

Abstract
This review summarizes three different approaches to engineering systems for the solar-driven evolution of hydrogen fuel from water: molecular, nanomaterials and biomolecular. Molecular systems have the advantage of being highly amenable to modification and detailed study and have provided great insight into photophysics, electron transfer and catalytic mechanism. However, they tend to display poor stability. Systems based on nanomaterials are more robust but also are more difficult to synthesize in a controlled manner and to modify and study in detail. Biomolecular systems share many properties with molecular systems and have the advantage of displaying inherently high efficiencies for light absorption, electron–hole separation and catalysis. However, biological systems must be engineered to couple modules that capture and convert solar photons to modules that produce hydrogen fuel. Furthermore, biological systems are prone to degradation when employed in vitro. Advances that use combinations of these three tactics also are described. Multidisciplinary approaches to this problem allow scientists to take advantage of the best features of biological, molecular and nanomaterials systems provided that the components can be coupled for efficient function.
Keywords: artificial photosynthesis, hydrogen evolution, energy conversion
1. Introduction
A key challenge facing society is the provision of energy to support economic prosperity with responsible environmental stewardship [1]. In 2011, 81% of all energy use worldwide originated from fossil fuels [2]. As world population grows and economies develop in the non-legacy world, energy demands will increase and the energy source predicted to show the most growth is coal [3–5]. The reliance on fossil fuels for energy has led to increases in worldwide atmospheric CO2 levels, which rose from 339 ppm in 1980 to surpass 400 ppm in 2014 [6]. Looking ahead, continuing to meet our energy demand with fossil fuels is predicted to increase atmospheric CO2 to 900–1100 ppm by 2100 [7]. Although the consequences of this increase in CO2 levels cannot be precisely predicted, there is scientific consensus that worldwide climate is already being impacted [8]. To mitigate future impacts of rising CO2 levels while maintaining world economic growth, development and wide implementation of carbon-neutral energy sources are necessary.
Of the alternatives to fossil fuels, solar energy is the most abundant, and is truly renewable and carbon-free. The harvestable solar energy received by the Earth far exceeds current and future energy needs, and exceeds all other renewable energy sources combined [9,10]. Although electricity generation from the Sun using photovoltaic cells has seen robust growth in recent years, it is predicted to remain a small share of global electricity generation, increasing from 0.4% to 2.6% in 2035 [11]. A barrier to wider use of solar energy is that photovoltaic cells that convert sunlight into electricity are relatively expensive [12]. Fuelling the recent growth in use of solar energy, however, is a substantial decrease in the cost of photovoltaic cells in recent years [11]. Nevertheless, there are structural barriers to the broad implementation of solar energy. The daily and seasonable variation in solar flux means that availability cannot be controlled and is not linked to demand. Furthermore, solar energy is diffuse and varies with location [13]. Meeting these challenges requires the development of methods for storing solar energy so that it can be dispatched according to demand.
Nature handles the solar energy storage problem through photosynthesis. In oxygenic photosynthesis, the overall light-dependent reaction is
| 1.1 |
where Pi indicates inorganic phosphate. The energy-storing molecules produced in this light-driven reaction are NADPH and ATP. NADPH provides reducing equivalents used in the Calvin–Benson–Bassham cycle that converts CO2 into triose phosphates (sugars).
Inspired by natural photosynthesis, scientists have been working toward developing systems for artificial photosynthesis. In artificial photosynthesis, an energy-requiring chemical reaction is driven by light to produce a fuel. While there are many reactions that could be used including CO2 and N2 reduction, much of the focus has been on the water-splitting reaction, which stores 1.23 V of energy:
| 1.2 |
The resulting hydrogen fuel has the highest specific energy of combustion per gram of any chemical fuel; however, its volume energy density is less competitive [14]. Nevertheless, the volume energy density of hydrogen exceeds that of any existing batteries and major commercial efforts are underway to make hydrogen fuel for automotive transportation based on fuel cells [15,16]. However, hydrogen used in this application is produced mainly from natural gas, with CO2 as one of the by-products. The increasing viability of hydrogen as a usable fuel drives home the need to develop systems for its production through artificial photosynthesis.
Accomplishing water splitting requires the capture and conversion of light energy sufficient to drive the reaction and catalysis of two half-reactions, the oxidation of water to yield oxygen and the reduction of protons to yield hydrogen:
| 1.3 |
and
| 1.4 |
Successfully deriving solar hydrogen from water will require progress on both of these two reactions and means by which to couple them. The water oxidation reaction (1.3) has proved to be a particularly challenging target [17]. It requires oxidation by four electrons per O2 molecule evolved, formation of an O–O bond and highly oxidizing conditions, even for catalysts that perform close to the thermodynamic potential of the reaction. As a result, homogeneous catalysts that contain organic ligands are typically oxidatively deactivated. More successful are heterogeneous inorganic catalysts [18,19], but they are challenging to prepare in a controlled manner and to study and modify systematically. Another barrier to performing full water splitting is the need to deliver the electrons and protons produced from the oxidation reaction to the hydrogen evolution catalyst in a way that does not interfere with the chemistry on either side and that yields an overall rapid and efficient solar-driven process.
As a result of the complications inherent in performing full water splitting, on-going research in hydrogen evolution from water often uses sacrificial chemical electron donors, focusing on the reductive side of water splitting (equation (1.4)). This approach avoids the problems with water oxidation catalysts and the need to couple the oxidation and reduction reactions. Ultimately, however, hydrogen production from water for widespread use will require either full water splitting or development of an alternative sustainable electron source. An example of an alternative approach is the implementation of photocathodes that replace the sacrificial electron donor with electrons derived electrochemically in a photoelectrosynthetic cell [14].
The general strategy for the reductive side of water splitting outlined in figure 1 can be guided by the key steps and requirements of natural photosynthesis [20]. (i) Solar photons must be efficiently captured by antennae and/or a charge-transfer chromophore. (ii) The chromophore, upon excitation directly and/or by energy transfer, forms an electron–hole pair, which must be separated through charge transfer with minimal energy loss. (iii) Charge accumulation takes place for the energy-storing steps that follow. This step may occur at a charge-transfer module and/or the catalyst. (iv) Catalysis of the energy-storing bond-forming reaction takes place. The electrons must be energetic enough to supply the reducing power needed according to the overpotential of the catalyst. (v) Quenching of the oxidizing equivalents with a sacrificial electron donor resets the system. The nature of the process as outlined by these steps highlights the possibility of taking a modular and an interdisciplinary approach to the development of photosensitizers, charge-transfer modules and catalysts. Working across disciplines allows the integration of results from fields as disparate as biology and solid-state chemistry and physics. Of course, while work on the modules may take place independently, the components must form a functional integrated system, and they must be compatible when it comes to materials, conditions, means of coupling, rates of the key steps and energetics.
Figure 1.
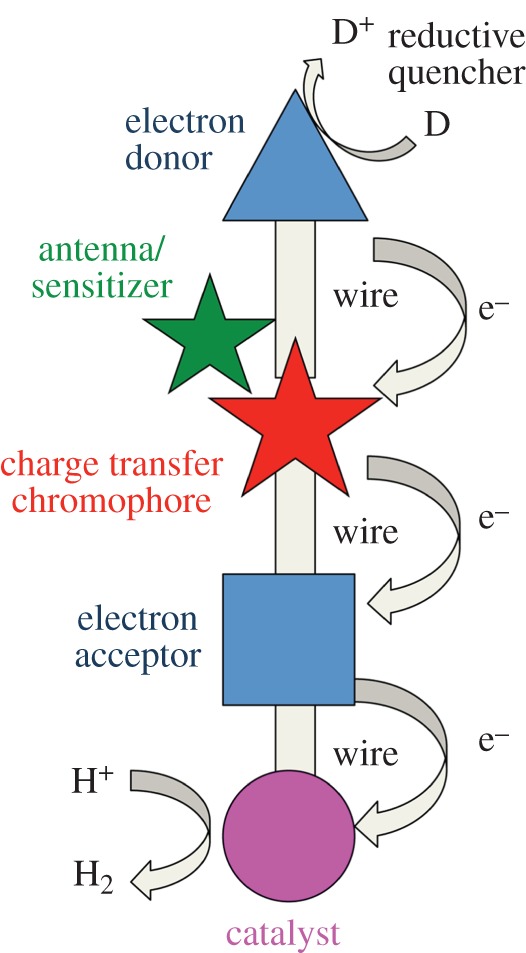
General scheme for solar hydrogen production using the reductive side of water splitting. A photosensitizer is photoexcited followed by energy transfer to a charge-transfer chromophore. The charge-transfer chromophore may also be directly excited. (Online version in colour.)
This review summarizes advances in molecular, nanomaterials, biomolecular and biological approaches to the development of photosensitizers, charge-transfer modules and hydrogen-evolution catalysts, with a goal of highlighting the different ways in which these approaches contribute to the understanding and development of systems for solar hydrogen. Examples of combinations of these modules to provide functional systems also are presented. The successes seen in the development of assemblies for solar hydrogen generation that draw components from different fields highlight the value of a multidisciplinary approach to this important problem.
2. Molecular components for solar hydrogen generation
The development of molecular systems for solar hydrogen generation aims to build modules or entire artificial photosynthetic systems on the molecular level. Molecular systems can be subjected to an exquisitely detailed level of design and modification through chemical synthesis. Furthermore, their photophysics and mechanisms of action can be probed in depth through spectroscopic and kinetics measurements performed on series of judiciously chosen derivatives. While molecular systems are ideal subjects for detailed study, those that are complex can be synthetically burdensome, and as a result not amenable to scale-up. Furthermore, molecular systems and particularly molecular chromophores are subject to degradation, thus making them less robust than nanoscale and solid-state systems (§3) [21,22]. However, the ability to study molecular systems in depth has been invaluable in the development and understanding of the fundamental processes in artificial photosynthesis.
In the field of molecular hydrogen-evolution catalysts, an emphasis has been on developing inexpensive alternatives to platinum [23,24]. Thus, natural systems, which consist only of inexpensive elements, have been an inspiration for the development of synthetic molecular catalysts. Some have taken the approach of making functional models that structurally resemble the active site of a hydrogenase (hydrogen-evolving) enzyme (figure 2a), which is made possible by the availability of high-resolution structures of different hydrogenases (active sites are given in figure 3) [25,33]. This approach has been shown to be effective in an integrated system in which a functional mimic is paired with a photosensitizer [34]. Other functional hydrogenase mimics may incorporate metals such as Fe and Ni used for proton reduction by nature, but not resemble enzyme active sites otherwise (figure 2b,c) [26]. On the other hand, catalysts incorporating inexpensive metals that are not found in natural hydrogenases such as cobalt [28,35–38] (figure 2d,e) and molybdenum [39] have been successful. Though many metal complexes are able to reduce protons to hydrogen, most of these catalysts have limitations. Often, potentials much lower than the thermodynamic potential for the reaction are required, representing a limitation on the photoreductants that can be used and an inefficient use of energy. Historically, most catalysts were limited to use in organic solvents, were sensitive to oxygen and operated at very high overpotentials (over 500 mV and approaching 1 V). However, significant improvements in molecular catalysts have been seen recently as a result of our ability to access a wide range of different molecular structures through synthesis and our increased understanding of mechanism. Molecular catalysts with longevities of over 60 h [29], turnover frequencies over 100 000 s−1 [40,41], and overpotentials of only a few hundred millivolts or less [41] have resulted. There also has been recent progress on molecular catalysts that are active in water–solvent mixtures [27,40,42] and even in neutral pH water [29,39]. However, a stable molecular catalyst that functions in water near neutral pH, displays a low overpotential, is not sensitive to oxygen and evolves hydrogen at a rapid rate has not yet been discovered.
Figure 2.
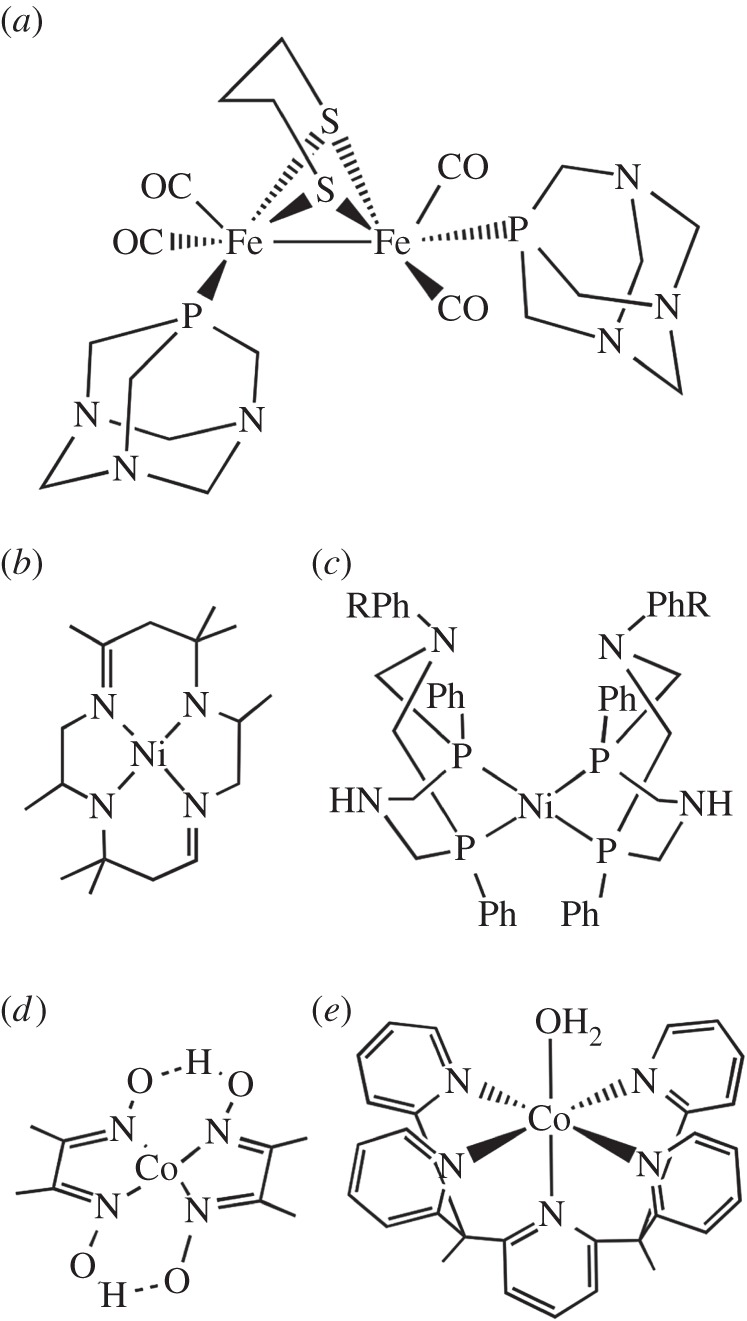
Examples of molecular catalysts for solar hydrogen evolution. (a) Functional mimic of [FeFe] hydrogenase active site [25]. (b) Early bioinspired catalyst for reduction of both CO2 and H+ [26]. (c) Nickel catalyst with ligand that can provide second-sphere interactions [27]. (d) Cobalt diglyoxime catalyst first reported in the 1980s and used in many laboratories until the present day [28]. (e) Robust cobalt pentapyridine catalyst that functions in water at neutral pH [29].
Figure 3.

Chemical structures of the active sites of hydrogenase enzymes: (a) [NiFe] hydrogenase active site [30], (b) [FeFe] hydrogenase H-cluster [31] and (c) Fe-GP cofactor of the [Fe] hydrogenase [32].
To construct a molecular system for artificial photosynthesis, chromophores and charge-transfer modules (if used) need to be paired with catalysts. As with molecular catalysts, many molecular chromophores for light absorption, energy transfer and charge transfer are inspired by nature's structures. Molecular synthetic approaches have been essential in developing our understanding of how to construct systems that yield the long-lived charge-separated states needed to provide chemical potential for solar fuel synthesis. Principles learned through the study of natural photosynthetic systems have been tested and incorporated in these assemblies [43–47]. There is an extensive literature on photoinduced charge separation by molecular chromophores, but a successful chromophore for artificial photosynthesis is defined by whether it functions in energy conversion when coupled with an energy-storing catalyst.
Molecular photochemical systems for hydrogen evolution may be assembled into a structurally defined system via covalent or non-covalent interactions (figure 1). Alternatively, the chromophore and catalyst may be allowed to interact in a bimolecular fashion through diffusion, or via an electron shuttle. While simple in concept, many processes must function well together for success. Ideally, these systems will absorb light broadly across the solar spectrum and convert a high proportion of that light energy into chemical potential sufficient for the catalyst of choice to reduce protons. Furthermore, the resulting energetic electron(s) must be delivered rapidly to the catalyst, which will require two electrons for the hydrogen-evolution reaction. With these multiple steps required, there are many opportunities to waste energy through back electron transfer, through the need for downhill electron transfer steps to acceptors or electron shuttles and ultimately the catalyst, and by the catalyst overpotential.
Although there has been extensive development of organic charge-transfer chromophores through the years, the first photosensitizers successfully paired with molecular hydrogen-evolution catalysts were metal complexes of noble metals that access charge-transfer excited states, such as bipyridyl complexes of Ru(II), Ir(III) and Rh(III) [48,49] and Pt(II) bipyridine or terpyridine acetylide complexes [50]. In addition to using noble metals, these photosensitizers exhibit a number of limitations including sometimes requiring high-energy light and exhibiting relatively low molar absorptivities [14]. Ruthenium(II) trisbipyridine and related compounds have remained popular chromophores in this application and have been paired with a variety of hydrogen-evolution catalysts in a bimolecular fashion and in tethered unimolecular systems [37,51]. With the recent emphasis on developing sustainable systems for solar fuels came the demonstration of the use of organic dyes as photosensitizers for hydrogen production. Paired with a cobalt diglyoxime catalyst (figure 2), the organic dye eosin was shown to serve as a photosensitizer to provide hydrogen for over 10 h, yielding 900 turnovers if excess ligand was provided to regenerate catalyst lost through ligand dissociation [52]. While this was the first noble metal-free system for solar hydrogen evolution, a number of other noble metal-free molecular systems have followed [14].
Through the study of molecular systems for hydrogen evolution, considerable gains have been made in understanding key system components and steps [14,37,53,54]. Although molecular systems have provided insights into mechanism and have shown vast improvements in performance and in the use of sustainable elements, they suffer from limitations in performance. Molecular chromophores are prone to photodegradation [21], and molecular catalysts tend to also undergo degradation over many turnovers [14], thus limiting longevity of the system. Furthermore, a greater efficiency of excited state quenching, a decrease in the unproductive back-reaction, and improvement in the efficiency of the dark energy-storing reaction are still required to yield a highly efficient system. Despite the increased understanding of each step in the overall process, an efficient and practical molecule-based system for visible light-driven hydrogen production from water remains elusive.
3. Nanoscale components for solar hydrogen generation
A complementary approach to the use of molecular systems for artificial photosynthesis is to use nanoscale systems. The major advantage of these systems is that they are usually much more robust than molecular systems upon illumination and during turnover. Nanoscale or solid-state photosensitizers have seen significant development with the explosion of interest in nanoscience in recent years. These systems, however, do pose their own challenges. For one, characterizing these materials in detail is significantly more difficult than it is for molecular systems. As a result, reproducibly synthesizing and characterizing components and systems can be problematic, and mechanistic details often are elusive. Another challenge is the management of light absorption and charge-transfer reactions. While semiconductor nanocrystals have attractive light-absorbing properties, it can be difficult to assemble arrays of nanocrystals to achieve efficient and directional electron transfer within a device. Furthermore, attaching nanomaterial-based photosensitizers to appropriate electron acceptors in a structurally and chemically defined fashion is not straightforward. Nevertheless, the area of nanoscale systems for solar hydrogen has seen incredible growth and many exciting developments.
In nano- and mesoscale catalysts for hydrogen evolution, reactions occur on metal surfaces, semiconductors or metal oxides [55]. In early studies, colloidal metal (usually platinum or palladium) catalysts were paired with a molecular photosensitizer ([Ru(bpy)3]2+ being most popular), an electron shuttle and a sacrificial electron donor to drive the production of hydrogen from water [56–59]. Later, platinized TiO2 was used along with a molecular platinum-based photosensitizer [60]. Although platinum and palladium offer a low overpotential for proton reduction, they are rare and expensive. More recently, with the move toward sustainable catalysts, there has been interest in the development of similarly robust systems consisting of Earth-abundant elements. Successful inorganic heterogeneous catalysts include nickel phosphide or cobalt phosphide nanoparticles, molybdenum and tungsten sulfides, and mixed metal solids [24,61–65]. Mixed metal solids consisting of nickel and molybdenum have been studied for years and show excellent performance, with low overpotentials and high exchange current densities. Furthermore, photocathodes consisting of alloys of nickel and molybdenum electrodeposited onto silicon substrates have been developed for light-driven H2 production [24,66]. An advantage of these solid-state and nanoscale systems is that they have the potential of being durable, low-cost and amenable to scale-up [67,68].
Nanostructured or mesoscopic materials are attractive light-absorbing and electron-transfer components of systems for solar hydrogen [67,69–72]. Early studies used wide band gap semiconductors such as TiO2 and UV radiation to drive the full water-splitting reaction [69,73–80]. More recently, nanostructured semiconductors have been used. For example, branched TiO2 nanorods achieve high photon-to-current conversion efficiencies and drive hydrogen evolution using a platinum catalyst. Nanostructures are proposed to improve efficiency because their small diameter enhances charge separation and transport and their large surface area facilitates hole transfer [81]. Nanostructured Si light absorbers also have been successfully used as photosensitizers for hydrogen evolution [82,83]. Another attractive material is Cu2O, a p-type semiconductor that has been employed as a photocathode in hydrogen production [84,85].
Semiconductor quantum dots (QDs) are an attractive alternative photosensitizer receiving significant recent attention that potentially combine the positive qualities of crystalline semiconductors and small molecules [86,87]. For example, QDs can be manipulated in solution, but they are inherently crystalline and thus are more robust than molecules. QDs also have a much larger absorption cross section and greater photostability than organic dyes [88–94]. QDs also have size-tunable qualities, with smaller QDs exhibiting higher band gaps as a result of quantum confinement effects [95]. Thus, the excited state of a QD material is expected to be more reducing than that of the bulk material. QDs have been demonstrated to be highly robust photosensitizers for the hydrogen-evolution reaction (figure 4). In a seminal study, it was shown that the irradiation of a system consisting of dihydrolipoic acid-capped CdSe QDs, Ni(NO3)2 precatalyst and the electron donor ascorbic acid in water evolves hydrogen for over 360 h, giving over 600 000 turnovers [96]. The quantum yield for hydrogen production was over 38% in water and over 66% in a 1 : 1 water–ethanol mixture. The turnover frequency was also exceptionally high; over 7000 turnovers per hour were achieved, which is comparable with the activity of noble metal nanoparticle catalysts. While this result represents an outstanding system in terms of performance, there remain many questions about how it functions including what state serves to donate the electron and what the active form of the catalyst is. Other applications of semiconductor QDs as photoexcitable donors for small-molecule catalysts have been reported, but most have catalyst lifetimes that are fairly short (less than 24 h) and/or turnover numbers per catalyst that are not greatly improved over leading organic dye photosensitizers (less than 10 000) [97–99]. A recent exception is a report in which CdTe QDs were combined with CoCl2 to form a catalyst in situ [100]. This system was active for 70 h and achieved over 59 000 turnovers with respect to a single catalyst molecule. However, the turnover frequency was modest, around 1000 turnovers per hour.
Figure 4.
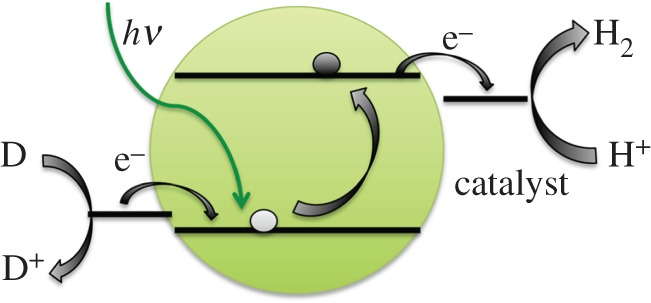
Illustration of a semiconductor nanoparticle photosensitizer for hydrogen evolution. Absorption of light excites an electron from the conduction band to the valence band, providing the electron with enough energy for the hydrogen-evolution catalyst. The hole is filled by a sacrificial electron donor (D). (Online version in colour.)
In addition to serving as photosensitizers, nanostructured materials also have been used to enhance photoinduced charge separation. While TiO2 requires UV excitation, the use of CdS nanowires is able to drive hydrogen generation using visible light [101]. ZnO nanowires are able to carry a photocurrent that could be used for hydrogen evolution when sensitized by CdSe QDs [102]. Carbon nanotubes (NTs) provide an attractive structure for separating charge and transporting electrons. NTs are chemically robust, making them stable supports for catalysts, and they also transfer electrons over large distances with minimal resistance [103]. As a result NTs have been shown to be effective electron acceptors in applications for energy conversion [104–106]. It has been shown that photoexcited porphyrins inject electrons into NTs in porphyrin–NT conjugates [107], and that these assemblies produce long-lived electron–hole pairs on the NT surface [108,109]. NTs also have been reported to produce hydrogen with dye sensitizers using all reaction components in a single solution [110]. The attachment of biomimetic molecular hydrogen-evolution catalysts to carbon NTs has been shown to result in a long-lived material for hydrogen evolution [111]. Nanomaterials including NTs and QDs also have been interfaced with biological molecules for hydrogen production, as discussed in the following section. However, NTs have not yet been successfully used as components in a system for artificial photosynthesis that is structurally well defined and well characterized.
Nanoscale components of systems for artificial photosynthesis show significant advantages over molecular systems in terms of durability. On the downside, these systems are relatively difficult to assemble in a controlled manner and to characterize in detail. As a result, there remain many questions about how to engineer these systems to optimize and tune their functions. Nevertheless, the durability and high activity offered by nanoscale systems indicate that they will remain a focus of research in artificial photosynthesis going forward.
4. Biological components for solar hydrogen generation
Nature's biomolecular structures for photosynthesis and proton reduction inspire many of the molecular systems described above. In the light reactions of photosynthesis, biological molecules make use of macrocyclic conjugated chromophores such as porphyrins and chlorins (along with other pigments and metal ions) for light absorption, energy transfer, generation of a charge-separated state and subsequent energy-storing electron transfers [112]. Furthermore, the inspiration for many of the molecular catalysts for hydrogen evolution comes from the structure and function of natural hydrogenases [33,113–115]. Recently, chemists have turned to concepts from biology such as the use of proton relays to deliver substrate to the active site, resulting in an increase in the efficiency of molecular catalysts [40,41]. Rather than working to mimic biology, another approach is to use nature's highly evolved biomolecular structures directly in assemblies for artificial photosynthesis. For solar hydrogen production, photosystem I (PSI) and hydrogenase enzymes are the most relevant biological structures, and will be the focus herein.
PSI is an integral membrane protein complex that carries out photosynthetic light reactions in algae, plants and some bacteria (figure 5) [117]. It absorbs visible light and generates a charge-separated state, transferring the electron to a ferredoxin, which donates electrons to the enzyme ferredoxin-NADP+ oxidoreductase that catalyses the two-electron reduction of NADP+ to NADPH, a biological reductant used in a number of processes including CO2 fixation. The hole generated in this process is filled by a soluble biomolecular electron donor such as the haemeprotein cytochrome c6 or the copper protein plastocyanin. By interfacing PSI with an appropriate catalyst in place of its biological acceptor, PSI can be used as a module for converting light energy to chemical potential that is stored in a fuel.
Figure 5.
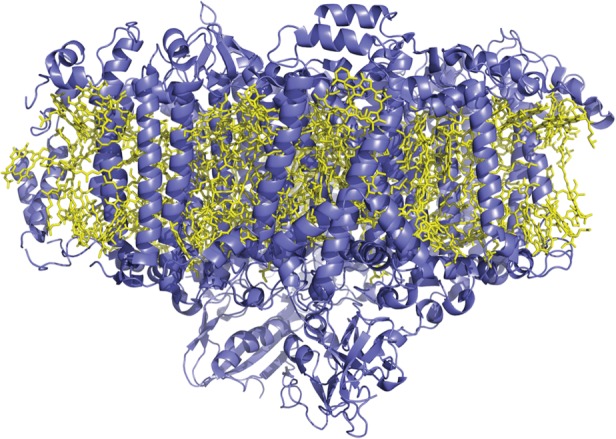
Structure of photosystem I from the cyanobacterium Synechococcus elongatus. The polypeptide is shown in ribbon format, and the bound chromophores as sticks [116]. (Online version in colour.)
The earliest systems that made use of nature's light absorption and charge-transfer machinery in solar hydrogen generation consisted of chloroplasts that were modified by precipitation of platinum [118]. Similarly, PSI modified with platinum [119] or electrostatically attached platinum nanoparticles [120,121] produces hydrogen photocatalytically. A more structurally defined system was created by covalent tethering of a gold or platinum nanoparticle to the terminal (FB) iron–sulfur cluster of PSI [122]. These systems use ascorbate as a sacrificial electron donor and biological electron shuttles such as cytochrome c6 or plastocyanin to reduce PSI. The use of the biological electron-transfer partners ensures that the electrons are delivered efficiently to the correct site on the macromolecular complex. Getting away from using noble metal catalysts, a complex between a cobaloxime catalyst and PSI has been prepared by self-assembly and shown to drive light-driven hydrogen evolution with a respectable turnover number of 5200 mol H2 (mol PSI)−1. The activity of this system levels off after 1.5 h [123]. In comparison, the PSI–platinum system yields less than 80 000 mol hydrogen (mol PSI)−1 in 5 h. The development of catalysts using Earth-abundant materials that provide activity that rivals platinum in these systems still is needed. At the same time, even the system using the platinum catalyst lasts a relatively short period of 5 h. A challenge with the use of nature's photosystems in assemblies or devices is that they are damaged by sunlight. In vivo, repair mechanisms are in place [124], but in vitro and in devices, photodamage is a major limitation. For this reason, as noted above, more robust photosensitizers consisting of nanomaterials have come into greater favour.
Nature provides not only elegant systems for light absorption and charge transfer but also highly efficient catalysts for hydrogen evolution. Hydrogenases are enzymes that catalyse the reversible reduction of protons to hydrogen [125]. They are remarkable for using Earth-abundant metals in their active sites and displaying an activity comparable with that of platinum [126]. They have low overpotentials and very high activities, producing 100–10 000 moles of hydrogen per enzyme molecule per second [125,127–129]. There are three main families of hydrogenases. The [FeFe] hydrogenases have a diiron active site, the [NiFe] hydrogenases have a dinuclear nickel–iron active site and the [Fe] hydrogenases have a mononuclear iron active site (figure 3). Many hydrogenases are quite oxygen sensitive, but some are oxygen tolerant and some have been engineered to increase oxygen tolerance [130].
Hydrogenases have been coupled to semiconductor surfaces or colloids with hydrogen production induced by irradiating the semiconductor, causing the transfer of an energetic electron to the hydrogenase to drive the proton reduction reaction. For example, hydrogenase from a hyperthermophilic organism was used in a device in which anodized tubular TiO2 electrodes were derivatized with the enzyme to evolve hydrogen upon UV illumination [131,132]. In order to use visible light, similar constructs have used either ruthenium-based [133] or porphyrin [134] photosensitizers on TiO2 nanoparticles coupled to a hydrogenase. To prepare assemblies driven by visible light that use robust nanoparticle photodonors while avoiding the need for a molecular photosensitizer, semiconductor QDs and related structures are attractive. For example, hydrogenase has been attached to CdS nanorods or nanocrystalline CdTe for photocatalytic reduction of protons to hydrogen upon illumination at 405 nm [135,136]. While combining QDs with hydrogenases arguably brings together a highly robust photosensitizer and an extremely efficient catalyst, results on integrated systems have been disappointing. While these systems can have high initial turnover rates [135], they generally show poor longevity [135,136] and can have low overall turnover numbers [137]. Possible weak links in the nanocrystal–hydrogenase system are a poor yield of electron injection into the hydrogenase and a loss of hydrogenase activity when modified with the nanomaterial. These results are an example of how using individually optimized components of an assembly for artificial photosynthesis may not yield an efficient system. The linkage between the components also has to be considered, along with the energies they require and the rates for the processes that they carry out.
One way to improve the electrical contact from photodonor to hydrogenase catalyst is to engineer a molecular wire from the electron donor to the hydrogenase. These kinds of highly engineered systems have been created by linking PSI to a hydrogenase, thus using biomolecules for both light-induced electron–hole separation and for catalysis. The approach is to add a synthetic linker that coordinates the FB iron–sulfur cluster on the surface of PSI directly to the distal iron–sulfur cluster of a hydrogenase [138]. A [FeFe] hydrogenase was attached in this way to PSI to yield an artificial biomolecular photocatalytic assembly. Upon illumination with broad-spectrum light in the presence of ascorbate as an electron donor and soluble cytochrome c6 as an electron shuttle, hydrogen was produced at a rate of 30 μmol per mg of chlorophyll per hour. Notably, the construct showed little loss of activity after storage for up to 64 days [138]. The rate was increased further to 2200 μmol per mg of chlorophyll per hour by tethering the cytochrome c6 to PSI. The authors note that the corresponding electron-transfer rate is greater than overall electron-transfer rate through PSI in cyanobacteria [139].
An alternative to the use of nature's hydrogenases is to engineer non-hydrogenase biomolecules to take on hydrogenase activity. This approach requires introducing a novel function to a biomolecule and is less likely to result in the efficient systems that evolution yields, but also allows the researcher to use a robust and easily engineered biochemical architecture for the catalytic site. This protein engineering approach also allows the testing of hypotheses regarding how active site structure influences function. The engineering of artificial hydrogenases is at an early stage, but there are a few examples in the literature. One method is to introduce a known molecular catalyst into a biomolecular architecture. For example, a cobalt diglyoxime catalyst has been introduced into the haeme-binding pocket of myoglobin to yield hydrogen-evolution activity under neutral aqueous conditions [140]. Alternatively, complexes between catalysts and peptides have been created. The advantage of this approach is that the peptides can enhance water solubility and provide second-sphere interactions with the catalyst active site [141]. Using this approach, nickel catalysts have been modified with peptides to facilitate the introduction of second-sphere interactions and proton relays [142]. One peptide–catalyst model system was formed by attaching a peptide to iron carbonyl thiolate resembling the hydrogenase active site [143]. Another catalyst–peptide complex was prepared by digestion of horse cytochrome c to yield the 11-residue haeme peptide known as microperoxidase-11 followed by substitution of iron with cobalt. The resulting catalyst has a turnover number of more than 20 000 for hydrogen evolution and functions in neutral water in the presence of oxygen [144].
The use of biomolecules in assemblies for photocatalytic hydrogen evolution not only has a long history but also is seeing rapid recent development, in particular, in the area of engineered hydrogenases. With the great interest in developing environment friendly, non-toxic, water-soluble systems for artificial photosynthesis, biomolecular approaches are expected to continue to be of increasing interest to researchers. Another advantage of biomolecular systems that has only begun to be explored is the potential to engineer substrate channels and specific interactions with the catalytic active site through judicious choice of biomolecular scaffold for the catalyst and by engineering that scaffold. At the same time, biomolecular systems offer challenges. It is difficult to compete with nanoscale and solid-state systems when it comes to durability, and the preparation and modification of biomolecules can be more challenging in some cases than chemical synthesis of catalysts or photosensitizers. Nevertheless, the area of engineered biomolecular systems for solar hydrogen is expected to continue to grow.
Another biology-based approach to energy conversion is to make use of biological molecules or microbes in bioelectrochemical systems in which bacteria interact with electrodes to donate or accept electrons. Electron shuttles may be excreted by the bacterium, or added to the system. Also, the bacteria may interact with the electrode through diffusion or by forming a biofilm on the electrode. A number of different electron acceptors can typically be used by electrogenic bacteria, and thus the bacteria can be compatible with a range of cell designs and electrode materials [145]. In one class of these systems, microbial electrolysis cells, hydrogen evolution is driven in a system in which microbes supply electrons to the anode, and a fuel such as hydrogen is evolved at the cathode, which is typically platinum. A supplemental voltage is applied to overcome the barrier for hydrogen formation (figure 6) [146–148]. This set-up does not convert solar energy, but could be modified to do so. An example of a microbial electrolysis set-up that could store light energy in the form of hydrogen upon modification makes use of a nanowire array as a photocathode and a film of electrogenic bacteria on the anode. This cell produces a current upon irradiation with white light, and the authors note that if protons served as electron acceptors at the cathode of their device, hydrogen would be produced [149]. Subsequently, a microbial fuel cell has been coupled to a photoelectrochemical cell. In this configuration, the microbial fuel cell provides electrons to the photoelectrochemical cell in which irradiation of the TiO2 photoanode drives water splitting [150]. Another interesting bioelectrocatalysis approach for solar H2 would incorporate both H2 production and consumption in one unit to produce electricity. A precedent is given by the successful use of an H2-consuming hydrogenase enzyme electrode for production of electricity [151]. In the future, a system could be imagined that would use light to drive H2 production by cyanobacteria and direct the resulting solar H2 to a hydrogenase-based fuel cell to produce electricity. The use of microbes to generate hydrogen, to assist with light conversion and/or electron-transfer steps, and also to convert hydrogen to electricity is a flourishing area that is expected to show significant future growth.
Figure 6.
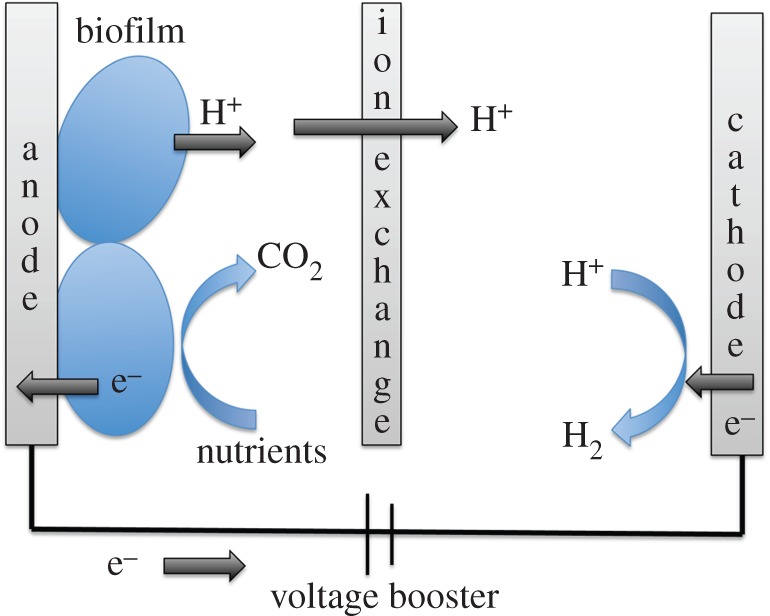
Schematic of a microbial electrolysis cell. Respiring microbes provide electrons to the anode, which are delivered to the cathode for hydrogen evolution. A voltage boost provides additional energy needed. The microbes serve to lower the voltage required to drive the reaction. (Online version in colour.)
Biomolecular systems for solar hydrogen may be considered a subset of molecular systems, and thus they carry many of the same positive features and drawbacks, while offering unique opportunities and challenges. For one, both biomolecular and molecular chromophores are prone to photodegradation. The biological systems have the advantage that they have evolved repair mechanisms to provide the needed longevity. However, these systems are not in place when the isolated photosystems are incorporated into an engineered system. One solution to this problem is to make use of organisms rather than isolated biomolecules so that the elegant biological repair systems remain in place, as is seen in some bio-photoelectrochemical systems. Identifying novel ways to make use of biology's ‘self-healing’ capabilities is a direction that requires additional effort. Another advantage of biomolecular systems that is shared with molecular systems is that they may be manipulated and studied at atomic-level resolution. Understanding systems at this level of detail, however, is more challenging for biomolecules given their larger size. Furthermore, synthetic approaches that are commonly used to modify molecular catalysts or chromophores to understand detailed structure–reactivity relationships are rarely applicable to biological systems. An exception is when synthetic molecules are incorporated into biomolecular frameworks; in this case, a series of changes in the catalyst may be made before constructing the ‘holo-protein’ hybrid construct. While small-molecule catalysts have the advantage of being amenable to detailed synthetic modifications, biomolecular frameworks provide an abundance of pathways for the engineering of second-sphere interactions and substrate-recognition sites. Although these kinds of interactions have been built into synthetic catalysts, introducing and controlling such complex structures can require elaborate ligand designs, and may not place functional groups where intended.
5. The case for a global artificial photosynthesis effort
Some of the most exciting efforts in the development of systems for solar hydrogen are seen at the interface between fields. Interactions of microbes with electrode materials, the modification of nanostructures with biomolecules, and the attachment of synthetic molecules to nanocrystals are examples of advances at the interface. Thus, developing efficient, robust and environmentally benign systems for artificial photosynthesis will require the continued advancement of research in these diverse fields as well as interactions between groups with different areas of expertise. Importantly, results on integrated systems for artificial photosynthesis underline that the real challenge lies in effectively coupling the components. The development of efficient and robust systems for solar hydrogen generation and for artificial photosynthesis more broadly is most likely to succeed if we can capitalize on the diverse expertise of research groups across the globe, and encourage and support exchange of ideas and collaborations between these groups.
A key question is how to establish, encourage, and support a global artificial photosynthesis (GAP) effort toward developing efficient, inexpensive and environmentally benign artificial photosynthetic systems. Such a project would need to raise the public profile of the importance of artificial photosynthesis and address legal issues as well as support and develop the science [152,153]. In this review, the advantages and disadvantages of approaches to one form of artificial photosynthesis, storing solar energy in the form of hydrogen, spanning a wide range of scientific disciplines have been highlighted. A conclusion is that since each approach offers unique pluses and minuses, any major GAP effort must encompass a wide range of approaches represented by but not limited to those described in this review. The nature of the modern scientist is to be specialized, a necessity that comes from ever-increasing depth of knowledge and technological developments. While having a specialty does not bar any researcher or research group from tackling a system from multiple angles, any one researcher or group will usually have an area with which they are most experienced. A GAP project will benefit from capitalizing on this experience and depth while also building bridges between researchers to allow development of science at the interface. Thus, a proposal is that a global system for supporting artificial photosynthesis research would fund single-principal investigator (PI) grants, multi- (approx. two to five) PI grants and centres of excellence. The single-PI grants would be distributed to fund focused research on the individual components using specific approaches to capitalize on the recognized expertise of individual PIs and their laboratories. The multi-PI grants would fund focused efforts at the interface between areas. As has been illustrated a number of times in this review, the fact that individual components function well does not mean that they will function well in combination, and thus efforts that develop the photosynthetic modules in parallel while testing their compatibility and performance are needed. Finally, the centres of excellence would organize the development, testing and scale-up of the most successful integrated systems for artificial photosynthesis that arise from the multi-PI efforts.
A GAP project would require funds drawn internationally and distributed internationally. Any system accomplishing this would necessarily be complex and cannot be fully outlined in a short review. However, a few general principles will be outlined here. Firstly, the establishment of an international fund for the support of GAP research would need to draw on governmental funds. A model for such a funding system that spans multiple countries is the European Research Council. Alternatively, funds might be solicited from philanthropic organizations. A challenge with the philanthropic route is that these organizations may not be willing to give up control of the distribution of the funds in order for them to be used in the most effective way. With government funding, the participating governments would agree that the funds would be used for this purpose and distributed broadly among the participating countries. This model would support the ethical principle that natural photosynthesis is a common heritage of humanity that should be administered through an agency that sees that photosynthesis-related technology is developed for the good of humanity. The distribution of the funds would be administered by an international governing board advised by peer reviewers. Funding would consider diversity and quality in science and also geography. In order to encourage participation by individual countries, a promise of direct benefit at home would be made by linking geographical distribution of funds to some extent to the amount of funds provided by the participating country. Furthermore, the funding would be set up to encourage collaboration and the development of full functional systems rather than decades-long study on a single isolated component. The single-PI grants would have a limited time frame (e.g. a 3-year grant that can be renewed only once), after which these PIs would be eligible to apply only under the multi-PI mechanism. The idea is that individual PIs would come together to propose integrated systems for funding going forward. The centres of excellence would serve as resources for all participants by providing specialized equipment, technical expertise and by performing benchmarking. To highlight these centres and to encourage collaboration, the centres would hold annual conferences for current PIs and other interested researchers.
Funding statement
The author's work on solar hydrogen generation is supported by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, US Department of Energy, grant no. DE-FG02–09ER16121.
References
- 1.Lewis NS, Nocera DG. 2006. Powering the planet: chemical challenges in solar energy utilization. Proc. Natl Acad. Sci. USA 103, 15 729–15 735. ( 10.1073/pnas.0603395103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International Energy Agency. 2014. IEA Statistics. See http://www.iea.org/stats/index.asp (accessed October 2014).
- 3.U.S. Energy Information Administration. 2011. Annual Energy Outlook 2011, DOE/EIA-0383(2011).
- 4.U.S. Energy Information Administration. 2011. AEO2011 National Energy Modeling System, run REF2011.D020911A. See http://www.eia.gov/aeo.
- 5.Kintisch E. 2006. Climate change—along the road from Kyoto—global greenhouse gas emissions keep rising. Science 311, 1702–1703. ( 10.1126/science.311.5768.1702) [DOI] [PubMed] [Google Scholar]
- 6.NOAA. 2014. Global greenhouse gas reference network trends in carbon dioxide. See http://www.esrl.noaa.gov/gmd/ccgg/trends/ (accessed October 2014).
- 7.Intergovernmental Panel on Climate Change. 2007. Climate change 2007: mitigation of climate change. IPCC Working Group III Fourth Assessment Report, Geneva.
- 8.Parry ML, Canziani OF, Palutikof JP, van der Linden PJ, Hanson CE. 2007. Climate change 2007: impacts, adaptations and vulnerability. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 9.Jacobson MZ. 2009. Review of solutions to global warming, air pollution, and energy security. Energy Environ. Sci. 2, 148–173. ( 10.1039/B809990C) [DOI] [Google Scholar]
- 10.Barber J. 2009. Photosynthetic energy conversion: natural and artificial. Chem. Soc. Rev. 38, 185–196. ( 10.1039/b802262n) [DOI] [PubMed] [Google Scholar]
- 11.International Energy Agency. 2013. World energy outlook. See http://www.worldenergyoutlook.org/publications/weo-2013/.
- 12.For an industry view, see Sunpower's. 2010. annual report. See http://investors.sunpowercorp.com/annuals.cfm. (accessed October 2013).
- 13.Kulheim C, Agren J, Jansson S. 2002. Rapid regulation of light harvesting and plant fitness in the field. Science 297, 91–93. ( 10.1126/science.1072359) [DOI] [PubMed] [Google Scholar]
- 14.Han Z, Eisenberg R. 2014. Fuel from water: the photochemical generation of hydrogen from water. Acc. Chem. Res. 47, 2537–2544. ( 10.1021/ar5001605) [DOI] [PubMed] [Google Scholar]
- 15.Pagliaro M, Konstandopoulos AG, Ciriminna R, Palmisano G. 2010. Solar hydrogen: fuel of the near future. Energy Environ. Sci. 3, 279–287. ( 10.1039/b923793n) [DOI] [Google Scholar]
- 16.California fuel cell partnership: driving for the future. See http://cafcp.org (accessed October 2014).
- 17.Eisenberg R, Gray HB. 2008. Preface on making oxygen. Inorg. Chem. 47, 1697–1699. ( 10.1021/ic800155g) [DOI] [PubMed] [Google Scholar]
- 18.Kanan MW, Nocera DG. 2008. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 321, 1072–1075. ( 10.1126/science.1162018) [DOI] [PubMed] [Google Scholar]
- 19.Yin Q, Tan JM, Besson C, Geletii YV, Musaev DG, Kuznetsov AE, Luo Z, Hardcastle KI, Hill CL. 2010. A fast soluble carbon-free molecular water oxidation catalyst based on abundant metals. Science 328, 342–345. ( 10.1126/science.1185372) [DOI] [PubMed] [Google Scholar]
- 20.Alstrum-Acevedo JH, Brennaman MK, Meyer TJ. 2005. Chemical approaches to artificial photosynthesis. Inorg. Chem. 44, 6802–6827. ( 10.1021/ic050904r) [DOI] [PubMed] [Google Scholar]
- 21.Esswein AJ, Nocera DG. 2007. Hydrogen production by molecular photocatalysis. Chem. Rev. 107, 4022–4047. ( 10.1021/cr050193e) [DOI] [PubMed] [Google Scholar]
- 22.McCormick TM, Calitree BD, Orchard A, Kraut ND, Bright FV, Detty MR, Eisenberg R. 2010. Reductive side of water splitting in artificial photosynthesis: new homogeneous photosystems of great activity and mechanistic insight. J. Am. Chem. Soc. 132, 15 480–15 483. ( 10.1021/ja1057357) [DOI] [PubMed] [Google Scholar]
- 23.Thoi VS, Sun Y, Long JR, Chang CJ. 2013. Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions. Chem. Soc. Rev. 42, 2388–2400. ( 10.1039/c2cs35272a) [DOI] [PubMed] [Google Scholar]
- 24.McKone JR, Marinescu SC, Brunschwig BS, Winkler JR, Gray HB. 2014. Earth-abundant hydrogen evolution electrocatalysts. Chem. Sci. 5, 865–878. ( 10.1039/c3sc51711j) [DOI] [Google Scholar]
- 25.Mejia-Rodriguez R, Chong DS, Reibenspies JH, Soriaga MP, Darensbourg MY. 2004. The hydrophilic phosphatriazaadamantane ligand in the development of H2 production electrocatalysts: iron hydrogenase model complexes. J. Am. Chem. Soc. 126, 12 004–12 014. ( 10.1021/ja039394v) [DOI] [PubMed] [Google Scholar]
- 26.Fisher B, Eisenberg R. 1980. Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 102, 7361–7363. ( 10.1021/ja00544a035) [DOI] [Google Scholar]
- 27.Kilgore UJ, et al. 2011. [Ni((P2N2C6H4)-N-Ph)2]2+ complexes as electrocatalysts for H2 production: effect of substituents, acids, and water on catalytic rates. J. Am. Chem. Soc. 133, 5861–5872. ( 10.1021/Ja109755f) [DOI] [PubMed] [Google Scholar]
- 28.Hawecker J, Lehn JM, Ziessel R. 1983. Efficient homogeneous photochemical hydrogen generation and water reduction mediated by cobaloxime or macrocyclic cobalt complexes. Nouv. J. Chim. 7, 271–277. [Google Scholar]
- 29.Sun YJ, Bigi JP, Piro NA, Tang ML, Long JR, Chang CJ. 2011. Molecular cobalt pentapyridine catalysts for generating hydrogen from water. J. Am. Chem. Soc. 133, 9212–9215. ( 10.1021/ja202743r) [DOI] [PubMed] [Google Scholar]
- 30.Volbeda A, Charon MH, Piras C, Hatchikian EC, Frey M, Fontecillacamps JC. 1995. Crystal-structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 373, 580–587. ( 10.1038/373580a0) [DOI] [PubMed] [Google Scholar]
- 31.Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. 1998. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Å resolution. Science 282, 1853–1858. ( 10.1126/science.282.5395.1853) [DOI] [PubMed] [Google Scholar]
- 32.Shima S, Pilak O, Vogt S, Schick M, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer RK, Ermler U. 2008. The crystal structure of Fe-hydrogenase reveals the geometry of the active site. Science 321, 572–575. ( 10.1126/science.1158978) [DOI] [PubMed] [Google Scholar]
- 33.Gloaguen F, Lawrence JD, Rauchfuss TB. 2001. Biomimetic hydrogen evolution catalyzed by an iron carbonyl thiolate. J. Am. Chem. Soc. 123, 9476–9477. ( 10.1021/Ja016516f) [DOI] [PubMed] [Google Scholar]
- 34.Li C, Wang M, Pan JX, Zhang P, Zhang R, Sun LC. 2009. Photochemical hydrogen production catalyzed by polypyridyl ruthenium-cobaloxime heterobinuclear complexes with different bridges. J. Organomet. Chem. 694, 2814–2819. ( 10.1016/j.jorganchem.2009.04.041) [DOI] [Google Scholar]
- 35.Kellett RM, Spiro TG. 1985. Cobalt(I) porphyrin catalysts of hydrogen production from water. Inorg. Chem. 24, 2373–2377. ( 10.1021/ic00209a011) [DOI] [Google Scholar]
- 36.Artero V, Chavarot-Kerlidou M, Fontecave M. 2011. Splitting water with cobalt. Angew. Chem. Int. Ed. Engl. 50, 7238–7266. ( 10.1002/anie.201007987) [DOI] [PubMed] [Google Scholar]
- 37.Dempsey JL, Brunschwig BS, Winkler JR, Gray HB. 2009. Hydrogen evolution catalyzed by cobaloximes. Acc. Chem. Res. 42, 1995–2004. ( 10.1021/ar900253e) [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Wang M, Han K, Zhang P, Gloaguen F, Sun L. 2014. A super-efficient cobalt catalyst for electrochemical hydrogen production from neutral water with 80 mV overpotential. Energy Environ. Sci. 7, 329–334. ( 10.1039/c3ee42194e) [DOI] [Google Scholar]
- 39.Karunadasa HI, Chang CJ, Long JR. 2010. A molecular molybdenum-oxo catalyst for generating hydrogen from water. Nature 464, 1329–1333. ( 10.1038/nature08969) [DOI] [PubMed] [Google Scholar]
- 40.Helm ML, Stewart MP, Bullock RM, Rakowski DuBois M, DuBois DL. 2011. A synthetic nickel electrocatalyst with a turnover frequency above 100 000 s−1 for H2 production. Science 333, 863–866. ( 10.1126/science.1205864) [DOI] [PubMed] [Google Scholar]
- 41.Hoffert WA, Roberts JAS, Bullock RM, Helm ML. 2013. Production of H2 at fast rates using a nickel electrocatalyst in water–acetonitrile solutions. Chem. Commun. 49, 7767–7769. ( 10.1039/c3cc43203c) [DOI] [PubMed] [Google Scholar]
- 42.Bigi JP, Hanna TE, Harman WH, Chang A, Chang CJ. 2010. Electrocatalytic reduction of protons to hydrogen by a water-compatible cobalt polypyridyl platform. Chem. Commun. 46, 958–960. ( 10.1039/B915846d) [DOI] [PubMed] [Google Scholar]
- 43.Gust D, et al. 1990. Efficient multistep photoinitiated electron-transfer in a molecular pentad. Science 248, 199–201. ( 10.1126/science.248.4952.199) [DOI] [PubMed] [Google Scholar]
- 44.Gust D, Moore TA, Moore AL. 2009. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 42, 1890–1898. ( 10.1021/ar900209b) [DOI] [PubMed] [Google Scholar]
- 45.Wasielewski MR. 2009. Self-assembly strategies for integrating light harvesting and charge separation in artificial photosynthetic systems. Acc. Chem. Res. 42, 1910–1921. ( 10.1021/ar9001735) [DOI] [PubMed] [Google Scholar]
- 46.Kang YK, Iovine PM, Therien MJ. 2011. Electron transfer reactions of rigid, cofacially compressed, pi-stacked porphyrin-bridge-quinone systems. Coord. Chem. Rev. 255, 804–824. ( 10.1016/j.ccr.2010.12.011) [DOI] [PubMed] [Google Scholar]
- 47.Fukuzumi S, Ohkubo K, Suenobu T. 2014. Long-lived charge separation and applications in artificial photosynthesis. Acc. Chem. Res. 47, 1455–1464. ( 10.1021/ar400200u) [DOI] [PubMed] [Google Scholar]
- 48.Cline ED, Adamson SE, Bernhard S. 2008. Homogeneous catalytic system for photoinduced hydrogen production utilizing iridium and rhodium complexes. Inorg. Chem. 47, 10 378–10 388. ( 10.1021/ic800988b) [DOI] [PubMed] [Google Scholar]
- 49.Meyer TJ. 1989. Chemical approaches to artificial photosynthesis. Acc. Chem. Res. 22, 163–170. ( 10.1021/ar00161a001) [DOI] [Google Scholar]
- 50.Du PW, Schneider J, Jarosz P, Eisenberg R. 2006. Photocatalytic generation of hydrogen from water using a platinum(II) terpyridyl acetylide chromophore. J. Am. Chem. Soc. 128, 7726–7727. ( 10.1021/ja0610683) [DOI] [PubMed] [Google Scholar]
- 51.Magnuson A, et al. 2009. Biomimetic and microbial approaches to solar fuel generation. Acc. Chem. Res. 42, 1899–1909. ( 10.1021/ar900127h) [DOI] [PubMed] [Google Scholar]
- 52.Lazarides T, McCormick T, Du P, Luo G, Lindley B, Eisenberg R. 2009. Making hydrogen from water using a homogeneous system without noble metals. J. Am. Chem. Soc. 131, 9192–9194. ( 10.1021/ja903044n) [DOI] [PubMed] [Google Scholar]
- 53.Wasielewski MR. 1992. Photoinduced electron-transfer in supramolecular systems for artificial photosynthesis. Chem. Rev. 92, 435–461. ( 10.1021/cr00011a005) [DOI] [Google Scholar]
- 54.Balzani V, Juris A, Venturi M, Campagna S, Serroni S. 1996. Luminescent and redox-active polynuclear transition metal complexes. Chem. Rev. 96, 759–833. ( 10.1021/cr941154y) [DOI] [PubMed] [Google Scholar]
- 55.McKone JR, Lewis NS, Gray HB. 2014. Will solar-driven water-splitting devices see the light of day? Chem. Mater. 26, 407–414. ( 10.1021/cm4021518) [DOI] [Google Scholar]
- 56.Lehn JM, Sauvage JP. 1977. Chemical storage of light energy—catalytic generation of hydrogen by visible-light or sunlight—irradiation of neutral aqueous-solutions. New J. Chem. 1, 449–451. [Google Scholar]
- 57.Kalyanasundaram K, Kiwi J, Grätzel M. 1978. Hydrogen evolution from water by visible-light, a homogeneous 3-component test system for redox catalysis. Helv. Chim. Acta 61, 2720–2730. ( 10.1002/hlca.19780610740) [DOI] [Google Scholar]
- 58.Miller DS, Bard AJ, McLendon G, Ferguson J. 1981. Catalytic water reduction at colloidal metal microelectrodes. 2. Theory and experiment. J. Am. Chem. Soc. 103, 5336–5341. ( 10.1021/ja00408a010) [DOI] [Google Scholar]
- 59.Kiwi J, Grätzel M. 1979. Protection, size factors, and reaction dynamics of colloidal redox catalysts mediating light-induced hydrogen evolution from water. J. Am. Chem. Soc. 101, 7214–7217. ( 10.1021/ja00518a015) [DOI] [Google Scholar]
- 60.Zhang J, Du PW, Schneider J, Jarosz P, Eisenberg R. 2007. Photogeneration of hydrogen from water using an integrated system based on TiO2 and platinum(II) diimine dithiolate sensitizers. J. Am. Chem. Soc. 129, 7726–7727. ( 10.1021/ja071789h) [DOI] [PubMed] [Google Scholar]
- 61.Popczun EJ, McKone JR, Read CG, Biacchi AJ, Wiltrout AM, Lewis NS, Schaak RE. 2013. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 135, 9267–9270. ( 10.1021/ja403440e) [DOI] [PubMed] [Google Scholar]
- 62.Jaramillo TF, Jorgensen KP, Bonde J, Nielsen JH, Horch S, Chorkendorff I. 2007. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 317, 100–102. ( 10.1126/science.1141483) [DOI] [PubMed] [Google Scholar]
- 63.Merki D, Hu X. 2011. Recent developments of molybdenum and tungsten sulfides as hydrogen evolution catalysts. Energy Environ. Sci. 4, 3878–3888. ( 10.1039/c1ee01970h) [DOI] [Google Scholar]
- 64.Tran PD, Wong LH, Barber J, Loo JSC. 2012. Recent advances in hybrid photocatalysts for solar fuel production. Energy Environ. Sci. 5, 5902–5918. ( 10.1039/c2ee02849b) [DOI] [Google Scholar]
- 65.Popczun EJ, Read CG, Roske CW, Lewis NS, Schaak RE. 2014. Highly active electrocatalysis of the hydrogen evolution reaction by cobalt phosphide nanoparticles. Angew. Chem. Int. Ed. Engl. 53, 5427–5430. ( 10.1002/anie.201402646) [DOI] [PubMed] [Google Scholar]
- 66.McKone JR, Warren EL, Bierman MJ, Boettcher SW, Brunschwig BS, Lewis NS, Gray HB. 2011. Evaluation of Pt, Ni, and Ni-Mo electrocatalysts for hydrogen evolution on crystalline Si electrodes. Energy Environ. Sci. 4, 3573–3583. ( 10.1039/c1ee01488a) [DOI] [Google Scholar]
- 67.Cook TR, Dogutan DK, Reece SY, Surendranath Y, Teets TS, Nocera DG. 2010. Solar energy supply and storage for the legacy and non legacy worlds. Chem. Rev. 110, 6474–6502. ( 10.1021/cr100246c) [DOI] [PubMed] [Google Scholar]
- 68.Grätzel M. 2009. Recent advances in sensitized mesoscopic solar cells. Acc. Chem. Res. 42, 1788–1798. ( 10.1021/ar900141y) [DOI] [PubMed] [Google Scholar]
- 69.Walter MG, Warren EL, McKone JR, Boettcher SW, Mi QX, Santori EA, Lewis NS. 2010. Solar water splitting cells. Chem. Rev. 110, 6446–6473. ( 10.1021/cr1002326) [DOI] [PubMed] [Google Scholar]
- 70.Chen X, Shen S, Guo L, Mao SS. 2010. Semiconductor-based photocatalytic hydrogen generation. Chem. Rev. 110, 6503–6570. ( 10.1021/cr1001645) [DOI] [PubMed] [Google Scholar]
- 71.Hochbaum AI, Yang P. 2010. Semiconductor nanowires for energy conversion. Chem. Rev. 110, 527–546. ( 10.1021/cr900075v) [DOI] [PubMed] [Google Scholar]
- 72.Youngblood WJ, Lee S-HA, Maeda K, Mallouk TE. 2009. Visible light water splitting using dye-sensitized oxide semiconductors. Acc. Chem. Res. 42, 1966–1973. ( 10.1021/ar9002398) [DOI] [PubMed] [Google Scholar]
- 73.Fujishima A, Kohayakawa K, Honda K. 1975. Formation of hydrogen gas with an electrochemical photo-cell. Bull. Chem. Soc. Jpn 48, 1041–1042. ( 10.1246/bcsj.48.1041) [DOI] [Google Scholar]
- 74.Desilvestro J, Grätzel M, Kavan L, Moser J, Augustynski J. 1985. Highly efficient sensitization of titanium dioxide. J. Am. Chem. Soc. 107, 2988–2990. ( 10.1021/ja00296a035) [DOI] [Google Scholar]
- 75.Oregan B, Grätzel M. 1991. A low-cost, high-efficiency solar-cell based on dye-sensitized colloidal TiO2 films. Nature 353, 737–740. ( 10.1038/353737a0) [DOI] [Google Scholar]
- 76.Wrighton MS, Ginley DS, Wolczanski PT, Ellis AB, Morse DL, Linz A. 1975. Photoassisted electrolysis of water by irradiation of a titanium dioxide electrode. Proc. Natl Acad. Sci. USA 72, 1518–1522. ( 10.1073/pnas.72.4.1518) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bard AJ, Fox MA. 1995. Artificial photosynthesis—solar splitting of water to hydrogen and oxygen. Acc. Chem. Res. 28, 141–145. ( 10.1021/ar00051a007) [DOI] [Google Scholar]
- 78.Wrighton MS. 1979. Photoelectrochemical conversion of optical energy to electricity and fuels. Acc. Chem. Res. 12, 303–310. ( 10.1021/ar50141a001) [DOI] [Google Scholar]
- 79.Heller A. 1981. Conversion of sunlight into electrical power and photoassisted electrolysis of water in photoelectrochemical cells. Acc. Chem. Res. 14, 154–162. ( 10.1021/ar00065a004) [DOI] [Google Scholar]
- 80.Borja M, Dutta PK. 1993. Storage of light energy by photoelectron transfer across a sensitized zeolite solution interface. Nature 362, 43–45. ( 10.1038/362043a0) [DOI] [Google Scholar]
- 81.Cho IS, Chen Z, Forman AJ, Kim DR, Rao PM, Jaramillo TF, Zheng X. 2011. Branched TiO2 nanorods for photoelectrochemical hydrogen production. Nano Lett. 11, 4978–4984. ( 10.1021/nl2029392) [DOI] [PubMed] [Google Scholar]
- 82.Oh J, Deutsch TG, Yuan H-C, Branz HM. 2011. Nanoporous black silicon photocathode for H2 production by photoelectrochemical water splitting. Energy Environ. Sci. 4, 1690–1694. ( 10.1039/c1ee01124c) [DOI] [Google Scholar]
- 83.Zang G-L, Sheng G-P, Shi C, Wang Y-K, Li W-W, Yu H-Q. 2014. A bio-photoelectrochemical cell with a MoS3-modified silicon nanowire photocathode for hydrogen and electricity production. Energy Environ. Sci. 7, 3033–3039. ( 10.1039/c4ee00654b) [DOI] [Google Scholar]
- 84.Paracchino A, Laporte V, Sivula K, Graetzel M, Thimsen E. 2011. Highly active oxide photocathode for photoelectrochemical water reduction. Nat. Mater. 10, 456–461. ( 10.1038/nmat3017) [DOI] [PubMed] [Google Scholar]
- 85.Barreca D, Fornasiero P, Gasparotto A, Gombac V, Maccato C, Montini T, Tondello E. 2009. The potential of supported Cu2O and CuO nanosystems in photocatalytic H2 production. ChemSusChem 2, 230–233. ( 10.1002/cssc.200900032) [DOI] [PubMed] [Google Scholar]
- 86.Murray CB, Kagan CR, Bawendi MG. 2000. Synthesis and characterization of monodisperse nanocrystals and close-packed nanocrystal assemblies. Ann. Rev. Mater. Sci. 30, 545–610. ( 10.1146/annurev.matsci.30.1.545) [DOI] [Google Scholar]
- 87.Alivisatos AP. 1996. Perspectives on the physical chemistry of semiconductor nanocrystals. J. Phys. Chem. 100, 13 226–13 239. ( 10.1021/jp9535506) [DOI] [Google Scholar]
- 88.Hahn MA, Tabb JS, Krauss TD. 2005. Detection of single bacterial pathogens with semiconductor quantum dots. Anal. Chem. 77, 4861–4869. ( 10.1021/ac050641i) [DOI] [PubMed] [Google Scholar]
- 89.Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. 2002. In vivo imaging of quantum dots encapsulated in phospholipid micelles. Science 298, 1759–1762. ( 10.1126/science.1077194) [DOI] [PubMed] [Google Scholar]
- 90.Chan WCW, Nie SM. 1998. Quantum dot bioconjugates for ultrasensitive nonisotopic detection. Science 281, 2016–2018. ( 10.1126/science.281.5385.2016) [DOI] [PubMed] [Google Scholar]
- 91.Bruchez M, Moronne M, Gin P, Weiss S, Alivisatos AP. 1998. Semiconductor nanocrystals as fluorescent biological labels. Science 281, 2013–2016. ( 10.1126/science.281.5385.2013) [DOI] [PubMed] [Google Scholar]
- 92.Frame FA, Carroll EC, Larsen DS, Sarahan M, Browning ND, Osterloh FE. 2008. First demonstration of CdSe as a photocatalyst for hydrogen evolution from water under UV and visible light. Chem. Commun. 2206–2208. ( 10.1039/B718796c) [DOI] [PubMed] [Google Scholar]
- 93.Huang Y-X, et al. 2011. A new cathodic electrode deposit with palladium nanoparticles for cost-effective hydrogen production in a microbial electrolysis cell. Int. J. Hydrog. Energy 36, 2773–2776. ( 10.1016/j.ijhydene.2010.11.114) [DOI] [Google Scholar]
- 94.Holmes MA, Townsend TK, Osterloh FE. 2012. Quantum confinement controlled photocatalytic water splitting by suspended CdSe nanocrystals. Chem. Commun. 48, 371–373. ( 10.1039/c1cc16082f) [DOI] [PubMed] [Google Scholar]
- 95.Alivisatos AP. 1996. Semiconductor clusters, nanocrystals, and quantum dots. Science 271, 933–937. ( 10.1126/science.271.5251.933) [DOI] [Google Scholar]
- 96.Han Z, Qiu F, Eisenberg R, Holland PL, Krauss TD. 2012. Robust photogeneration of H2 in water using semiconductor nanocrystals and a nickel catalyst. Science 338, 1321–1324. ( 10.1126/science.1227775) [DOI] [PubMed] [Google Scholar]
- 97.Huang J, Mulfort KL, Du P, Chen LX. 2012. Photodriven charge separation dynamics in CdSe/ZnS core/shell quantum dot/cobaloxime hybrid for efficient hydrogen production. J. Am. Chem. Soc. 134, 16 472–16 475. ( 10.1021/ja3062584) [DOI] [PubMed] [Google Scholar]
- 98.Wang F, Wang WG, Wang XJ, Wang HY, Tung CH, Wu LZ. 2011. A highly efficient photocatalytic system for hydrogen production by a robust hydrogenase mimic in an aqueous solution. Angew. Chem. Int. Ed. Engl. 50, 3193–3197. ( 10.1002/anie.201006352) [DOI] [PubMed] [Google Scholar]
- 99.Lakadamyali F, Kato M, Reisner E. 2012. Colloidal metal oxide particles loaded with synthetic catalysts for solar H2 production. Faraday Discuss. 155, 191–205. ( 10.1039/c1fd00077b) [DOI] [PubMed] [Google Scholar]
- 100.Li Z-J, Li X-B, Wang J-J, Yu S, Li C-B, Tung C-H, Wu L-Z. 2013. A robust ‘artificial catalyst’ in situ formed from CdTe QDs and inorganic cobalt salts for photocatalytic hydrogen evolution. Energy Environ. Sci. 6, 465–469. ( 10.1039/c2ee23898e) [DOI] [Google Scholar]
- 101.Jang JS, Joshi UA, Lee JS. 2007. Solvothermal synthesis of CdS nanowires for photocatalytic hydrogen and electricity production. J. Phys. Chem. C 111, 13 280–13 287. ( 10.1021/jp072683b) [DOI] [Google Scholar]
- 102.Wang G, Yang X, Qian F, Zhang JZ, Li Y. 2010. Double-sided CdS and CdSe quantum dot co-sensitized ZnO nanowire arrays for photoelectrochemical hydrogen generation. Nano Lett. 10, 1088–1092. ( 10.1021/nl100250z) [DOI] [PubMed] [Google Scholar]
- 103.Frank S, Poncharal P, Wang ZL, deHeer WA. 1998. Carbon nanotube quantum resistors. Science 280, 1744–1746. ( 10.1126/science.280.5370.1744) [DOI] [PubMed] [Google Scholar]
- 104.Guldi DM, et al. 2006. Versatile coordination chemistry towards multifunctional carbon nanotube nanohybrids. Chem. Eur. J. 12, 2152–2161. ( 10.1002/chem.200500933) [DOI] [PubMed] [Google Scholar]
- 105.Guldi DM, Rahman GMA, Zerbetto F, Prato M. 2005. Carbon nanotubes in electron donor-acceptor nanocomposites. Acc. Chem. Res. 38, 871–878. ( 10.1021/ar040238i) [DOI] [PubMed] [Google Scholar]
- 106.Sgobba V, Guldi DM. 2009. Carbon nanotubes—electronic/electrochemical properties and application for nanoelectronics and photonics. Chem. Soc. Rev. 38, 165–184. ( 10.1039/B802652C) [DOI] [PubMed] [Google Scholar]
- 107.D'Souza F, Chitta R, Sandanayaka ASD, Subbaiyan NK, D'Souza L, Araki Y, Ito O. 2007. Supramolecular carbon nanotube-fullerene donor-acceptor hybrids for photoinduced electron transfer. J. Am. Chem. Soc. 129, 15 865–15 871. ( 10.1021/ja073773x) [DOI] [PubMed] [Google Scholar]
- 108.Guldi DM, Rahman GMA, Prato M, Jux N, Qin SH, Ford W. 2005. Single-wall carbon nanotubes as integrative building blocks for solar-energy conversion. Angew. Chem. Int. Ed. Engl. 44, 2015–2018. ( 10.1002/anie.200462416) [DOI] [PubMed] [Google Scholar]
- 109.Guldi DM, et al. 2004. Donor-acceptor nanoensembles of soluble carbon nanotubes. Chem. Commun. 2034–2035. ( 10.1039/B406933A) [DOI] [PubMed] [Google Scholar]
- 110.Li Q, Chen L, Lu G. 2007. Visible-light-induced photocatalytic hydrogen generation on dye-sensitized multiwalled carbon nanotube/Pt catalyst. J. Phys. Chem. C 111, 11 494–11 499. ( 10.1021/jp072520n) [DOI] [Google Scholar]
- 111.Le Goff A, Artero V, Jousselme B, Tran PD, Guillet N, Metaye R, Fihri A, Palacin S, Fontecave M. 2009. From hydrogenases to noble metal-free catalytic nanomaterials for H2 production and uptake. Science 326, 1384–1387. ( 10.1126/science.1179773) [DOI] [PubMed] [Google Scholar]
- 112.Croce R, van Amerongen H. 2014. Natural strategies for photosynthetic light harvesting. Nat. Chem. Biol. 10, 492–501. ( 10.1038/nchembio.1555) [DOI] [PubMed] [Google Scholar]
- 113.Barton BE, Whaley CM, Rauchfuss TB, Gray DL. 2009. Nickel-iron dithiolato hydrides relevant to the [NiFe]-hydrogenase active site. J. Am. Chem. Soc. 131, 6942–6943. ( 10.1021/Ja902570u) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Camara JM, Rauchfuss TB. 2011. Mild redox complementation enables H2 activation by [FeFe]-hydrogenase models. J. Am. Chem. Soc. 133, 8098–8101. ( 10.1021/ja201731q) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carroll ME, Barton BE, Gray DL, Mack AE, Rauchfuss TB. 2011. Active-site models for the nickel-iron hydrogenases: effects of ligands on reactivity and catalytic properties. Inorg. Chem. 50, 9554–9563. ( 10.1021/ic2012759) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krauss N. 2001. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature 411, 909–917. ( 10.1038/35082000) [DOI] [PubMed] [Google Scholar]
- 117.Croce R, van Amerongen H. 2013. Light-harvesting in photosystem I. Photosynth. Res. 116, 153–166. ( 10.1007/s11120-013-9838-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Greenbaum E. 1985. Platinized chloroplasts—a novel photocatalytic material. Science 230, 1373–1375. ( 10.1126/science.230.4732.1373) [DOI] [PubMed] [Google Scholar]
- 119.Evans BR, O'Neill HM, Hutchens SA, Bruce BD, Greenbaum E. 2004. Enhanced photocatalytic hydrogen evolution by covalent attachment of plastocyanin to photosystem I. Nano Lett. 4, 1815–1819. ( 10.1021/nl0493388) [DOI] [Google Scholar]
- 120.Utschig LM, Dimitrijevic NM, Poluektov OG, Chemerisov SD, Mulfort KL, Tiede DM. 2011. Photocatalytic hydrogen production from noncovalent biohybrid photosystem I/Pt nanoparticle complexes. J. Phys. Chem. Lett. 2, 236–241. ( 10.1021/jz101728v) [DOI] [Google Scholar]
- 121.Iwuchukwu IJ, Vaughn M, Myers N, O'Neill H, Frymier P, Bruce BD. 2010. Self-organized photosynthetic nanoparticle for cell-free hydrogen production. Nat. Nanotechnol. 5, 73–79. ( 10.1038/nnano.2009.315) [DOI] [PubMed] [Google Scholar]
- 122.Grimme RA, Lubner CE, Bryant DA, Golbeck JH. 2008. Photosystem I/molecular wire/metal nanoparticle bioconjugates for the photocatalytic production of H2. J. Am. Chem. Soc. 130, 6308–6309. ( 10.1021/ja800923y) [DOI] [PubMed] [Google Scholar]
- 123.Utschig LM, Silver SC, Mulfort KL, Tiede DM. 2011. Nature-driven photochemistry for catalytic solar hydrogen production: a photosystem I-transition metal catalyst hybrid. J. Am. Chem. Soc. 133, 16 334–16 337. ( 10.1021/ja206012r) [DOI] [PubMed] [Google Scholar]
- 124.Takahashi S, Badger MR. 2011. Photoprotection in plants: a new light on photosystem II damage. Trends Plant Sci. 16, 53–60. ( 10.1016/j.tplants.2010.10.001) [DOI] [PubMed] [Google Scholar]
- 125.Lubitz W, Ogata H, Ruediger O, Reijerse E. 2014. Hydrogenases. Chem. Rev. 114, 4081–4148. ( 10.1021/cr4005814) [DOI] [PubMed] [Google Scholar]
- 126.Jones AK, Sillery E, Albracht SPJ, Armstrong FA. 2002. Direct comparison of the electrocatalytic oxidation of hydrogen by an enzyme and a platinum catalyst. Chem. Commun. 866–867. ( 10.1039/b201337a) [DOI] [PubMed] [Google Scholar]
- 127.Frey M. 2002. Hydrogenases: hydrogen-activating enzymes. ChemBioChem 3, 153–160. () [DOI] [PubMed] [Google Scholar]
- 128.Evans DJ, Pickett CJ. 2003. Chemistry and the hydrogenases. Chem. Soc. Rev. 32, 268–275. ( 10.1039/b201317g) [DOI] [PubMed] [Google Scholar]
- 129.Cammack R, et al. 2001. Hydrogenases and their activities. In Hydrogen as a fuel: learning from nature (eds Cammack R, Frey M, Robson R.), pp. 73–92, 238–261. London, UK: Taylor & Francis. [Google Scholar]
- 130.Liebgott P-P, et al. 2011. Original design of an oxygen-tolerant NiFe hydrogenase: major effect of a valine-to-cysteine mutation near the active site. J. Am. Chem. Soc. 133, 986–997. ( 10.1021/ja108787s) [DOI] [PubMed] [Google Scholar]
- 131.Bae S, Shim E, Yoon J, Joo H. 2008. Photoanodic and cathodic role of anodized tubular titania in light-sensitized enzymatic hydrogen production. J. Power Sources 185, 439–444. ( 10.1016/j.jpowsour.2008.06.094) [DOI] [Google Scholar]
- 132.Yoon J, Bae S, Shim E, Joo H. 2009. Pyrococcus furiosus-immobilized anodized tubular titania cathode in a hydrogen production system. J. Power Sources 189, 1296–1301. ( 10.1016/j.jpowsour.2008.12.072) [DOI] [Google Scholar]
- 133.Reisner E, Powell DJ, Cavazza C, Fontecilla-Camps JC, Armstrong FA. 2009. Visible light-driven H2 production by hydrogenases attached to dye-sensitized TiO2 nanoparticles. J. Am. Chem. Soc. 131, 18 457–18 466. ( 10.1021/ja907923r) [DOI] [PubMed] [Google Scholar]
- 134.Hambourger M, Gervaldo M, Svedruzic D, King PW, Gust D, Ghirardi M, Moore AL, Moore TA. 2008. FeFe -hydrogenase-catalyzed H2 production in a photoelectrochemical biofuel cell. J. Am. Chem. Soc. 130, 2015–2022. ( 10.1021/ja077691k) [DOI] [PubMed] [Google Scholar]
- 135.Brown KA, Wilker MB, Boehm M, Dukovic G, King PW. 2012. Characterization of photochemical processes for H2 production by CdS nanorod-[FeFe] hydrogenase complexes. J. Am. Chem. Soc. 134, 5627–5636. ( 10.1021/ja2116348) [DOI] [PubMed] [Google Scholar]
- 136.Brown KA, Dayal S, Ai X, Rumbles G, King PW. 2010. Controlled assembly of hydrogenase-CdTe nanocrystal hybrids for solar hydrogen production. J. Am. Chem. Soc. 132, 9672–9680. ( 10.1021/ja101031r) [DOI] [PubMed] [Google Scholar]
- 137.Greene BL, Joseph CA, Maroney MJ, Dyer RB. 2012. Direct evidence of active-site reduction and photodriven catalysis in sensitized hydrogenase assemblies. J. Am. Chem. Soc. 134, 11 108–11 111. ( 10.1021/ja3042367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lubner CE, Knorzer P, Silva PJN, Vincent KA, Happe T, Bryant DA, Golbeck JH. 2010. Wiring an [FeFe]-hydrogenase with photosystem I for light-induced hydrogen production. Biochemistry 49, 10 264–10 266. ( 10.1021/bi1016167) [DOI] [PubMed] [Google Scholar]
- 139.Lubner CE, Applegate AM, Knoerzer P, Ganago A, Bryant DA, Happe T, Golbeck JH. 2011. Solar hydrogen-producing bionanodevice outperforms natural photosynthesis. Proc. Natl Acad. Sci. USA 108, 20 988–20 991. ( 10.1073/pnas.1114660108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bacchi M, et al. 2014. Cobaloxime-based artificial hydrogenases. Inorg. Chem. 53, 8071–8082. ( 10.1021/ic501014c) [DOI] [PubMed] [Google Scholar]
- 141.Ginovska-Pangovska B, Dutta A, Reback ML, Linehan JC, Shaw WJ. 2014. Beyond the active site: the impact of the outer coordination sphere on electrocatalysts for hydrogen production and oxidation. Acc. Chem. Res. 47, 2621–2630. ( 10.1021/ar5001742) [DOI] [PubMed] [Google Scholar]
- 142.Jain A, Lense S, Linehan JC, Raugei S, Cho H, DuBois DL, Shaw WJ. 2011. Incorporating peptides in the outer-coordination sphere of bioinspired electrocatalysts for hydrogen production. Inorg. Chem. 50, 4073–4085. ( 10.1021/ic1025872) [DOI] [PubMed] [Google Scholar]
- 143.Jones AK, Lichtenstein BR, Dutta A, Gordon G, Dutton PL. 2007. Synthetic hydrogenases: incorporation of an iron carbonyl thiolate into a designed peptide. J. Am. Chem. Soc. 129, 14 844–14 845. ( 10.1021/ja075116a) [DOI] [PubMed] [Google Scholar]
- 144.Kleingardner JG, Kandemir B, Bren KL. 2014. Hydrogen evolution from neutral water under aerobic conditions catalyzed by cobalt microperoxidase-11. J. Am. Chem. Soc. 136, 4–7. ( 10.1021/ja406818h) [DOI] [PubMed] [Google Scholar]
- 145.Gralnick JA, Newman DK. 2007. Extracellular respiration. Mol. Microbiol. 65, 1–11. ( 10.1111/j.1365-2958.2007.05778.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Selembo PA, Merrill MD, Logan BE. 2010. Hydrogen production with nickel powder cathode catalysts in microbial electrolysis cells. Int. J. Hydrog. Energy 35, 428–437. ( 10.1016/j.ijhydene.2009.11.014) [DOI] [Google Scholar]
- 147.Lee H-S, Vermaas WFJ, Rittmann BE. 2010. Biological hydrogen production: prospects and challenges. Trends Biotechnol. 28, 262–271. ( 10.1016/j.tibtech.2010.01.007) [DOI] [PubMed] [Google Scholar]
- 148.Cheng S, Logan BE. 2007. Sustainable and efficient biohydrogen production via electrohydrogenesis. Proc. Natl Acad. Sci. USA 104, 18 871–18 873. ( 10.1073/pnas.0706379104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Qian F, Wang G, Li Y. 2010. Solar-driven microbial photoelectrochemical cells with a nanowire photocathode. Nano Lett. 10, 4686–4691. ( 10.1021/n110277n) [DOI] [PubMed] [Google Scholar]
- 150.Wang HY, Qian F, Li Y. 2014. Solar-assisted microbial fuel cells for bioelectricity and chemical fuel generation. Nano Energy 8, 264–273. ( 10.1016/j.nanoen.2014.06.004) [DOI] [Google Scholar]
- 151.Shastik ES, Vokhmyanina DV, Zorin NA, Voronin OG, Karyakin AA, Tsygankov AA. 2011. Demonstration of hydrogenase electrode operation in a bioreactor. Enzyme Microb. Technol. 49, 453–458. ( 10.1016/j.enzmictec.2011.08.007) [DOI] [PubMed] [Google Scholar]
- 152.Faunce T. 2011. Global artificial photosynthesis project: a scientific and legal introduction. J. Law Med. 19, 275–281. [PubMed] [Google Scholar]
- 153.Faunce TA, et al. 2013. Energy and environment policy case for a global project on artificial photosynthesis. Energy Environ. Sci. 6, 695–698. ( 10.1039/c3ee00063j) [DOI] [Google Scholar]