

Abstract
Moving from homogeneous water-splitting photocatalytic systems to photoelectrochemical devices requires the preparation and evaluation of novel p-type transparent conductive photoelectrode substrates. We report here on the sensitization of polystyrene-block-poly-(2-vinylpyridine) (PS-b-P2VP) diblock copolymer-templated NiO films with an organic push–pull dye. The potential of these new templated NiO film preparations for photoelectrochemical applications is compared with NiO material templated by F108 triblock copolymers. We conclude that NiO films are promising materials for the construction of dye-sensitized photocathodes to be inserted into photoelectrochemical (PEC) cells. However, a combined effort at the interface between materials science and molecular chemistry, ideally funded within a Global Artificial Photosynthesis Project, is still needed to improve the overall performance of the photoelectrodes and progress towards economically viable PEC devices.
Keywords: artificial photosynthesis, photocatalysis, surface chemistry, functional materials
1. Introduction
The production of fuels through light-driven processes is a promising solution for the durable storage of solar energy [1–3]. For example, molecular hydrogen can be produced from water splitting with O2 being generated as a side product. Alternatively, coupling photocatalytic water oxidation with the reduction of CO2 produces carbon-based fuels, with a net zero-carbon footprint. A number of molecular and supramolecular photocatalytic systems for H2 evolution, CO2 reduction or water oxidation have been reported during the last decade [4–20]. These systems however only achieve one-half of the targeted process and require the use of sacrificial components to furnish or accept the electrons required or produced by the catalytic reaction, respectively. Most attempts to couple light-driven oxidative and reductive processes in homogeneous phase have not been successful so far. A solution to this issue consists of separating these processes into two compartments of a photoelectrochemical (PEC) cell [21]. This approach has the obvious advantage of avoiding the production of potentially exploding mixtures. It, however, requires a fine regulation of photon, electron and proton management between both photocatalytic systems, which can be achieved through the grafting of the active components at the surface of transparent conductive electrode substrates [22]. Extending the n-type dye-sensitized solar cell (DSSC) technology, significant achievements in this direction have been reported recently regarding the preparation of molecular-based photoanodes for water oxidation [23–29]. Co-grafting of a water-oxidizing catalyst with a molecular dye on mesoscopic TiO2 substrates yielded electrodes able to deliver up to 2 mA cm−2 photocurrent corresponding to O2 evolution under visible irradiation [27]. Of note, such a current density corresponds to approximately 20% of the targeted performance for implementation in a PEC device with 10% solar to fuel efficiency [30]. By contrast, the preparation of molecular photocathodes with similar architectures is strongly hampered by the lack of a suitable p-type transparent electrode substrate [31]. Despite its inherently low electronic conductivity, nickel oxide (NiO) currently stands as the benchmark of such materials. Efforts have targeted the preparation of nanostructured NiO films to increase the active surface area of the electrode, and thus to optimize the loading in photocatalytic units [32–37]. A facile synthetic route to nanostructured NiO films with a Brunauer–Emmett–Teller (BET) area of about 50 m2 g−1 has been recently described based on the use of amphiphilic polystyrene-block-poly-(2-vinylpyridine) (PS-b-P2VP) diblock copolymers as templates [38]. We report here on the sensitization of such a material with an organic push–pull dye and demonstrate the potential of templated NiO film preparation for photoelectrochemical applications.
2. Results and discussion
2.1. NiO film preparation
Porous nanostructured NiO films were deposited onto fluorine-doped tin oxide (FTO)-coated glass substrates according to a procedure previously described for deposition on glass substrates using amphiphilic polystyrene-block-poly-(2-vinylpyridine) (PS75-b-P2VP25) diblock copolymers—the subscripts denote the weight fraction of the corresponding block [38]. The procedure was repeated three times to obtain three-layer films with homogeneous film thicknesses of 150–200 nm as measured by scanning electron microscopy (figure 1). These films are almost transparent in the visible region (400–800 nm; see below). In this study, we compared PS-b-P2VP-templated films with previously reported F108-templated films [35,39]. These films (figure 1) have thicknesses ranging from 300–400 nm for mono-layered substrates to 900–1000 nm for three-layer films.
Figure 1.

Scanning electron micrographs from secondary electrons (SEI) (top view and inset: cross-section) of NiO films. (a) A triple layer of NiO synthesized with PS-b-P2VP as template; (b--d) F108-templated NiO with increasing number of layers from 1 to 3.
2.2. Synthesis and characterization of a push–pull organic dye
Donor-π-acceptor (push–pull) organic dyes have shown relatively high performances in NiO-based p-type DSSCs because of their large molar extinction coefficients [40]. Compound 1 (figure 2 and table 1), containing a triarylamine electron-donor part and an ethyl cyanoacetate electron-acceptor part separated by a thiophene unit, has been prepared in two steps from previously described di-tert-butyl 4,4′-((4-(5-formylthiophen-2-yl)phenyl)azanediyl)dibenzoate [41]. The UV–visible absorption spectrum of 1 in CH3CN solution displays a typical charge-transfer (CT) band centred at 436 nm (29 300 M−1 cm−1). Upon excitation at 420 nm, emission of 1 is observed with maximum intensity at 572 nm. The cyclic voltammogram of 1 in CH3CN displays a reversible oxidation signal at +0.72 V versus Fc+/Fc and an irreversible reduction wave at Epc = –1.57 V versus Fc+/Fc (figure 2). From these values, we could determine potential values of +1.24 V versus the normal hydrogen electrode (NHE) and –1.14 V versus NHE for the 1/1+ and 1*/1+ couples, respectively, 1* representing the excited state of 1 (table 1). These potentials can be approximated to the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels, respectively (figure 2b), calculated using quantum chemistry (density functional theory (DFT)) to be separated by 2.1 eV. This value was determined as the lowest energy from the UV–vis spectrum evaluated using a time-dependent DFT method [42]. Plots of the HOMO and LUMO are shown in figure 2b, whereas the calculated spectrum is shown in the electronic supplementary material. The distributions of HOMO and LUMO of 1 evidence another advantage of push–pull dyes achieving spatial charge separation in the excited state. Indeed, the donor part, where the HOMO is centred and a hole located in the excited state, was designed to support the anchor groups. It is thus connected to the electrode which optimizes hole injection. By contrast, the acceptor moiety, where the LUMO is centred and an electron is located in the excited state, is located away from the electrode surface, which both limits recombination with the p-type semi-conductor material and fosters electron transfer towards an electron acceptor in solution. The HOMO energy level in 1 is lower than the top of the valence band (VB) in NiO (0.54 V versus NHE at pH 7) [43] (table 1), in good agreement with hole injection from photo-excited 1 to the NiO VB (figure 2b). In that context, the presence of two anchoring carboxylate groups attached on the triarylamine moiety favours both strong binding of 1 to NiO and electronic coupling optimization between the HOMO in 1 and the NiO VB orbitals.
Figure 2.

(a) Structure of 1. (b) Energy-level diagram of a p-type NiO photocathode sensitized with 1, in the presence of [Co(NH3)5Cl]Cl2 as the irreversible electron acceptor (IEA) in solution. (c) UV–vis (black line) and normalized fluorescence (red line, excitation at 420 nm) spectra of 1 measured in CH3CN at a concentration of 5 μM. (d) Cyclic voltammogram of compound 1 (1 mM) recorded at a scan rate of 100 mV s−1 on a glassy carbon electrode in CH3CN containing 0.1 M n-Bu4NPF6 as supporting electrolyte. (Online version in colour.)
Table 1.
Spectroscopic and energetic data for compound 1.
|
λmax(Abs)a |
λmax(Em)a |
ɛa |
E0–0b |
EHOMO |
ELUMO | ||
|---|---|---|---|---|---|---|---|
| nm | nm | M−1 cm−1 | eV | (V versus NHE)c | eVd | (V versus NHE)e | eVe |
| 436 | 572 | 29 300 | 2.39 | +1.24 | −5.82 | −1.14 | −3.43 |
aSpectra measured at 5 μM in CH3CN.
b0–0 transition energy, E0–0, estimated from the intercept of the normalized absorption and emission spectra in CH3CN.
cEstimated HOMO energy, EHOMO, obtained from the ground state oxidation potential.
dHOMO energy in eV obtained from the redox potential related to Fc+/Fc with a calculated absolute energy of −0.51 eV [44].
eEstimated LUMO energy, ELUMO, obtained from the estimated HOMO energy by adding the 0–0 transition energy, E0–0.
2.3. NiO film sensitization and characterization
The different NiO substrates were sensitized through soaking in a CH3CN solution of dye 1 for 24 h. The samples were rinsed with CH3CN and dried in air before characterization. Figure 3 displays typical absorption spectra of the dye-sensitized electrodes showing new features as compared with the spectra of pure NiO samples. Subtraction of the absorption of the blank NiO substrate (blanks were measured on the same sample before sensitization) reveals the absorption spectrum of dye 1 grafted onto the NiO surface. For all samples, a spectral broadening associated with a small red shift (ca 10 nm) of the absorption band is observed. These modifications could result from interactions of the dye molecules with the NiO substrate through grafting and possibly from intermolecular interactions between adjacent grafted dyes. Assuming that these interactions do not significantly modify the molar absorption coefficient ɛmax of the dye, we estimated the surface concentration of 1 using equation (2.1). We give in table 2 the ranges of dye surface and volume concentrations obtained for the various substrates using equations (2.1) and (2.2),
| 2.1 |
and
| 2.2 |
As expected, the surface concentration of the F108-templated substrates varies almost linearly with the thickness of the films. The surface concentration of PS-b-P2VP-templated three-layer films is slightly lower than the one obtained for the mono-layer F108-templated films, in good agreement with the reduced thickness of these films. Accordingly, the calculated dye volume concentration is almost identical in all dye-sensitized films.
Figure 3.
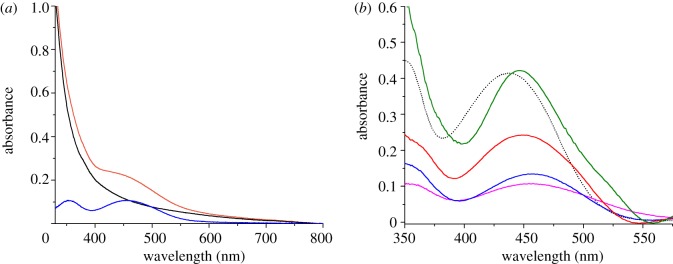
(a) Absorption spectra of a blank PS-b-P2VP-templated three-layer NiO electrode (black line) and of the same electrode after sensitization with 1 (orange line). The difference between these two spectra is shown as a blue line. (b) Comparison of corrected spectra recorded on a dye-sensitized PS-b-P2VP-templated three-layer NiO film (blue line), dye-sensitized F108-templated NiO films (one layer: fuchsia line; two layers: red line; three layers: green line) and CH3CN (5 µM) solution spectrum of 1 (dotted line).
Table 2.
Surface and volume concentrations of 1 grafted on different substrates.
| substrate | thickness (nm) | dye surface conc. (mol cm−2) | dye volume conc. (mol cm−3) |
|---|---|---|---|
| PS-b-P2VP-templated three-layer films | 150–200 | 3 × 10−9–5 × 10−9 | 14 × 10−5 |
| F108-templated one-layer films | 300–400 | 4 × 10−9–5 × 10−9 | 12 × 10−5 |
| F108-templated two-layer films | 500–600 | 7 × 10−9–9 × 10−9 | 13 × 10−5 |
| F108-templated three-layer films | 900–1000 | 9 × 10−9–1.5 × 10−8 | 14 × 10−5 |
In order to detail the electron-density changes within the dye on the NiO surface, which are associated with photoabsorption, resonance Raman (rR) measurements have been carried out. Furthermore, rR mapping allows for visualizing the distribution of the dye on the NiO surface. The rR spectra in solution (figure 4a) and on NiO (figure 4b,c) are identical except for the fluorescence background (see below) and the Raman bands of the solvent. All of them are in good agreement with the DFT-calculated Raman spectra after application of a 0.98 scaling factor (figure 4d). This means that binding to the surface does not influence the nature of the initial photoexcitation in 1. Upon changing the excitation wavelength from 413 to 476 nm the overall pattern of the rR spectrum of 1 is preserved except for slight alterations of band intensities: bands at 1440, 1526 and the three bands at about 1200 cm−1 gain intensity, while the band at 1584 cm−1 drops in intensity. The rR map (figure 5) shows a homogeneous distribution of the dye on the surface since most pixels of the map exhibit integrals of the band at 1440 cm−1, which correlates with the dye concentrations between 3500 and 4500 counts cm−1. Nonetheless, some regions of higher dye concentration (up to ca 6500 counts cm−1) on the surface are observed. These might originate from inhomogeneities in the NiO surface introduced upon the deposition of the copolymer–Ni reaction mixture during the doctor blading.
Figure 4.

Resonance Raman spectra of 1 in CH3CN at an excitation wavelength of 413 nm (a); 1 on PS-b-P2VP-templated three-layer NiO film at 413 nm (b) and 476 nm (c) excitation wavelengths; DFT-calculated Raman spectrum of 1 after application of a 0.98 scaling factor (d).
Figure 5.
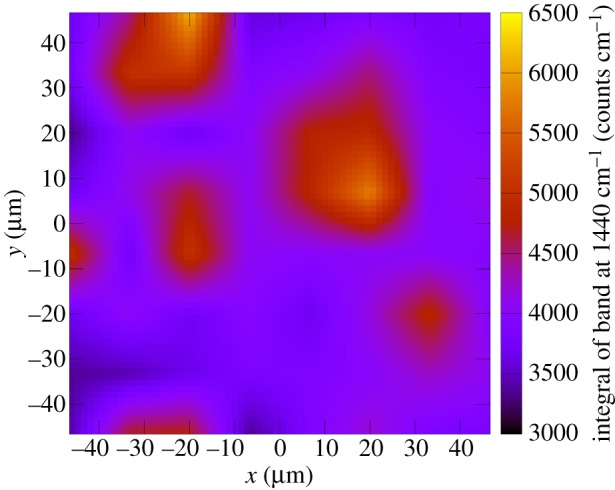
Resonance Raman map of 1 on a PS-b-P2VP-templated three-layer NiO film. (Online version in colour.)
2.4. Photoelectrochemical properties of dye-sensitized NiO films
We then investigated the PEC properties of these dye-sensitized NiO films. The substrates were used as the working electrode in a three-electrode configuration purpose-designed cell. Aqueous sodium acetate buffer (pH 4.5; 0.1 M) was first chosen as the electrolyte and the current was recorded under chopped irradiation conditions (400–800 nm filtered Xe lamp light; 100 mW cm–2; two suns) at various potentials applied to the working electrode in the presence of [Co(NH3)5Cl]Cl2 as an irreversible electron acceptor (IEA in figure 2b) in the solution. The linear sweep voltammogram recorded under chopped light (figure 6) shows the establishment of a photocurrent with onset at +0.61 V versus NHE, in good agreement with the potential of the VB edge of NiO (figure 2), and maximum photocurrent densities at +0.20 V versus NHE (0 V versus Ag/AgCl). We therefore used this potential for the whole series of experiments described below. In the absence of any electron acceptor in solution, small cathodic photocurrents (ca 10 μA cm−2) were recorded. This phenomenon has been previously observed and assigned to H2 evolution [45] although we could not detect any hydrogen in the headspace of the cell after 12 h of continuous photoelectrolysis. We thus believe that a significant part of this photocurrent is due to the reduction of residual traces of O2 present in the electrolyte or trapped within the nickel oxide films. Nevertheless, such a behaviour is directly related to the presence of 1 at the surface of the films since non-sensitized NiO films do not show any photocurrent under the same conditions.
Figure 6.
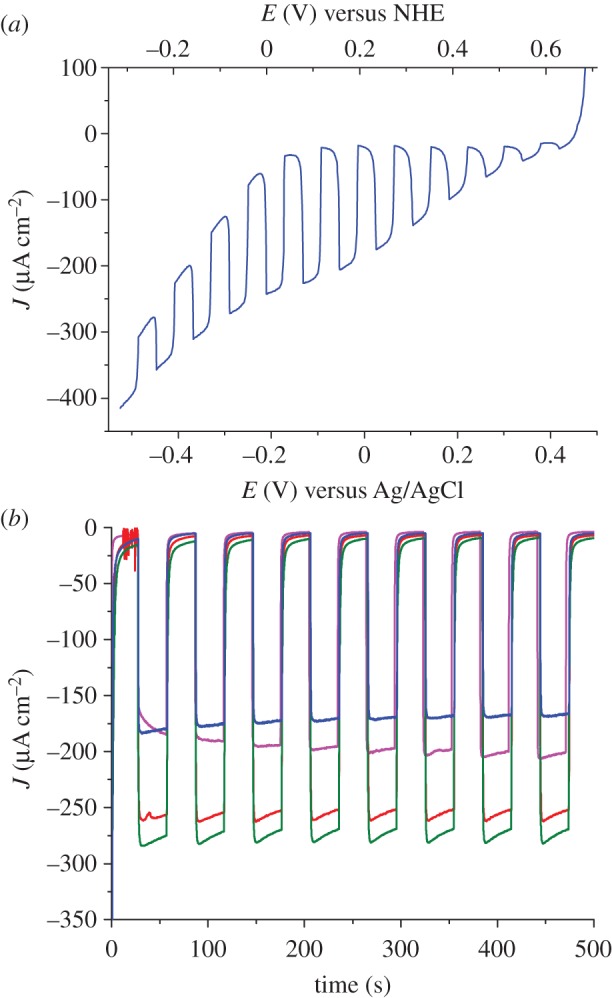
(a) Linear sweep voltammogram (10 mV s−1) recorded under chopped light on a PS-b-P2VP-templated three-layer NiO film sensitized with 1 in [Co(NH3)5Cl]Cl2 (10 mM)/acetate buffer (pH 4.5; 0.1 M). (b) Cathodic photocurrent measurements recorded on NiO electrodes sensitized with 1 in [Co(NH3)5Cl]Cl2 (10 mM)/acetate buffer (pH 4.5; 0.1 M) with 0 V versus Ag/AgCl applied potential: PS-b-P2VP-templated three-layer NiO film (blue trace) and F108-templated NiO films (one layer: fuchsia trace; two layers: red trace; three layers: green trace).
Photocurrent enhancement observed in the presence of IEA (figure 6) is attributed to the establishment of photo-induced electron transfers from NiO to IEA, mediated by the excited dye. Two mechanisms are possible: in the oxidative quenching process, a photo-induced electron transfer occurs from photo-excited 1 to the Co(III) acceptor in solution, the ground state of 1 being subsequently regenerated through hole transfer to the conducting band of NiO. In the alternative reductive quenching process, hole injection from the excited state of 1 to NiO occurs first, producing surface immobilized 1−, which in turns reduces the Co(III) complex and regenerates the ground state of 1. The latter mechanism is supported by the strongly reduced fluorescent background observed during rR spectra of 1 when shifting from solution onto NiO (see electronic supplementary material). We have studied the variation of the photocurrent value with the concentration of the Co(III) electron acceptor in solution. Figure 7 shows that the photocurrent value does not vary much for cobalt concentrations superior to 10 mM. There are two possibilities for the rate-determining step in this experiment. First, diffusion of the Co(III) electron acceptor within the porous film can limit the reaction. Second, under the assumption that diffusion does not influence photocurrents, hole injection can be rate-determining. The probability that diffusion is dominating this process is higher as hole injection is generally reported to be very fast with time constants of hundreds of femto-seconds to a few pico-seconds for organic dyes [46–50]. Photocurrent densities as high as 270 μA cm−2 are obtained for F108-templated three-layer NiO films (figure 6). Interestingly, 1-sensitized PS-b-P2VP-templated three-layer films display photocurrent values as high as those obtained for thicker F108-templated one-layer films. This indicates that the PS-b-P2VP-templated formulation proves superior to the thicker F108-templated one for such PEC applications. For thicker films, however, the photocurrent density does not linearly vary with the film thickness or the surface concentration of the dye. Such a current plateauing may come from limited diffusion of the Co(III) complex within the films.
Figure 7.
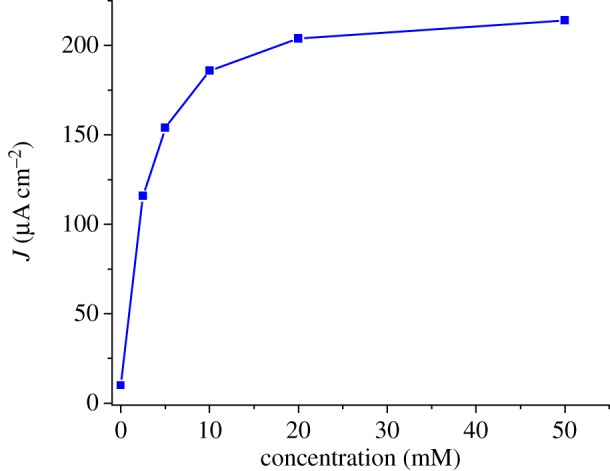
Variation of the stabilized photocurrent with the concentration of [Co(NH3)5Cl]Cl2, measured on a PS-b-P2VP-templated three-layer film sensitized with 1, in acetate buffer (0.1 M, pH 4.5) at an applied potential of 0 V versus Ag/AgCl. (Online version in colour.)
Nevertheless, in all cases, the photocurrents proved very stable with time. We thus decided to compare the aqueous acetate buffer conditions examined here with phosphate buffer conditions more typically used in the literature [27,51,52]. Figure 8 compares the PEC performance of the same electrode recorded first in acetate buffer (pH 4.5, black trace) and then in phosphate buffer (pH 7, red trace). Photocurrents measured in phosphate buffer are clearly less stable with time than those measured in the presence of acetate buffer as the electrolyte. After 10 min, they usually stabilize to values between 40 and 60 µA cm−2. This is likely to be due to the fact that phosphate ions interact more strongly with the NiO surface than the carboxylate anchors in 1, so that some dye leaches from the surface. We observe a lag phase to this phenomenon which may correspond to the time required to displace surface-anchored acetate anions which are more tightly bound than dye 1.
Figure 8.
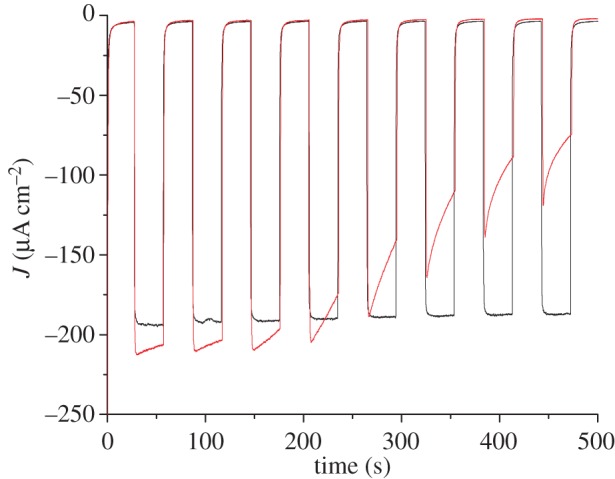
Cathodic photocurrent measurements recorded on PS-b-P2VP-templated three-layer NiO electrodes sensitized with 1 in [Co(NH3)5Cl]Cl2 (10 mM) with 0 V versus Ag/AgCl applied potential. The measurement was first carried out in acetate buffer pH 4.5 (black trace). Then the electrolyte was changed for phosphate buffer pH 7 and a new measurement was performed (red trace).
We finally compared the push–pull dye 1 with the Ru-based dye [Ru(bpy)2(4,4′-(CH2PO3H2)2-bpy)]Cl2 (2) containing two phosphonic acid functions as anchoring groups. This dye also absorbs visible light around 450 nm but with a lower molar absorptivity (ɛ = 11 000 M−1 cm−1) than 1. PS-b-P2VP-templated NiO films sensitized with 2 display similar surface concentrations in the 1.5–2.8 nmol cm−2 range. A maximum photocurrent density value of 15 µA cm−2 could be measured at 0 V versus Ag/AgCl for 20 mM concentrations of [Co(NH3)5Cl]Cl2 in either acetate or phosphate buffer. This value is 10-fold lower than those obtained with 1-sensitized films under similar conditions. This value is also lower than that (50 μA cm−2) measured for a 500-nm-thick polymer-templated nano-ITO film sensitized with the same ruthenium-based dye 2 and a similar surface concentration (4.4 nmol cm−2) [51]. This comparison supports the combination of a p-type NiO electrode substrate with push–pull organic dyes with enhanced light absorptivity as optimal architecture for the design of molecular-based photoelectrodes.
3. Conclusion
This study shows that state-of-the-art NiO films are suitable materials for the construction of dye-sensitized photocathodes with current densities up to 300 μA cm−2. Such photocurrent densities can be achieved with simple and easily accessible push–pull organic dyes displaying high molar absorptivities in the visible region. Further developments in materials chemistry are needed to increase these values to a few mA cm−2 and to comply with the requirement of economically viable PEC devices [30]. Nevertheless, state-of-the-art H2-evolving molecular photocathodes still exhibit 10-fold lower photocurrents, indicating that efforts should now be directed towards the optimization of directional electron transfer from the grafted dye (either in the photoexcited or reduced state) to the catalytic unit and/or the development and integration of faster multi-electron reduction catalysts. The stable photocurrents obtained in pH 4.5 acetate buffer electrolyte, i.e. conditions where cobalt-based catalysts have proved to be stable and active for H2 evolution [53,54], hold promise for the development of functional photoelectrodes. This study resulting from the combined efforts of four distinct groups from France and Germany demonstrates the relevance of a Global Artificial Photosynthesis Project promoting interactions at the interface between various fields such as materials science, molecular chemistry, theoretical chemistry and physical chemistry. Such a global enterprise must not only be continued, but also supported and funded, at the international level.
3.1. Experimental section
All chemical reagents were purchased from Sigma sources and used as received. Di-tert-butyl-4,4′-((4-(5-formylthiophen-2-yl)phenyl)azanediyl)dibenzoate was synthesized as previously described [41]. FTO-coated glass substrates (1.1 mm thickness; 80 Ω □−1) were purchased from SOLEMS S.A., Palaiseau, France. 1H and 13C NMR analyses were performed on a Bruker Avance III 300 spectrometer. Chemical shift values are given in ppm with reference to solvent residual signals [55]. Fourier transform infrared spectra were recorded on a Perkin-Elmer Spectrum 100 spectrometer using a Pike Miracle single reflection attenuated total reflectance (ATR) sampling accessory equipped with a Ge crystal. UV–visible absorption and emission spectra were recorded on an Agilent Cary 60 UV–Vis spectrometer and a Jasco FP-8500 fluorimeter, respectively. Cyclic voltammograms were recorded with a Bio-logic SP300 potentiostat under nitrogen at room temperature. A standard three-electrode configuration was used consisting of a glassy carbon working electrode, an auxiliary platinum wire and an Ag/AgCl/aqueous AgClsat + KCl 3 mol l−1 (denoted as Ag/AgCl throughout this text) reference electrode closed by a Vycor frit and dipped directly into the solution. The [Fe(CN)6]3−/[Fe(CN)6]4− couple (E0 = +0.215 V versus Ag/AgCl; referred at +0.425 V versus NHE in 0.1 M potassium phosphate buffer at pH = 7) [56] was then used for the standardization of the measurements in aqueous solution [56]. The Fc+/Fc couple (measured at E0 = +0.43 V versus Ag/AgCl) was used as a reference for CV measurements in acetonitrile (potentials measured versus Fc+/Fc and converted to NHE by addition of +0.53 V [57]).
The ORCA program [58] (v. 3.0.2) was employed for all DFT calculations. Geometry optimizations were performed using the B3LYP functional [59,60] and the Ahlrichs TZVP basis set, which is of triple-zeta quality [61], augmented by extra polarization and diffuse functions (ORCA keyword ACCOPT). Calculations were done both in vacuum and with an implicit solvent model of acetonitrile, using ORCA's version of the COSMO method [62]. After geometry optimization, time-dependent DFT calculations were carried out to determine the UV–vis spectra and the HOMO–LUMO gap [42], together with harmonic frequency calculations to evaluate the IR and Raman spectra. The plots of the HOMO and LUMO orbitals were made with the VMD program [63].
Scanning electron microscopy was performed on a JSM-6300F (JEOL, Tokyo, Japan) field-emission microscope (FE-SEM). The lateral resolving power of the system is specified to be approximately 3 nm at 15 keV. An Everhardt–Thornley-type detector was used for imaging for secondary electrons (SEI). If needed the specimen stage was tilted (up to 60°) to enhance the topographic visibility. The Ni2+-loaded material was deposited onto glass substrates. In order to avoid charging effects all samples were coated with approximately 20 nm carbon. Cross-sectional preparations were performed after short-term LN2-cooling in order to reduce the degree of ductility and to achieve plain true-edge fracture patterns.
For the rR microspectroscopy measurements a conventional micro-Raman set-up (Labram HR, Horiba Jobin Yvon) was used. The spectrometer was equipped with a 600 line mm−1 grating and combined with an Olympus inverse microscope IX71. The excitation wavelengths were delivered by a krypton-ion laser (Model Coherent Innova 301C; λ = 413 and 476 nm). For the measurements, the laser was focused onto the dye-sensitized NiO surface using an Olympus 20× UPlan FLN microscope objective. The incident laser power was 1 mW at the sample, the spot diameter was 1 µm. The spectra were recorded with an integration time of 2 s per spectrum. All spectra presented in this paper are mean spectra of a quadratic map (60 µm edge length, 10 × 10 spectra), which builds a profound and statistically reliable basis for the comparison of rR intensities. The mean spectra were processed by the statistics-sensitive nonlinear iterative peak-clipping (SNIP) algorithm for background correction using Gnu R [64] and the library ‘Peaks’ with 20 iterations [65].
The synthetic procedure for the preparation of templated NiO films followed that described in the literature [38]. Polystyrene-block-poly(2-vinylpyridine) (PS-b-P2VP) was synthesized via sequential anionic polymerization using a procedure adopted from the literature [66]. The mixture of block copolymer and Ni was deposited through doctor blading for three times on FTO-coated glass substrates with 5 min in a furnace at 450°C after deposition of the first and second layer and 25 min in the furnace after the third deposition step.
3.1.1. Di-tert-butyl-(E)-4,4′-((4-(5-(2-cyano-3-ethoxy-3-oxoprop-1-en-1-yl)thiophen-2-yl)phenyl)azanediyl)dibenzoate
To a solution of di-tert-butyl-4,4′-((4-(5-formylthiophen-2-yl)phenyl)azanediyl)dibenzoate (1 eq, 0.2 mmol, 110 mg) and ethyl cyanoacetate (95 eq, 19 mmol, 2 ml) in 5 ml of dry CHCl3 was added piperidine (25 eq, 5 mmol, 0.5 ml). The reaction mixture was stirred at room temperature for 12 h under argon and protected from sunlight. The mixture was diluted with CHCl3 and washed with 1 M aqueous HCl then water. After removal of the solvent, the crude product was purified on a silica gel column using pentane/ethyl acetate (8/2) as eluent to give the di-tert-butylester as an orange solid (88 mg; 68%). 1H NMR (CDCl3, 300 MHz): δ (ppm) 8.27 (s, 1H), 7.90 (d, J = 8.6 Hz, 4H), 7.73 (d, J = 4 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H), 7.35 (d, J = 4 Hz, 1H), 7.11 (m, 6H), 4.35 (q, J = 7.1 Hz, 2H), 1.58 (s, 18H), 1.38 (t, J = 7.1 Hz, 3H). 13C NMR (CDCl3, 75 MHz): δ (ppm) 165.3, 163.1, 154, 150.2, 147.8, 146.5, 139.3, 134.8, 131.1, 128.6, 127.8, 127.2, 125.3, 124.1, 123.5, 116.2, 97.9, 81, 62.5, 28.3, 14.3. HR-MS (ESI+): m/z calcd for C38H39N2O6S 651.2523; found 651.2517 [M + H]+.
3.1.2. (E)-4,4′-((4-(5-(2-cyano-3-ethoxy-3-oxoprop-1-en-1-yl) thiophen-2-yl)phenyl)azanediyl)dibenzoic acid (1)
To a solution of di-tert-butylester (1 eq, 35 mg. 53.8 µmol) in 3.5 ml of CH2Cl2 was added trifluoroacetic acid (10 eq, 0.4 ml, 0.52 mmol). The reaction mixture was stirred for 5 h at room temperature. After removal of the solvent, the crude product was purified by precipitation from a CH2Cl2/cyclohexane mixture. The resulting orange powder was filtered, washed with pentane and dried under a vacuum to afford pure 1 (28 mg; 97%). 1H NMR (CDCl3 + 10% MeOD, 300 MHz): δ 8.28 (s, 1H), 7.97 (d, J = 8.1 Hz, 4H), 7.74 (d, J = 3.3 Hz, 2H), 7.63 (d, J = 8.1 Hz, 2H), 7.36 (d, J = 3.3 Hz, 1H), 7.15 (m, 6H), 4.35 (q, J = 7 Hz, 2H), 1.38 (t, J = 7 Hz, 3H). 13C NMR (CDCl3 + 10% MeOD, 75 MHz): δ 168.4, 163.2, 154, 150.6, 147.5, 146.7, 139.5, 134.7, 131.6, 128.9, 127.9, 125.7, 125.3, 124.2, 123.3, 116.1, 97.7, 62.6, 14.2. HR-MS (ESI+): m/z calcd for C30H23N2O6S 539.1271; found 539.1268 [M + H]+.
3.2. Film sensitization
The variously prepared NiO electrodes were soaked in a 0.5 mM solution of 1 in CH3CN for 24 h on an orbital stirring table. The electrodes were rinsed with CH3CN and dried in air.
3.3. Photoelectrochemical measurements
Photocurrent measurements were performed in a specific cell in a three-electrode configuration. The FTO-coated glass substrate (onto which the NiO film had been deposited) was clamped on the cell, serving as both working electrode and window. The surface of the working electrode in contact with the electrolyte was 0.42 cm2. Ti wire and Ag/AgCl have been used as counter and reference electrodes, respectively. We used sodium acetate buffer (0.1 M, pH = 4.5) and potassium phosphate buffer (0.1 M, pH = 7) as electrolytes and [Co(NH3)5Cl]Cl2 (10 mM) as the electron acceptor in solution.
Photoelectrodes were back-illuminated with a 300 W ozone-free xenon lamp (Newport) operated at 280 W coupled to a water-filled Spectra-Physics 6123NS liquid filter for elimination of IR radiation and a Spectra-Physics 59472 UV cut-off filter (λ > 400 nm). Irradiance at the substrate surface was measured to 100 mW cm−2 (ca two suns) using the Newport PM1918-R power-meter.
Funding statement
M.B. gratefully acknowledges a PhD fellowship from the Studienstiftung des deutschen Volkes. This work was supported by the COST Action CM1202 PERSPECT-H2O, the French National Research Agency (Labex program, ARCANE, ANR-11-LABX-0003-01), the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC grant agreement no. 306398 and the Life Science Division of CEA (2011 DSV-Energy program). Funding from the Royal Society related to the Chicheley Hall meeting on artificial photosynthesis (July 2014) is gratefully acknowledged.
References
- 1.Faunce TA, et al. 2013. Energy and environment policy case for a global project on artificial photosynthesis. Energy Environ. Sci. 6, 695–698. ( 10.1039/c3ee00063j) [DOI] [Google Scholar]
- 2.Faunce T, et al. 2013. Artificial photosynthesis as a frontier technology for energy sustainability. Energy Environ. Sci. 6, 1074–1076. ( 10.1039/c3ee40534f) [DOI] [Google Scholar]
- 3.Thapper A, et al. 2013. Artificial photosynthesis for solar fuels—an evolving research field within AMPEA, a Joint Programme of the European Energy Research Alliance. Green 3, 43–57. ( 10.1515/green-2013-0007) [DOI] [Google Scholar]
- 4.Artero V, Chavarot-Kerlidou M, Fontecave M. 2011. Splitting water with cobalt. Angew. Chem. Int. Ed. 50, 7238–7266. ( 10.1002/anie.201007987) [DOI] [PubMed] [Google Scholar]
- 5.Andreiadis ES, Chavarot-Kerlidou M, Fontecave M, Artero V. 2011. Artificial photosynthesis: from molecular catalysts for light-driven water splitting to photoelectrochemical cells. Photochem. Photobiol. 87, 946–964. ( 10.1111/j.1751-1097.2011.00966.x) [DOI] [PubMed] [Google Scholar]
- 6.Eckenhoff WT, Eisenberg R. 2012. Molecular systems for light driven hydrogen production. Dalton Trans. 41, 13 004–13 021. ( 10.1039/c2dt30823a) [DOI] [PubMed] [Google Scholar]
- 7.Krassen H, Ott S, Heberle J. 2011. In vitro hydrogen production—using energy from the sun. Phys. Chem. Chem. Phys. 13, 47–57. ( 10.1039/C0CP01163K) [DOI] [PubMed] [Google Scholar]
- 8.Schulz M, Karnahl M, Schwalbe M, Vos JG. 2012. The role of the bridging ligand in photocatalytic supramolecular assemblies for the reduction of protons and carbon dioxide. Coord. Chem. Rev. 256, 1682–1705. ( 10.1016/j.ccr.2012.02.016) [DOI] [Google Scholar]
- 9.Thoi VS, Sun Y, Long JR, Chang CJ. 2013. Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions. Chem. Soc. Rev. 42, 2388–2400. ( 10.1039/C2CS35272A) [DOI] [PubMed] [Google Scholar]
- 10.Halpin Y, Pryce MT, Rau S, Dini D, Vos JG. 2013. Recent progress in the development of bimetallic photocatalysts for hydrogen generation. Dalton Trans. 42, 16 243–16 254. ( 10.1039/c3dt52319e) [DOI] [PubMed] [Google Scholar]
- 11.McKone JR, Marinescu SC, Brunschwig BS, Winkler JR, Gray HB. 2014. Earth-abundant hydrogen evolution electrocatalysts. Chem. Sci. 5, 865–878. ( 10.1039/C3SC51711J) [DOI] [Google Scholar]
- 12.Appel AM, et al. 2013. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 113, 6621–6658. ( 10.1021/cr300463y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris AJ, Meyer GJ, Fujita E. 2009. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc. Chem. Res. 42, 1983–1994. ( 10.1021/ar9001679) [DOI] [PubMed] [Google Scholar]
- 14.Reithmeier R, Bruckmeier C, Rieger B. 2012. Conversion of CO2 via visible light promoted homogeneous redox catalysis. Catalysts 2, 544–571. ( 10.3390/catal2040544) [DOI] [Google Scholar]
- 15.Finn C, Schnittger S, Yellowlees LJ, Love JB. 2012. Molecular approaches to the electrochemical reduction of carbon dioxide. Chem. Commun. 48, 1392–1399. ( 10.1039/C1CC15393E) [DOI] [PubMed] [Google Scholar]
- 16.Kondratenko EV, Mul G, Baltrusaitis J, Larrazabal GO, Perez-Ramirez J. 2013. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 6, 3112–3135. ( 10.1039/c3ee41272e) [DOI] [Google Scholar]
- 17.Qiao JL, Liu YY, Hong F, Zhang JJ. 2014. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675. ( 10.1039/C3CS60323G) [DOI] [PubMed] [Google Scholar]
- 18.Romain S, Vigara L, Llobet A. 2009. Oxygen−oxygen bond formation pathways promoted by ruthenium complexes. Acc. Chem. Res. 42, 1944–1953. ( 10.1021/ar900240w) [DOI] [PubMed] [Google Scholar]
- 19.Sala X, Romero I, Rodriguez M, Escriche L, Llobet A. 2009. Molecular catalysts that oxidize water to dioxygen. Angew. Chem. Int. Ed. 48, 2842–2852. ( 10.1002/anie.200802659) [DOI] [PubMed] [Google Scholar]
- 20.Herrero C, Quaranta A, Leibl W, Rutherford AW, Aukauloo A. 2011. Artificial photosynthetic systems. Using light and water to provide electrons and protons for the synthesis of a fuel. Energy Environ. Sci. 4, 2353–2365. ( 10.1039/c0ee00645a) [DOI] [Google Scholar]
- 21.Meyer TJ. 2011. Catalysis: oxidizing water two ways. Nat. Chem. 3, 757–758. ( 10.1038/nchem.1161) [DOI] [PubMed] [Google Scholar]
- 22.Artero V, Fontecave M. 2011. Light-driven bioinspired water splitting: recent developments in photoelectrode materials. C. R. Chim. 14, 799–810. ( 10.1016/j.crci.2011.06.004) [DOI] [Google Scholar]
- 23.Hocking RK, Brimblecombe R, Chang L-Y, Singh A, Cheah MH, Glover C, Casey WH, Spiccia L. 2011. Water-oxidation catalysis by manganese in a geochemical-like cycle. Nat. Chem. 3, 461–466. [DOI] [PubMed] [Google Scholar]
- 24.Brimblecombe R, Koo A, Dismukes GC, Swiegers GF, Spiccia L. 2010. Solar driven water oxidation by a bioinspired manganese molecular catalyst. J. Am. Chem. Soc. 132, 2892–2894. ( 10.1021/ja910055a) [DOI] [PubMed] [Google Scholar]
- 25.Youngblood WJ, Lee SHA, Kobayashi Y, Hernandez-Pagan EA, Hoertz PG, Moore TA, Moore AL, Gust D, Mallouk TE. 2009. Photoassisted overall water splitting in a visible light-absorbing dye-sensitized photoelectrochemical cell. J. Am. Chem. Soc. 131, 926–927. ( 10.1021/ja809108y) [DOI] [PubMed] [Google Scholar]
- 26.Zhao YX, et al. 2012. Improving the efficiency of water splitting in dye-sensitized solar cells by using a biomimetic electron transfer mediator. Proc. Natl Acad. Sci. USA 109, 15 612–15 616. ( 10.1073/pnas.1118339109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Y, Ding X, Liu JH, Wang L, Lu ZK, Li L, Sun LC. 2013. Visible light driven water splitting in a molecular device with unprecedentedly high photocurrent density.. J. Am. Chem. Soc. 135, 4219–4222. ( 10.1021/ja400402d) [DOI] [PubMed] [Google Scholar]
- 28.Alibabaei L, et al. 2013. Solar water splitting in a molecular photoelectrochemical cell. Proc. Natl Acad. Sci. USA 110, 20 008–20 013. ( 10.1073/pnas.1319628110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding X, Gao Y, Zhang L, Yu Z, Liu J, Sun L. 2014. Visible light-driven water splitting in photoelectrochemical cells with supramolecular catalysts on photoanodes. ACS Catalysis 4, 2347–2350. ( 10.1021/cs500518k) [DOI] [Google Scholar]
- 30.Walter MG, Warren EL, McKone JR, Boettcher SW, Mi Q, Santori EA, Lewis NS. 2010. Solar water splitting cells. Chem. Rev. 110, 6446–6473. ( 10.1021/cr1002326) [DOI] [PubMed] [Google Scholar]
- 31.Odobel F, Pellegrin Y. 2013. Advances in the sensitization of wide-band-gap nanostructured p-type semiconductors. Photovoltaic and photocatalytic applications. J. Phys. Chem. Lett. 4, 2551–2564. ( 10.1021/jz400861v) [DOI] [Google Scholar]
- 32.He JJ, Lindstrom H, Hagfeldt A, Lindquist SE. 1999. Dye-sensitized nanostructured p-type nickel oxide film as a photocathode for a solar cell. J. Phys. Chem. B 103, 8940–8943. ( 10.1021/jp991681r) [DOI] [Google Scholar]
- 33.Li L, Gibson EA, Qin P, Boschloo G, Gorlov M, Hagfeldt A, Sun LC. 2010. Double-layered NiO photocathodes for p-type DSSCs with record IPCE. Adv. Mater. 22, 1759–1762. ( 10.1002/adma.200903151) [DOI] [PubMed] [Google Scholar]
- 34.Nattestad A, Ferguson M, Kerr R, Cheng YB, Bach U. 2008. Dye-sensitized nickel(II)oxide photocathodes for tandem solar cell applications. Nanotechnology 19, 295304 ( 10.1088/0957-4484/19/29/295304) [DOI] [PubMed] [Google Scholar]
- 35.Sumikura S, Mori S, Shimizu S, Usami H, Suzuki E. 2008. Syntheses of NiO nanoporous films using nonionic triblock co-polymer templates and their application to photo-cathodes of p-type dye-sensitized solar cells. J. Photochem. Photobiol. A 199, 1–7. ( 10.1016/j.jphotochem.2008.04.007) [DOI] [Google Scholar]
- 36.Awais M, Gibson E, Vos JG, Dowling DP, Hagfeldt A, Dini D. 2014. Fabrication of efficient NiO photocathodes prepared via RDS with novel routes of substrate processing for p-type dye-sensitized solar cells. Chemelectrochem 1, 384–391. ( 10.1002/celc.201300178) [DOI] [Google Scholar]
- 37.Gibson EA, Awais M, Dini D, Dowling DP, Pryce MT, Vos JG, Boschloo G, Hagfeldt A. 2013. Dye sensitised solar cells with nickel oxide photocathodes prepared via scalable microwave sintering. Phys. Chem. Chem. Phys. 15, 2411–2420. ( 10.1039/c2cp43592f) [DOI] [PubMed] [Google Scholar]
- 38.Bräutigam M, Weyell P, Rudolph T, Dellith J, Krieck S, Schmalz H, Schacher FH, Dietzek B. 2014. Porous NiOx nanostructures templated by polystyreneblock-poly(2-vinylpyridine) diblock copolymer micelles. J. Mater. Chem. A 2, 6158–6166. ( 10.1039/c3ta14890d) [DOI] [Google Scholar]
- 39.Ji Z, He M, Huang Z, Ozkan U, Wu Y. 2013. Photostable p-type dye-sensitized photoelectrochemical cells for water reduction. J. Am. Chem. Soc. 135, 11 696–11 699. ( 10.1021/ja404525e) [DOI] [PubMed] [Google Scholar]
- 40.Mishra A, Fischer MKR, Bauerle P. 2009. Metal-free organic dyes for dye-sensitized solar cells: from structure: property relationships to design rules. Angew. Chem. Int. Ed. 48, 2474–2499. ( 10.1002/anie.200804709) [DOI] [PubMed] [Google Scholar]
- 41.Ji Z, Natu G, Huang Z, Wu Y. 2011. Linker effect in organic donor-acceptor dyes for p-type NiO dye sensitized solar cells. Energy Environ. Sci. 4, 2818–2821. ( 10.1039/c1ee01527c) [DOI] [Google Scholar]
- 42.Zhang G, Musgrave CB. 2007. Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A 111, 1554–1561. ( 10.1021/jp061633o) [DOI] [PubMed] [Google Scholar]
- 43.Boschloo G, Hagfeldt A. 2001. Spectroelectrochemistry of nanostructured NiO. J. Phys. Chem. B 105, 3039–3044. ( 10.1021/jp003499s) [DOI] [Google Scholar]
- 44.Weidelener M, et al. 2014. Synthesis and characterization of organic dyes with various electron-accepting substituents for p-type dye-sensitized solar cells. Chem. Asian J. 9, 3251–3263. [DOI] [PubMed] [Google Scholar]
- 45.Tong L, et al. 2012. Sustained solar hydrogen generation using a dye-sensitised NiO photocathode/BiVO4 tandem photo-electrochemical device. Energy Environ. Sci. 5, 9472–9475. ( 10.1039/c2ee22866a) [DOI] [Google Scholar]
- 46.Qin P, Wiberg J, Gibson EA, Linder M, Li L, Brinck T, Hagfeldt A, Albinsson B, Sun L. 2010. Synthesis and mechanistic studies of organic chromophores with different energy levels for p-type dye-sensitized solar cells. J. Phys. Chem. C 114, 4738–4748. ( 10.1021/jp911091n) [DOI] [Google Scholar]
- 47.Gibson EA, et al. 2009. A p-type NiO-based dye-sensitized solar cell with an open-circuit voltage of 0.35 V. Angew. Chem. Int. Ed. 48, 4402–4405. ( 10.1002/anie.200900423) [DOI] [PubMed] [Google Scholar]
- 48.Morandeira A, Boschloo G, Hagfeldt A, Hammarström L. 2005. Photoinduced ultrafast dynamics of coumarin 343 sensitized p-type-nanostructured NiO films. J. Phys. Chem. B 109, 19 403–19 410. ( 10.1021/jp053230e) [DOI] [PubMed] [Google Scholar]
- 49.Le Pleux L, Smeigh AL, Gibson E, Pellegrin Y, Blart E, Boschloo G, Hagfeldt A, Hammarstrom L, Odobel F. 2011. Synthesis, photophysical and photovoltaic investigations of acceptor-functionalized perylene monoimide dyes for nickel oxide p-type dye-sensitized solar cells. Energy Environ. Sci. 4, 2075–2084. ( 10.1039/c1ee01148k) [DOI] [Google Scholar]
- 50.Morandeira A, Boschloo G, Hagfeldt A, Hammarström L. 2008. Coumarin 343−NiO films as nanostructured photocathodes in dye-sensitized solar cells: ultrafast electron transfer, effect of the I 3−/I−redox couple and mechanism of photocurrent generation. J. Phys. Chem. C 112, 9530–9537. ( 10.1021/jp800760q) [DOI] [Google Scholar]
- 51.Hamd W, et al. 2013. Dye-sensitized nanostructured crystalline mesoporous tin-doped indium oxide films with tunable thickness for photoelectrochemical applications. J. Mater. Chem. A 1, 8217–8225. ( 10.1039/c3ta10728k) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li L, Duan LL, Wen FY, Li C, Wang M, Hagfeld A, Sun LC. 2012. Visible light driven hydrogen production from a photo-active cathode based on a molecular catalyst and organic dye-sensitized p-type nanostructured NiO. Chem. Commun. 48, 988–990. ( 10.1039/C2CC16101J) [DOI] [PubMed] [Google Scholar]
- 53.Varma S, Castillo CE, Stoll T, Fortage J, Blackman AG, Molton F, Deronzier A, Collomb MN. 2013. Efficient photocatalytic hydrogen production in water using a cobalt(III) tetraaza-macrocyclic catalyst: electrochemical generation of the low-valent Co(I) species and its reactivity toward proton reduction. Phys. Chem. Chem. Phys. 15, 17 544–17 552. ( 10.1039/c3cp52641k) [DOI] [PubMed] [Google Scholar]
- 54.Andreiadis ES, et al. 2013. Molecular engineering of a cobalt-based electrocatalytic nanomaterial for H2 evolution under fully aqueous conditions. Nat. Chem. 5, 48–53. ( 10.1038/nchem.1481) [DOI] [PubMed] [Google Scholar]
- 55.Gottlieb HE, Kotlyar V, Nudelman A. 1997. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 62, 7512–7515. ( 10.1021/jo971176v) [DOI] [PubMed] [Google Scholar]
- 56.O'Reilly JE. 1973. Oxidation-reduction potential of the ferro-ferricyanide system in buffer solutions. Biochim. Biophys. Acta 292, 509–515. ( 10.1016/0005-2728(73)90001-7) [DOI] [PubMed] [Google Scholar]
- 57.Parker VD, Handoo KL, Roness F, Tilset M. 1991. Electrode potentials and the thermodynamics of isodesmic reactions. J. Am. Chem. Soc. 113, 7493–7498. ( 10.1021/ja00020a007) [DOI] [Google Scholar]
- 58.Neese F. 2012. The ORCA program system . WIREs Comput. Mol. Sci. 2, 73–78. ( 10.1002/wcms.81) [DOI] [Google Scholar]
- 59.Becke AD. 1988. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100. ( 10.1103/PhysRevA.38.3098) [DOI] [PubMed] [Google Scholar]
- 60.Lee C, Yang W, Parr RG. 1988. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. ( 10.1103/PhysRevB.37.785) [DOI] [PubMed] [Google Scholar]
- 61.Weigend F, Ahlrichs R. 2005. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. ( 10.1039/b508541a) [DOI] [PubMed] [Google Scholar]
- 62.Sinnecker S, Rajendran A, Klamt A, Diedenhofen M, Neese F. 2006. Calculation of solvent shifts on electronic g-tensors with the conductor-like screening model (COSMO) and its self-consistent generalization to real solvents (direct COSMO-RS). J. Phys. Chem. A 110, 2235–2245. ( 10.1021/jp056016z) [DOI] [PubMed] [Google Scholar]
- 63.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. ( 10.1016/0263-7855(96)00018-5) [DOI] [PubMed] [Google Scholar]
- 64.R Core Team. 2015. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See http://www.R-project.org. [Google Scholar]
- 65.Bräutigam M, Schulz M, Inglis J, Popp J, Vos JG, Dietzek B. 2012. Resonance-Raman microspectroscopy for quality assurance of dye-sensitized NiOx films with respect to dye desorption kinetics in water. Phys. Chem. Chem. Phys. 14, 15 185–15 190. ( 10.1039/c2cp42938a) [DOI] [PubMed] [Google Scholar]
- 66.Giebeler E, Stadler R. 1997. ABC triblock polyampholytes containing a neutral hydrophobic block, a polyacid and a polybase. Macromol. Chem. Phys. 198, 3815–3825. ( 10.1002/macp.1997.021981204) [DOI] [Google Scholar]