

Abstract
The gene gaoA encoding the copper-dependent enzyme galactose oxidase (GAO) from Fusarium graminearum PH-1 was cloned and successfully overexpressed in E. coli. Culture conditions for cultivations in shaken flasks were optimized, and optimal conditions were found to be double-strength LB medium, 0.5% lactose as inducer, and induction at the reduced temperature of 25°C. When using these cultivation conditions ~24 mg of active GAO could be produced in shaken flasks per litre medium. Addition of copper to the fermentation medium decreased the enzyme production significantly. The His-tagged recombinant enzyme could be purified conveniently with a single affinity chromatography step. The purified enzyme showed a single band on SDS–PAGE with an apparent molecular mass of 66 kDa and had kinetic properties similar to those of the fungal wild-type enzyme.
Keywords: Fusarium graminearum, E. coli, Galactose oxidase, Overexpression, Fermentation, Affinity chromatography
Introduction
Galactose oxidase (GAO; D-galactose:oxygen 6-oxidoreductase, E.C. 1.1.3.9) is a member of the free radical copper oxidase family, and contains a novel metalloradical complex (Whittaker 2005) consisting of a copper atom and a tyrosine residue covalently attached to the sulphur of a cystein. This enzyme is secreted by filamentous fungi such as Dactylium dendroides (Markus et al. 1965), Gibberella fujikuroi (Aisaka and Terada 1981) and Fusarium spp. (Barbosa-Tessmann et al. 2001). GAO catalyses the oxidation of primary alcohols to the corresponding aldehydes, while concomitantly reducing oxygen to hydrogen peroxide in its catalytic reaction (Whittaker and Whittaker 2000, 2001) (Fig. 1).
Fig. 1.
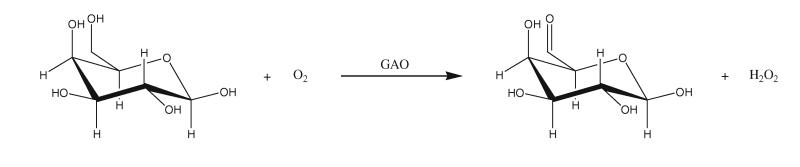
Reaction catalysed by GAO
GAO is strictly regioselective, and no secondary alcohol or other reducing group is oxidized. However, the enzyme exhibits a broad substrate spectrum ranging from monosaccharides and polysaccharides to aliphatic and aromatic alcohols and polyalcohols (Sun et al. 2001; Schlegel et al. 1968; Avigad et al. 1962; Bretting and Jacobs 1987; Mendonca and Zancan 1987). Interestingly, despite of its overall broad substrate spectrum GAO discriminates strongly between galactose and glucose (Siebum et al.2006), and the latter sugar is not accepted as a substrate by GAO.
GAO is used for various biomedical applications, including clinical assays for galactose in blood and other fluids (Karube et al. 1990), histochemical studies (Schulte and Spicer 1983), and early detection of cancer (Carter et al. 1997). GAO is a promising enzyme for the production of third-generation biosensors because of its ability for direct electron transfer (DET) to the electrode (Shleev et al. 2008), and could thus be attractive for applications in biofuel cells, especially when the substrate specificity of GAO could be broadened to other sugars, especially glucose. A prerequisite to enzyme evolution is a fast, reliable and simple expression system. To date wild-type GAO is produced recombinantly in fungal and yeast expression systems, which are not ideal for directed evolution studies.
Expression of functional GAO in E. coli was only possible as a lacZ fusion protein (Lis and Kuramitsu 1997), or after introduction of six mutations, which were identified in a directed evolutions study (Sun et al. 2001). In this paper we used a different approach, and report the enhancement of the expression of wild-type recombinant GOA in E. coli through improvement and optimization of the fermentation conditions.
Materials and methods
Materials
All chemicals used were of the highest grade available and were purchased from Sigma (St. Louis, MO) unless otherwise stated. 2,2′-Azino-bis (3-ethylbenzthiazoline-6-sulfonic acid (ABTS) was purchased from Amresco (Solon, OH). The Hisprep™ FF 16/10 column was from GE Healthcare Bioscience AB (Uppsala, Sweden). Restriction enzymes and ligase were obtained from Fermentas (Vilnius, Lithuania), while protein standards for SDS PAGE (Precision Plus Protein Dual Color Standard) was from BioRad (Herts, UK). F. graminearum strain PH-1 was kindly provided by Gerhard Adam (Department of Applied Genetics and Cell Biology, BOKU Vienna, Austria). E.coli strain BL21(DE3) and the pET21a cloning vector were from Novagen (Madison, WI).
Isolation and cloning of the gaoA gene
F. graminearum strain PH-1 was cultivated for 2 days using shaken flasks at 25°C and 120 rpm and Sabouraud medium (5 g/l peptone from casein, 5 g/l peptone from meat, 10 g/l glucose, 10 g/l maltose, 5 g/l yeast extract). Mycelia were harvested by centrifugation (4°C, 15 min and 5,000×g), washed and genomic DNA was isolated using the Wizard® SV Genomic DNA Purification Kit (Promega, Madison, WI). The gaoA gene including its prepro sequence was amplified by PCR using primers based on the published genome sequence (Broad Institute, Accession Number FGSG_11032.3) (GAO-for: 5′-GCCTCAGCACCTATCGGAAGCGCT-3′ and GAO-rev: 5′-TCACTGAGTAACGCGAATCGTCG-3′), and subsequently subcloned into the pJET 1.2 vector (Fermentas). Restriction sites were introduced by PCR using the following forward primers: 5′-AGGACATATGAAACACTTTTTATCATCT-3′ and 5′-CCTTCATATGGCCTCAGC-3′ for the gaoA gene with and without the prepro sequence, respectively, and 5′-GCCCTTGTCGACTCACTGAG-3′ as reverse primer. After gel purification and digestion with NdeI and SalI, the PCR product was ligated into the multiple cloning site of the pET21a vector that adds the sequence for a His-tag at the C terminus, and transformed into E. coli BL21 (DE3). DNA sequencing was performed as a commercial service (VBC Biotech, Vienna, Austria).
Optimization of expression conditions
In order to find optimal inducer concentrations a single colony from an overnight culture of E. coli was used to inoculate a 100-ml shaken flask containing 25 ml of LB medium, to which 50 mg of ampicillin was added per l, and shaking was continued at 37°C for 4 h. Aliquots (4 ml) were used to inoculate 1–1 shaken flasks containing 250 ml LB medium, and incubation was continued at 37°C until an OD600 of 0.4–0.6 was reached. Either IPTG or lactose were added as inducer at varying concentrations, and the incubation was then continued at 25°C for 16 h. Biomass was harvested by centrifugation (10 min at 6,000 g). To evaluate the influence of the medium composition on the expression of active GAO the following media were used: TB medium (12 g/l peptone from casein, 24 g/l yeast extract, 4 ml/l glycerol in 0.1 M potassium phosphate buffer, pH 7.5), MCH-Glyc medium (700 ml of 10 g/l peptone from casein, 10 g/l glycerol; 100 ml of 1 M CaCl2; 2 ml of 1 M MgSO4; and 200 ml of M9-Salts stock solution consisting of 64 g/l Na2HPO4.7H2O, 15 g/l KH2PO4, 2.5 g/l NaCl and 5 g/l NH4Cl; all medium components were autoclaved separately), LB medium (10 g/l peptone from casein, 5 g/yeast extract and 10 g/l NaCl), and double concentration LB medium (20 g/l peptone from casein, 10 g/l yeast extract and 10 g/l NaCl). All media contained 50 mg of ampicillin per litre to maintain the expression plasmid. Lactose (0.5%) was used as the inducer in this experiment.
Enzyme purification
Biomass was resuspended in phosphate buffer (50 mM, pH 7.0), and the cells were disrupted by a freezing/thawing step followed by the addition of lysozyme (1 mg/ml) and sonication. Cell debris was removed by centrifugation (15 min at 15,000×g). The clear supernatant was applied to a Hisprep™ FF 16/10 column previously equilibrated with phosphate buffer (50 mM, pH 7.0 containing 1 M NaCl and 20 mM imidazole). GAO was eluted using a linear gradient of 20–500 mM imidazole in 10 column volumes. Active fractions were pooled, concentrated and diafiltrated against phosphate buffer (50 mM, pH 7.0). Purified enzyme was aliquoted and stored at −30°C.
Enzyme assay
Enzyme samples were activated by incubation with 0.4 mM CuSO4 for 30 min at 25°C prior to activity measurements. GAO activity was determined spectrophotometrically at 420 nm and 30°C by continuously measuring the formation of H2O2 for 3 min using a peroxidase-coupled assay with ABTS as the chromogen (Leitner et al. 2001). The standard reaction mixture (total volume was 1 ml) contained 1 μmol of ABTS in 50 mM potassium phosphate buffer (pH 7.0), 2 U of horseradish peroxidase, 10 μmol of galactose, and a suitable amount of GAO. One unit of GAO activity was defined as the amount of enzyme necessary for the oxidation of 2 μmol ABTS (corresponding to the oxidation of 1 μmol galactose) per min under the conditions described above. Protein concentrations were determined by the dye-binding method of Bradford (1976) using BSA as standard. A substantially identical activity assay was used for determination of the substrate specificity of GAO, however, galactose was replaced by 10 μmol of the respective substrate in the assay mixture.
Results and discussion
The gene for GAO was successfully amplified comprising its prepro sequence from the genomic DNA of F. graminearum strain PH-1. The gao sequence determined showed an exact match with the published sequence (Gene ID: 2792888 FG11032.1), and did not contain any introns. After introducing suitable restriction sites the gene was cloned with and without its prepro sequence into the expression vector pET21a, which adds a C-terminal His-tag to the protein. After transformation into E. coli BL21(DE3) no activity was found when a standard expression protocol (1 mM IPTG in LB medium at 37°C for 4 h) was used. Decreasing the induction temperature to 25°C and increasing the cultivation time to 16 h yielded an activity of 15.6 and 16.2 U/l medium for GAO with and without its prepro sequence, respectively. This amount of recombinant protein (~0.25 mg/l GAO) is similar to published results for the expression of wild-type enzyme in E. coli at 30°C (Sun et al. 2001). A further decrease in the induction temperature to 16°C almost doubled the cultivation time necessary to 30 h without increasing the GAO activity yield (data not shown).
In a second step different concentrations of IPTG and lactose were compared with respect to their effect on GAO activity levels obtained. As shown in Table 1 decreasing the IPTG concentration also decreased the expression levels of active GAO. A significant fraction of GAO was found in inclusion bodies when IPTG was the inducer, especially when 1 mM IPTG was used as the inducer, as was confirmed by SDS PAGE analysis after solubilization of the complete biomass with 6 M urea. The T7 promoter used in the pET system is a very strong promoter. This fact combined with the strong inducer IPTG and the complex active site of GAO presumably leads to an overproduction of protein that cannot be processed correctly by E. coli. Using a weaker inducer should therefore slow the protein production and thus increase the time for the correct folding of GAO. In fact, the expression of soluble, active GAO was increased dramatically when using lactose as inducer. The optimal concentration for lactose found was 0.5% for this purpose, yielding 302 and 1,300 U/l for GAO with and without its prepro sequence, respectively. The specific activity of GAO also increased to 1.23 and 5.39 U/mg, respectively, which corresponds to approximately 8% of the total soluble protein in the cell. Biomass yields were also increased slightly since E. coli can utilise lactose as a carbon source.
Table 1.
Comparison of the influence of the inducer concentration on the expression of GAO with (GAO + pp) and without (GAO − pp) prepro sequence
| Inducer | Biomass (g/l) |
Enzyme activity (U/l) |
Specific activity (U/mg) |
|||
|---|---|---|---|---|---|---|
| GAO − pp | GAO + pp | GAO − pp | GAO + pp | GAO − pp | GAO + pp | |
| No inducer | 15.3 | 12.5 | 1.50 | 1.60 | 0.01 | 0.02 |
| IPTG (mM) | ||||||
| −0.01 | 16.4 | 13.5 | 1.36 | 1.17 | 0.03 | 0.03 |
| −0.1 | 15.6 | 13.6 | 10.3 | 12.9 | 0.05 | 0.09 |
| −1.0 | 10.8 | 11.4 | 16.2 | 15.6 | 0.07 | 0.06 |
| Lactose (%) | ||||||
| −0.1 | 15.6 | 14.8 | 545 | 163 | 3.45 | 0.57 |
| −0.5 | 17.7 | 15.6 | 1303 | 302 | 5.39 | 1.23 |
| −2.5 | 20.7 | 17.0 | 793 | 56.6 | 4.10 | 0.34 |
To analyze the influence of the fermentation medium on GAO without its prepro sequence production four different media were compared. Increasing the concentration of the nutrients in LB medium increased the enzyme yield to 1,540 U/l. This yield is more than twice the amount of enzyme that has previously been published for the mutated enzyme A3.E7, which contains six mutations and which was obtained in a directed evolution approach aiming at increased expression (Sun et al. 2001). Furthermore, this expression yields are comparable to the expression of native GAO in P. pastoris (Whittaker and Whittaker 2000), however, cultivation times of P. pastoris were 120 h compared to 16 h of cultivation in this paper. Using a rich medium like TB increased the biomass production but reduced the enzyme yield to 436 U/l. Interestingly, the use of MCH-Gly medium almost completely abolished the production of GAO (27 U/l). The reason for this could be the high concentration of glycerol in the medium, since glycerol also acts as substrate for GAO. H2O2, which is formed during the oxidation of glycerol, could hamper and negatively affect enzyme production.
The results obtained for GAO with its prepro sequence were essentially similar, albeit the maximum yield was 486 U of GAO activity per l medium for double LB medium, and therefore significantly lower than those obtained for GAO without the prepro sequence. Addition of copper to the medium (concentration varying from 1 to 10 mM) decreased the production of enzyme by a factor of 5–10 (results not shown). Because of the threefold higher yields, GAO without its prepro sequence was used for the following experiments in this study.
The His-tagged enzyme could be conveniently purified by a simple, single affinity chromatography step to apparent homogeneity as judged by SDS PAGE (Fig. 2). The enzyme preparation thus obtained had a specific activity of 63.9 U/mg, and was purified with a yield of 86% and a purification factor of 13.2 (Table 2). The molecular mass of the enzyme was 66 kDa as analysed by SDS PAGE, which is in agreement with published data (Alberton et al. 2007; Ricardo de Biazio et al. 2008; Wilkinson et al. 2004). Data on the substrate specificity of the recombinant enzyme are summarized in Table 3. The addition of the His-tag did not change the broad substrate specificity of the enzyme, and—as expected—no activity was found with glucose. Determination of kinetic constants for the standard substrate galactose gave a Km value of 34.7 mM and a turnover number kcat of 554 s−1. These values are lower than previously published data for the wild-type enzyme (Wilkinson et al. 2004).
Fig. 2.

SDS-PAGE of recombinant GAO without prepro sequence. Lane 1 Molecular weight marker. Lane 2 crude extract. Lane 3 and 4 purified enzyme in different concentrations
Table 2.
Purification of recombinant GAO without prepro sequence
| Protein (mg) | Total activity (U) | Specific activity (U/mg) | Purification (fold) | Yield (%) | |
|---|---|---|---|---|---|
| Crude-extract | 844 | 4,084 | 4.84 | 1 | 100 |
| Affinity chromatography | 55 | 3,512 | 63.9 | 13.2 | 86 |
Table 3.
Relative activity of GAO toward different substrates
| Substrate | Relative activity (%) |
|---|---|
| D-galactose | 100 |
| Methyl-β-d-galactopyranoside | 126 |
| Dihydroxyacetone | 163 |
| Lactose | 8 |
| Raffinose | 130 |
| Melibiose | 115 |
| Lactobionic acid | 3 |
In conclusion, by simple optimization of the fermentation/expression conditions we were able to increase the yields of recombinant wild-type GAO in E. coli by a factor of ~100, obtaining approximately 24 mg of recombinant, active GAO per l of medium. An improved one-step purification procedure based on the added His-tag resulted in 20 mg of purified enzyme per l of cultivation medium, this corresponds to a space–time yield of 1.25 mg/l h for the purified protein.
Acknowledgments
This project was supported by a grant from the Austrian Science Foundation (FWF project L 504-B11).
References
- Aisaka K, Terada O. Production of galactose oxidase by Gibberella fujikuroi. Agric Biol Chem. 1981;10:2311–2316. [Google Scholar]
- Alberton D, De Oliveira LS, Peralta RM, Barbosa-Tessmann IP. Production, purification, and characterization of a novel galactose oxidase from Fusarium acuminatum. J Basic Microbiol. 2007;47:203–212. doi: 10.1002/jobm.200610290. [DOI] [PubMed] [Google Scholar]
- Avigad G, Amaral D, Asensio C, Horecker BL. The D-galactose oxidase of Polyporus circinatus. J Biol Chem. 1962;237:2736–2743. [PubMed] [Google Scholar]
- Barbosa-Tessmann IP, Da Silva DA, Peralta RM, Kemmelmeier C. A new species of Fusarium producer of galactose oxidase. J Basic Microbiol. 2001;3-4:143–148. doi: 10.1002/1521-4028(200107)41:3/4<143::aid-jobm143>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bretting H, Jacobs G. The reactivity of galactose oxidase with snail galactans, galactosides and D-galactose-composed oligosaccharides. Biochim Biophys Acta. 1987;913:342–348. doi: 10.1016/0167-4838(87)90145-2. [DOI] [PubMed] [Google Scholar]
- Carter JH, Deddens JA, Pullman JL, Colligan BM, Whiteley LO, Carter HW. Validation of the galactose-oxidase-Schiff’s reagent sequence for early detection and prognosis in human colorectal adenocarcinoma. Clin Cancer Res. 1997;3:1479–1489. [PubMed] [Google Scholar]
- Karube I, Kimura J, Yokoyama K, Tamiya E. Integrated microbiosensors for clinical diagnosis. Anal NY Acad Sci. 1990;613:385–389. doi: 10.1111/j.1749-6632.1990.tb18183.x. [DOI] [PubMed] [Google Scholar]
- Leitner C, Volc J, Haltrich D. Purification and characterization of pyranose oxidase from the white rot fungus Trametes multicolor. Appl Environ Microbiol. 2001;67:3636–3644. doi: 10.1128/AEM.67.8.3636-3644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis M, Kuramitsu HK. Galactose oxidase-glucan binding domain fusion protein as targeting inhibitors of dental plaque bacteria. Antimicrob Agents Chemother. 1997;41:999–1003. doi: 10.1128/aac.41.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus Z, Miller G, Avigad G. Effect of culture condtions on the production of D-galactose oxidase by Dactylium dendroides. Appl Microbiol. 1965;13:686–693. doi: 10.1128/am.13.5.686-693.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonca MH, Zancan GT. Purification and characterization of intracellular galactose oxidase from Dactylium dendroides. Arch Biochem Biophys. 1987;252:507–514. doi: 10.1016/0003-9861(87)90058-0. [DOI] [PubMed] [Google Scholar]
- Ricardo de Biazio G, Leite GGS, Tessmann DJ, Barbosa-Tessmann IP. A new PCR approach for the identification of Fusarium graminearum. Braz J Microbiol. 2008;39:554–560. doi: 10.1590/S1517-838220080003000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel RA, Gerbeck CM, Montgomery R. Substrate specificity of D-galactose oxidase. Carbohydr Res. 1968;7:193–199. [Google Scholar]
- Schulte BA, Spicer SS. Light microscopic histochemical detection of sugar residues in secretory glycoproteins of rodent and human tracheal glands with lectin-horseradish peroxidase conjugates and the galactose oxidase-Schiff sequence. J Histochem Cytochem. 1983;31:391–403. doi: 10.1177/31.3.6186733. [DOI] [PubMed] [Google Scholar]
- Shleev S, Wang Y, Gorbacheva M, Christenson A, Haltrich D, Ludwig R, Ruzgas T, Gorton L. Direct heterogeneous electron transfer reactions of Bacillus halodurans bacterial blue multicopper oxidase. Electroanalysis. 2008;20:963–969. [Google Scholar]
- Siebum A, Wijk AV, Schoevaart R, Kieboom T. Galactose oxidase and alcohol oxidase: scope and limitations for the enzymatic synthesis of aldehydes. J Mol Cat B Enzymatic. 2006;41:141–145. [Google Scholar]
- Sun L, Petrounia IP, Yagasaki M, Bandara G, Arnold FH. Expression and stabilization of galactose oxidase in Escherichia coli by directed evolution. Protein Eng. 2001;14:699–704. doi: 10.1093/protein/14.9.699. [DOI] [PubMed] [Google Scholar]
- Whittaker JW. The radical chemistry of galactose oxidase: minireview. Arch Biochem Biophys. 2005;433:227–239. doi: 10.1016/j.abb.2004.08.034. [DOI] [PubMed] [Google Scholar]
- Whittaker MM, Whittaker JW. Expression of recombinant galactose oxidase by Pichia pastoris. Protein Expr Purif. 2000;20:105–111. doi: 10.1006/prep.2000.1287. [DOI] [PubMed] [Google Scholar]
- Whittaker MM, Whittaker JW. Catalytic reaction profile for alcohol oxidation by galactose oxidase. Biochemistry. 2001;40:7140–7148. doi: 10.1021/bi010303l. [DOI] [PubMed] [Google Scholar]
- Wilkinson D, Akumanyi N, Hurtado-Guerreiro R, Dawkes H, Knowles PF, Phillips SEV, McPherson MJ. Structural and kinetic studies of a series of mutants of galactose oxidase identified by directed evolution. Protein Eng. 2004;17:141–147. doi: 10.1093/protein/gzh018. [DOI] [PubMed] [Google Scholar]