. 2013;14:2383. doi: 10.1021/bm400501c. [DOI] [PubMed] [Google Scholar]
- 44.Wu CL, Belenda C, Leroux JC, Gauthier MA. Chem.-Eur. J. 2011;17:10064. doi: 10.1002/chem.201101024. [DOI] [PubMed] [Google Scholar]
- 45.Lyles MM, Gilbert HF. J. Biol. Chem. 1994;269:30946. [PubMed] [Google Scholar]
- 46.Ferruti P. J. Pol. Sci. Pol. Chem. 2013;51:2319. [Google Scholar]
- 47.Cavalli R, Bisazza A, Sessa R, Primo L, Fenili F, Manfredi A, Ranucci E, Ferruti P. Biomacromolecules. 2010;11:2667. doi: 10.1021/bm100685t. [DOI] [PubMed] [Google Scholar]
- 48.Franchini J, Ranucci E, Ferruti P, Rossi M, Cavalli R. Biomacromolecules. 2006;7:1215. doi: 10.1021/bm060054m. [DOI] [PubMed] [Google Scholar]
- 49.Ferruti P, Mauro N, Falciola L, Pifferi V, Bartoli C, Gazzarri M, Chiellini F, Ranucci E. Macromol. Biosci. 2013 doi: 10.1002/mabi.201300387. [DOI] [PubMed] [Google Scholar]
- 50.Piest M, Lin C, Mateos-Timoneda MA, Lok MC, Hennink WE, Feijen J, Engbersen JFJ. J. Controlled Rel. 2008;130:38. doi: 10.1016/j.jconrel.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 51.Christensen LV, Chang CW, Kim WJ, Kim SW, Zhong ZY, Lin C, Engbersen JFJ, Feijen J. Bioconjugate Chem. 2006;17:1233. doi: 10.1021/bc0602026. [DOI] [PubMed] [Google Scholar]
- 52.Emilitri E, Ranucci E, Ferruti P. J. Polym. Sci. Pol. Chem. 2005;43:1404. [Google Scholar]
- 53.Ranucci E, Ferruti P, Lattanzio E, Manfredi A, Rossi M, Mussini PR, Chiellini F, Bartoli C. J. Pol. Sci. Pol. Chem. 2009;47:6977. [Google Scholar]
- 54.Malgesini B, Verpilio I, Duncan R, Ferruti P. Macromol. Biosci. 2003;3:59. doi: 10.1002/mabi.200400093. [DOI] [PubMed] [Google Scholar]
- 55.Lin C, Blaauboer C-J, Timoneda MM, Lok MC, van Steenbergen M, Hennink WE, Zhong Z, Feijen J, Engbersen JFJ. J. Controlled Rel. 2008;126:166. doi: 10.1016/j.jconrel.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 56.Blacklock J, You YZ, Zhou QH, Mao G, Oupicky D. Biomaterials. 2009;30:939. doi: 10.1016/j.biomaterials.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu DC, Liu Y, Chen L, He CB, Chung TS, Goh SH. Macromolecules. 2005;38:5519. [Google Scholar]
- 58.Hong CY, You YZ, Wu DC, Liu Y, Pan CY. J. Am. Chem. Soc. 2007;129:5354. doi: 10.1021/ja070871+. [DOI] [PubMed] [Google Scholar]
- 59.Li J, Zhu Y, Hazeldine ST, Firestine SM, Oupicky D. Biomacromolecules. 2012;13:3220. doi: 10.1021/bm3009999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J, Zhu Y, Hazeldine ST, Li C, Oupicky D. Angew. Chem. Int. Ed. Engl. 2012;51:8740. doi: 10.1002/anie.201203463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gosselin MA, Guo WJ, Lee RJ. Bioconjugate Chem. 2001;12:989. doi: 10.1021/bc0100455. [DOI] [PubMed] [Google Scholar]
- 62.Kloeckner J, Wagner E, Ogris M. Eur. J. Pharm. Sci. 2006;29:414. doi: 10.1016/j.ejps.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 63.Xia W, Wang P, Lin C, Li Z, Gao X, Wang G, Zhao X. J. Controlled Rel. 2012;157:427. doi: 10.1016/j.jconrel.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 64.Bonner DK, Zhao X, Buss H, Langer R, Hammond PT. J. Controlled Rel. 2013;167:101. doi: 10.1016/j.jconrel.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trubetskoy VS, Loomis A, Slattum PM, Hagstrom JE, Budker VG, Wolff JA. Bioconjugate Chem. 1999;10:624. doi: 10.1021/bc9801530. [DOI] [PubMed] [Google Scholar]
- 66.Oupicky D, Carlisle RC, Seymour LW. Gene Ther. 2001;8:713. doi: 10.1038/sj.gt.3301446. [DOI] [PubMed] [Google Scholar]
- 67.Neu M, Sitterberg J, Bakowsky U, Kissel T. Biomacromolecules. 2006;7:3428. doi: 10.1021/bm060788z. [DOI] [PubMed] [Google Scholar]
- 68.Namgung R, Kim WJ. Small. 2012;8:3209. doi: 10.1002/smll.201200496. [DOI] [PubMed] [Google Scholar]
- 69.Lee D, Kim D, Mok H, Jeong JH, Choi D, Kim SH. Pharm. Res. 2012;29:2213. doi: 10.1007/s11095-012-0750-4. [DOI] [PubMed] [Google Scholar]
- 70.Neu M, Germershaus O, Mao S, Voigt KH, Behe M, Kissel T. J. Controlled Rel. 2007;118:370. doi: 10.1016/j.jconrel.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 71.Park K, Hong SW, Hur W, Lee M-Y, Yang J-A, Kim SW, Yoon SK, Hahn SK. Biomaterials. 2011;32:4951. doi: 10.1016/j.biomaterials.2011.03.044. [DOI] [PubMed] [Google Scholar]
- 72.Park K, Lee M-Y, Kim KS, Hahn SK. Biomaterials. 2010;31:5258. doi: 10.1016/j.biomaterials.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 73.Wang Z-H, Zhu Y, Chai M-Y, Yang W-T, Xu F-J. Biomaterials. 2012;33:1873. doi: 10.1016/j.biomaterials.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 74.Li RQ, Hu Y, Yu BR, Zhao NN, Xu FJ. Bioconjugate Chem. 2013 doi: 10.1021/bc400467h. [DOI] [PubMed] [Google Scholar]
- 75.Zhang G, Liu J, Yang Q, Zhuo R, Jiang X. Bioconjugate Chem. 2012;23:1290. doi: 10.1021/bc300133r. [DOI] [PubMed] [Google Scholar]
- 76.Piao J-G, Yan J-J, Wang M-Z, Wu D-C, You Y-Z. Biomaterials Sci. 2014 [Google Scholar]
- 77.McKenzie DL, Smiley E, Kwok KY, Rice KG. Bioconjugate Chem. 2000;11:901. doi: 10.1021/bc000056i. [DOI] [PubMed] [Google Scholar]
- 78.Oupicky D, Parker AL, Seymour LW. J. Am. Chem. Soc. 2002;124:8. doi: 10.1021/ja016440n. [DOI] [PubMed] [Google Scholar]
- 79.Read ML, Bremner KH, Oupicky D, Green NK, Searle PF, Seymour LW, Gene Med J. 2003;5:232. doi: 10.1002/jgm.331. [DOI] [PubMed] [Google Scholar]
- 80.Read ML, Singh S, Ahmed Z, Stevenson M, Briggs SS, Oupicky D, Barrett LB, Spice R, Kendall M, Berry M, Preece JA, Logan A, Seymour LW. Nucleic Acids Res. 2005;33:e86. doi: 10.1093/nar/gni085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Manickam DS, Bisht HS, Wan L, Mao G, Oupicky D. J. Controlled Rel. 2005;102:293. doi: 10.1016/j.jconrel.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 82.Haider M, Megeed Z, Ghandehari H. J. Controlled Rel. 2004;95:1. doi: 10.1016/j.jconrel.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 83.Kwok KY, Park Y, Yang Y, McKenzie DL, Liu Y, Rice KG. J. Pharm. Sci. 2003;92:1174. doi: 10.1002/jps.10384. [DOI] [PubMed] [Google Scholar]
- 84.Chen S, Han K, Yang J, Lei Q, Zhuo R-X, Zhang X-Z. Pharm. Res. 2013:1. doi: 10.1007/s11095-013-1040-5. [DOI] [PubMed] [Google Scholar]
- 85.Ogris M, Carlisle RC, Bettinger T, Seymour LW. J. Biol. Chem. 2001;276:47550. doi: 10.1074/jbc.M108331200. [DOI] [PubMed] [Google Scholar]
- 86.Zanta MA, Belguise-Valladier P, Behr JP. Proc. Natl. Acad. Sci. U. S. A. 1999;96:91. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang J, Lei Q, Han K, Gong Y-H, Chen S, Cheng H, Cheng S-X, Zhuo R-X, Zhang X-Z. J. Mater. Chem. 2012;22:13591. [Google Scholar]
- 88.Manickam DS, Oupicky D. Bioconjugate Chem. 2006;17:1395. doi: 10.1021/bc060104k. [DOI] [PubMed] [Google Scholar]
- 89.Park Y, Kwok KY, Boukarim C, Rice KG. Bioconjugate Chem. 2002;13:232. doi: 10.1021/bc010070a. [DOI] [PubMed] [Google Scholar]
- 90.Trentin D, Hall H, Wechsler S, Hubbell JA. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2506. doi: 10.1073/pnas.0505964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Trentin D, Hubbell J, Hall H. J. Controlled Rel. 2005;102:263. doi: 10.1016/j.jconrel.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 92.Ericson MD, Rice KG. Tetrahedron Lett. 2013;54:3440. doi: 10.1016/j.tetlet.2013.04.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ericson MD, Rice KG. Peptide Sci. 2012;98:510. doi: 10.1002/bip.22117. [DOI] [PubMed] [Google Scholar]
- 94.McKenzie DL, Kwok KY, Rice KG. J. Biol. Chem. 2000;275:9970. doi: 10.1074/jbc.275.14.9970. [DOI] [PubMed] [Google Scholar]
- 95.Bauhuber S, Hozsa C, Breunig M, Gopferich A. Adv. Mater. 2009;21:3286. doi: 10.1002/adma.200802453. [DOI] [PubMed] [Google Scholar]
- 96.Kang HC, Kang H-J, Bae YH. Biomaterials. 2011;32:1193. doi: 10.1016/j.biomaterials.2010.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tian L, Kang HC, Bae YH. Biomacromolecules. 2013 doi: 10.1021/bm400337f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Oliveira H, Fernandez R, Pires LR, Martins MCL, Simões S, Barbosa MA, Pêgo AP. J. Controlled Rel. 2010;143:350. doi: 10.1016/j.jconrel.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 99.Matsumoto S, Christie RJ, Nishiyama N, Miyata K, Ishii A, Oba M, Koyama H, Yamasaki Y, Kataoka K. Biomacromolecules. 2008;10:119. doi: 10.1021/bm800985e. [DOI] [PubMed] [Google Scholar]
- 100.Peng Q, Hu C, Cheng J, Zhong Z, Zhuo R. Bioconjugate Chem. 2009;20:340. doi: 10.1021/bc800451j. [DOI] [PubMed] [Google Scholar]
- 101.Peng Q, Zhong Z, Zhuo R. Bioconjugate Chem. 2008;19:499. doi: 10.1021/bc7003236. [DOI] [PubMed] [Google Scholar]
- 102.Lee Y, Mo H, Koo H, Park JY, Cho MY, Jin GW, Park JS. Bioconjugate Chem. 2007;18:13. doi: 10.1021/bc060113t. [DOI] [PubMed] [Google Scholar]
- 103.You YZ, Manickam DS, Zhou QH, Oupicky D. J. Controlled Rel. 2007;122:217. doi: 10.1016/j.jconrel.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.You YZ, Manickam DS, Zhou QH, Oupicky D. Biomacromolecules. 2007;8:2038. doi: 10.1021/bm0702049. [DOI] [PubMed] [Google Scholar]
- 105.You YZ, Zhou QH, Manickam DS, Wan L, Mao GZ, Oupicky D. Macromolecules. 2007;40:8617. doi: 10.1021/ma071176p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carmine A, Domoto Y, Sakai N, Matile S. Chem. – Eur. J. 2013;19:11558. doi: 10.1002/chem.201301567. [DOI] [PubMed] [Google Scholar]
- 107.Balakirev M, Schoehn G, Chroboczek J. Chem. Biol. 2000;7:813. doi: 10.1016/s1074-5521(00)00030-2. [DOI] [PubMed] [Google Scholar]
- 108.Chung YC, Regen SL. Macromolecules. 1991;24:5738. [Google Scholar]
- 109.Bang E-K, Gasparini G, Molinard G, Roux A, Sakai N, Matile S. J. Am. Chem. Soc. 2013;135:2088. doi: 10.1021/ja311961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Drake CR, Aissaoui A, Argyros O, Serginson JM, Monnery BD, Thanou M, Steinke JHG, Miller AD. Mol. Pharm. 2010;7:2040. doi: 10.1021/mp9002249. [DOI] [PubMed] [Google Scholar]
- 111.Pack DW, Hoffman AS, Pun S, Stayton PS. Nat. Rev. Drug Discov. 2005;4:581. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 112.Scholz C, Wagner E. J. Controlled Rel. 2012;161:554. doi: 10.1016/j.jconrel.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 113.Khalil IA, Kogure K, Akita H, Harashima H. Pharmacol. Rev. 2006;58:32. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 114.Payne CK, Jones SA, Chen C, Zhuang X. Traffic. 2007;8:389. doi: 10.1111/j.1600-0854.2007.00540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Blacklock J, Handa H, Manickam DS, Mao G, Mukhopadhyay A, Oupicky D. Biomaterials. 2007;28:117. doi: 10.1016/j.biomaterials.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 116.Blacklock J, Mao GZ, Oupicky D, Mohwald H. Langmuir. 2010;26:8597. doi: 10.1021/la904673r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Blacklock J, Sievers TK, Handa H, You YZ, Oupicky D, Mao GZ, Mohwald H. J. Phys. Chem. B. 2010;114:5283. doi: 10.1021/jp100486h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Blacklock J, Vetter A, Lankenau A, Oupický D, Möhwald H. Biomaterials. 2010;31:7167. doi: 10.1016/j.biomaterials.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li J, Manickam DS, Chen J, Oupicky D. Eur. J. Pharm. Sci. 2012;46:173. doi: 10.1016/j.ejps.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brulisauer L, Kathriner N, Prenrecaj M, Gauthier MA, Leroux JC. Angew. Chem. Int. Ed. Engl. 2012;51:12454. doi: 10.1002/anie.201207070. [DOI] [PubMed] [Google Scholar]
- 121.Kim TI, Ou M, Lee M, Kim SW. Biomaterials. 2009;30:658. doi: 10.1016/j.biomaterials.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim T.-i., Lee M, Kim SW. Biomaterials. 2010;31:1798. doi: 10.1016/j.biomaterials.2009.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Takae S, Miyata K, Oba M, Ishii T, Nishiyama N, Itaka K, Yamasaki Y, Koyama H, Kataoka K. J. Am. Chem. Soc. 2008;130:6001. doi: 10.1021/ja800336v. [DOI] [PubMed] [Google Scholar]
- 124.Vercauteren D, Piest M, van der Aa LJ, Al Soraj M, Jones AT, Engbersen JFJ, De Smedt SC, Braeckmans K. Biomaterials. 2011;32:3072. doi: 10.1016/j.biomaterials.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 125.Vercauteren D, Deschout H, Remaut K, Engbersen JFJ, Jones AT, Demeester J, De Smedt SC, Braeckmans K. ACS Nano. 2011;5:7874. doi: 10.1021/nn2020858. [DOI] [PubMed] [Google Scholar]
- 126.Zhang B, Ma X, Murdoch W, Radosz M, Shen Y. Biotechnol. Bioeng. 2013;110:990. doi: 10.1002/bit.24772. [DOI] [PubMed] [Google Scholar]
- 127.Kim SH, Ou M, Bull DA, Kim SW. Macromol. Biosci. 2010;10:898. doi: 10.1002/mabi.200900482. [DOI] [PubMed] [Google Scholar]
- 128.Kim SH, Jeong JH, Kim T.-i., Kim SW, Bull DA. Mol. Pharm. 2009;6:718. doi: 10.1021/mp800161e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang H, Vinogradov SV. J. Controlled Rel. 2010;143:359. doi: 10.1016/j.jconrel.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen HH, Ho Y-P, Jiang X, Mao H-Q, Wang T-H, Leong KW. Mol. Ther. 2008;16:324. doi: 10.1038/sj.mt.6300392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grigsby CL, Ho Y-P, Lin C, Engbersen JF, Leong KW. Sci. Reports. 2013:3. doi: 10.1038/srep03155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Manickam DS, Li J, Putt DA, Zhou QH, Wu C, Lash LH, Oupicky D. J. Controlled Rel. 2010;141:77. doi: 10.1016/j.jconrel.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lin C, Zhong Z, Lok MC, Jiang X, Hennink WE, Feijen J, Engbersen JF. J. Controlled Rel. 2006;116:130. doi: 10.1016/j.jconrel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 134.Yue Y, Jin F, Deng R, Cai J, Chen Y, Lin M, Kung H-F, Wu C. J. Controlled Rel. 2011;155:67. doi: 10.1016/j.jconrel.2010.10.028. [DOI] [PubMed] [Google Scholar]
- 135.Nguyen J, Szoka FC. Acc. Chem. Res. 2012;45:1153. doi: 10.1021/ar3000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Godbey WT, Wu KK, Mikos AG. Biomaterials. 2001;22:471. doi: 10.1016/s0142-9612(00)00203-9. [DOI] [PubMed] [Google Scholar]
- 137.Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A, Moghimi SM. FEBS Lett. 2005;579:6191. doi: 10.1016/j.febslet.2005.09.092. [DOI] [PubMed] [Google Scholar]
- 138.Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. Mol. Ther. 2005;11:990. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 139.Yang J, Hendricks W, Liu G, McCaffery JM, Kinzler KW, Huso DL, Vogelstein B, Zhou S. Proc. Nat. Acad. Sci. U.S.A. 2013 doi: 10.1073/pnas.1313330110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Maksimenko A, Dosio F, Mougin J, Ferrero A, Wack S, Reddy LH, Weyn A-A, Lepeltier E, Bourgaux C, Stella B, Cattel L, Couvreur P. Proc. Nat. Acad. Sci. U.S.A. 2014;111:E217. doi: 10.1073/pnas.1313459110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Proc. Nat. Acad. Sci. U.S.A. 2011;108:1850. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Parhamifar L, Larsen AK, Hunter AC, Andresen TL, Moghimi SM. Soft Matter. 2010;6:4001. [Google Scholar]
- 143.Hong S, Leroueil PR, Janus EK, Peters JL, Kober M-M, Islam MT, Orr BG, Baker JR, Banaszak Holl MM. Bioconjugate Chem. 2006;17:728. doi: 10.1021/bc060077y. [DOI] [PubMed] [Google Scholar]
- 144.Chen J, Hessler JA, Putchakayala K, Panama BK, Khan DP, Hong S, Mullen DG, DiMaggio SC, Som A, Tew GN, Lopatin AN, Baker JR, Holl MMB, Orr BG. J. Phys. Chem. B. 2009;113:11179. doi: 10.1021/jp9033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Merkel OM, Beyerle A, Beckmann BM, Zheng M, Hartmann RK, Stöger T, Kissel TH. Biomaterials. 2011;32:2388. doi: 10.1016/j.biomaterials.2010.11.081. [DOI] [PubMed] [Google Scholar]
- 146.Hunter AC, Moghimi SM. Biochim. Biophys. Acta-Bioenerg. 2010;1797:1203. doi: 10.1016/j.bbabio.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 147.Grandinetti G, Ingle NP, Reineke TM. Mol. Pharm. 2011;8:1709. doi: 10.1021/mp200078n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Klemm AR, Young D, Lloyd JB. Biochem. Pharmacol. 1998;56:41. doi: 10.1016/s0006-2952(98)00098-7. [DOI] [PubMed] [Google Scholar]
- 149.Sun W, Davis PB. J. Controlled Rel. 2010;146:118. doi: 10.1016/j.jconrel.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lin C, Zhong Z, Lok MC, Jiang X, Hennink WE, Feijen J, Engbersen JF. Bioconjugate Chem. 2007;18:138. doi: 10.1021/bc060200l. [DOI] [PubMed] [Google Scholar]
- 151.Wu C, Li J, Zhu Y, Chen J, Oupický D. Biomaterials. 2013;34:8843. doi: 10.1016/j.biomaterials.2013.07.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhou QH, Miller DL, Carlisle RC, Seymour LW, Oupicky D. J. Controlled Rel. 2005;106:416. doi: 10.1016/j.jconrel.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 153.Zhou Q, Wu C, Manickam DS, Oupicky D. Pharm. Res. 2009;26:1581. doi: 10.1007/s11095-009-9847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Brumbach JH, Lee YW, Kim SW, Yockman JW. J. Controlled Rel. 2012;159:111. doi: 10.1016/j.jconrel.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gao L-Y, Liu X-Y, Chen C-J, Wang J-C, Feng Q, Yu M-Z, Ma X-F, Pei X-W, Niu Y-J, Qiu C, Pang W-H, Zhang Q. Biomaterials. 2014;35:2066. doi: 10.1016/j.biomaterials.2013.11.046. [DOI] [PubMed] [Google Scholar]
- 156.Russ V, Frohlich T, Li YQ, Halama A, Ogris M, Wagner E. J. Gene Med. 2010;12:180. doi: 10.1002/jgm.1430. [DOI] [PubMed] [Google Scholar]
- 157.Christensen LV, Chang CW, Yockman JW, Conners R, Jackson H, Zhong Z, Feijen J, Bull DA, Kim SW. J. Controlled Rel. 2007;118:254. doi: 10.1016/j.jconrel.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kim P-H, Lee M, Kim SW. J. Controlled Rel. 2012;162:9. doi: 10.1016/j.jconrel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lee Y, Nam HY, Kim J, Lee M, Yockman JW, Shin SK, Kim SW. Mol. Ther. 2012;20:1360. doi: 10.1038/mt.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lee Y, McGinn AN, Olsen CD, Nam K, Lee M, Shin SK, Kim SW. J. Controlled Rel. 2013;171:24. doi: 10.1016/j.jconrel.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hyun H, Won YW, Kim KM, Lee J, Le M, Kim YH. Biomaterials. 2010;31:9128. doi: 10.1016/j.biomaterials.2010.08.038. [DOI] [PubMed] [Google Scholar]
- 162.Won Y-W, Kim K-M, An SS, Lee M, Ha Y, Kim Y-H. Biomaterials. 2011;32:9766. doi: 10.1016/j.biomaterials.2011.08.089. [DOI] [PubMed] [Google Scholar]
- 163.Chen CJ, Wang JC, Zhao EY, Gao LY, Feng Q, Liu XY, Zhao ZX, Ma XF, Hou WJ, Zhang LR, Lu WL, Zhang Q. Biomaterials. 2013;34:5303. doi: 10.1016/j.biomaterials.2013.03.056. [DOI] [PubMed] [Google Scholar]
- 164.Kim S-J, Ise H, Kim E, Goto M, Akaike T, Chung BH. Biomaterials. 2013 doi: 10.1016/j.biomaterials.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 165.Won YW, Yoon SM, Lee KM, Kim YH. Mol. Ther. 2011;19:372. doi: 10.1038/mt.2010.242. [DOI] [PMC free article] [PubMed] [Google Scholar]