

Young α-satellite tandem dimers dominate functional human centromeres and precisely position pairs of 100-bp CENP-A nucleosomes.
Abstract
The intractability of homogeneous α-satellite arrays has impeded understanding of human centromeres. Artificial centromeres are produced from higher-order repeats (HORs) present at centromere edges, although the exact sequences and chromatin conformations of centromere cores remain unknown. We use high-resolution chromatin immunoprecipitation (ChIP) of centromere components followed by clustering of sequence data as an unbiased approach to identify functional centromere sequences. We find that specific dimeric α-satellite units shared by multiple individuals dominate functional human centromeres. We identify two recently homogenized α-satellite dimers that are occupied by precisely positioned CENP-A (cenH3) nucleosomes with two ~100–base pair (bp) DNA wraps in tandem separated by a CENP-B/CENP-C–containing linker, whereas pericentromeric HORs show diffuse positioning. Precise positioning is largely maintained, whereas abundance decreases exponentially with divergence, which suggests that young α-satellite dimers with paired ~100-bp particles mediate evolution of functional human centromeres. Our unbiased strategy for identifying functional centromeric sequences should be generally applicable to tandem repeat arrays that dominate the centromeres of most eukaryotes.
INTRODUCTION
Human centromeres are deeply embedded within tandemly repetitive α-satellite DNA, presenting severe challenges for understanding centromere identity, function, and evolution and for finishing the human genome project. Centromeres lie within the most homogeneous α-satellite arrays (1). At present, only the most distal portions of some of the α-satellite arrays are annotated as being contiguous with chromosome arms, with other arrays comprising sequence contigs that have not been placed on particular chromosomes or not annotated at all (2).
Annotated α-satellite arrays are dominated by higher-order repeats (HORs). By definition, an α-satellite HOR comprises multiple tandem copies of an α-satellite array, which itself consists of multiple tandem ~170–base pair (bp) units that are diverged from one another. For example, the DXZ1 HOR on the X chromosome consists of tandem copies of a 12-copy array, with each array comprising 171-bp repeat units that are on average only 77% identical in pairwise alignments (3). Divergence of copies within the HOR implies that the ancestral DXZ1 171-bp unit had already duplicated and diverged for a long evolutionary period before the DXZ1 HOR evolved, and similar divergence is seen for other HORs that have been mapped to the most proximal regions of chromosome arms (1). Because α-satellite is estimated to make up 2 to 3% of the human genome, or ~500,000 copies of ~170-bp tandem repeats per haploid genome (4), there should be on average ~20,000 α-satellite copies in more proximal regions of each chromosome, a gap that is almost two orders of magnitude larger than the α-satellite HORs that have been uniquely mapped to the proximal edges of current sequence assemblies. Thus, only the minor pericentric fraction of centromeric α-satellite sequence space has been assembled on the human genome reference sequence, and the situation is even more ambiguous for other eukaryotic genomes, most of which have centromeres that are dominated by tandem repeats (5).
Sequence homogeneity of tandem repeats is thought to be actively maintained in evolution by premeiotic unequal crossing over between sister chromatids or homologs (3, 6, 7). An inevitable consequence of unequal crossing over within a tandem array is that repeat units that become homogenized will follow a separate mutational trajectory from units at the edge of the array. Thus, present-day sequences at the array edge might be too diverged from those undergoing homogenization to be useful in identifying more centromere-proximal sequences. This divergence between repeat units implies that events subsequent to the appearance of an HOR might not be inferable from sequences in current genome assemblies. Given that current sequencing technologies cannot assemble perfectly homogeneous α-satellite arrays, we reasoned that isolation of sequences by centromere function might reveal “young” (recently homogenized) sequences.
To identify functional centromere sequences in the human genome, we took an unbiased approach. We used chromatin immunoprecipitation (ChIP) of centromere proteins to identify the most abundantly enriched sequences, and found them to be dominated by two distantly related tandem dimers of 340 and 342 bp that are present in longer arrays. These dimers precisely position two CENP-A nucleosomal particles of ~100 bp that flank a CENP-B/CENP-C–containing particle, whereas HORs show no distinct positioning.
RESULTS
CENP-A and CENP-C enrichment decreases with α-satellite divergence in pericentric heterochromatin
We used micrococcal nuclease (MNase) digestion of nuclei for native high-resolution ChIP (8) of the HuRef lymphoblastoid line (9) and recovered fragments as small as 25 bp, which we subjected to Illumina library preparation (fig. S1) and 100 × 100–bp paired-end sequencing. Merging overlapping paired-end reads resulted in a collection of 25- to 185-bp fragments that we mapped to several α-satellite–containing bacterial artificial chromosomes (BACs). For a proximal Xp DXZ1–containing BAC, we observed dense enrichment for CENP-A and CENP-C ChIP over the most homogeneous sequences and depletion of histone H3, diminishing with distance from the centromere-proximal end where the HOR diverges (Fig. 1). The proximal-to-distal reduction in CENP-A and CENP-C enrichment and H3 depletion corresponds to divergence of the DXZ1 HOR from 98 to 99% identity between the 12-copy α-satellite repeat array to ~70% identity over ~40 kb (3).
Fig. 1. CENP-A and CENP-C enrichment decreases with α-satellite divergence in pericentric heterochromatin.
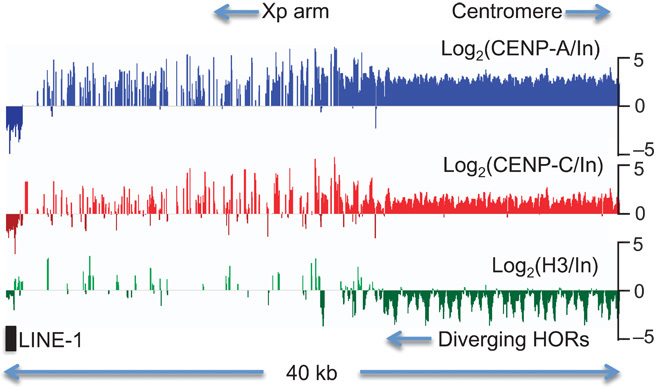
Log-ratio CENP-A, CENP-C, and H3 enrichment profiles spanning the 40-kb most proximal annotated segment of chromosome arm Xp, which spans the DXZ1 α-satellite HOR gradient (3). Dense CENP-A and CENP-C enrichment diminishes with distance from the centromere-proximal edge, and depletion of H3 diminishes ~20 kb from the edge. Diverged α-satellite occupies the Xp arm punctuated by LINE-1 and other elements where centromere protein enrichment is low.
Enrichment of CENP-A and CENP-C and depletion of H3 were also seen for other annotated HORs. For the DYZ3 Y-chromosome–specific HOR, we observed robust CENP-A and CENP-C enrichment for HuRef, which derives from a male, but only background ChIP for HeLa from a female (fig. S2). For the D17Z1B, D11Z1, and D7Z1 HORs, we observed strong enrichment similar to that for DXZ1, but only over some repeat units within each HOR (Fig. 2). Variable enrichment of units within an HOR suggests that subsets of repeat units are differentially amplified at unknown locations within the genome, which can account for the up to ~1000-fold difference in abundance between the different HOR profiles. The D19Z1 HOR showed no CENP-A or CENP-C ChIP enrichment, but this was also the only HOR tested by Hayden et al. (4) that failed to show activity in an artificial chromosome assay. The D5Z1 HOR also showed no CENP-A or CENP-C ChIP enrichment, but this HOR was found to map away from CENP-A foci by cytological analysis, in contrast to the D5Z2 HOR, which overlapped CENP-A foci by fluorescence in situ hybridization (FISH) and showed some repeat units to be strongly enriched (10). Thus, our CENP-A and CENP-C ChIP sequences are enriched for the subsets of α-satellite that likely correspond to functional centromeres.
Fig. 2. Variable CENP-A, CENP-C, and H3 occupancies at annotated α-satellite arrays.
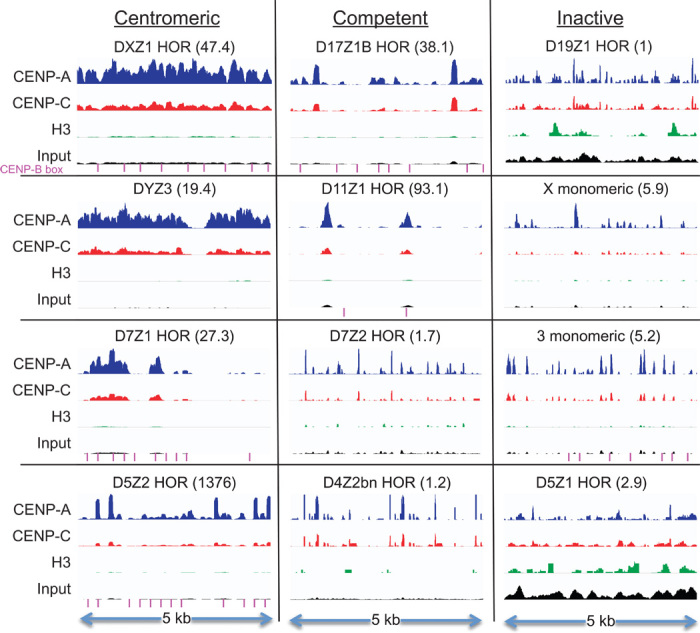
Occupancy profiles for the most centromere-proximal 5-kb regions of eight HORs and monomeric α-satellite arrays present on BAC clones that have been tested for artificial centromere function (4), and for four selected HORs from the hg38 genomic assembly (2). The DXZ1 profile represents an enlargement of the rightmost 5 kb of Xp shown in Fig. 1. HORs are classified on the basis of localization by FISH (centromeric) (10, 35) or by an artificial chromosome assay (competent or inactive) (4). Within each segment, normalized count occupancies were scaled to the maximum occupancy of CENP-A ChIP using the IGV Genome Browser (46). The number in parentheses indicates the fold enrichment of the maximum relative to that of the D19Z1 HOR, which is set at 1, such that the maximum (CENP-A) peak in the D5Z2 HOR is 1376-fold higher than the maximum (H3) peak in the D19Z1 HOR, and the maximum (CENP-A) peak in the D11Z1 HOR is 93.1-fold higher than that in the D19Z1 HOR. Significant BLAST matches to the 17-bp CENP-B box consensus sequence (CTTCGTTGGAAACGGAA) are indicated (magenta lines).
CENP-A ChIP identifies functional centromeric sequences
Sequence assembly programs fail on the most homogeneous α-satellite arrays, because successive copies are too similar to one another to distinguish alternative tandem registers. However, sequences recovered by CENP-A or CENP-C ChIP are enriched for functional centromere sequences without being biased by current assemblies, which are limited to chromosomal regions that are sufficiently diverged to allow for assembly programs to piece together adjacent copies of tandem arrays. If we consider an MNase-protected CENP-A nucleosome to be a centromeric unit, native ChIP-seq reads that recover CENP-A nucleosomes can provide a means of annotating human centromeres de novo.
With Illumina PE100 sequencing, CENP-A nucleosomes have been recovered and mapped to previously assembled HORs as reference sequences (11, 12), but we can also use individual merged PE100 nucleosome sequences themselves as reference sequences, where each covers about a single ~171-bp α-satellite repeat unit. Because assembly programs are designed to extend contigs, but not to identify abundant short repeated sequences, we chose a clustering approach. We aimed to identify the collection of centromere-specific nucleosome-associated reference sequences that represent functional centromeric chromatin, as defined by CENP-A or CENP-C enrichment. We applied the same procedures to each independent data set and then used phylogenetic analysis to determine the degree to which the data sets correspond to one another. Noncorrespondence might be attributable to technical differences between samples, laboratories, or clustering methods or to biological differences. Conversely, close correspondence between phylogenies implies that there are no important technical or biological differences between data sets. Specific sequences identified in this way were used to search existing annotations to identify arrays of these sequences that have undergone homogenization by unequal crossing over in the more recent past (6).
To determine whether the correspondence between centromere protein ChIP enrichment and sequence homogeneity at pericentric HORs generalizes to more proximal sequences, we identified the most abundant individual sequences in our ChIP data sets using two cluster-based analyses (Fig. 3A). We clustered distinct CENP-A ChIP merged pairs from HuRef samples and also clustered a filtered subset of input merged pairs (see Materials and Methods for details). Both clustering strategies returned similar sets of the most abundant reference sequences (Fig. 3B). We also clustered published CENP-A ChIP data sets from four other human individuals (PDNC4, IMS13q, MS4221, and HeLa) and constructed phylogenies from the most abundant sequences in all data sets. All major branches were shared, with two leaves on separate branches especially well represented from nearly all individuals (Fig. 3C and fig. S3). Thus, despite biological differences between individuals and experimental differences between ChIP protocols and antibodies, similar diverse sequences are found in the most abundant α-satellite–containing CENP-A nucleosomes in the human population.
Fig. 3. Centromere proteins from multiple human individuals occupy the same subsets of α-satellite units.
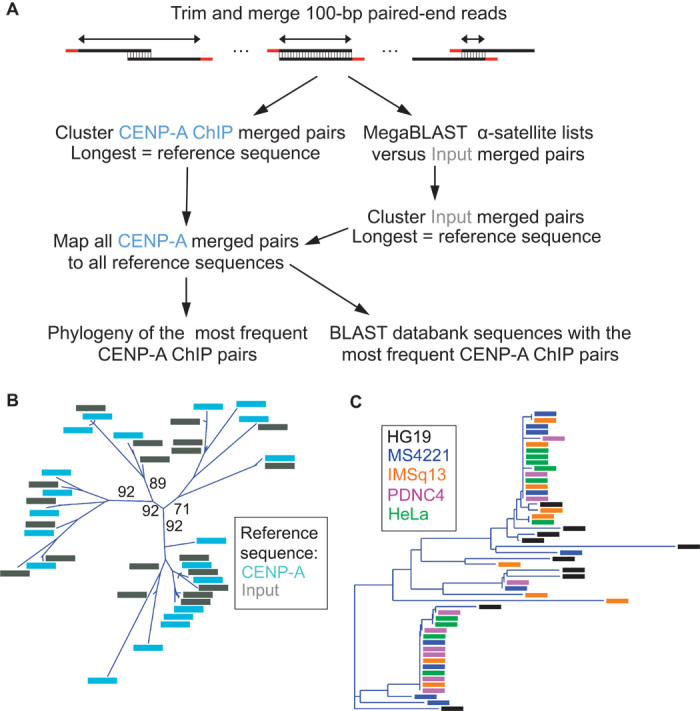
(A) Clustering strategies for identifying the most abundant CENP-A ChIP-enriched sequences. (B) Phylogenetic tree representing the 20 ChIP and input reference sequences that were most abundantly enriched for CENP-A ChIP. Bootstrap percentages are shown for the earliest divergences, defining four branches on the basis of a 70% bootstrap threshold. The same four branches were obtained using only ChIP or only input reference sequences in the alignment. (C) Phylogeny representing the 10 most abundant CENP-A ChIP reference sequences from each of five individuals.
Two α-satellite dimeric units dominate CENP-A ChIP
To determine the spatial distribution of these most abundant ChIP-enriched α-satellite sequences, we searched GenBank (2) using BLAST (13). As queries, we used each of the 20 most abundant reference sequences from a native CENP-A ChIP data set, representing four major clades (Fig. 4A). Sequences from two clades matched sequences within α-satellite arrays annotated as being chromosomes 1, 5, and 19 α-satellite repeats (Cen1-like) (14), and sequences from the other two clades matched those annotated as being from chromosomes 13, 14, 21, and 22 (Cen13-like). We also recovered four unplaced clones from the HG19 reference genome with 99 to 100% identity to several of our ChIP-enriched sequences. Alignment of each sequence with these clones revealed a repeating dimeric pattern of sequence similarity (15), where reference sequences from one clade alternated with reference sequences from the other clade (Fig. 4B, top, and fig. S4). Each dimeric unit was associated with a 15-bp exact match to the consensus CENP-B box, the binding site of the only known sequence-specific mammalian centromere protein (16). Overall, 11 reference sequences aligned on average at 11 positions with ~95% identity to a 4-kb clone (NW_001839579.1) with CENP-B boxes 338 to 340 bp apart (Fig. 4B, top). The unplaced clones therefore represent dimeric α-satellite arrays that likely originated from regions that have undergone homogenization more recently than those from HORs on centromere edges.
Fig. 4. Young α-satellite dimers are the basic units of expansion and homogenization.
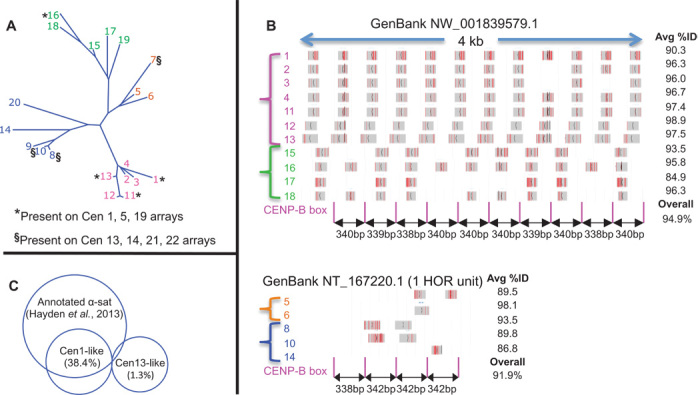
(A) Phylogenetic tree of the 20 most abundantly CENP-A-enriched input sequences, numbered by decreasing abundance and color-coded by clade. (B) Top: MegaBLAST alignments of 11 reference sequences to GenBank NW_001835979.1, where gray horizontal bars represent 100% identity and vertical red lines represent mismatches. Bottom: Same as top except for one of 11 HOR units of NT_167220.1. Numbers on the left are color-coded to correspond to clades in (A). (C) Overlaps of Cen-like and annotated α-satellites for CENP-A ChIP merged pairs.
BLAST also identified a 15-kb homogeneous clone (NT_167220.1) that closely matched Cen13-like sequences comprising ~11 copies of a four-dimer HOR. This clone showed ~92% dimer homogeneity, with a precisely repeating 338-342-342-342–bp pattern of CENP-B distances (bottom, Fig. 4B). Both Cen1-like and Cen13-like sequences appear to account for a large percentage of our α-satellite–specific ChIP libraries because the total number of CENP-A ChIP fragments mapping to concatenated arrays of the two dimeric units was more than half that mapping to annotated sets of concatenated arrays of presumably all α-satellites. Whereas most of the Cen1-like sequences overlapped the annotated sequences, accounting for 38.4% of our α-satellite–specific CENP-A ChIP fragments, the Cen13-like sequences were almost completely absent from annotated sequences, indicating that our unbiased method is able to identify uncatalogued repeats (Fig. 4C and Table 1). Thus, of the ~500,000 α-satellite repeats in the haploid human genome, only few distinct variants dominate CENP-bound and presumably functional centromeres. These functional variants comprise sequences that are younger than those at the edges of genomic assemblies.
Table 1. Merged pairs mapping to annotated α-satellites [chromosome-specific α-satellite units catalogued by Hayden et al. (4)].
Merged pairs aligned with multiple sites were counted only once. Intersection percentages are of the catalogued α-satellite.
| No. of merged pairs | CENP-A | CENP-C | Input |
| Total merged pairs | 3,652,730 | 4,432,991 | 21,929,193 |
| Catalogued α-satellite* | 539,990 | 165,420 | 194,925 |
| Cen1-like† | 277,248 | 78,886 | 87,758 |
| Cen1-like intersecting α-sat |
207,267 (38.4%) | 54,910 (33.2%) | 61,567 (31.5%) |
| Cen13-like‡ | 148,958 | 40,747 | 48,062 |
| Cen13-like intersecting α-sat | 7064 (1.3%) | 1834 (1.2%) | 1737 (0.9%) |
| Cen1-like intersecting Cen13-like |
62 (0.01%) | 40 (0.02%) | 54 (0.03%) |
| All three intersecting | 61 (0.01%) | 39 (0.02%) | 53 (0.03%) |
*Total merged pairs mapping to concatenated α-satellite units in the catalog.
†Total merged pairs mapping to the 38-mer concatenated array.
‡Total merged pairs mapping to the 16-mer concatenated array.
To confirm the long tandem arrangement of Cen1-like sequences, we used BLAST to search published PacBio reads representing an unamplified sample of a human genome derived from a hydatidiform mole (17). We identified many single DNA molecules with tandem copies of a Cen1-like consensus sequence that densely tiled as much as ~30 kb, on the basis of low-stringency BLAST mapping (Fig. 5A). Although these single raw PacBio reads suffered from an ~15% error rate dominated by indels (18), alignment of successive reads to yield consensus sequences and phylogenetic analysis of these consensus sequences (Fig. 5B) indicated that all single-molecule reads identified in this way were derived from a repeat unit that is closely related to the 340-bp Cen1-like consensus (Fig. 5C). On the basis of conservative analysis of PacBio sequencing data, we estimate that there are ~430 Cen1-like dimers per human chromosome (see Materials and Methods). It has recently been estimated that there are ~400 CENP-A molecules per chromosome (19). Assuming that there are two to four CENP-A molecules per dimeric unit (8), 25 to 50% of total centromeric sequence could be Cen1-like. Despite multiple uncertainties in these estimates, the abundance of Cen1-like sequences based on PacBio reads is close to our estimate of 38.4% of annotated α-satellites based on the relative abundance of Cen1-like sequence in our CENP-A ChIP data (Fig. 4C).
Fig. 5. Long tandem repeats of the Cen1-like consensus are detected in PacBio single sequence reads.

(A) Maps of BLASTN hits (boxes, where gray horizontal bars represent 100% identity, vertical red lines represent mismatches, and vertical black lines represent indels) in raw PacBio reads. Displayed are the 10 PacBio single sequence reads (indicated by their sequence read identifier) with the highest bit scores in a MegaBLAST search of SRR1304331 using the Cen1-like 340-bp query. Alternating hits are shown in two tiers for visual clarity. We attribute gaps in the array to the ~15% mostly indel error rate characteristic of PacBio raw data, an interpretation that is supported by the near-perfect alignment of BLAST hits to the 340-bp tiling shown as tandem black diamonds at bottom. (B) A consensus sequence was derived for each of the raw sequences indicated in (A) by automated alignment of the tandem BLAST hits, and a dendrogram was produced, rooting the tree with the Cen1-like consensus. (C) Alignment of the Cen1-like consensus (top sequence) identifies 44 ambiguous residues (indicated as “u” or “s”) and six indels (indicated as dashes) in the overall PacBio-derived consensus (bottom sequence) over the 340-bp sequence.
A unique chromatin conformation characterizes young α-satellite dimers
For unplaced clones tiled by Cen1-like and Cen13-like sequences, we generated a simple consensus by choosing the most frequent base pair in a multiple alignment of dimeric α-satellite units (fig. S5). Phylogenetic analysis indicated that each half of the Cen1-like consensus clustered with the two best-represented branches of the tree that represents the most frequent 20 sequences from five individuals (fig. S3), and each half of the Cen13-like consensus clustered with two other well-represented branches. When we aligned ChIP reads to the Cen1-like consensus, we observed striking CENP-A occupancy patterns, with two precisely positioned ~100-bp “pillars” on either side of the CENP-B box (Fig. 6A). A similar pattern was seen for CENP-C occupancy, with the CENP-B box at the center of a sharply defined MNase-protected “pedestal” ~20 bp wide. In contrast, the most highly represented α-satellite dimer found by CENP-A ChIP among previously annotated and tested HORs (Fig. 2) showed a single poorly defined particle, and CENP-C ChIP revealed it to be adjacent to a well-defined CENP-B pedestal (Fig. 6B). We conclude that different CENP-A– and CENP-C–containing particles occupy young dimeric and old HOR α-satellites.
Fig. 6. Two 100-bp CENP-A nucleosomes are precisely positioned over young, but not old, α-satellite units.
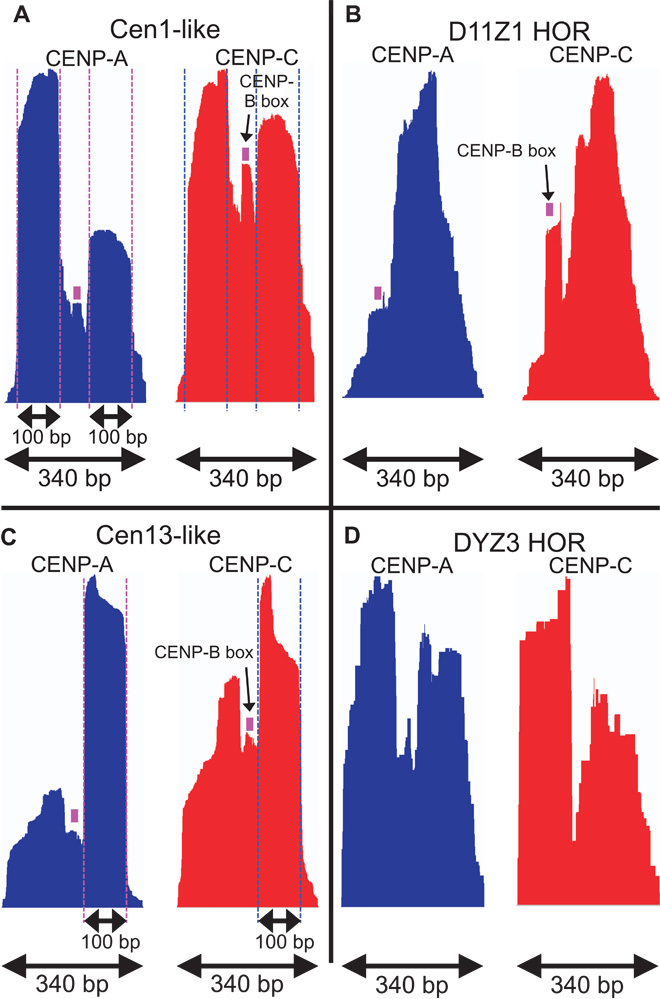
(A) Normalized count profiles of CENP-A and CENP-C ChIP occupancies mapped to the 340-bp Cen1-like consensus. (B) Same as (A) except mapped to the most abundantly enriched 340-bp noncentromeric α-satellite dimer derived from a centromere-competent chromosome 11 HOR (Fig. 2). (C) Same as (A) except for a Cen13-like dimer. (D) Same as (A) except for a Y-chromosome dimer, which lacks a CENP-B box.
CENP-A and CENP-C ChIP sequences mapping to both the Cen13-like dimeric unit and a Y-chromosome dimer also showed the twin ~100-bp pillar/pedestal conformation (Fig. 6, C and D). Because Y-chromosome–derived α-satellite sequences lack CENP-B boxes, it seems likely that another sequence-specific DNA binding protein is responsible for the pedestal-shaped feature. However, motif searches failed to identify a candidate DNA binding protein (S.K., data not shown).
Dimeric sequence abundance decreases exponentially with divergence
To trace back the specific evolutionary trajectories that resulted in recently homogenized dimers, we aligned them with α-satellite arrays from current genomic assemblies. Using BLAST, we identified the closest dimers to both the Cen1-like and Cen13-like consensus sequences within α-satellite arrays. We then mapped CENP-A, CENP-C, and input merged pairs to each dimeric unit and plotted the percent abundance as a function of the percent divergence from the consensus. For both Cen1-like and Cen13-like sequences, we observed a log-linear relationship and strong anti-correlations (r = −0.85 to −0.95), which imply that CENP-A and CENP-C chromatin abundance decreases exponentially with divergence from the consensus (Fig. 7, A and C). Analogous to radioactive decay, exponential divergence of sequence by base substitution implies a stochastic time-dependent process and provides compelling evidence that the two young Cen-like dimers have left behind diverged copies of themselves. Similar exponential decay is seen for ChIP input, which implies that the youngest Cen1-like and Cen13-like α-satellite dimers are also the most abundant, decreasing exponentially in abundance with age.
Fig. 7. Two distinct chromatin complexes occupy specific α-satellite arrays of human centromeres.
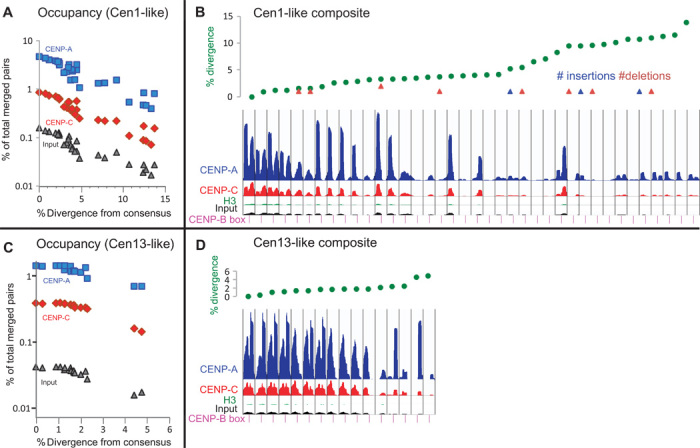
(A) Sequence divergence of selected dimeric units relative to the Cen1-like consensus dimers. (B) ChIP occupancy profiles for a composite 38-mer with dimers rank-ordered by divergence (green dots with indels indicated as triangles). (C) Same as (A) except for Cen13-like dimers. (D) Same as (C) except for a 16-mer Cen13-like composite sequence.
We next asked whether divergence from the consensus has consequences for centromere protein particle conformation. For each Cen-like sequence family, we rank-ordered dimers by increasing divergence. As a reference sequence for mapping merged pairs, we constructed a single composite sequence from the rank-ordered set of dimers. In both cases, we observed gradual reductions in abundance down to 2% divergence, followed by variable loss of one or both 100-bp peaks, followed by more consistent reduction in abundance of both peaks beyond ~10% divergence (Fig. 7, B and D). Despite the gradual loss of abundance, twin-pillar positioning was observed even in a dimer that is only 86% identical to the Cen1-like consensus. We conclude that sharp 100-bp CENP-A and CENP-C positioning is a feature that is shared by Cen1-like and Cen13-like units even as they age.
Young α-satellite dimers precisely position ~100-bp CENP-A nucleosomes
Close examination of the size distributions of the ~100-bp particles revealed a marked regularity: Most of the fragments mapping to the Cen1-like composite sequence showed a sawtooth pattern of lengths 97, 99, 101, and 103 bp (Fig. 8A). The Cen13-like composite also showed a sawtooth pattern, but it was of lengths 98, 100, and 102 bp (Fig. 8B). The CENP-B box spacing differences between Cen1-like (340 bp) and Cen13-like (mostly 342 bp) sequences provide a simple explanation for this difference: with one additional base pair on either side of the central CENP-B box of the Cen13-like dimer, there is 1 bp more DNA for wrapping each twin pillar particle. Consistent with this scenario, we note that there is a missing CENP-B box in the third of 10 dimeric units in clone NW_001839622.1, and this unit is almost entirely devoid of CENP-A and CENP-C occupancy and input abundance (fig. S6). In contrast to the precise ~100-bp size distributions seen for ChIP of young α-satellite dimers, ChIP profiles of the DXZ1 HOR showed a much broader distribution including a major peak at ~130 bp (Fig. 8C), similar to particles previously described as likely octameric CENP-A or CENP-A/H3.3 nucleosomes (11, 20).
Fig. 8. Young α-satellite dimers precisely position ~100-bp CENP-A nucleosomes.
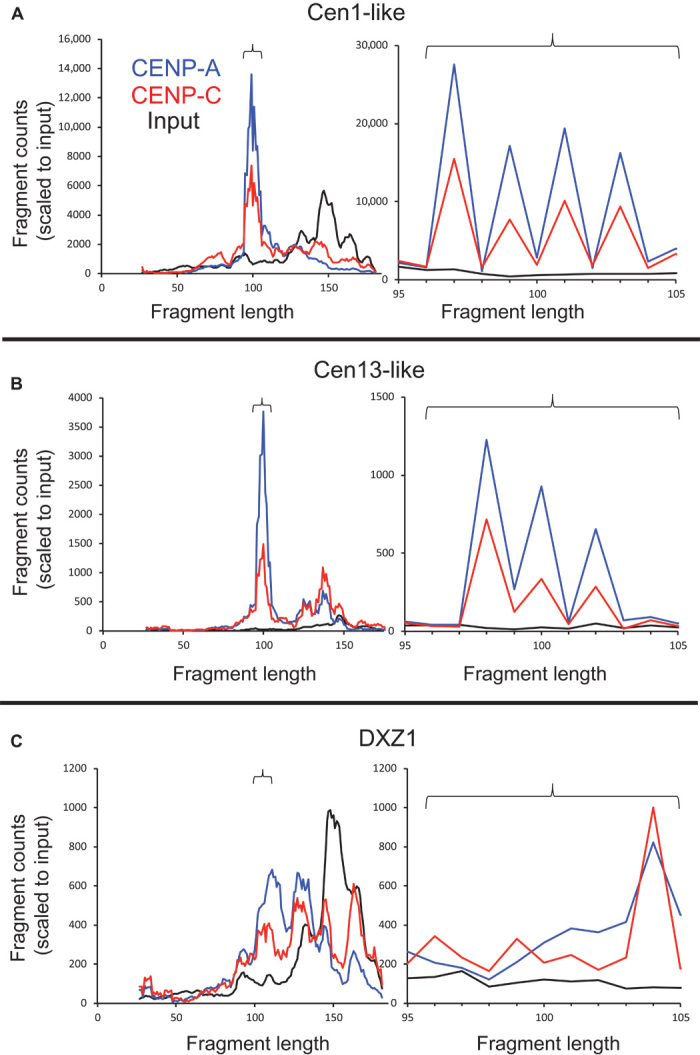
(A to C) Size distributions of fragments mapping to the Cen1-like (A) and Cen13-like (B) composites and the most proximal 6-kb region of DXZ1 (C). Graphs on the right are expansions of graphs on the left (indicated by brackets). The y-axis scale is for input normalized counts, and the areas under the other curves were equalized to that for input.
DISCUSSION
We have introduced an unbiased computational strategy to identify functional human centromeric repeats based on the identification of the most abundant sequences in CENP-A ChIP libraries. We found that two dimeric α-satellite sequences dominate the ChIP signal in the human reference genome, which was the source of our ChIP library, as well as in ChIP libraries from four unrelated female and male individuals from other studies (11, 12). Therefore, these two dimeric units, which are phylogenetically highly diverged from one another, would appear to make up most functional human centromeric chromatin. Our findings support a model in which HORs evolved from ancestral tandem α-satellite dimers that diverged from one another before undergoing the long-period unequal crossing-over events (1). These findings might seem unexpected, insofar as most discussions of human centromeres have focused on HORs (4, 21), which dominate human pericentromeres. However, dimeric α-satellite repeat units have previously been identified in the human genome, including the abundant D5Z2 subset of the Cen1-like consensus (14, 22), which has been mapped by FISH to the centromere of chromosome 5 (10). Also, the two dominant dimeric units identified in our study correspond to the two major α-satellite homology groups defined by Alexandrov and co-workers on the basis of in situ hybridization with abundant α-satellite repeat probes (1). Specifically, suprachromosomal family 1 of Alexandrov et al. corresponds to our Cen1-like dimer and is found on chromosomes 1, 3, 5, 6, 7, 10, 12, 16, and 19, and suprachromosomal family 2 corresponds to our Cen13-like dimer and is found on chromosomes 2, 4, 8, 9, 13, 14, 15, 18, 20, 21, and 22. Therefore, the dominance of Cen1-like and Cen13-like α-satellite dimers in ChIP data fits well with cytological evidence that most human centromeres fall into these two phylogenetically defined classes.
In contrast to the general perception that HORs dominate the most homogeneous α-satellite arrays (3, 23), our findings point to young homogeneous α-satellite dimers as being the fundamental units of centromere evolution. Our findings also can help reconcile the evidence for HORs consisting of diverged α-satellite units at the boundaries of chromosome arms with the abundance of young α-satellite dimers more proximally. Indeed, a widely accepted model for evolution of tandem repeats by unequal crossing over (6) predicts precisely this situation: Repeat units toward the middle of a tandem array will undergo out-of-register pairing, unequal crossing over, and homogenization, whereas repeat units at array edges will be prevented from pairing out of register because they are adjacent to nonhomologous sequences (Fig. 9). Over time, mutations will accumulate on array edges, whereas recurrent duplication/deletion events toward the middle of an array will result in successive homogenization events and random sampling of new mutations. Thus, from the time of the original expansion of a tandem array until the present day, repeat units that accumulate mutations at the edge will have followed an evolutionary trajectory that is independent of that for units that have undergone homogenization of successive mutations.
Fig. 9. Satellite DNA evolution by mutation and unequal crossing over [based on (6) and (47)].
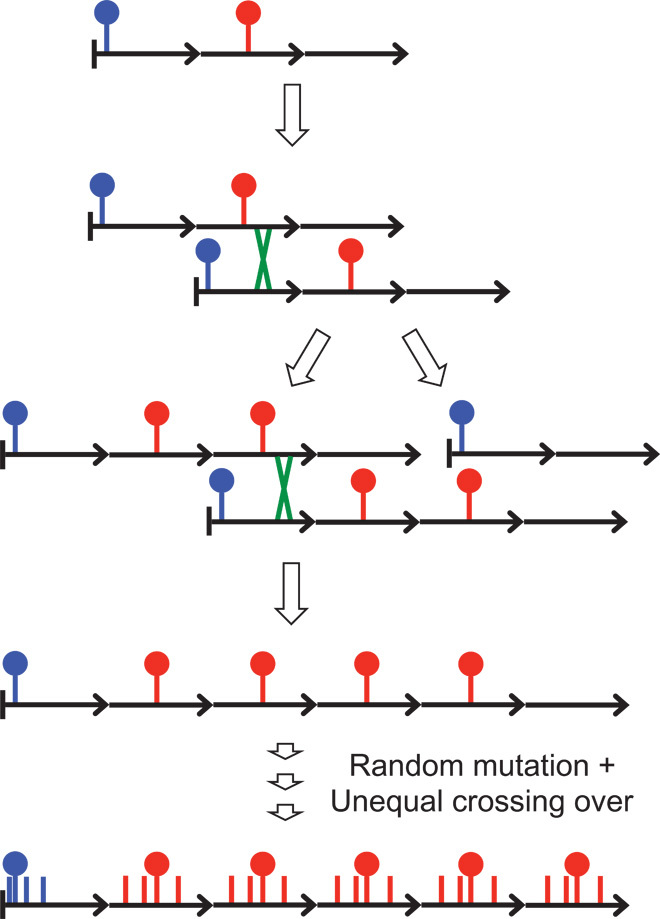
In this toy example, a three-unit tandem array undergoes an out-of-register pairing event and unequal crossing over to produce a four-unit duplication and a two-unit deletion. Because the blue mutation is close to the left edge of the array, crossing-over events are most likely to occur to its right, and it will be inherited in both the duplication and deletion daughter chromosomes, whereas the red mutation is near the middle, and so it will be duplicated and deleted with similar expected frequencies. Further unequal crossing-over events within the four-unit array will result in expansion and contraction of the array, with corresponding gains and losses of the red mutation, leading to homogenization, but without consequence for the blue mutation. Other mutations that arise near the middle of the array will undergo homogenization like the red mutation, and those that arise near the edge will accumulate without gain or loss like the blue mutation. Over evolutionary time, the edges of the array will diverge, and longer-period out-of-register pairing and crossing-over events will result in HORs encompassing multiple tandem repeat units that are diverged from one another (3). Successive mutations and homogenization events in the middle of the array will result in divergence of homogeneous satellite sequences from the ancestral repeat unit.
Not only are Cen1-like and Cen13-like sequences the most abundant sequences in CENP-A ChIP libraries, but they also appear to be the most abundant α-satellite units in input libraries, which is consistent with selection for some adaptive property. It is possible that this property is CENP-A nucleosome positioning, which is markedly precise for the Cen1-like, Cen13-like, and DYZ3 sequences, in contrast to the relative lack of positioning for HORs. However, these young α-satellite dimers are occupied by CENP-A and CENP-C at levels that are similar to occupancies seen at many HORs, which suggests that nucleosome assembly or stability is not directly responsible for the success of these α-satellite dimers relative to HORs. Another possibility is that precise positioning of CENP-A nucleosomes on either side of the CENP-B box places the CENP-B protein, which is related to the Pogo transposases (24), in a conformation that might facilitate recombination and satellite repeat expansion (25). Consistent with this possibility, CENP-A has been implicated in double-strand break and repair processes (26). The homogeneity of young tandem repeats might also facilitate their expansion to achieve a favored orientation toward the egg pole during female meiosis, resulting in centromere drive (27–29). These alternative evolutionary scenarios are not mutually exclusive.
The existence of CENP-A nucleosomes protecting ~100 bp of α-satellite has been previously described (11), although no sequence specificity of this prominent CENP-A ChIP size class was reported in that study. In rice, the 155-bp CentO satellite was found to precisely position a cenH3-containing particle of ~100 bp in monomeric arrays (30), which suggests that nucleosomal particles that wrap less DNA than has been observed by X-ray crystallography of reconstituted CENP-A–containing octameric nucleosomes (>120 bp) (20, 31) are common in satellite-containing centromeres. Well-phased single-wrap cenH3 nucleosome particles have also been observed in Caenorhabditis elegans holocentromeres (32) and budding yeast “point” centromeres (33). Therefore, the well-phased pillar conformations that we observed in young α-satellite units would appear to be a general feature of centromeres of many different types.
The identification of a twin pillar conformation with a CENP-B pedestal over the 340- to 342-bp dimeric repeat units in young α-satellites places human centromeres in the same category as budding yeast point centromeres (34), which are determined by DNA sequence and are occupied by precisely positioned single-wrap tetrameric nucleosomes (33). In contrast, the poorly phased particles found on older dimeric units likely provide weak centromere function that is detected in artificial chromosome assays (4, 35, 36). Because several HORs are found to be competent on the basis of this assay, we expect the same to hold for the young α-satellite dimers that we have described. However, the lack of correlation between competence in the assay and CENP-A abundance (4), and the nonessentiality of CENP-B in vivo (37) but the requirement for both CENP-B protein and CENP-B boxes in artificial chromosome assays (38, 39), suggests that more reliable assays will be required to settle this issue.
Precise phasing of single-wrap particles has obvious implications for centromere function in organisms as distant as yeast and humans, where near-perfect segregation is required at every cell cycle. Our findings provide a rational basis for artificial chromosome design based on sequence specificity that should be applicable not only to humans but also to most eukaryotes with centromeres that are embedded in satellite repeats.
MATERIALS AND METHODS
Nuclei preparation
The HuRef lymphoblastoid and HeLa cell lines were grown in RPMI and Dulbecco’s modified Eagle’s medium, respectively, using standard protocols. Antibodies used in this study were purchased from Abcam. Nuclei were prepared following a previously published protocol (11). Briefly, for each immunoprecipitation, 40 to 60 million cells were collected and washed with 1× phosphate-buffered saline. Cells were resuspended in ice-cold buffer I [0.32 M sucrose, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 15 mM tris (pH 7.5), 0.5 mM dithiothreitol (DTT), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor] at a density equal to ~25 million cells/ml. An equal volume of ice-cold buffer I supplemented with 0.1% NP-40 was added, and samples were incubated on ice for 10 min. The nuclei suspension was layered on ice-cold buffer III [1.2 M sucrose, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 15 mM tris (pH 7.5), 0.5 mM DTT, 0.1 mM PMSF, and protease inhibitor] and centrifuged at 10,000g for 20 min at 4°C. The pellet was resuspended in buffer A [0.34 M sucrose, 15 mM Hepes (pH 7.4), 15 mM NaCl, 60 mM KCl, 4 mM MgCl2, 1 mM DTT, 0.1 mM PMSF, 3 mM CaCl2, and protease inhibitor] at a density equal to ~32.5 million cells/ml.
MNase digestion, needle extraction, and immunoprecipitation
MNase (Sigma, cat. no. N3755) was added at ~2.5 U/ml, and digestion was carried out at 37°C for 5 min. Reactions were stopped by addition of EGTA to a final concentration of 20 mM, and EDTA to 10 mM. The final NaCl concentration was adjusted to 215 mM, and needle extraction was performed as described previously (8) to enhance solubility of the kinetochore complex. The resulting solution was incubated overnight at 4°C on a nutator. Soluble chromatin was collected by centrifuging the mixture at 13,400g at 4°C for 8 min.
Soluble chromatin was diluted three times with 20 mM tris (pH 8.0), 5 mM EDTA, and 200 mM NaCl, and Triton-X was added at a final concentration of 0.1% (v/v). Next, the chromatin solution was precleared using protein A/G fast-flow Sepharose beads for 20 min at 4°C, and 15 μg of antibody [Abcam, anti–CENP-A (Ab13939), anti–CENP-C (cat. no. 33034), and anti-H3 (Ab1791)] was added per ChIP sample and incubated overnight at 4°C. Dynabeads were added to the samples, and the mixture was incubated at 4°C for 2 hours. Immunoprecipitated complexes were washed six times with 50 mM phosphate buffer (pH 7.4), 5 mM EDTA, 200 mM NaCl, and DNA was extracted from Dynabeads and from soluble chromatin as described (8), producing respectively ChIP and input DNA.
Illumina sequencing and data processing
Solexa library construction was performed as described (33, 40) on CENP-A and CENP-C ChIP and input DNA for two HuRef biological replicates and for CENP-A and input HeLa DNA, and 100-bp paired-end reads were obtained by Illumina HiSeq 2500 sequencing. Paired reads were merged using SeqPrep (https://github.com/jstjohn/SeqPrep, 20 Feb 2014) with parameters −q 30 (quality) and −L 25 (minimum merged pair length). SeqPrep also removed adaptors and low-quality reads. All alignments were done using BWA version 0.7.5 (http://bio-bwa.sourceforge.net/bwa.shtml). Single-end alignments were done on the merged pairs using the aln and samse algorithms in BWA. Default parameters were used, except up to 10 alignments were saved per merged pair (parameter −n 10) unless otherwise noted. MegaBLAST version 2.2.26 (www.ncbi.nlm.nih.gov/blast/html/megablast.html) with all default options (for example, word size = 28) was used to compare input merged pairs with the catalogued α-satellite sequences (4).
Cluster-based analyses of ChIP data
Merged pairs produced by SeqPrep were ≤185 bp. Read pairs that did not get merged were discarded to ensure that most of the α-satellite merged pairs were derived from a single central unit. Given that our input libraries were dominated by mononucleosomes (fig. S1), there should be few, if any, false merges between dimers or trimers. We counted identical merged pairs to give a comprehensive set of distinct merged pairs, each with a frequency. In analyses that counted merged pairs mapped to a reference sequence, each matching merged pair contributed a value equal to its frequency.
We then used two different methods to obtain reference sequences. There were millions of distinct merged pairs for each ChIP sample, more than the estimated total number of α-satellite units in the haploid genome. Thus, we clustered them on the basis of sequence identity using CD-HIT-EST (41, 42) (http://cd-hit.org, version 4.6.1). CD-HIT-EST uses a fast, greedy incremental clustering algorithm that avoids making all pairwise comparisons by applying a short word filter. We used a word size of 10 (parameter −n 10) and a default identity threshold of 0.90 (parameter −c 0.90). For the first method, we clustered the distinct CENP-A merged pairs. CD-HIT-EST reports a representative longest sequence for each cluster, and this was used as a reference sequence. We aligned all CENP-A merged pairs with these CENP-A–derived reference sequences using BWA. We then rank-ordered the reference sequences by the total number of alignments so that those with the most alignments represented the most abundant CENP-A nucleosomes. For phylogenetic analysis, only the 10 and 20 most abundant reference sequences were used to make trees that could be practically displayed on a page.
A possible drawback to clustering based on CENP-A merged pairs is that it strongly favors the most abundantly amplified sequences whether or not they are the most enriched, and highly CENP-A enriched sequences of moderate or low abundance might pass below the threshold, and in aggregate these sequences might make up a large part of human functional centromeres. Further exacerbating this potential problem are “jackpots” in which polymerase chain reaction amplification for library preparation results in an artifactual abundance of the same starting fragment. This is a potentially severe problem for α-satellite sequences that are very unequally represented in the genome, and where native ChIP typically results in low amounts of starting material, because kinetochore complexes are largely insoluble (8). To address these issues, for our second method, we used an indirect strategy based on clustering input rather than CENP-A ChIP data to define reference sequences. Because the amounts of starting DNA available for input samples are typically high enough to reduce the jackpot problem, this approach may result in a more democratic representation of starting MNase-protected fragments. Our input data sets are large and include many reads unrelated to α-satellite. To reduce the amount of data while retaining α-satellite reads, we first used MegaBLAST to filter the distinct merged input pairs using sets of concatenated α-satellite units comprehensively catalogued by Hayden et al. (4) as queries, saving all those with a significant BLAST score. We then clustered with CD-HIT-EST as above and aligned all CENP-A merged pairs with these input-derived reference sequences using BWA, saving up to 10 alignments per merged pair.
We first clustered distinct CENP-A ChIP merged pairs and obtained >200,000 clusters. We also selected α-satellite from input sequences by searching comprehensive sets of concatenated α-satellite sequences (4), followed by clustering, which yielded ~100,000 clusters. In both cases, we used the representative longest sequence from each cluster as a reference sequence for single-end mapping of ChIP merged pairs using BWA (43) and rank-ordered reference sequences on the basis of the frequency of merged pairs mapping to them. We applied these same two clustering procedures to data from other laboratories to construct additional sequences for phylogenetic analysis from several human individuals. The following data sets were downloaded from the GenBank Short Read Archive and analyzed as described above: CENP-A ChIP, SRR766736 (PDNC4), SRR766737 (IMS13q), SRR766738 (MS4221), and SRR633614-5 (HeLa); input: SRR766739 (PDNC4), SRR766740 (IMS13q), SRR766741 (MS4221), and SRR633612-3 (HeLa).
Phylogenetic analysis
To compare the results of applying this analysis pipeline to different ChIP and input data sets, we used MAFFT (44) and associated tools for automated multiple alignment of reference sequences with default parameters. We constructed phylogenies by neighbor-joining (45) using the resulting distance matrix to assign percent divergence values. Similar topologies were obtained using different alignment parameters. To confirm the similarity between phylogenies made from the two sets (CENP-A–derived and input-derived) of the 20 most abundant reference sequences from a mixture of all data sets, we performed the same procedure on the 10 most abundant sequences from each clustering strategy. This phylogeny showed only nonsignificant differences in the population of the four major branches with respect to clustering strategy (P > 0.45). Likewise, data sets from five human individuals showed only nonsignificant differences between branches (P > 0.16). Indeed, some leaves of the tree contained multiple reference sequences from different clustering strategies and different human individuals, reflecting sequence identity over the central MNase-protected segment of the reference sequences. The close correspondence between phylogenies suggests that our “bottom-up” approach to identifying centromeric α-satellite units is robust to variations in the source of reference sequences (CENP-A ChIP or filtered input), to variations in the protocols used for sample preparation in MNase (native) ChIP in three different laboratories, and to variations between human individuals.
Comparison with annotated clones
We downloaded annotated HORs and BACs and mapped our input and ChIP merged pairs to them using the aln and samse algorithms of BWA and saving up to 10 alignments per merged pair. In most cases, this procedure resulted in saving all alignments (BWA does not have an option to save all alignments). For each base pair (i) in the reference sequence, the number of merged pairs aligned over it (ni) was counted and normalized by dividing by the total number of merged pairs (N) and multiplying by the annotated human genome size [= (ni/N) × 3,095,693,983].
Identification of homogeneous arrays
We searched the most abundant reference sequences against the National Center for Biotechnology Information (NCBI) DNA databases using BLAST to identify unplaced genomic sequences. All merged pairs from input and ChIP samples were aligned with the identified sequences and constructed consensus sequences using BWA, saving up to 10 alignments per merged pair. Abundance was estimated as the number of alignments. For each base pair in the reference sequence, we counted the number of merged pairs aligned over it. We normalized base pair counts as described above and made track files for the IGV Genome Browser (46), which was used to further scale and display occupancy.
Analysis of PacBio SMRT single molecule real-time sequencing data
To obtain a random sample of a long, unamplified, and uncloned human genome sequence that spans multiple Cen1-like dimers, we used MegaBLAST with default parameters to search the 340-bp Cen1-like consensus sequence as query of a sample of 163,482 single molecule real-time sequencing reads (NCBI Sequence Read Archive, accession SRR1304331). This corresponded to a search of 21% of the human genome and resulted in 2981 hits. Using a conservative threshold of at least eight alignments per read, we scored 2192 total alignments, or on average ~430 [= (2192/0.21)/24] alignments per chromosome. Sequences were rank-ordered by total BLAST score, and the NCBI Genomic Workbench was used to automatically parse repeats within the top-scoring reads. Phylogenies were constructed using MAFFT with default parameters. The EBI MView sequence editor was used for alignment display.
Supplementary Material
Acknowledgments
We thank H. Malik, P. Green, P. Talbert, A. Drinnenberg, and F. Steiner for discussions and critical comments. Funding: Supported by NIH R01 ES020116 and the Howard Hughes Medical Institute. Author contributions: J.G.H. conceived of the project and performed the analyses; J.T. performed the ChIP experiments; S.K. performed the PacBio analysis; S.H. conceived of the project and performed the analyses. Data and materials availability: Sequence data are available from GEO (GSE60951).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/ content/full/1/1/e1400234/DC1
Fig. S1. Size distribution of input library fragments.
Fig. S2. CENP occupancies in a male and female cell line.
Fig. S3. Joint phylogeny of the most frequent CENP-A ChIP sequences for five human individuals.
Fig. S4. Cen1-like repeat units in an unplaced clone.
Fig. S5. Cen1-like and Cen13-like alignments.
Fig. S6. Normalized count profiles mapped to individual clones.
References and notes
- 1.Alexandrov I., Kazakov A., Tumeneva I, Shepelev V., Yurov Y., Alpha-satellite DNA of primates: Old and new families. Chromosoma 110, 253–266 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Benson D. A., Clark K., Karsch-Mizrachi I., Lipman D. J., Ostell J., Sayers E. W., GenBank. Nucleic Acids Res. 42, D32–D37 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schueler M. G., Higgins A. W., Rudd M. K., Gustashaw K., Willard H. F., Genomic and genetic definition of a functional human centromere. Science 294, 109–115 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Hayden K. E., Strome E. D., Merrett S. L., Lee H.-R., Rudd M. K., Willard H. F., Sequences associated with centromere competency in the human genome. Mol. Cell. Biol. 33, 763–772 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melters D. P., Bradnam K. R., Young H. A., Telis N., May M. R., Ruby J. G., Sebra R., Peluso P., Eid J., Rank D., Garcia J. F., DeRisi J. L., Smith T., Tobias C., Ross-Ibarra J., Korf I., Chan S. W. L., Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 14, R10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith G. P., Evolution of repeated DNA sequences by unequal crossover. Science 191, 528–535 (1976). [DOI] [PubMed] [Google Scholar]
- 7.Henikoff S., Ahmad K., Malik H. S., The centromere paradox: Stable inheritance with rapidly evolving DNA. Science 293, 1098–1102 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Krassovsky K., Henikoff J. G., Henikoff S., Tripartite organization of centromeric chromatin in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 109, 243–248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy S., Sutton G., Ng P. C., Feuk L., Halpern A. L., Walenz B. P., Axelrod N., Huang J., Kirkness E. F., Denisov G., Lin Y., MacDonald J. R., Pang A. W., Shago M., Stockwell T. B., Tsiamouri A., Bafna V., Bansal V., Kravitz S. A., Busam D. A., Beeson K. Y., McIntosh T. C., Remington K. A., Abril J. F., Gill J., Borman J., Rogers Y. H., Frazier M. E., Scherer S. W., Strausberg R. L., Venter J. C., The diploid genome sequence of an individual human. PLOS Biol. 5, e254 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slee R. B., Steiner C. M., Herbert B. S., Vance G. H., Hickey R. J., Schwarz T., Christan S., Radovich M., Schneider B. P., Schindelhauer D., Grimes B. R., Cancer-associated alteration of pericentromeric heterochromatin may contribute to chromosome instability. Oncogene 31, 3244–3253 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Hasson D., Panchenko T., Salimian K. J., Salman M. U., Sekulic N., Alonso A., Warburton P. E., Black B. E., The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat. Struct. Mol. Biol. 20, 687–695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacoste N., Woolfe A., Tachiwana H., Garea A. V., Barth T., Cantaloube S., Kurumizaka H., Imhof A., Almouzni G., Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol. Cell 53, 631–644 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 14.Baldini A., Smith D. I., Rocchi M., Miller O. J., Miller D. A., A human alphoid DNA clone from the EcoRI dimeric family: Genomic and internal organization and chromosomal assignment. Genomics 5, 822–828 (1989). [DOI] [PubMed] [Google Scholar]
- 15.Waye J. S., Willard H. F., Human beta satellite DNA: Genomic organization and sequence definition of a class of highly repetitive tandem DNA. Chromosoma 97, 6250–6254 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masumoto H., Masukata H., Muro Y., Nozaki N., Okazaki T., A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol. 109, 1963–1973 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaisson M. J. P., Huddleston J., Dennis M. Y., Sudmant P. H., Malig M., Hormozdiari F., Antonacci F., Surti U., Sandstrom R., Boitano M., Landolin J. M., Stamatoyannopoulos J. A., Hunkapiller M. W., Korlach J., Eichler E. E., Resolving the complexity of the human genome using single-molecule sequencing. Nature (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koren S., Schatz M. C., Walenz B. P., Martin J., Howard J. T., Ganapathy G., Wang Z., Rasko D. A., McCombie W. R., Jarvis E. D., Phillippy A. M, Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30, 693–700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bodor D. L., Mata J. F., Sergeev M., David A. F., Salimian K. J., Panchenko T., Cleveland T. W., Black B. E., Shah J. V., Jansen L. E., The quantitative architecture of centromeric chromatin. eLife 3, e02137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arimura Y., Shirayama K., Horikoshi N., Fujita R., Taguchi H., Kagawa W., Fukagawa T., Almouzni G., Kurumizaka H., Crystal structure and stable property of the cancer-associated heterotypic nucleosome containing CENP-A and H3.3. Sci. Rep. 4, 7115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schueler M. G., Swanson W., Thomas P. J., Grxeen E. D., Adaptive evolution of foundation kinetochore proteins in primates. Mol. Biol. Evol. 27, 1585–1597 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greig G. M., England S. B., Bedford H. M., Willard H. F., Chromosome-specific alpha satellite DNA from the centromere of human chromosome 16. Am. J. Hum. Genet. 45, 862–872 (1989). [PMC free article] [PubMed] [Google Scholar]
- 23.Plohl M., Mestrovic N., Mravinac B., Centromere identity from the DNA point of view. Chromosoma 123, 313–325 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mateo L., Gonzalez J., Pogo-like transposases have been repeatedly domesticated into CENP-B-related proteins. Genome Biol. Evol. 6, 2008–2016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warburton P. E., Waye J. S., Willard H. F., Nonrandom localization of recombination events in human alpha satellite repeat unit variants: Implications for higher-order structural characteristics within centromeric heterochromatin. Mol. Cell. Biol. 13, 6520–6529 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talbert P. B., Henikoff S., Environmental responses mediated by histone variants. Trends Cell Biol. 24, 642–650 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Brown J. D., O'Neill R. J., Chromosomes, conflict, and epigenetics: Chromosomal speciation revisited. Annu. Rev. Genomics Hum. Genet. 11, 291–316 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Malik H. S., Henikoff S., Major evolutionary transitions in centromere complexity. Cell 138, 1067–1082 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Chmátal L., Gabriel S. I., Mitsainas G. P., Martínez-Vargas J., Ventura J., Searle J. B., Schultz R. M., Lampson M. A., Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution in mice. Curr. Biol. 24, 2295–2300 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang T., Talbert P. B., Zhang W., Wu Y., Yang Z., Henikoff J. G., Henikoff S., Jiang J., The CentO satellite confers translational and rotational phasing on cenH3 nucleosomes in rice centromeres. Proc. Natl. Acad. Sci. U.S.A. 110, E4875–E4883 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tachiwana H., Kagawa W., Shiga T., Osakabe A., Miya Y., Saito K., Hayashi-Takanaka Y., Oda T., Sato M., Park S. Y., Kimura H., Kurumizaka H., Crystal structure of the human centromeric nucleosome containing CENP-A. Nature 476, 7115 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Steiner F. A., Henikoff S., Holocentromeres are dispersed point centromeres localized at transcription factor hotspots. eLife 3, e02025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henikoff S., Ramachandran S., Krassovsky K., Bryson T. D., Codomo C. A., Brogaard K., Widom J., Wang J. P., Henikoff J. G., The budding yeast Centromere DNA Element II wraps a stable Cse4 hemisome in either orientation in vivo. eLife 3, e01861 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clarke L., Carbon J., Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature 287, 504–509 (1980). [DOI] [PubMed] [Google Scholar]
- 35.Maloney K. A., Sullivan L. L., Matheny J. E., Strome E. D., Merrett S. L., Ferris A., Sullivan B. A., Functional epialleles at an endogenous human centromere. Proc. Natl. Acad. Sci. U.S.A. 109, 13704–13709 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrington J. J., Van Bokkelen G., Mays R. W., Gustashaw K., Willard H. F., Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet. 15, 345–355 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Kapoor M., Montes de Oca Luna R., Liu G., Lozano G., Cummings C., Mancini M., Ouspenski I., Brinkley B. R., May G. S., The cenpB gene is not essential in mice. Chromosoma 107, 570–576 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Okada T., Ohzeki J., Nakano M., Yoda K., Brinkley W. R., Larionov V., Masumoto H., CENP-B controls centromere formation depending on the chromatin context. Cell 131, 1287–1300 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Ohzeki J., Nakano M., Okada T., Masumoto H., CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. J. Cell Biol. 159, 765–775 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henikoff J. G., Belsky J. A., Krassovsky K., Macalpine D. M., Henikoff S., Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. U.S.A. 108, 18318–18323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li W., Godzik A., Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Fu L., Niu B., Zhu Z., Wu S., Li W., Cd-hit: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katoh K., Standley D. M., MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saitou N., Nei M., The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987). [DOI] [PubMed] [Google Scholar]
- 46.Thorvaldsdottir H., Robinson J. T., Mesirov J. P., Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henikoff S., Near the edge of a chromosome's “black hole.” Trends Genet. 18, 165–167 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/ content/full/1/1/e1400234/DC1
Fig. S1. Size distribution of input library fragments.
Fig. S2. CENP occupancies in a male and female cell line.
Fig. S3. Joint phylogeny of the most frequent CENP-A ChIP sequences for five human individuals.
Fig. S4. Cen1-like repeat units in an unplaced clone.
Fig. S5. Cen1-like and Cen13-like alignments.
Fig. S6. Normalized count profiles mapped to individual clones.