

Abstract
Nitric oxide synthase (NOS) catalyzes the conversion of l-arginine to l-citrulline and nitric oxide. N5-(1-Iminoethyl)-L-ornithine (L-NIO), an amidine-containing molecule, is a natural product known to be an inactivator of inducible NOS (iNOS). Because of the presence of the amidine methyl group in place of the guanidine amino group of substrate l-arginine, the active site heme peroxy intermediate sometimes cannot be protonated, thereby preventing its conversion to the heme oxo intermediate; instead, a heme oxygenase type mechanism occurs, leading to conversion of the heme to biliverdin. This might be a new and general inactivation mechanism for heme-containing enzymes. In the studies described here, we attempted to provide support for amidines as substrates and inactivators of iNOS by the design and synthesis of amidine analogues of l-NIO having groups other than the amidine methyl group. No nitric oxide or enzyme-catalyzed products could be detected by incubation of these amidines with iNOS. Although none of the l-NIO analogues acted as substrates, they all inhibited iNOS; increased inhibitory potency correlated with decreased substituent size. Computer modeling and molecular dynamics simulations were run on 10 and 11 to rationalize why these compounds do not act as substrates. Unlike the methyl amidine (L-NIO), the other alkyl groups block O2 binding at the heme iron. Compounds 8, 9, and 11 were inactivators, but no heme was lost and no biliverdin was formed. No kinetic isotope effect on inactivation was observed with perdeuterated ethyl 8. A small amount of dimer disruption occurred by these inactivators, although the amount would not account for complete enzyme inactivation. The l-NIO analogues inactivate iNOS by a yet unknown mechanism, but it is different from that of l-NIO, and the inactivation mechanism previously reported for l-NIO appears to be unique to methyl amidines.
Nitric oxide (NO) is an important cell-signaling agent, which is synthesized in vivo from oxygen and l-arginine by the enzyme called nitric oxide synthase (NOS, EC 1.14.13.39).1 The importance of understanding the chemistry and biochemistry of NO has been highlighted in fields as diverse as immunology and reproductive technology, while the inhibition of NO biosynthesis has been an important approach for the design of drugs to treat septic shock, rheumatoid arthritis, stroke, Alzheimer's disease, and Parkinson’s disease.2, 3, 4, 5 There are three mammalian NOS isoforms: neuronal nitric oxide synthase (nNOS), endothelial nitric oxide synthase (eNOS), and inducible nitric oxide synthase (iNOS), which exhibit 50-60% sequence identity and share identical overall architecture.6, 7. The active sites of NOS isozymes are highly conserved; 16 of 18 residues within 6 Å of the substrate binding site of nNOS and eNOS are identical, and the side chain of one of the two dissimilar amino acids points out of the substrate-binding site.8 Therefore, there is only a one amino acid difference to take advantage of in the active sites of these two isoforms.
However, since NOS inhibition may also be detrimental to the essential functions of NO, selective inhibition of individual NOS isoforms is important for the development of therapeutics.
In the presence of O2, NOS converts l-arginine (1) to l-citrulline (3) and NO with concomitant oxidation of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) (Scheme 1). Numerous cofactors bind to NOS, including flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), NADPH, protoporphyrin IX, and tetrahydrobioperin (H4B). Each NOS isoform is calmodulin (CaM) dependent, although iNOS has been shown to bind CaM very tightly regardless of change in cellular Ca2+ levels;9 therefore, iNOS is kinetically independent of Ca2+ concentration. NOS is active only as a homodimer,10 with each monomer consisting of two subdomains. Inspection of the peptide sequence of NOS and the crystal structure of iNOS11 reveals a C-terminal and an N-terminal domain. Sequences in the C-terminal domain of NOS contain binding sites for FMN, FAD, and NADPH.12 The C-terminal domain serves to shuttle electrons from the two-electron donor, NADPH, through the one or two electron acceptor/donors, FMN and FAD, to the H4B13,14 and heme cofactors in the N-terminal domain of the other monomer.15 The H4B is believed to be involved in electron donation to the heme, 16, 17, 18 where substrate oxidation occurs; 19, 20 during the catalytic cycle the oxidized H4B becomes rereduced.21,22 Calmodulin and Ca2+ are required to enable the electron transfer between the domains.23,24
Scheme 1.
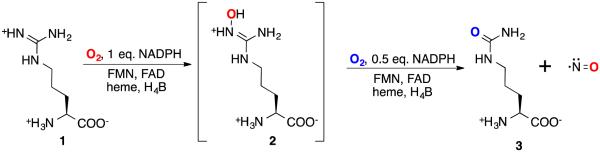
Nitric oxide synthase-catalyzed conversion of l-arginine (1) to l-citrulline (3) and NO
Fast et al.25 reported that the natural product N5-(1-iminoethyl)-l-ornithine (l-NIO) (4) is a time-, concentration-, and NADPH-dependent iNOS inactivator, and it acted as a substrate as well as an inactivator of iNOS. Incubation of iNOS with l-NIO leads to loss of the heme cofactor from the enzyme and the formation of biliverdin, as monitored by HPLC and LC/MS analysis. The same conversion of heme cofactor to biliverdin was identified by Zhu et al.26 from the inactivation of iNOS by N-(3-(aminomethyl)benzyl)acetamidine (1400W) (5). It was proposed that l-NIO, 1400W, and possibly all amidine-containing
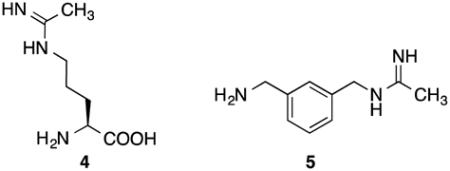
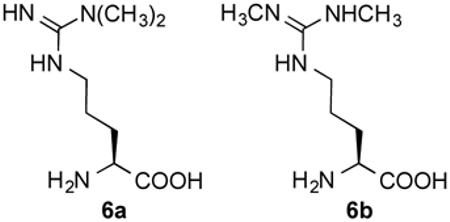
inactivators of NOS, inactivate the enzyme by preventing protonation of the heme peroxide intermediate, which is believed to occur by the guanidinium group of l-arginine, thereby preventing the formation of the heme iron-oxo species and the normal substrate oxygenation (Scheme 2). Instead, a self-oxidation and irreversible inactivation of NOS occurs with conversion of its heme cofactor into biliverdin, along with the production of carbon monoxide (Scheme 3). This is a unique example of inactivation of an enzyme, because the inactivator is not modified during inactivation phase. On the basis of the results that l-NIO acts both as a substrate and inactivator, we hypothesized that l-NIO might bind with either the CH3 or the NH2+ adjacent to the catalytic heme; when the NH2+ group is rotated over the heme, oxygenation to N-hydroxy-l-arginine occurs (2, Scheme 1), but when the methyl group is rotated toward the heme, inactivation occurs (Scheme 3). This is a reasonable hypothesis because interchange of the CH3 and the NH2+ groups in l-NIO would not cause any steric problems in the NOS active site. However, it is known that Nω, Nωω-dimethylarginine (6a) is an endogenous NOS inhibitor, but Nω,N’ω-dimethylarginine (6b) is inactive,27 indicating that the binding of 6b is intolerable in the NOS pocket which is too
Scheme 2.
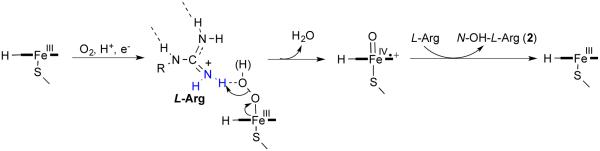
General mechanism for the first step of the NOS reaction
Scheme 3.
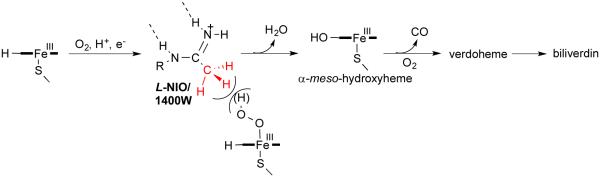
The proposed mechanism of l-NIO and 1400W inactivation with iNOS.26
narrow to allow the methyl group on both terminal guanidine nitrogens. We, therefore, designed and synthesized analogues of l-NIO with variable groups (R) at the terminus (Figure 1 to determine whether rotation of the amidine bond is indeed affected by R group size, and if it determines substrate turnover compared with inactivation. The results of these studies are described here.
Figure 1.
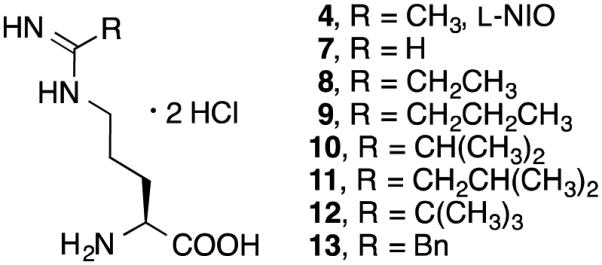
Analogues of l-NIO
EXPERIMENTAL PROCEDURES
General methods
All syntheses were conducted under anhydrous conditions in an atmosphere of argon, using flame-dried apparatus and employing standard techniques in handling air-sensitive materials. Non-synthesized reagents were purchased from Sigma – Aldrich Co. and TCI, and were used without further purification. Propanenitrile-d5 was purchased from CDN Isotopes. 1H NMR spectra were recorded on 500 MHz Varian or Bruker AVANCE spectrometers. Data are presented as follows: chemical shift (in ppm on the δscale relative to δ = 0.00 ppm for the protons in TMS), integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant (J/Hz). Coupling constants were taken directly from the spectra and are uncorrected. 13C NMR spectra were recorded at 125 MHz, and all chemical shift values are reported in ppm on the δ scale. All ion exchange chromatography was performed using Dowex 50WX8-200 resin. Chemical shifts are reported as δ values in parts per million with the CDCl3, D2O, and CD3OD peaks set at 7.26, 4.80, and 3.31 ppm, respectively. An Orion research model 701H pH meter with a general combination electrode was used for pH measurements. LC–MS (ESI) was conducted on an Agilent MSD mass spectrometer. High-resolution mass spectra (HRMS) were measured with an Agilent 6210 LC-TOF (ESI) mass spectrometer. Optical rotations were measured on a PerkinElmer Model 341 digital readout polarimeter. The enzyme assay was monitored on a BioTek Synergy 4 microplate reader. A Beckman System Gold 125P solvent module was used to run HPLC.
Enzyme Expression and Purification
The expression and purification of recombinant murine iNOS from Escherichia coli was performed according to the methods reported by Stuehr and Ikeda-Saito.28
Hemoglobin Assay for iNOS Activity
The production of NO was measured by the rapid oxidation of oxyHb to metHb by nitric oxide.28 The assay mixture contained l-arginine (10 µM), NADPH (100 µM), oxyhemoglobin (0.125 mg/mL), tetrahydrobiopterin (10 µM), and different amounts of inhibitors. The final volume was adjusted to 600 µL with 100 mM Hepes buffer, pH 7.4. The enzymatic reaction was initiated by addition of 10 µL of iNOS stock, and the rate of NO production was monitored by the change in absorbance at 401 nm in the initial 60 s on a spectrophotometer at 37 °C. Curves were fit using the Michaelis– Menten equation in GraphPad Prism 5.0 (GraphPad Software, Inc.). For Ki determinations, IC50 values were calculated using nonlinear regressions (dose-response inhibition, four-parameter variable slope). Subsequent Ki values were calculated using the Cheng-Prusoff relationship: Ki = IC50/(1 + [S]/Km) (Km for murine iNOS is 8.3 µM).29
HPLC of Amino Acid Metabolites
The inactivation mixture was derivatized with o-phthalaldehyde and 2-mercaptoethanol. Inactivation mixtures contained 11.2 µL of 4, 7-13 (2 mM), 6.7 µL of Hepes buffer (100 mM, pH 7.4), 3.3 µL of catalase (3.42 mg/249 µL), 13.8 µL glycerol, 32 µL NADPH (39 mM), 15.7 µL H4B (8 mM), and 64 µL of iNOS stock. The mixture was incubated at 37 °C for 3 h until completion of inactivation. Aliquots (10 µL) were removed and added to 20 µL of o-phthalaldehyde /2-mercaptoethanol (20:1, v/v) reagent. The sample was injected onto an Econosil C18 HPLC column (Alltech, 10 µm, 250 mm × 4.6 mm). A Beckman System Gold 125P solvent module was used to control the gradient elution as follows: Isocratic elution with 90% solvent A: 10% solvent B was carried out for 5 min. The contents of solvent B were then increased linearly to 100% over a period of 10 min. 100% solvent B was carried out for 10 min. Solvent A was 50 mM sodium acetate, pH 5.9. Solvent B was 80% (v/v) methanol and 20% (v/v) solvent A. Sample elution was detected by absorbance at 340 nm with a flow rate of 1.0 mL/min.
Irreversible Inhibition Kinetics
The same inactivation mixtures were prepared as noted in HPLC of Amino Acid Metabolites section. The reactions were initiated by the addition of enzyme, and 10 µL aliquots were removed at specified time points and tested via the initial velocity assay. Controls were performed by replacing the inhibitor solution with Hepes buffer. KI and kinact values were determined by the method of Kitz and Wilson.30
Activity Recovery Determination
The incubation mixture for enzyme activity recovery determination contains 70 µM of potential inactivator. When the incubation mixture showed an obvious activity loss after 16 h at 0 °C, microdialysis was performed over 16 h. The total incubation volume was 100 µL, which was dialyzed in a volume of 100 mL Hepes buffer, pH 7.4. The Hepes buffer was changed two times, every 5 h. All of the incubation mixtures with different compounds and the control were distributed into separate flasks to avoid any cross-contamination. The enzyme activity recovery was checked after overnight dialysis.
HPLC and LC/MS Determination of Biliverdin
Biliverdin determination after treatment of iNOS with 4, 7-13 was carried out by LC/MS with diode array detection, using a C18 reversed-phase column (Vydac, 218TP54, 5 µm, 4.6 × 250 mm) at 401 nm with 60% H2O (0.1% TFA) and 40% CH3CN (0.1% TFA) at a flow rate of 1.0 mL/min. Injections were made after the incubation mixture containing compounds 4, 8, 9, or 11 no longer showed iNOS activity. A gradient method was used: solvent B began at 40% and was then increased linearly to 60% over a period of 10 min. 60% solvent B was decreased to 40% B in 2 min and 40% B was carried out for 3 min.
Computer Modeling of 10 and 11 in iNOS
Models of 10 and 11 were manually built according to the binding model of substrate (L-Arg) and docked to the murine iNOS structure (IN2N). Crystallographic waters were retained, and the system was further solvated in an octahedral unit cell with a 10 Å cushion. Sodium ions were added to maintain net neutrality giving a total of 75,195 and 75,201 atoms for the 10 and 11 complex, respectively. The Amber 12.0 suite (http://ambermd.org/) was used for all calculations.31 The ff10 force field provided with the Amber 12.0 package was used for the protein, while the heme-Cys ligand parameters were taken from Shahrokh et al.32 Parameters for 10 and 11 were derived with antechamber and the gaff force field33 using the BCC charging scheme.34,35
The structure was prepared for production molecular dynamics runs by first energy minimization for 1000 cycles with all heavy atoms except water molecules fixed in position followed by another 1000 cycles where only the inhibitor atoms were allowed to move. In the final 1000 cycles all atoms were allowed to move. Production runs were carried out with a 1 fs time step and coordinates saved every 10 ps. Temperature and pressure were held constant through weak coupling with a 1 ps pressure relaxation time and Langevin dynamics using a collision frequency of 1 ps−1. Periodic boundary conditions were used with a Particle Mesh Ewald implementation of the Ewald sum for the description of long-range electrostatic interactions.36 A spherical cutoff of 10.0 Å was used for nonbonded interactions. Bonds involving hydrogen atoms were constrained using SETTLE.37 Analysis was carried out using the Amber software suite.
RESULTS
Syntheses of 4, 7-13, 22, and 26
Compounds 4, 7-13 (Figure 1) were synthesized as shown in Scheme 4. The synthesis of all of the l-NIO analogues started by alcoholysis of nitriles 14, converting them to the corresponding imidic esters (15).25,38,39 Boc-protected l-NIO analogues (16) were produced by condensation of Boc-l-Orn-OH and 15. After removal of the Boc group by treatment with 4 M aqueous HCl, compounds 4, 7-13 were obtained.
Scheme 4.
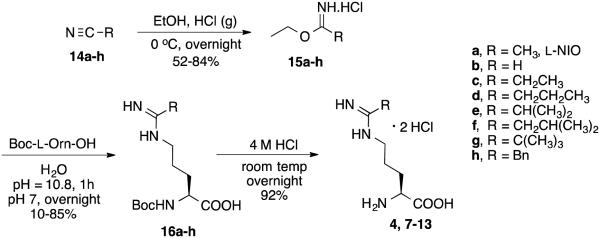
Synthesis of compounds 4, 7-13
The reactions producing 10-12 did not proceed to completion, despite extended reaction times, and the unreacted Boc-l-Orn-OH was difficult to remove by chromatography. Therefore, to remove 17 the reaction mixture was subjected to N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) lactamization (Scheme 5),40 to form lactam 18, which was easily removed by extraction.
Scheme 5.
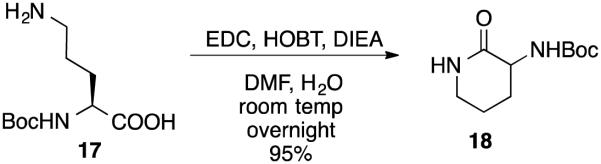
Method to remove 17 from the reaction mixture
Compound 22, the potential hydroxylation metabolite of 8, was synthesized as shown in Scheme 6; oxime 20 was made from propanal (19).41 Compound 22 was obtained after removal of the Boc protecting group.
Scheme 6.
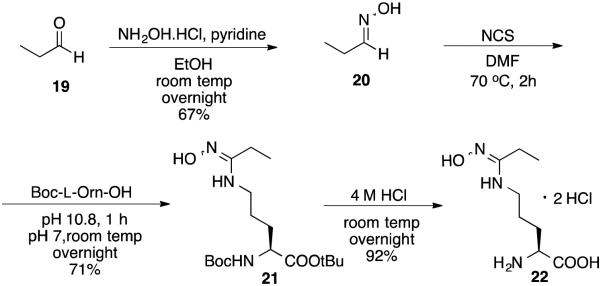
Synthesis of compound 22
Compound 26 was successfully prepared following the synthetic route below (Scheme 7). Starting with propanenitrile-d5 (23), a Pinner reaction was used to make imidic ester 24. Substitution with Boc-l-Orn-OH gave 25, and the Boc group was removed in 4 M HCl to give 26.
Scheme 7.
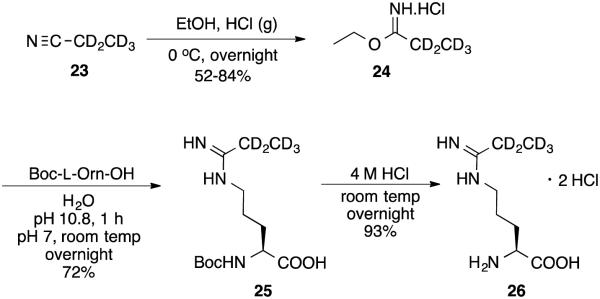
Synthesis of 26
Determination of Substrate Activity, Inhibition Constants (IC50), and Inhibition Type
None of the l-NIO analogues generated any NO at concentrations up to 2 mM. Inhibition experiments were performed by measuring the velocity of NO generation after using different concentrations of the inhibitor and different concentrations of substrate l-arginine. Inhibition of iNOS decreased as the alkyl group became more bulky (Table 1). The only exception is the reversal of Ki values for 10 and 11. l-NIO and all analogues except 12 were found to be competitive inhibitors; 12 was a noncompetitive inhibitor.
Table 1.
IC50 and Ki values for compounds 4, 7-13 against iNOS
| Compound | IC50(μM)a | Ki(μM)b |
|---|---|---|
| 4 (L-NIO) | 8.1 | 3.65 |
| 7 | 0.4 | 0.18 |
| 8 | 16.1 | 7.25 |
| 9 | 19.1 | 8.61 |
| 10 | 138 | 65.18 |
| 11 | 68.7 | 30.95 |
| 12 | 323 | 146 |
| 13 | 5333 | 2402 |
The apparent IC50 values are represented as the mean from two or more independent experiments performed in duplicate with five or six data points each and correlation coefficients of 0.92-0.99. The experimental standard deviations were less than 10%.
The apparent Ki values were obtained by measuring the percent enzyme inhibition in the presence of 10 μM l-arginine with at least five concentrations of inhibitor. The parameters of the following inhibition equation42 were fitted to the initial velocity data: % inhibition = 100[I]/([I]+Ki(1+[S]/Km)). Km value for l-arginine was 8.2 μM (iNOS).
Inactivation of iNOS by 8, 9, and 11
Compounds 8, 9, and 11 (Figure 1) were inactivators of iNOS, and their dissociation constants were measured. 43 Inactivation experiments were performed by measuring the enzyme activity remaining after incubation with different concentrations of the inactivator with the cofactors and iNOS. As shown in Table 2, l-NIO (4) is a more efficient inactivator than 8, 9, and 11, although the inactivation rate constants (kinact) for 8, 9, and 11 are greater than that for 4.
Table 2.
Kinetic constants for 4, 8, 9, and 11
| l-NIO (4) | 8 | 9 | 11 | |
|---|---|---|---|---|
| kinact(min−1) | 0.073 ± 0.003 | 0.0743 ± 0.0073 | 0.131 ± 0.0095 | 0.347 ± 0.006 |
| KI(μM) | 13.7 ± 1.6 | 243 ± 31.4 | 674 ± 80 | 4009 ± 426 |
|
kinact/KI
(μM−1.min−1) |
0.0053 ± 0.0007 | 0.00031 ± 0.000028 | 0.00020 ± 0.0000096 | 0.000087 ± 0.000011 |
Metabolites from Inactivation of iNOS by 8, 9, and 11
HPLC analysis of the amino acid metabolites from 8, 9, and 11 inactivation using o-phthaldialdehyde (OPA) and 2-mercaptoethanol (MCE) was conducted.25, 44, 45 LC/MS was employed to characterize any metabolites formed; however, none was detected. Compound 22 was synthesized as a standard for the potential N-hydroxylated metabolite of 8; no 22 was observed.
Kinetic Isotope Effect on Inactivation of iNOS by 8 and 26
Inactivation of iNOS with (S)-2-amino-5-(propanimidamido-2,2,3,3,3-d5)pentanoic acid (26) showed no kinetic isotope effect on Hkinact/Dkinact (1.025 (±0.071)) or on H(kinact/KI)/D(kinact/KI) (1.159 (±0.108)) (Table 3).
Table 3.
Kinetic Isotope Effect on iNOS inactivation by 8 and 26
| 8 | 26 | H/D | |
|---|---|---|---|
| kinact(min−1) | 0.0743 ± 0.0073 | 0.073 ± 0.0063 | 1.025 ± 0.071 |
| KI(μM) | 243 ± 31.4 | 278 ± 27.7 | 0.884 ± 0.068 |
|
kinact/KI
(μM−1.min−1) |
0.00031 ± 0.000028 | 0.000265 ± 0.000015 | 1.159 ± 0.108 |
Detection of Heme and Biliverdin from Inactivation of iNOS by 8, 9 and 11
l-NIO and 1400W both convert the active site heme in iNOS to biliverdin IXα, determined by LC-electrospray mass spectrometry and by HPLC analysis.26 No apparent heme loss or biliverdin formation was observed from incubation of iNOS with 8, 9, or 11.
LT-PAGE Assay
To determine whether 8, 9, and 11 inactivate iNOS by disruption of the homodimer structure to monomers, iNOS was incubated with l-NIO (4), 8, 9, and 11, and low temperature SDS-polyacrylamide gel electrophoresis (LT-PAGE) was performed. The gels were stained with SYPRO tangerine protein gel stain and imaged for protein quantitation in a Typhoon 9400 imager with a blue laser beam at 488 nm wavelength. It was reported that the addition of 1 µM l-Arg and 0.2 mM H4B produced a stable and noncovalently linked iNOS dimer;46 both the iNOS monomer and dimer were present on the gel. Less dimer was detected after incubation with l-NIO (4), 8, 9, and 11 (Figure 2). The amounts of dimer and monomer remaining after inactivation are shown in Table 4.
Figure 2.

LT-PAGE gel of iNOS with l-NIO (4), 8, 9, and 11. Lane 2, native iNOS; lane 3, 4; lane 4, 8; lane 5, 9; lane 6, 11, lane 8, denatured iNOS
Table 4.
Amount of dimer remaining after inactivation
| Lane | % iNOS Dimer | |
|---|---|---|
| 2 | Native iNOS | 65 ± 5 |
| 3 | 4 with iNOS | 51 ± 4 |
| 4 | 8 with iNOS | 46 ± 2 |
| 5 | 9 with iNOS | 47 ± 5 |
| 6 | 11 with iNOS | 49 ± 5 |
| 8 | Denatured iNOS | <2 |
DISCUSSION
On the basis of no nitric oxide having been produced and no products detected by LC/MS from incubation of iNOS with up to 2 mM 7-13, it can be concluded that these compounds do not act as substrates, unlike l-NIO (4).
Compounds 7-13 all inhibited iNOS activity; all are competitive reversible inhibitors except 12, which is a noncompetitive reversible inhibitor, and 8, 9, and 11, which are irreversible inhibitors. The inhibitor potency decreases as the amidine alkyl group becomes larger; 7, which has the smallest sized R group (H), was the most potent inhibitor of iNOS, with a Ki = 0.18 µM. This implies that the active site pocket of iNOS has sufficient room to fit a bulky group at the amidine terminus, but the larger the substituent, the greater the steric hindrance with surrounding residues. The only reversal in apparent size is 10 and 11, with the Ki for the isobutyl analogue (11) being half of that for the isopropyl analogue (10).
Since crystal structures of these l-NIO analogues bound to iNOS were not available, computer models of 10 and 11 were manually built according to the binding model of substrate (L-Arg) and docked into the murine iNOS structure (IN2N).47 The docked models were then energy minimized and subjected to molecular dynamic simulations. The models right after the initial minimization are shown in Figure 3 and those of the final frame of the simulation in Figure 4. Both 10 and 11 remain stably bound throughout the 1 ns MD simulation. The side chain isopropyl and isobutyl groups of 10 and 11, respectively, are tucked into a hydrophobic pocket lined by Pro344, Val346, and Phe363.
Figure 3.
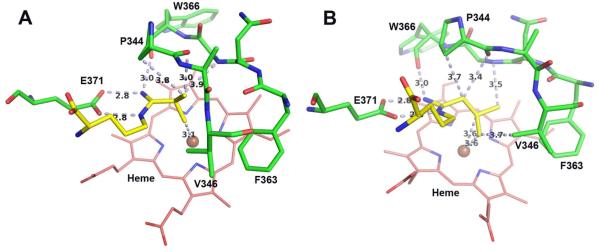
Models of (A) 10 and (B) 11 right after the initial minimization. Major distances less than 4.0 Å are marked in Å including the three H-bonds that anchor the inhibitor to the NOS active site glutamate. The figure was prepared with PyMol (www.pymol.org).
Figure 4.
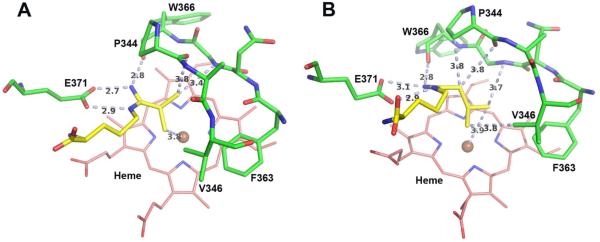
Models of (A) 10 and (B) 11 in the final frame of the simulation. Major distances less than 4.0 Å are marked in Å including the three H-bonds that anchor the inhibitor to the NOS active site glutamate. The figure was prepared with PyMol (www.pymol.org).
In the computer model of 10 after minimization (Figure 3A), tight contacts are shown between the isopropyl group and the Pro344 carbonyl as well as the heme iron, whereas 11 does not have those tight contacts (Figure 3B). MD calculations relax all of the tight contacts but at the cost of distortion of the ligand’s geometry, especially around the amino acid moiety, which departs from what is known about how l-Arg binds (Figure 4). Although just computer models, it is interesting that after minimization they are more agreeable with the Ki measurements, where 10 has poorer binding affinity than 11. This may result from steric crowding of 10; 11 has a longer, and therefore more flexible, isobutyl tail, which can avoid unfavorable steric clashes and instead make more favorable nonbonded contacts (Figure 3).
Over the course of the simulation the distance between the center of mass of the alkyl groups and iron is 4.49 ± 0.30 Å and 3.90 ± 0.17 Å for 10 and 11, respectively, with the alkyl groups remaining positioned directly over the iron. The low standard deviation indicates that the alkyl groups are quite stable and remain close to the heme iron. Such a close approach directly over the iron leaves insufficient room for O2 to bind, which would preclude their substrate activity, as observed. In sharp contrast, l-NIO is isosteric with l-Arg, and the crystal structure of the eNOS-l-NIO complex (a structure in iNOS is not available)48 shows that there is sufficient room for O2 to bind to the heme iron. Moreover, the comparable size of NH2 and CH3 of the acetamidino of l-NIO allows either the protonated imino group to be available to donate a proton to the heme peroxide and act as a substrate or the methyl group to block proton donation and lead to inactivation, as observed. In contrast, the l-NIO analogues with larger alkyl group cannot rotate, because of the limited space in the NOS active site (Figure 3), to bring the imino group closer to the iron. Therefore, blockage of O2 binding to heme and the lack of a close proton donor are the reasons why compounds 8-13 do not act as substrates. The only exception is 7 (R = H), which also should allow O2 to bind to the heme iron because it is smaller than l-NIO and should be able to rotate, but 7 does not act as a substrate or as an inactivator. Although the hydrogen of the formamidino group of 7 can presumably rotate toward the heme, unlike l-NIO, whose methyl group can fill the void left by the rotated imino group, the hydrogen of 7 cannot, and the protonated imino group of 7 is not aligned properly to protonate the heme peroxide. Therefore, 7 does not act as a substrate or inactivator, as observed.
Compounds 8, 9, and 11 exhibit weak irreversible inhibition of iNOS at much higher concentrations than l-NIO (4); although the KI increases with increasing size, the kinact also increases, but the overall efficiency (kinact/KI) diminishes with size, and that for l-NIO is 17-61 times greater than the other three compounds. No inactivation was observed if the R group of the amidine was branched at the α-carbon atom, replacing one or both of the α-protons, as in 10 and 12. Compound 13 (R = Bn) also has a secondary carbon at that position but it is not an irreversible inhibitor, probably because its binding is so weak (Ki 2.4 mM).
Fast et al. reported that l-NIO is both an inactivator and a substrate of iNOS;23 incubation of iNOS with l-NIO gives Nω-hydroxy-l-NIO, which inactivates iNOS, although it is not on the inactivation pathway from l-NIO. Compound 22 was designed as the potential hydroxylation product of 8, but no 22 was observed, again supporting the lack of detection of nitric oxide from incubation of 8 with iNOS. Furthermore, 22 did not inactivate iNOS.
Compound 8 is a time-, concentration-, and NADPH-dependent inhibitor of iNOS. To determine if inactivation proceeds by hydrogen atom abstraction from the amidino ethyl group, (S)-2-amino-5-(propanimidamido-2,2,3,3,3-d5)pentanoic acid (26) was synthesized. Although it inactivated iNOS, it did so with no kinetic deuterium isotope effect on kinact or kinact/KI (Table 3), consistent with an inactivation mechanism that does not involve amidine modification.
One other possible inactivation mechanism was tested: iNOS dimer disruption. The results, demonstrated by low temperature PAGE in the presence of H4B and l-arginine (Figure 2 and Table 4), indicated that of the 65% SDS-resistant iNOS dimer that appeared in the control, only 16-19% was converted to iNOS monomer after inactivation with compounds l-NIO, 8, 9, and 11. However, this does not account for complete loss of enzyme activity, although it could be one of the mechanisms of inactivation.
l-NIO25 and 1400W26 appear to inactivate iNOS by conversion of their heme cofactor to biliverdin, which requires molecular oxygen. With 8, 9, and 11, which our computer model suggests prohibit oxygen binding, no biliverdin could be detected by HPLC with either UV or MS detection. Also, <5% of heme was lost by incubation of iNOS with 8, 9, or 11. Therefore, the mechanism of inactivation of iNOS by these compounds cannot yet be determined but is definitely different from that by l-NIO,25 where substrate turnover via N-hydroxylation; and methyl amidines l-NIO25 and 1400W,26 in which subsequent heme degradation were previously described.
CONCLUSIONS
A series of l-NIO analogues was synthesized to determine if the dual substrate/inactivation properties of l-NIO occurred throughout the series and if the size of the alkyl group determined the ratio of turnover to inactivation, as was observed for l-NIO25 and 1400W26. None of the analogues acted as substrates for iNOS. All were inhibitors, with inhibitory activity diminishing with increased size of the amidine substituent, but only analogues 8 (R = Et), 9 (R = n-Pr), and 11 (R = i-Bu) produced irreversible inhibition. Although 7 (R = H) was the most potent inhibitor, even more potent than l-NIO, it did not inactivate iNOS. One common feature of the compounds that produced irreversible inhibition was the primary or secondary carbon adjacent to the imino carbon of the amidine group. Some iNOS dimer was lost (16-19%) with inactivation by 8, 9, and 11, but that was not much different from the amount that was lost by l-NIO inactivation. However, unlike l-NIO, no nitric oxide was formed, no N-hydroxylated product was formed, no heme was lost, and no biliverdin was formed upon inactivation of iNOS by 8, 9, and 11. Therefore, the mechanism of inactivation of iNOS by 8, 9, and 11 is different from that with l-NIO and 1400W, but that mechanism is still unknown.
Supplementary Material
Acknowledgments
Funding
The authors are grateful for financial support from the National Institutes of Health (GM049725 to R.B.S. and GM057353 to T.L.P.).
Footnotes
Supporting Information
Kinetic data for 8, 9, 11, and 26, HPLC of control, 4, 8, 9, and 11 with iNOS, preparation and characterization of compounds, and 1H NMR and 13C NMR spectra of inhibitors. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Kerwin JF, Heller M. The arginine-nitric oxide pathway: a target for new drugs. Med. Res. Rev. 1994;14:23–74. doi: 10.1002/med.2610140103. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura T, Lipton SA. Emerging roles of S-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox Signal. 2008;10:87–101. doi: 10.1089/ars.2007.1858. [DOI] [PubMed] [Google Scholar]
- 3.Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog. Brain Res. 1998;118:215–229. doi: 10.1016/s0079-6123(08)63210-0. [DOI] [PubMed] [Google Scholar]
- 4.Vallance PP, Leiper JJ. Blocking NO synthesis: how, where and why? Nat. Rev. Drug Discov. 2002;1:939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- 5.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AMG. Nitric oxide in the CNS: Neuroprotection versus neurotoxicity. Nat. Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 6.Hobbs AJ, Higgs A, Moncada S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annu. Rev. Pharmacol. Toxicol. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]
- 7.Kerwin JF, Lancaster JR, Feldman PL. Nitric oxide: A new paradigm for secnd messengers. J. Med. Chem. 1995;38:4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 8.Ji H, Stanton BZ, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Minimal pharmacophoric elements and fragment hopping, an approach directed at molecular diversity and isozyme selectivity. Design of selective neuronal nitric oxide synthase inhibitors. J. Am. Chem. Soc. 2008;130(12):3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinert JR, Chernova T, Forsythe ID. Nitric oxide in brain function, dysfunction and dementia. The Neuroscientist. 2010;16:435–452. doi: 10.1177/1073858410366481. [DOI] [PubMed] [Google Scholar]
- 10.Daff S. Nitric oxide synthase: Structures and mechanisms. Nitric Oxide: Biology and Chemistry. 2010;23:1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Crane BR, Arval AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 12.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: Structure, function, and inhibition. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambert LE, French JF, Whitten JP, Baron BM, McDonald IA. Characterization of cell selectivity of two novel inhibitors of nitric oxide synthesis. Eur. J. Pharmacol. 1992;216:131–134. doi: 10.1016/0014-2999(92)90221-o. [DOI] [PubMed] [Google Scholar]
- 14.Adak S, Sharma M, Meade AL, Stuehr DJ. A conserved flavin-shielding residue regulates NO synthase electron transfer and nicotinamide coenzyme specificity. Proc. Natl. Acad. Sci. USA. 2002;99:13516–13521. doi: 10.1073/pnas.192283399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siddhanta U, Presta A, Fan BC, Wolan D, Rousseau DL, Stuehr DJ. Domain swapping in inducible nitric-oxide synthase - electron transfer occurs between flavin and heme groups located on adjacent subunits in the dimer. J. Biol. Chem. 1998;273:18950–18958. doi: 10.1074/jbc.273.30.18950. [DOI] [PubMed] [Google Scholar]
- 16.Hurshman AR, Krebs C, Edmondson DE, Huynh BH, Marletta MA. Formation of a pterin radical in the reaction of the heme domain of inducible nitric oxide synthase with oxygen. Biochemistry. 1999;38:15689–15696. doi: 10.1021/bi992026c. [DOI] [PubMed] [Google Scholar]
- 17.Wei C-C, Wang Z-Q, Wang Q, Meade AL, Hemann C, Hille R, Stuehr DJ. Rapid kinetic studies link tetrahydrobiopterin radical formation to heme-dioxy reduction and arginine hydroxylation in inducible nitric oxide synthase. J. Biol. Chem. 2001;276:315–219. doi: 10.1074/jbc.M008441200. [DOI] [PubMed] [Google Scholar]
- 18.Wei C-C, Crane BR, Stuehr DJ. Tetrahydrobiopterin radical enzymology. Chem. Rev. 2003;103:2365–2384. doi: 10.1021/cr0204350. [DOI] [PubMed] [Google Scholar]
- 19.Stuehr DJ, Wei C-C, Wang ZQ, Hille R. Exploring the redox reactions between heme and tetrahydrobiopterin in the nitric oxide synthases. Dalton Trans. 2005;21:3427–3435. doi: 10.1039/b506355h. [DOI] [PubMed] [Google Scholar]
- 20.Stuehr DJ, Wei CC, Santolini J, Wang Z-Q, Aoyagi M, Getzoff ED. Radical Reactions of Nitric Oxide Synthases. Biochem Soc. Symp. 2004;71:39–49. doi: 10.1042/bss0710039. [DOI] [PubMed] [Google Scholar]
- 21.Woodward JJ, NejatyJahromy Y, Britt RD, Marletta MA. Pterin- centered radical as a mechanistic probe of the second step of nitric oxide synthase. J. Am. Chem. Soc. 2010;132:5105–5113. doi: 10.1021/ja909378n. [DOI] [PubMed] [Google Scholar]
- 22.Wei C-C, Wang Z-Q, Tejero J, Yang Y-P, Hemann C, Hille R, Stuehr DJ. Catalytic reduction of a tetrahydrobiopterin radical within nitric oxide synthase. J. Biol. Chem. 2008;283:11734–11742. doi: 10.1074/jbc.M709250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abu-Soud HM, Yoho LL, Stuehr DJ. Calmodulin controls neuronal nitric oxide synthase by a dual mechanism. Activation of intra- and interdomain electron transfer. J. Biol. Chem. 1994;269:32047–32050. [PubMed] [Google Scholar]
- 24.Sheta EA, McMillan K, Siler-Masters BS. Evidence for a bidomain structure of constitutive cerebellar nitric oxide synthase. J. Biol. Chem. 1994;169:15147–15153. [PubMed] [Google Scholar]
- 25.Fast W, Nikolic D, Van Breemen RB, Silverman RB, Mechanistic studies of the inactivation of inducible nitric oxide synthase by N5-(1-iminoethyl)L-ornithine (l-NIO) J. Am. Chem. Soc. 1999;121:903–916. [Google Scholar]
- 26.Zhu Y, Nikolic D, Silverman RB, Mechanism of Inactivation of Inducible Nitric Oxide Synthase by Amidines. Irreversible Enzyme Inactivation without Inactivator Modification. J. Am. Chem. Soc. 2005;127:858–868. doi: 10.1021/ja0445645. [DOI] [PubMed] [Google Scholar]
- 27.Böger RH, Bode-Böger SM. Asymmetric dimethylarginine, derangements of the endothelial nitric oxide synthase pathway, and cardiovascular diseases. Seminar Thromb. Homeostasis. 2000;26:539–545. doi: 10.1055/s-2000-13210. [DOI] [PubMed] [Google Scholar]
- 28.Stuehr DJ, Ikeda-Saito M. Spectral characterization of brain and macrophage nitric oxide synthases. Cytochrome P-450-like hemeproteins that contain a flavin semiquinone radical. J. Biol. Chem. 1992;267:20547–20550. [PubMed] [Google Scholar]
- 29.Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 30.Kitz R, Wilson IB. Esters of Methanesulfonic Acid as Irreversible Inhibitors of Acetylcholinesterase. J. Biol. Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 31.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J. Comp. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shahrokh K, Orendt A, Yost GS, Cheatham TE., 3rd Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J Comput Chem. 2012;33:119–133. doi: 10.1002/jcc.21922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case D. Development and testing of a general Amber force field. J. Amer. Chem. Soc. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 34.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atom charges. AM1-BCC model: I. Method. J. Comp. Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 35.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atom charges. AM1-BCC model: II. Parameterization and validation. J. Comp. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 36.Darden T, York D, Pedersen L. Particle mesh Ewald-an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;103:8577–8593. [Google Scholar]
- 37.Miyamoto S, Kollman PA. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water molecules. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 38.Litzinger EA, Martasek P, Roman LJ, Silverman RB. Design, synthesis, and biological testing of potential heme-coordinating nitric oxide synthase inhibitors. Bioorg. Med. Chem. 2006;14:3185–3198. doi: 10.1016/j.bmc.2005.12.043. [DOI] [PubMed] [Google Scholar]
- 39.Moore WM, Webber RK, Jerome GM, Tjoeng FS, Misko TP, Currie MG. l-N6-(1-Iminoethyl)lysine: A selective inhibitor of inducible nitric Ooxide synthase. J. Med. Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]
- 40.Hah J, Roman LJ, Martasek P, Silverman RB, Reduced amide bond peptidomimetics (4S)-N-(4-Amino-5-[aminoalkyl]aminopentyl)-N‘-nitroguanidines, potent and highly selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- 41.Li D, Shi F, Deng Y. One-step C=N, C=O bonds cleavage and C=O, C=N bonds formation over supported ionic liquid in water. Tetrahedron Lett. 2004;45:6791–6794. [Google Scholar]
- 42.Segel IH. Enzyme Kinetics. John Wiley & Sons; New York: 1975. p. 105. [Google Scholar]
- 43.Hevel JM, Marletta MA. Nitric oxide synthase assays. Meth. Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 44.Huang H, Hah J, Silverman RB. Mechanism of nitric oxide synthase. Evidence that direct hydrogen atom abstraction from the O−H bond of NG-hydroxyarginine is not relevant to the mechanism. J. Am. Chem. Soc. 2001;123:2674–2676. doi: 10.1021/ja005900u. [DOI] [PubMed] [Google Scholar]
- 45.Aswad DW. Determination of d- and l-aspartate in amino acid mixtures by high- performance liquid chromatography after derivatization with a chiral adduct of o-phthaldialdehyde. Anal. Biochem. 1984;137:405–409. doi: 10.1016/0003-2697(84)90106-4. [DOI] [PubMed] [Google Scholar]
- 46.Klatt P, Schmidt K, Lehner D, Glatter O, Bächinger HP, Mayer B. Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and l-arginine in the formation of an SDS-resistant dimer. EMBO J. 1995;14:3687. doi: 10.1002/j.1460-2075.1995.tb00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fedorov R, Ghosh DK, Schlichting I. Crystal structures of cyanide complexes of P450cam and the oxygenase domain of inducible nitric oxide synthase-structural models of the short-lived oxygen complexes. Arch. Biochem. Biophys. 2003;409:25–31. doi: 10.1016/s0003-9861(02)00555-6. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Roman CS, Martásek P, Masters BSS, Poulos TL. Crystallographic Studies on Endothelial Nitric Oxide Synthase Complexed with Nitric Oxide and Mechanism-Based Inhibitors. Biochemistry. 2001;40:5399. doi: 10.1021/bi002658v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.