

Abstract
The identification of disulfide bonds provides critical information regarding the structure and function of a protein and is a key aspect in understanding signaling cascades in biological systems. Recent proteomic approaches using digestion enzymes have facilitated the characterization of disulfide-bonds and/or oxidized products from cysteine residues, although these methods have limitations in the application of MS/MS. For example, protein digestion to obtain the native form of disulfide bonds results in short lengths of amino acids, which can cause ambiguous MS/MS analysis due to false positive identifications. In this study we propose a new approach, termed planned digestion, to obtain sufficient amino acid lengths after cleavage for proteomic approaches. Application of DBond software to planned digestion of specific proteins accurately identified disulfide-linked peptides. RNase A was used as a model protein in this study because the disulfide bonds of this protein have been well characterized. Application of this approach to peptides digested with Asp-N/C (chemical digestion) and trypsin under acidic hydrolysis conditions identified the four native disulfide bonds of RNase A. Missed cleavages introduced by trypsin treatment for only 3 hours generated sufficient lengths of amino acids for identification of the disulfide bonds. Analysis by MS/MS successfully showed additional fragmentation patterns that are cleavage products of S-S and C-S bonds of disulfide-linkage peptides. These fragmentation patterns generate thioaldehydes, persulfide, and dehydroalanine. This approach of planned digestion with missed cleavages using the DBond algorithm could be applied to other proteins to determine their disulfide linkage and oxidation patterns of cysteine residues.
Introduction
Proper folding of proteins is critical for their specific and programmed functions in both eukaryotes and prokaryotes.1–4 In many cases, the generation of disulfide bonds in proteins controls the folding and stability needed for the activation of their functional roles.2, 4–6 The detailed mechanisms of thiol-oxidation from cysteine residues are being extensively studied to understand the biological reactions and/or roles of oxidation.4, 7 These mechanisms are catalyzed by oxidoreductases or isomerases. The reduced cysteine thiolate (S-) and oxidized disulfide (S-S) in many enzymes undergo a series of exchange reactions that are required for enzymatic activity.8, 9 Protein folding, for instance, requires the accurate formation of disulfide bonds because disulfide bridges stabilize the formation of β-strands and α-helices.8–10 This stabilization of protein folding through disulfide bonds formation can control substrate access or protein-protein interactions that are needed for the catalytic activities of enzymes.
The recently introduced powerful tools of mass spectrometry (MS) have focused on the identification of disulfide bonds to understand their mechanisms via structural analysis.11–14 This is of interest as the formation of disulfide linkages not only contributes to the stability of a protein’s tertiary structure, but is also known to regulate various biological pathways in the cell.15–17 Enzymatic digestion of disulfide bonds has extensive applications in proteomic studies.11, 18 Digested disulfide-linked peptides can be subjected to liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) and their spectra can be analyzed to determine the formation of disulfide linkages. The products of the protein digestion can include three types of disulfide bonds: inter-peptide (one S-S bond from two different peptides after digestion, Figure 1a), intra-peptide (one or more S-S bonds from a single peptide, Figure 1b), and inter- and intra-peptide bonds (Figure 1c). Assuming that disulfide-linked peptides include up to two disulfide bonds, various forms of possible linkages may exist, as summarized in Figure 1.5, 8, 15, 19 Although all possible forms of disulfide-linked peptides can be discovered through informatics methods, peptides with more than two disulfide bonds are difficult to characterize by MS/MS because of their poor fragmentation. The simplest example of this can be seen with an intra-peptide bond (Figure 1b). If fragmentation is observed between two cysteines that form the disulfide bond, the mass of the resulting fragments would be equal to the mass of the precursor as the fragments are still linked by this bond, thus little information can be derived from this experiment.
Figure 1.
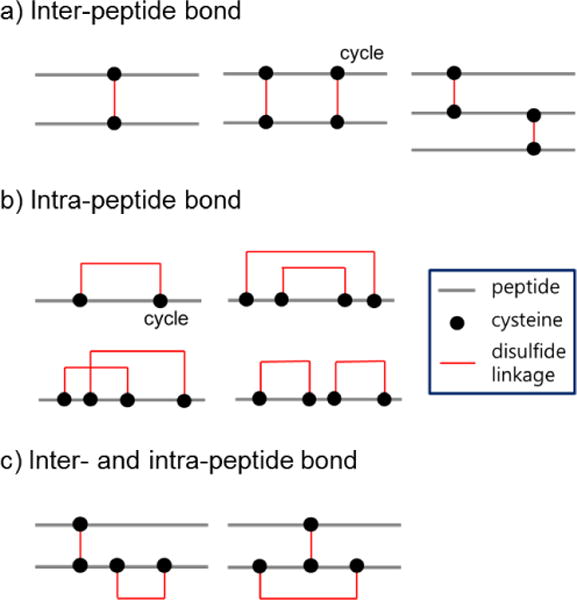
Types of disulfide-linked peptides. Peptides that contain a single disulfide bond are called simple forms; other forms are referred to as complex. a) A simple form of inter-disulfide–linked peptide is termed “desirable” when the lengths of its component peptides are longer than five amino acids and the peptide mass is less than 5000 Da. b) Intra-peptide bonds and c) inter- and intra-peptide bonds can be generated from the oxidation of cysteine thiols.
Disulfide-linked peptides can produce multiple and overlapping fragment ions from each component peptide, thus generating multiplexed MS/MS spectra that are not readily interpretable by conventional search algorithms. Dedicated software has been introduced for analysis of the MS/MS data of disulfide-linked peptides and usually considers a disulfide-linked peptide as a peptide pair, although more peptides can be involved.20, 21 To aid in MS/MS followed by informatics analysis, complex disulfide-linked forms can be separated into simpler forms by digesting a protein into shorter segments, often by combining two proteases such as trypsin and chymotrypsin. However, multiple digestions do not always yield better results as MS/MS fragments ions from these smaller peptides. If the fragments are too short, sequence-specific information cannot be used for analysis. Most false positive identifications arise from cases in which one peptide is correctly identified but the second peptide is given an incorrect assignment.22 The proportion of such cases increases if one peptide is very short. Short peptides can cause inaccurate results, particularly in the case of ion trap instruments, because ions are typically not detectable at both terminal ends of an MS/MS spectrum. In addition, if the terminal residues of both peptides are identical, the ions may not confirm the sequence identity. Furthermore, if the peptide sequence has repeats in a database it cannot be unambiguously assigned, and a peptide with a shorter length has a higher possibility of repetition in the database, even when the size of the database is very small.
The question arises of whether one can obtain desirable forms of disulfide-linked peptides for MS/MS analysis. The forms of disulfide bonds are highly dependent on the digestion method, and with the recent increase in the availability of various proteases we aimed to identify the optimal digestion scheme for a target protein such that it can be digested into desirable peptides that are readily amenable to MS/MS analysis in terms of mass, size, and amino acid composition. For example, if a protein includes multiple Lys and Arg residues, trypsin digestion would produce very short peptides, so Lys-C may be a better choice for the digestion. In contrast, if a protein includes only a few Lys and Arg residues, other proteases can be combined with trypsin for optimal digestion. Alternatively, missed cleavages can be induced by reducing the incubation time of trypsin digestion. An optimal digestion scheme for MS/MS analysis of disulfide-linked peptides can be selected to preclude undesired peptides and any short component peptides. This can be achieved by analysis of in silico digestions of a protein. This analysis will identify desirable peptides that have only one cysteine from each peptide, are more than five amino acids in length, and have a sequence that is unique within a database. This will ensure that all disulfide bonds of the protein can be identified, including only one disulfide bond, and their site assignments can be confidently determined. MS/MS would also provide sufficient sequencing information, improving confidence in the result. Overall, optimal digestion schemes can reduce the complexity of MS/MS-based analysis of disulfide-linked proteins. If it is inevitable that a peptide will include two cysteine residues, which may form a cycle, the distance between the two cysteines can be optimized to be as short as possible. This concept can also be extended to MS/MS-based analyses of chemically cross-linked proteins or protein complexes to analyze protein conformations and protein-protein interactions.22
Ribonuclease A (RNase A) is one of the most extensively studied enzymes that contain disulfide linkages.14, 17 The disulfide bonds of this protein are required for proper RNA cleavage mechanisms and are so well studied that they are used as a model for studying enzymatic stability. Early X-ray diffraction analysis and NMR studies have investigated these important structural features at the atomic level, and more than 70 sets of coordinates for RNase A have been deposited.17, 23, 24 These structures show that four disulfide bonds are generated from 8 cysteine residues in the native state of RNase A. The thermal stability of this protein is decreased when these cysteine residues are replaced with a pair of alanine or serine residues, indicating that disulfide formation improves the rigidity of protein folding, which is required for enzymatic activity.5, 25, 26 Proper folding of this protein is necessary as it improves substrate specificity through S-S bridges in cysteine residues. Additionally, preliminary results have shown that these bridges are crucial for the binding of RNase A to RNA. This interaction between RNase A and RNA requires a number of hydrogen bonds that selectively increase the binding ability and enzymatic activity.27, 28
In this study, the concept of designed digestion was applied to MS/MS-based analysis of disulfide bonds in bovine pancreatic RNase A. It has been shown that proteins with multiple disulfide linkages are more likely to be poorly digested because of limited access of the digestion enzyme. Therefore, chemical digestion using Asp-N/C was proposed to solve this technical problem.29, 30 In addition to this chemical digestion, missed trypsin cleavages were induced to produce peptides of appropriate lengths by regulating the digestion time.31 This missed cleavage generates undigested peptides that have the proper lengths, thus increasing the confidence of S-S bond identification. DBond software was used to assign disulfide bond sites in RNase A thorough analysis of MS/MS spectra of disulfide-linked peptides.20 DBond was originally designed to recognize diagnostic fragment ions specifically from disulfide linkages. In this study, the DBond algorithm was improved by incorporating fragmentation patterns of ion trap data into the scoring function. We used RNase A as a model protein to demonstrate the utility of planned digestion and the upgraded DBond software, which successfully identified the four known disulfide bonds as inter-peptide bonds. The spectra also characterize the products from S-S and C-S bond cleavages, and the results confirmed that the disulfide-linked peptides are generated from the simple formation of a disulfide bond.
Result and discussion
Digestion scheme
RNase A was chosen as a model protein in these studies because it has four disulfide bonds in its native state. These disulfide bonds have been extensively characterized, making this protein an appropriate choice for validation of the described analytical tools. For example, preliminary studies have demonstrated that the bond between Cys65-Cys72 is 3.5 times more stable than that of Cys58-Cys110, even though Cys58 and Cys72 are a similar distance from Cys65. Despite this extensive characterization, trypsin is not ideal for the identification of all of these four disulfide bonds therefore we applied a planned digestion method for RNase A and demonstrated its effectiveness in identifying all four of these bonds.
RNase A is a relatively small protein of 13,686 Da consisting of 124 amino acids, and tryptophan is the only amino acid not present in the protein (Figure 2, RNase A sequence). RNase A has been shown to contain a total of four disulfide bonds in its native state (Cys26–Cys84, Cys40–Cys95, Cys58–Cys110, and Cys65–Cys72) and Figure 2a shows the predicted disulfide-linked peptides after trypsin digestion of RNase A. Even in a theoretical digestion, trypsin does not cut each disulfide bond separately. Instead, it produces disulfide-linked peptides containing two disulfide linkages involving four cysteines. Using in silico digestion, RNase A was digested using a combination of chymotrypsin and trypsin as shown in Figure 2b. Chymotrypsin is sensitive to various residues (Phe, Tyr, Trp, Leu, and Met) and successfully separates each disulfide bond, but it produces short disulfide-linked peptides that may not provide the analytical information necessary to confirm the sequence of each peptide by MS/MS due to the difficulty of assignments.
Figure 2.

Known disulfide bonds (linked by red line) in the RNase A protein sequence. Based on the known disulfide bonds, four disulfide-linked products were digested in silico using a) trypsin; b) chymotrypsin + trypsin; c) Asp-N/C + LysC; d) Asp-N/C + trypsin.
The optimal digestion scheme for RNase A requires proteases sensitive to Asp and Lys residues that will generate simple single inter-disulfide-linked peptides with a length appropriate for analysis by MS/MS. A combination of Asp-N/C (a chemical reagent) and Lys-C would meet this requirement (Figure 2c); however, considering the digestion yield and specificity, a combination of Asp-N/C and trypsin was selected. Figure 2d shows the disulfide-linked peptides after Asp-N/C and Trypsin digestion. Because the length of the peptide that includes Cys84 is too short for analysis, we introduced missed cleavage for this peptide. Digestion by Asp-N/C was conducted first, followed by treatment with trypsin for 3 hrs (high missed cleavage), 6 hrs, or 24 hrs (low missed cleavage).
In Silico missed cleavage
We validated the role of these induced missed cleavages in creating the appropriate peptides by first conducting in silico experiments. Annotated disulfide bonds were obtained from the Swiss-Prot Human database (release 2011.08), excluding predicted bonds. A total of 2,612 disulfide bonds corresponding to 704 proteins were obtained. These proteins were analyzed to determine whether they can be identified when missed cleavages are introduced. In this initial test, each disulfide bond was regarded as interpretable when (1) in silico digestion of the protein resulted in inter-peptide bonds linking two digested peptides that do not include other disulfide bonds (simple forms), and (2) the lengths of the peptides are greater than five residues, with molecular weights of the disulfide-linked peptides being less than 5,000 Da (and thus amenable to MS/MS). Each protein was regarded as interpretable when all of its disulfide bonds were interpretable.
Table 1 shows the number of proteins that were interpretable for different numbers of allowed missed cleavages in several digestion schemes. The ‘Any length’ column shows the number of proteins when there is no constraint on the length of a peptide. This column represents the maximum number of proteins interpretable from simple forms of disulfide-linked peptides when each digestion scheme is adopted. In Asp-N/C and trypsin digestion with no missed cleavage, 167 proteins are interpretable based on resulting disulfide-linked peptides. In other words, 143 proteins (the difference between the ‘Any length’ and ‘MC=0’ column) are interpretable from peptides of lengths less than four amino acids, which may lead to ambiguous identifications. This proportion is more significant in chymotrypsin plus trypsin digestion. In contrast, 98% of proteins, on average, can be interpreted by desirable disulfide-linked peptides for all the digestion schemes with more than two missed cleavages. These results validate the use of missed cleavages in creating desirable peptides that will result in more reliable disulfide bond identification.
Table 1.
The number of proteins, based on the 704 disulfide-annotated proteins in the Swiss-Prot Human database, in which all disulfide bonds can be completely analyzed by simple or desirable disulfide-linked peptides. The ‘Any length’ column represents no constraint on the length of disulfide-linked peptides, whereas other columns represent a given constraint (≥5) on the length and correspond to the number of missed cleavages (MC).
| Any length | Both peptides’ lengths ≥ 5
|
|||||
|---|---|---|---|---|---|---|
| MC=0 | MC=1 | MC=2 | MC=3 | MC=4 | ||
| Asp-N/C+Trypsin | 310 | 167 | 266 | 300 | 304 | 304 |
| Chymotrypsin+Trypsin | 420 | 79 | 315 | 407 | 413 | 413 |
| CNBr+Trypsin | 229 | 160 | 221 | 226 | 226 | 226 |
| Glu-C+Trypsin | 378 | 152 | 344 | 370 | 371 | 371 |
| Asp-N+Trypsin | 309 | 176 | 289 | 299 | 299 | 299 |
| PepsinA+Trypsin | 385 | 126 | 351 | 378 | 382 | 382 |
| Trypsin only | 182 | 140 | 176 | 179 | 179 | 179 |
| Chymotrypsin only | 349 | 143 | 333 | 344 | 344 | 344 |
| Asp-N/C+Lys-C | 178 | 122 | 155 | 176 | 176 | 176 |
| Asp-N/C+Arg-C | 195 | 128 | 179 | 192 | 192 | 192 |
| Asp-N/C+Chymotrypsin | 400 | 98 | 309 | 379 | 394 | 395 |
Identification of disulfide bonds
DBond successfully identified all four native disulfide bonds of RNase A, as summarized in Table 2. The annotated MS/MS spectra are shown in Supplementary Information (Supplementary figures S1 – S26). Each identified bond is supported by several disulfide-linked peptides including only inter-peptide bonds. For the Cys26–Cys84 bond, two disulfide-linked peptides, ‘SSTSAASSSNYC26NQMMK–C84R’ and ‘SSTSAASSSNYC26NQMMK–C84RETGSSK’ from Asp- N/C and trypsin digestion were identified. The latter was identified only in samples with trypsin treatment for 3 hrs (high missed cleavage) and 6 hrs, but not in samples treated for 24 hrs (low missed cleavage), although low missed cleavage generated sufficient lengths of peptides for MS/MS identification (Table 2). Figure 3 compares MS/MS spectra of the two fragmentations from these digestions. Few fragment ions are observed in the MS/MS spectrum with the ‘CR’ peptide (Figure 3a), but fragment ions (underlined) are observed in the fragmentation from ‘CRETGSSK’ with digestion for 3 hrs, as seen in Figure 3b. This demonstrates that desirable peptides can be obtained using missed cleavage, and that this was in fact necessary to obtain proper fragment ions. MS/MS data confirmed that single forms of disulfide-linked peptides from Cys40–Cys 95, Cys65–Cys72, and Cys58–Cys110 successfully induce missed cleavage. Thus, careful design of planned digestions and missed cleavage reactions can lead to improved confidence in the results.
Table 2.
Disulfide-linked peptides from RNase A identified in this study. The “No. of matches” column represents the number of unique disulfide-linked peptides that support each disulfide bond. The numbers of spectra matched to each peptide are shown according to trypsin treatment time (3, 6, and 24 hrs). For the Cys65–Cys72 bond, intra-disulfide linked peptides were observed including missed cleavages in 3-hr and 6-hr trypsin samples.
| Disulfide bond | No. of matches | Disulfide–linked peptide | MW calculat ed | No. of spectra
|
||
|---|---|---|---|---|---|---|
| Tryp 3hrs | Tryp 6hrs | Tryp 24hrs | ||||
| C26–C84 | 4 | SSTSAASSSNYCNQMMK – CR | 2070.8228 | 2 | 6 | 7 |
| QHMDSSTSAASSSNYCNQMMK – CR | 2582.0077 | 2 | 2 | – | ||
| SSTSAASSSNYCNQMMK – CRETGSSK | 2660.0935 | 1 | 1 | – | ||
| QHMDSSTSAASSSNYCNQMMK – CRETGSSK | 3171.2784 | 1 | – | – | ||
| C40–C95 | 9 | CKPVNTFVHESLA – YPNCAYK | 2299.0765 | 10 | 10 | 11 |
| CKPVNTFVHESLAD – YPNCAYK | 2414.1035 | 6 | 8 | 9 | ||
| RCKPVNTFVHESLA – YPNCAYK | 2455.1776 | 6 | 6 | 6 | ||
| CK – YPNCAYK | 1104.4732 | 2 | 3 | 3 | ||
| RCK – YPNCAYK | 1260.5743 | 3 | 3 | 3 | ||
| DRCK – YPNCAYK | 1375.6013 | 4 | 4 | 1 | ||
| CKPVNTFVHESLA – ETGSSKYPNCAYK | 2888.3473 | 1 | – | – | ||
| CK – YPNCAYKTTQANK | 1747.8022 | – | – | 3 | ||
| CKPVNTFVHESLA – YPNCAYKTTQANK | 2942.4055 | – | 1 | – | ||
| C65–C72 | 4 | NVACK – NGQTNCYQSYSTMSIT | 2327.9821 | 8 | 6 | 5 |
| NVACK – NGQTNCYQSYSTMSITD | 2443.009 | 7 | 7 | 10 | ||
| NVACKNGQTNCYQSYSTMSIT | 2309.9715 | 3 | 2 | – | ||
| NVACKNGQTNCYQSYSTMSITD | 2424.9985 | 1 | 2 | – | ||
| C58–C110 | 9 | VQAVCSQK – HIIVACEGNPYVPVHF | 2653.3145 | 21 | 27 | 46 |
| VQAVCSQK – HIIVACEGNPYVPVHFD | 2768.3414 | 23 | 35 | 65 | ||
| VQAVCSQK – HIIVACEGNPYVPVHFDASV | 3025.4789 | 13 | 23 | 42 | ||
| VQAVCSQK – TTQANKHIIVACEGNPYVPVHF | 3296.6434 | 7 | 5 | 1 | ||
| VQAVCSQK – TTQANKHIIVACEGNPYVPVHFD | 3411.6703 | 10 | 10 | 2 | ||
| DVQAVCSQK – HIIVACEGNPYVPVHFD | 2883.3683 | 1 | – | 3 | ||
| PVNTFVHESLADVQAVCSQK – HIIVACEGNPYVPVHF | 3962.9447 | 9 | 12 | 6 | ||
| PVNTFVHESLADVQAVCSQK – HIIVACEGNPYVPVHFD | 4077.9716 | 10 | 10 | 13 | ||
| PVNTFVHESLADVQAVCSQK – HIIVACEGNPYVPVHFDASV | 4335.1092 | 4 | 9 | 12 | ||
Figure 3.
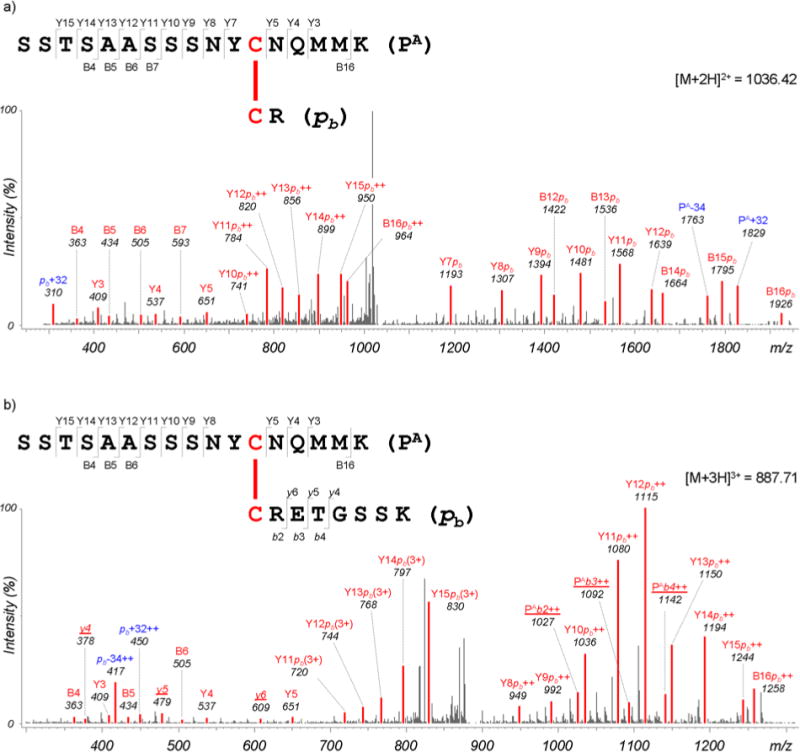
Annotated MS/MS spectra of (a) ‘SSTSAASSSNYCNQMMK–CR’ and (b) ‘SSTSAASSSNYCNQMMK–CRETGSSK’ disulfide-linked peptides. Matched peaks are shown in red. Ion annotations used: ‘++’ and ‘3+’, doubly and triply charged fragment ions, respectively; ‘PA’, one strand (top) of a dipeptide; ‘pb’, the other strand (bottom) of a dipeptide; uppercase letters, fragment ions from peptide PA; lowercase letters, fragment ions from peptide pb; ‘+32’ and ‘−34’, persulfide and dehydroalanine ions, respectively, formed by C–S bond cleavage reactions. In particular, fragment ions from peptide pb are underlined and are observed only for ‘CRETGSSK’.
Fragmentation patterns of disulfide-linked peptides
For the RNase A disulfide-linked peptides, two specific fragmentations at the disulfide bonds were observed (Figure 4). These fragmentation products are unusual at the peptide amide bonds. One product is an S–S bond fragmentation that occurs between the two sulfur atoms, resulting in two fragment ions, cysteine and cysteine thioaldehyde (−2 Da). The other is a C–S bond fragmentation that occurs between sulfur and carbon atoms of a cysteine residue involved in the disulfide linkage, resulting in two fragment ions, cysteine persulfide (+32 Da) and dehydroalanine (−34 Da).32 These ions represent characterization of disulfide-linked peptides that would not be detected in linear peptides. In previous work, we studied the fragmentation patterns of these ions based on Q-TOF data and applied them to a scoring model because cysteine and persulfide ions were frequently observed. Q-TOF data typically have a strong propensity to form one type of ion rather than simultaneously generating complementary ions. In addition, these ions themselves produce fragment ions via secondary fragmentation, forming an ion ladder in an MS/MS spectrum.
Figure 4.
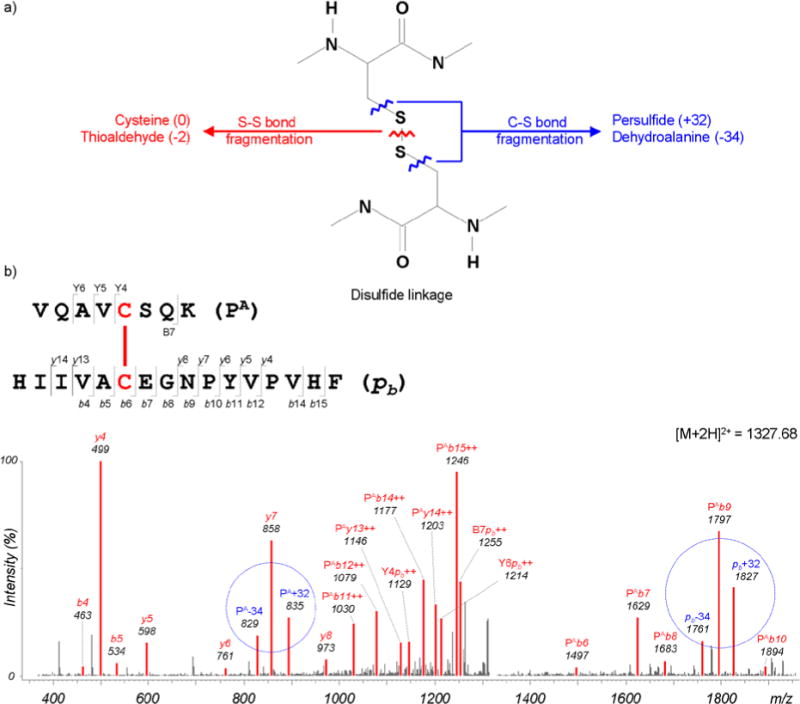
Additional fragmentations of C–S and S-S bond cleavages. a) Fragmentation patterns for persulfide and dehydroalanine fragment ions from C–S bond cleavage reactions, and cysteine and thioaldehyde fragment ions from S–S bond cleavage reactions. b) Observed persulfide (+32 Da) and dehydroalanine (−34 Da) ions are circled.
Ion trap data showed different fragmentation patterns in this planned digestion-tandem MS/MS from Q-TOF. First, cysteine persulfide and dehydroalanine ions produced by C–S bond fragmentation were frequently observed, but ions from S–S bond fragmentation were rarely found. The persulfide and dehydroalanine (underlined) ions are complementary and were almost always observed together, as shown in Figure 3. Few ions produced via secondary fragmentation were detected. These ion trap data cannot give information about the amino acid composition of a disulfide-linked peptide, but can provide molecular weights of component peptides. This information can be used to confirm peptide pairs as disulfide-linked peptide candidates and consequently reduce false positive rates. Given the differences in experimental outcomes of different types of MS, and based on the observations in Figure 4, a different fragmentation model was incorporated into the scoring model for DBond depending on the MS instrument type. This adjustment will aid in the accuracy of disulfide bond identification.
To test the performance of upgraded DBond, its search results were compared with the results from a MassMatrix21 search. The searches were conducted against the RNase A protein sequence (as a target) and a reversed version of Escherichia coli sequences (as a decoy) using the same parameters (more details is found in the Experimental section). The final identifications were obtained at a false-discovery rate of 1% using a target-decoy strategy.33 DBond identified 157, 204, and 267 peptide-spectrum matches (PSMs) from samples treated with trypsin for 3 hrs, 6 hrs, and 24 hrs, respectively. It should be noted that all known native disulfide bonds were observed in each sample, while only 4% of the identified PSMs corresponded to non-native bonds. On the other hand, MassMatrix identified 3, 3, and 2 PSMs from the three samples, respectively, and all the known bonds were not observed. This demonstrate well the utility of DBond for the identification of disulfide-linked peptides.
Applications of planned digestion and tandem MS/MS
Covalent inter- or intra-disulfide bonds are critical factors controlling the folding and stability of proteins, therefore protein engineering has focused on controlling disulfide interactions.34, 35 For example, membrane proteins are considered a major target because disulfide bonds occur widely in these proteins as a result of oxidation processes. It has been reported that disulfide bond exchanges drive major conformational changes of these membrane bound receptors even though they have structural rigidity conferred by the dense environments of the membrane.36 Although RNase A is a soluble protein, the digestive enzymes must cleave in limited spatial regions. The digestion sites of Asp-N/C and trypsin are mostly located on the surface, but cleavage is still limited by the presence of inaccessible sites within the folded protein. The disulfide bonds aid in the formation of very rigid secondary structures of RNase A, which has been shown to be three alpha helixes and long anti-parallel beta strands.5, 17, 25, 26, 37, 38 This well folded secondary structure is important for the precise functions of RNase A in binding to RNA, but limits enzyme accessibility. The Cys72 residue, one of the disulfide bond formation residues, is located around the alpha-helices and beta-strands, and may not be accessible to digestive enzymes. The results presented here demonstrate the validity of our novel method that eliminates the issue of digestive enzyme accessibility and prove the usefulness of this technique for analysis of membrane proteins and other complicated globular proteins.
The approach of acid hydrolysis and digestion with missed cleavage applied in this study shows that successful digestion can be accomplished using Asp-N/C with trypsin and generates proper fragments for the analysis of disulfide-linkage. A short reaction time (3 hrs) is required to induce missed cleavage, which produces fragments for proper mass analysis. Some proteins are not amenable to treatment with digestive enzymes for proteomic approaches because of the limitations of cleavage sites and the generation of short fragments that are not suitable for MS analysis. Induction of missed cleavages using a limited treatment time can be an alternative approach to overcome these barriers. The functional and structural consequences of disulfide bonds of such proteins can be easily determined using missed cleavage.
Conclusion
Although software analysis method that can process complex disulfide-linked peptide products involving more than two peptides would be useful, it might be difficult to reliably confirm the identity of certain disulfide products because those with complex forms are likely to include cycles with poor fragmentation. We propose use of an optimal digestion scheme for a given target protein such that the resulting products includes a single disulfide bond. At the same time, the scheme was designed such that the digested peptides are readily amenable to MS/MS-based analyses in terms of their amino acid length. These properties are important in order to reliably confirm the identity of the products and obtain confident results. If a protein includes many cysteine residues and is not digested into simple forms, a combination of several proteases (up to two in this study) may be required. If over-digestion leads to the problem of short peptides, missed cleavages can be induced. We developed a planned digestion scheme to alleviate the limitations of analysis using traditional digestion enzymes, such as trypsin, with subsequent application of MS/MS. This novel method led to successful disulfide bond identification in RNase A. Furthermore, we are currently developing appropriate algorithms to obtain desired cleavages for proteomic research in order to accurately identify disulfide bonds. This will aid in specific digestions based on the peptide sequences. Planned digestion will allow the identification of disulfide bonds from digested peptides with more than five amino acids in order to determine which enzymes are appropriate for analysis with MS/MS.
Experimental
Sample preparation and digestion
The acid hydrolysis solution was a 1:1 mixture of 30% (v/v) acetonitrile and 25% (v/v) acetic acid in water. These chemicals provide the water-miscible organic solvent needed to solubilize the proteins and result in cleavage at amino- or carboxyl-terminals of aspartyl residues. RNase A (2 nmole) was prepared in this acid hydrolysis solution at 95°C for 4 hrs. After the acid hydrolysis reactions, the dissolved proteins were cleaved to give a mixture of highly solubilized peptides without breaking the disulfide bonds (data not shown). The acid hydrolysis of RNase A was followed by the addition of trypsin in the first attempt at disulfide bond analysis for a high-quality MS/MS fragmented pattern. The post-acid hydrolysis reactants were cooled to room temperature and centrifuged at 12,000 rpm for 10 min to remove the non-solubilized pellets. The resulting peptide mixtures were dried under the freeze-dryer. To minimize the disulfide bond interchange, the acid hydrolyzed peptide mixture was digested in 25 mM phosphate buffer (pH = 6.0). The peptide mixture was incubated with trypsin (20 pmole, Promega, Madison, WI, USA) at 37°C for 3, 6, and 24 hrs respectively. The digested samples were dried in a SpeedVac and desalted using a solid phase Oasis HLB C18 microelution plate (Waters, Inc., Milford, MA, USA). The dried samples were stored at −80°C before being subjected to nano-LC-MS/MS analysis.
LC-MS/MS
For the separation and identification of peptides, a 7-Tesla LTQ-FT ICR-MS (Thermo, Waltham, MA, USA) equipped with a nano-ESI source was used as described below. The peptide mixture from the chemical-enzyme double digest was loaded on a home-made trapping column of 100 mm in length packed with C18 silica with a particle size of 5 μm for desalting. The flow-through peptides of the trapping column were directly applied onto a C18 column (75 μm × 150 mm, C18 silica of 5 μm particle size, LC Packings) After a washing step, peptides were eluted from the analytical column as follows: isocratic elution with 5% mobile phase B (97% acetonitrile in water with 0.1% formic acid) over 15 min, followed by a linear increase to 20% mobile phase B over 3 min, and then a more gradual linear increase to 60% mobile phase B for 45 min, and finally an increase to 95% mobile phase B over 2 min. The analytical column was washed with 100% mobile phase A (3% acetonitrile in water with 0.1% formic acid) over 15 min, followed by 5% mobile phase B over 10 min before the next run. ESI voltage was set at 2.5 kV. Data-dependent acquisition was selected for the 10 most abundant MS ions from MS spectra and MS/MS analysis was further performed for the fragmentation of selected peptide MS peaks.
DBond Software
DBond software was written in Java programming language (1.6.0_05), and was recently upgraded (version 3.02). The extended features of DBond v3.02 are as follows: supports user-defined cleavage rules (including cleavage at both or either side of amino acids) for flexibility of enzyme usage; allows disulfide linkages between different proteins; supports blocking of free cysteines that do not form disulfide bonds; and able to take approximately 10 modifications as input (by allowing one modification per peptide, disulfide-linked peptides can have up to two variable modifications). In addition, a spectral viewer program is provided to help users inspect MS/MS spectra manually. Search results of DBond v3.02 are provided as a XML file, which can be fed into the viewer program for visual inspection. Analysis using DBond took approximately 1 second per spectrum on a regular desktop Pentium IV PC when tested on Swiss-Prot human proteins (approximately 20,000 entries) using the following search parameters: trypsin as digestive enzyme, up to two missed cleavages, 0.5 Da precursor mass tolerance, and oxidation (Met) as variable modification. DBond software is available at http://prix.hanyang.ac.kr
DBond Search and Identification
The peak lists were generated using BioWorks software (version 3.3.1 Thermo Fisher, San Jose, CA), and then converted into mgf (mascot generic file) files. DBond search of the mgf files was conducted against the RNase A protein sequence (Swiss-Prot Number P61823) and a reversed version of Escherichia coli sequences (strain K12, Swiss-Prot release 2011.08, 4,430 entries) using the following parameters: ±15 ppm for mass accuracy of precursor ions, ± 0.8 Da mass tolerances of fragment ions, [DKR].* and *.[D] cleavage rules for Asp-N/C and trypsin, missed cleavage sites of up to 2, and oxidation of Met and deamidation of Asn as variable modifications. Among all search results, identifications were obtained with a false discovery rate (FDR) of 1% using a target-decoy approach.33 FDR is calculated as D / T, where T and D are the numbers of target and decoy hits above score threshold, respectively. For the performance comparison, a MassMatrix21 search was also conducted using the same parameters, and the pp score was used for the threshold in the FDR calculation.
Supplementary Material
Acknowledgments
This work was supported by the Proteogenomics Research Program through the National Research Foundation of Korea funded by the Korean Ministry of Education, Science and Technology (NRF-2012M3A9B9036676) and Ministry of Science, ICT & Future Planning (NRF-2012M3A9D1054452). The study was also supported by research funds of Chonbuk National University and the project fund (PBD083) from the R&D Convergence Program of Korea Research Council of Fundamental Science and Technology and a project fund (T34514) to J. S. Kwon from KBSI. S. Na was partially supported by the National Institutes of Health Grant 8P41 GM103485-05 from NIGMS.
Abbreviations
- MC
Missed Cleavage
- LC
Liquid Chromatography
- RNase A
Ribonuclease A
- FT ICR-MS
Fourier Transformed Ion Coupled Resonance Mass Spectrometry
- FDR
False Discovery Rate
- PA
one strand of dipeptide (top)
- Pb
one strand of dipeptide (bottom)
- MS/MS
Tandem Mass Spectrometry
- PSM
Peptide-Spectrum Match
References
- 1.Betz SF. Protein Sci. 1993;2:1551–1558. doi: 10.1002/pro.5560021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Craik DJ, Daly NL, Waine C. Toxicon. 2001;39:43–60. doi: 10.1016/s0041-0101(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 3.Filomeni G, Rotilio G, Ciriolo MR. Cell Death Differ. 2005;12:1555–1563. doi: 10.1038/sj.cdd.4401754. [DOI] [PubMed] [Google Scholar]
- 4.Hogg PJ. Trends Biochem Sci. 2003;28:210–214. doi: 10.1016/S0968-0004(03)00057-4. [DOI] [PubMed] [Google Scholar]
- 5.Klink TA, Woycechowsky KJ, Taylor KM, Raines RT. Eur J Biochem. 2000;267:566–572. doi: 10.1046/j.1432-1327.2000.01037.x. [DOI] [PubMed] [Google Scholar]
- 6.Wedemeyer WJ, Welker E, Narayan M, Scheraga HA. Biochemistry. 2000;39:7032–7032. doi: 10.1021/bi005111p. [DOI] [PubMed] [Google Scholar]
- 7.Toledano MB, Kumar C, Le Moan N, Spector D, Tacnet F. Febs Lett. 2007;581:4549–4549. doi: 10.1016/j.febslet.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Bulaj G. Biotechnol Adv. 2005;23:87–92. doi: 10.1016/j.biotechadv.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Sevier CS, Kaiser CA. Nat Rev Mol Cell Bio. 2002;3:836–847. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg ME, Guillou Y. Protein Sci. 1994;3:883–887. doi: 10.1002/pro.5560030603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbirz S, Jakob U, Glocker MO. J Biol Chem. 2000;275:18759–18766. doi: 10.1074/jbc.M001089200. [DOI] [PubMed] [Google Scholar]
- 12.Gorman JJ, Wallis TP, Pitt JJ. Mass Spectrom Rev. 2002;21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 13.Tsai PL, Chen SF, Huang SY. Rev Anal Chem. 2013;32:257–268. [Google Scholar]
- 14.Xu H, Freitas MA. Bmc Bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arolas JL, Aviles FX, Chang JY, Ventura S. Trends Biochem Sci. 2006;31:292–301. doi: 10.1016/j.tibs.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Frand AR, Cuozzo JW, Kaiser CA. Trends Cell Biol. 2000;10:203–210. doi: 10.1016/s0962-8924(00)01745-1. [DOI] [PubMed] [Google Scholar]
- 17.Raines RT. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Dewald HD, Chen H. J Proteome Res. 2011;10:1293–1304. doi: 10.1021/pr101053q. [DOI] [PubMed] [Google Scholar]
- 19.Woycechowsky KJ, Raines RT. Curr Opin Chem Biol. 2000;4:533–539. doi: 10.1016/s1367-5931(00)00128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi S, Jeong J, Na S, Lee HS, Kim HY, Lee KJ, Paek E. J Proteome Res. 2010;9:626–635. doi: 10.1021/pr900771r. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Zhang LW, Freitas MA. J Proteome Res. 2008;7:138–144. doi: 10.1021/pr070363z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anfinsen CB. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 24.Moore S, Stein WH. Science. 1973;180:458–464. doi: 10.1126/science.180.4085.458. [DOI] [PubMed] [Google Scholar]
- 25.Laity JH, Shimotakahara S, Scheraga HA. P Natl Acad Sci. 1993;90:615–619. doi: 10.1073/pnas.90.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimotakahara S, Rios CB, Laity JH, Zimmerman DE, Scheraga HA, Montelione GT. Biochemistry. 1997;36:6915–6929. doi: 10.1021/bi963024k. [DOI] [PubMed] [Google Scholar]
- 27.Wlodawer A, Miller M, Sjolin L. P Natl Acad Sci. 1983;80:3628–3631. doi: 10.1073/pnas.80.12.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zegers I, Maes D, Daothi MH, Poortmans F, Palmer R, Wyns L. Protein Sci. 1994;3:2322–2339. doi: 10.1002/pro.5560031217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon J, Oh J, Park C, Cho K, Kim SI, Kim S, Lee S, Bhak J, Norling B, Choi JS. J Chromatogr A. 2010;1217:285–293. doi: 10.1016/j.chroma.2009.11.045. [DOI] [PubMed] [Google Scholar]
- 30.Pisareva T, Kwon J, Oh J, Kim S, Ge CR, Wieslander A, Choi JS, Norling B. J Proteome Res. 2011;10:3617–3631. doi: 10.1021/pr200268r. [DOI] [PubMed] [Google Scholar]
- 31.Lawless C, Hubbard SJ. Omics. 2012;16:449–456. doi: 10.1089/omi.2011.0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thakur SS, Balaram P. J Am Soc Mass Spectr. 2008;19:358–366. doi: 10.1016/j.jasms.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Elias JE, Gygi SP. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 34.Sevier CS, Kaiser CA. Antioxid Redox Sign. 2006;8:797–811. doi: 10.1089/ars.2006.8.797. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Chou CP, Moo-Young M. Biotechnol Adv. 2011;29:923–929. doi: 10.1016/j.biotechadv.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 36.Scheraga HA, Wedemeyer WJ, Welker E. Method Enzymol. 2001;341:189–221. doi: 10.1016/s0076-6879(01)41153-0. [DOI] [PubMed] [Google Scholar]
- 37.Martelli PL, Fariselli P, Casadio R. Proteomics. 2004;4:1665–1671. doi: 10.1002/pmic.200300745. [DOI] [PubMed] [Google Scholar]
- 38.Meier S, Haussinger D, Pokidysheva E, Bachinger HP, Grzesiek S. Febs Lett. 2004;569:112–116. doi: 10.1016/j.febslet.2004.05.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.