

Abstract
In response to progressive nephron loss, volume and humoral signals in the circulation have increasing relevance. These signals, including plasma sodium, angiotensin II and those related to volume status, activate a slow neuromodulatory pathway within the central nervous system (CNS). The slow CNS pathway includes specific receptors for angiotensin II, mineralocorticoids, and endogenous ouabain (EO). Stimulation of the pathway leads to elevated sympathetic nervous system activity (SNA) and increased circulating EO. The sustained elevation of circulating EO (or ouabain) stimulates central and peripheral mechanisms that amplify the impact of SNA on vascular tone. These include: changes in synaptic plasticity in the brain and sympathetic ganglia that increase preganglionic tone and amplify ganglionic transmission, amplification of the impact of SNA on arterial tone in the vascular wall, and the reprogramming of calcium signaling proteins in arterial myocytes. These increase SNA, raise basal and evoked arterial tone, and elevate blood pressure (BP). In the setting of chronic kidney disease, we suggest that sustained elevation of the slow CNS pathway, plasma EO and the cardiotonic steroid marinobufagenin (MBG), comprise a feed-forward system that raises BP and accelerates kidney and cardiac damage. Block of the slow CNS pathway and/or circulating EO and MBG may reduce BP and slow the progression to end stage renal disease.
Keywords: Ouabain, marinobufagenin, brain, hypertension, renal, dialysis, hormones
Volume Expansion, Natriuretic Hormones and Sodium Pump Inhibitors in Kidney Disease
The kidney is critical to long term salt and water balance. In addition to renal perfusion pressure and an adequate glomerular filtration rate (GFR), salt balance involves well recognized hormones including aldosterone, the atrial peptides and angiotensin II. Moreover, as the number of functional nephrons progressively declines, the behavior of volume regulating hormones and extracellular fluid volume (ECFV) status in general becomes of increasing significance.
A progressive increase in ECFV becomes apparent as the number of filtering nephrons declines to a critically low number and profound adjustments in sodium regulating mechanisms have been suggested. In response to acute or chronic expansion of ECFV, a “third” factor other than aldosterone or GFR (or renal perfusion pressure) is required to explain sodium balance1. Numerous studies showed that the third factor was humoral and it induced natriuresis2. This so called natriuretic hormone (NH) has long been associated with the efferent natriuretic response to elevated plasma and intracerebroventricular sodium as well as intravascular volume expansion3. Hence, a significant portion of the pioneering work in the search for NHs employed human or experimental animals with chronic kidney disease (CKD) in whom ECFV is expanded. Some examples are presented in Table 1. The working assumption was that, in the progression to CKD, circulating factors that inhibit the tubular sodium pump were a compensatory mechanism that might help offset the decline in GFR29–32. Further, elevated levels of an NH might, as a side effect, augment vascular tone and raise blood pressure (BP). Indeed, increased circulating levels of a sodium pump inhibitor were readily detected in low renin (i.e., volume-expanded) hypertension33 and subsequent work demonstrated multiple natriuretic materials in the circulation and urine3. Moreover, in the last decade, several natriuretic materials have been isolated and identified. Some inhibit sodium transport and, like the classic cardiotonic steroids (CTS) ouabain and digoxin, inhibit sodium pumps. Yet other identified materials are natriuretic via secondary transport mechanisms3. Here, we focus on two endogenous CTS implicated in the pathogenesis of CKD and propose a feed-forward mechanism including specific elements by which one or both CTS become elevated in the circulation, raise BP, and promote renal and cardiac damage.
Table 1.
An Overview of Humoral CTS-like Factors in Kidney Failure
| Assay | Sample | Impact of CKF | Impact of Dialysis | Nature of Humoral Factor | Ref |
|---|---|---|---|---|---|
| RBC Na+ or Rb+ transport | Plasma or serum | ↑Inhibition of sodium pump mediated ion flux | Not tested No effect No effect |
Unknown |
4,5 6 7 |
| ↓ Inhibition of sodium pump mediated ion flux | ↓ pump activity | 8 | |||
| ↓ ouabain-insensitive sodium uptake | 9,10 | ||||
|
| |||||
| Thymocyte Na+ Pump Activity | ↑ Inhibition | Not tested | Unknown | 11 | |
|
| |||||
| Aortic Sodium Pump Activity | Hemodialysate | ↑ Inhibition | Not tested | Unknown, low MW | 12 |
|
| |||||
| Na,K-ATPase activity | Peritoneal dialysate, Plasma | ↑ Inhibition* | ↓ | Labile DLIF, Unknown |
13,14 15 |
|
| |||||
| Adipocyte sodium pump activity | Serum | No change | Not tested | Not applicable | 16 |
|
| |||||
| Skeletal myocyte Na+ pump activity | Serum | ↑ Inhibition | Not tested | NEFA? | 16 |
|
| |||||
| 3H-ouabain binding | Not relevant | ↓ Na pump sites adipocytes | Not tested | Not applicable | 16 |
| Plasma | ↑ competition | ↓ | Unknown, not from kidney | 17 | |
|
| |||||
| Digoxin Immunoassay† | Peritoneal dialysate | ↑ cross reactivity | Not stated, present in peritoneal dialysate | Labile DLIF, Unknown | 13,18 |
| Plasma | ↑ cross reactivity | 19 | |||
| Serum | ↑ cross reactivity | ↓ | 8 | ||
| Plasma | Not tested | 14,20–22 | |||
|
| |||||
| Ouabain RIA, LC-MSMS | Plasma | ↑ in plasma | No major effect on immunoreactivity | ir EO EO |
23 |
|
| |||||
| MBG Immunoassay, LC | Plasma | ↑ in plasma * | Not tested | ir MBG | 15 |
|
| |||||
| LC-MSMS for MBG and Telocinobufagin | Plasma | ↑ in plasma | Not tested | MBG, Telocinobufagin | 24 |
In most studies, CKD increases (worsens) the measured parameter. The identity of the measured humoral agents(s) is determined only in studies after 2005 that employ analytical instruments. Few studies report the impact of dialysis. RBC, red blood cell. NEFA, non-esterified fatty acids. RIA, radioimmunoassay. MBG, marinobufagenin. ir, immunoreactivity. LC, liquid chromatography. MS, mass spectrometry.
Effect is reversed by digoxin Fab fragments.
Endogenous Cardiotonic Steroids Implicated in Kidney Failure
Three endogenous CTS (eCTS) of relevance to ECFV balance and long-term BP control have been isolated and identified by analytical means in human plasma and/or urine24,34,35. The identified eCTS are ouabain, marinobufagenin (MBG), and telocinobufagin. In addition, there are strong indications that small amounts of digoxin, in addition to one or more digoxin-immunoreactive materials, are present in the circulation, urine and peritoneal dialysate of humans18,22,25,28,37. The presence of these multiple eCTS in mammals implies different signaling systems and functional effects38–45. The general evidence supporting the biosynthesis of eCTS is clear46–51 even though the specific details of the various pathway(s) remain to be elucidated. Nevertheless, as noted below, elevated circulating and or urinary levels of several eCTS are present in conditions associated with acute or chronic increases in ECFV. Further, these eCTS appear to contribute significantly to the underlying renal and cardiac pathology in renal failure and recent animal studies show that methods to control the negative effects of the eCTS are valuable. Indeed, one or more of these methods may warrant investigation as possible new therapeutic modalities in humans to delay dialysis and/or transplantation.
Endogenous CTS, Kidney Damage and Cardiac Fibrosis
Renal damage
The circulating levels of EO, MBG and telocinobufagin are increased dramatically in virtually all patients undergoing dialysis for kidney disease15,23,24. Further, in the rat, partial nephrectomy raises MBG52 and EO53. There is renal fibrosis associated with elevated MBG54 whereas the sustained elevation of circulating EO or ouabain causes podocyte damage and proteinuria and these effects can be blocked by the ouabain receptor antagonist, rostafuroxin55,56 which blocks ouabain binding to the Na+ pump without causing inhibition of ion transport. Thus far, there appear to be no large differences in circulating EO in CKD patients with or without HTN which may be explained by differences in the types and dosages of therapeutic agents. Further, as noted below, the relationship between EO and BP is complicated by lag phases and there are no studies that address these issues.
The mineralocorticoid receptor (MR) antagonists, long recognized for their beneficial effect in heart failure57 have, together with inhibitors of aldosterone biosynthesis, been suggested as a way to minimize renal fibrosis58,59. Indeed, spironolactone attenuates experimental uremic cardiomyopathy where MBG and EO are elevated60. It has been suggested that the basis for the beneficial effect of spironolactone is not solely related to MR blockade, but may also involve their ability to compete with MBG and EO and possibly other CTS for binding to the sodium pump61,62. Yet other mechanisms have been suggested for the beneficial actions of spironolactone63. Further, when given into the CNS in tiny amounts that have no effect when given peripherally, MR antagonists reduce sympathetic outflow, are antihypertensive64, and have a dramatic beneficial impact on cardiac function in heart failure65. Further, as an MR-dependent pathway in the brain controls circulating EO66, the CNS, not ordinarily recognized as a key factor in CKF, is likely to play an important role in the progression to end-stage renal disease (ESRD). In the latter context, nothing is known about the potential benefits of CNS MR blockade in experimental models of chronic renal failure. However, spironolactone is beneficial for the long-term treatment of resistant hypertension in patients with CKF and may improve kidney function67,68. The improved outcomes are reminiscent of the unanticipated beneficial effect of MR blockers in severe heart failure57; i.e., a condition where ECFV is often expanded, and there are also elevated levels of circulating EO and other CTS69–71.
Cardiac fibrosis
Patients with CKF progressively lose cardiac diastolic function and develop ventricular hypertrophy with various geometric manifestations72. Systolic dysfunction impacts 10–20% of patients with ESRD. It appears that the diastolic dysfunction and ventricular hypertrophy are not simply secondary to the hypertension and anemia observed in CKF73. Partially nephrectomized rats also develop diastolic dysfunction, ventricular hypertrophy, and cardiac fibrosis41,74,75. Further in rats, the prolonged infusion of MBG reproduces many features of uremic cardiomyopathy and MBG per se stimulates collagen formation in cardiac fibroblasts in cell culture41. More significantly, in partially nephrectomized rats, both active and passive immunization against MBG attenuates most of the cardiomyopathy75,76.
Recent insights into how EO raises vascular tone and blood pressure
Hypertension is a frequent and early component in CKF. Expansion of ECFV is associated with hypertension in ~75% of patients with chronic renal failure and typically can be controlled with hemodialysis; i.e., benefit reflects removal of fluid and not dialyzable vasopressor agents. Another significant cause of hypertension in uremic patients is hyperreninemia. The hypertension tends to be more severe, unresponsive to volume manipulation, and likely will require bilateral nephrectomy and/or transplant. There is a clear need for better control of hypertension in CRF. But what are the pressor pathway(s) in the volume- and renin-dependent patients?
EO, MBG and telocinobufagin are three known eCTS that circulate in elevated amounts in patients with CKF23,24,38. Although elevated EO is often observed in conditions where fluid volume is chronically expanded, it does not explain the acute “salt-sensitive” variations in BP that follow short term changes in salt intake77. However, the chronic elevation of EO and MBG typically generate sustained increases in BP in rodents75,78,79. It was initially suggested that the long-term pressor effect of EO involved interactions between the brain, arterial vasculature and the kidneys80. Subsequent studies in rats and transgenic mice have confirmed this hypothesis and elucidated many key events in the pressor mechanism of EO81, and also highlight the numerous gaps in knowledge that remain. The vasopressor effect of EO has acute and chronic facets. The acute pressor effect is generally believed to be mediated by inhibition of the Na+ pump and an indirect action that involves calcium entry mediated via the sodium-calcium exchanger that elevates myogenic and evoked tone82,83. The rise in intracellular calcium triggers increased contraction and, when short-term cardiovascular reflexes are blocked, raises BP. In response to sustained elevation of circulating EO, the chronic pressor effect is maintained by activation of a signaling pathway that upregulates expression of several key ion transport proteins in arterial myocytes. These proteins include the sodium calcium exchanger type 1 (NCX1), the sarcoplasmic reticulum calcium ATPase (SERCA) and the transient potential receptor canonical protein 6 (TRPC6). The upregulation of these proteins in arteries requires sustained occupation by circulating EO of the ouabain binding site on the alpha-2 isoform of the Na+ pump. The long-term binding of EO activates the protein kinase c-SRC and stimulates upregulation of the calcium transport proteins via unknown signaling events.
Further upstream, recent observations show that the CNS can control circulating EO. The CNS has a “slow neuromodulatory pathway”84 whose long term effects on BP and circulating EO can be blocked centrally by antagonists of aldosterone synthesis as well as MR66. The CNS slow pathway is overactive in salt- and volume- as well as angiotensin II-dependent forms of experimental hypertension where EO is elevated, as well as in heart failure64,65,69,70,85,86. The slow pathway components appear to constitute a major mechanism by which BP is elevated in many common disorders. Remarkably, the significance of this CNS pathway in renal failure is unknown. However, because the CNS receives volume, sodium and angiotensin II signals in various phases of CKF, we suggest that this brain pathway is likely to be fundamentally involved in raising BP and affecting progression. The overall pathway is summarized in Figure 1.
Figure 1. Proposed feed-forward pathway that raises vascular resistance and blood pressure in chronic kidney disease.

In CKD, progressive loss of nephrons generates humoral signals that activate a slow neuromodulatory pathway in the brain. The slow pathway raises preganglionic SNA and circulating EO. The sustained elevation of circulating EO (red dashed lines) amplifies ganglionic function [1] and raises postganglionic sympathetic nerve activity86, enhances the effect of sympathetic nerve activity [2] in the vascular wall87, and directly amplifies Ca2+ signaling [3] in arterial myocytes88. Block of angiotensin II receptors, or MR66, or EO in the brain may normalize downstream events. Elements of the pathway have been demonstrated in rodents and portions have been shown in human tissue89. The mechanism by which activation of the CNS slow pathway raises plasma EO may involve ACTH. The relative contribution of adrenal and brain EO to the elevated circulating EO in CKD is not known. Elevated circulating MBG is shown as secondary to volume expansion (no data demonstrate CNS control of MBG); renal ischemia may trigger MBG90. In the scheme presented, sustained increases in MBG and EO elevate BP and also promote renal and cardiac damage that enhances the progression of renal failure. The signaling pathways for the fibrotic effects have been described elsewhere41,91. This feed-forward mechanism provides further stimulus to circulating EO and MBG and may accelerate progression to complete failure. Maneuvers that block EO and MBG ameliorate this cycle of events56,60,75. Yellow horizontal bars: potential sites for acute antagonism by canrenone. White stars: sites of synaptic plasticity. The same overall pressor mechanism may be active among some patients with essential hypertension and in heart failure, and also where there are sustained increases in angiotensin II (e.g., low salt intake92).
Abbreviations: AT1R, angiotensin type I receptor. CKF, chronic kidney failure. CNS, central nervous system. EO, endogenous ouabain, GFR, glomerular filtration rate. MR, mineralocorticoid receptor. MT, myogenic tone. NCX1, sodium calcium exchanger type 1. SERCA, sarcoplasmic reticulum calcium ATPase. SNA, sympathetic nerve activity. TPVR, total peripheral vascular resistance. TRPC6, transient receptor potential cation channel, subfamily C, member 6.
Implications for Therapy
Clearance of Cardiac Glycosides in ESRD
The clearance of cardiac glycosides is one significant determinant of their circulating levels. Although digoxin and digitoxin are cleared primarily by the liver, renal clearance is significant, especially for the more polar digoxin93. The clearance of the highly polar EO is mediated to a large extent by the kidneys.
Various transport mechanisms recover cardiac glycosides from the tubular fluid. At the basolateral membrane of the proximal tubular cell, an organic anion transporting polypeptide (SLCO4C1) mediates recovery of digoxin and ouabain94. In addition, the apical secretion of cardiac glycosides is mediated by the P-glycoprotein (PGP) efflux transporter, a protein encoded by the multidrug resistance 1 gene (MDR1)95. Furthermore, among patients with EH, the MDR1 locus is related to elevated circulating EO and may reflect diminished apical secretion of EO96. In addition, those hypertensives with elevated circulating EO also exhibit estimated glomerular filtration rates (eGFR) that are reduced97.
In terms of cardiac glycoside therapy, exogenous ouabain is no longer widely used. Thus, a primary concern is when the declining renal function will impact circulating EO. Plasma EO rises with the progression of CKF and especially in ESRD. The early rise in EO is evident with stages III and IV (Fig 2) although the impact of the lower GFR is surprisingly modest at this level of impairment. One likely reason is that only a small fraction (<1–3%) of the normal filtered load of EO ordinarily appears in the urine92. Another reason is that as hyperfiltration develops, the increase in proximal tubular flow rate will diminish tubular recovery of EO and offset a large portion of the decline in the filtered load53. However, with the advent of end stage failure and near zero urinary excretion, plasma EO rises substantially into the low nanomolar range23. This scenario, analogous to unwanted and uncontrollable digitalization, is worrisome in terms of the potential for undesirable cardiac and vasopressor effects, and because neither hemodialysis nor peritoneal dialysis are effective in removing EO23,98. Further, among patients with ESRD in dialysis, plasma EO was strongly associated with left ventricular mass and geometry23. This association was independent of arterial pressure and other well-established determinants of left ventricular mass. Thus, in ESRD, left ventricular hypertrophy and high cardiovascular risk, including that from hypertension, represent additional multifactorial problems potentially related to EO.
Figure 2. Circulating EO as a function of estimated GFR in CKD (Stage III to V).
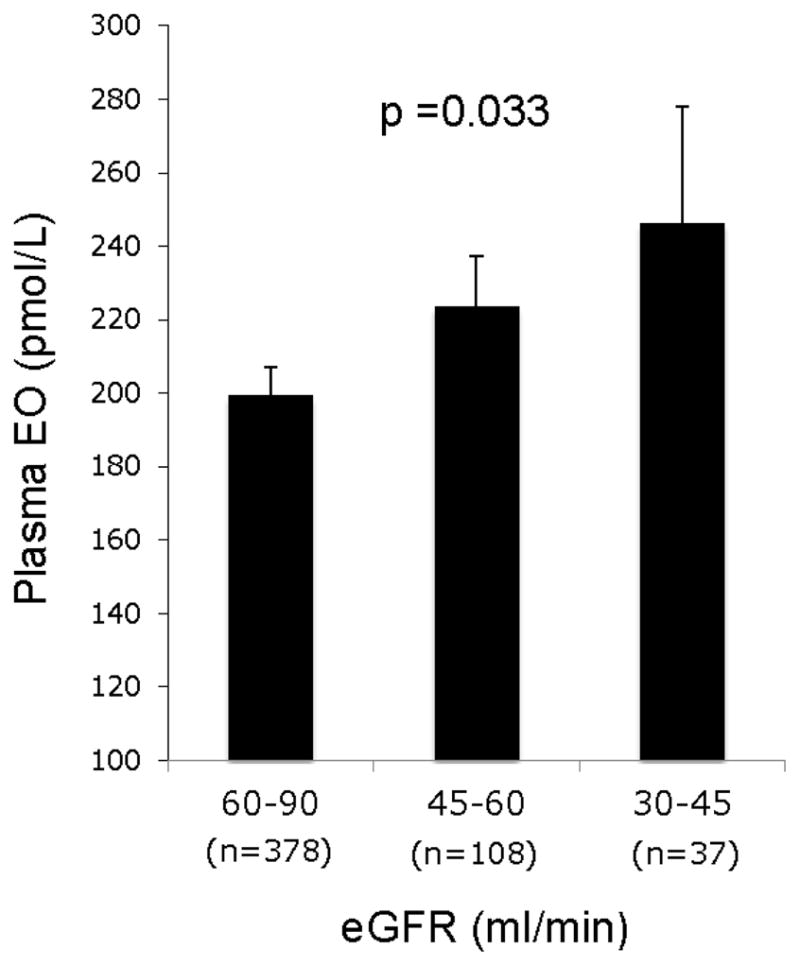
The trend to increasing EO as GFR declines is significant. Simonini and Manunta, unpublished observations.
In addition to raising BP, the elevated EO in kidney failure may directly damage podocytes55. In rats, the chronic elevation of circulating ouabain reduced creatinine clearance, increased urinary protein excretion, and reduced the expression of podocyte nephrin, a selective podocyte marker protein. This last finding was replicated ex vivo by incubating podocyte primary cell cultures with low-dose ouabain. Notably, the ouabain/EO antagonist rostafuroxin prevented the podocyte lesions and proteinuria56. These results suggest that rostafuroxin should be evaluated in patients that have progressive glomerular disease.
EO as a Biomarker for Acute Kidney Injury
The observation that ouabain foments kidney damage in the rat suggested a possible role of EO in acute kidney injury (AKI). Indeed, circulating EO rises with an interesting time course during the induction of anesthesia in patients about to undergo elective cardiac surgery98. In a follow up cross-sectional study, the preoperative plasma levels of EO were measured in 407 patients admitted for cardiac surgery and in a second (validation) population of 219 patients. Among the first group of patients, the incidence of acute kidney injury (2.8%, 8.3%, 20.3%, p<0.001) increased with each incremental preoperative EO tertile. Regression analyses revealed that preoperative circulating EO (and not the peak levels achieved during surgery) was the strongest predictor of AKI and this was confirmed also in the validation cohort. Most recently, we demonstrated the power of two clinical risk score models to predict AKI. Strikingly, the predictive power of both models was further and significantly improved by the addition of preoperative plasma EO levels99. Thus, preoperative EO appears to be a marker for preexisting renal injury and, as shown by the experimental studies, can be a direct cause of kidney damage. Accordingly, we have suggested that among patients that will need cardiac surgery, those with high EO have preexisting (subclinical) renal injury that is likely to be exacerbated by the stress of anesthesia and/or surgery. It appears that it is the combination of these stressors that augments the probability that the patient will develop AKI following cardiac surgery55. Fortunately, methods that lower circulating EO and/or that block its interaction with the Na,K-ATPase are available (see below) for clinical use and these should be tested for their potential to ameliorate or prevent AKI.
Rostafuroxin, a novel antihypertensive compound
Nanomolar concentrations of EO increase renotubular reabsorption of sodium and increase myogenic tone and arterial resistance81,83. Structure-activity relationships indicate that the chronic pressor effect is prominent for ouabain and ouabain-like steroids; the chronic infusion of digitalis preparations including digoxin and digitoxin does not lead to sustained increases in BP. Furthermore, in ouabain-dependent hypertension, the infusion of digitalis CTS such as digoxin or digitoxin normalized blood pressure77 and the effect with digitoxin, a more lipophilic agent likely to penetrate the CNS, was especially marked. Thus, digitalis glycosides have antihypertensive activity under conditions where the circulating levels of EO are elevated. These and other observations100 led to the development of rostafuroxin (formerly PST2238) (17β-(3-furyl)-5β-androstan-3β,14β,17α-triol), a digitoxigenin derivative that displaces ouabain from its binding sites on the Na+ pump (Na,K-ATPase) (IC50 1.7 × 10−6 M) without interacting with other receptors involved in blood pressure regulation or hormonal control101. In ouabain (and EO)-dependent models of hypertension, rostafuroxin dose-dependently (0.1 to 100 μg/kg p.o.,) lowers BP, and normalizes renal Na,K-ATPase activity101. In renal caveolae, subnanomolar ouabain concentrations switch on latent Na,K-ATPase units in the proximal tubular plasma membrane and activate a signaling pathway that involves the tyrosine-phosphorylation of c-Src, transactivation of the epidermal growth factor receptor103 and activation of cytosolic p42/44 MAPKl. Rostafuroxin at nanomolar concentrations antagonizes the ouabain-Na+ pump interaction and normalizes the above signaling events. Moreover, rostafuroxin (PST2238) blocked the ouabain activation of the cSRC-EGFr-ERK pathway and prevented ouabain-induced cardiac and renal hypertrophy103. Rostafuroxin is able to correct the abnormal upregulation of renal Na+ pump number in the plasma membrane without inhibiting renotubular transporters involved in sodium transport, as the diuretics do. The significance of this effect in terms of its ability to reduce BP is not known. It should be recalled that the kidney typically does not affect vascular tone directly. The mechanisms that generate most all of the vascular tone and that are responsible for increases in long-term increases in blood pressure reside in the central nervous system, the adrenal glands and in the vascular wall itself. Further, rostafuroxin blocks the vasopressor effect of ouabain in arteries104 and ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats105. As a digitalis derivative, rostafuroxin is likely (although not yet tested) to be an effective antagonist in the CNS at sites where brain EO augments sympathetic outflow85.
Clinical studies with Rostafuroxin
More than one-third of patients with essential hypertension (EH), most all patients with chronic renal failure, and a large portion of patients with heart failure have increased circulating levels of EO23,69,96,97,106,107. These are primary target populations for rostafuroxin therapy. However, in a trial of unselected patients with EH (i.e., in which ~70% of the patients would not have elevated EO), rostafuroxin had no overall impact on BP108. Instead, the BP lowering activity of rostafuroxin depended on genomic variation in factors related to the synthesis and clearance of EO and to cytoskeletal polymorphisms. In rostafuroxin-sensitive patients, the systolic BP declined 14 mm Hg after 4 weeks of treatment relative to controls108. The gene variants encode enzymes in steroid biosynthesis, transmembrane EO(ouabain) transport, and the cell cytoskeleton. This combination of gene variants occurred in 23% of the 196 (never treated) hypertensives in the study.
The antihypertensive effect of rostafuroxin was tested in never-treated, recently discovered hypertensive patients. The use of naïve patients may be important as even a 4-week period is not sufficient to wash-out the effects of prior treatment,107,109,110. For example, the stimulatory effect of diuretic treatment on the RAAS can last 6 months109,111. Thus, different enrollment criteria are required in pharmacogenomics studies. In addition, as one of us has noted elsewhere104, the maximal dose of rostafuroxin used thus far in clinical studies, even with drug-sensitive patients, may have been suboptimal by at least an order of magnitude or more. Data regarding the impact of rostafuroxin in treating heart failure and the hypertension in patients with end stage renal failure are not available. In the latter settings, circulating EO levels are considerably higher23,69 than in EH, and dramatically larger doses of rostafuroxin than those used in prior trials may be needed to demonstrate efficacy.
In view of the close structural similarity of rostafuroxin with the digitalis CTS, it is of interest that digoxin is associated with increased mortality in ESRD in one study112 but not another113. The potential for life-threatening effects of digitalis preparations is readily apparent in the setting of ESRD. However, rostafuroxin has no overt arrhythmogenic activity101 and thus is worth evaluating for its antihypertensive activity in the setting of CKD and in ESRD.
Volume Management in ESRD, CTS Levels and the Lag Phenomenon
It has been restated persuasively that the effective management of volume to “dry weight” is sufficient to restore normotension in the majority of dialysis patients114,115 and improve survival116. However, overly rapid fluid removal is not invariably synonymous with the restoration of normotension perhaps because it triggers short-term neurohumoral reflexes that attempt to maintain BP. More prolonged dialysis times with less abrupt fluid removal would be expected to minimize short-term reflex sympathetic responses117,118.
To our knowledge, there are no studies that address the impact of defined changes in volume in ESRD on the circulating levels of eCTS and their relationship with BP. And the design and execution of such studies is not trivial for reasons related to clearance and lag effects. For example, in rodents given steady infusions of ouabain, the elevated plasma ouabain takes nearly a week to begin to elevate BP, and several weeks are needed to achieve the maximal pressor effect78. In other words, ouabain (EO) is a slow pressor hormone and, therefore steady-state measurements are needed to reveal the relationship between circulating ouabain (EO) and BP. The slow pressor effect of ouabain involves peripheral and central components. In the periphery, there is functional reprogramming of the sympathetic ganglia and the arterial vasculature (2–3 days in the rat). With regard to the CNS, ouabain enters the brain slowly and is believed to contribute to the EO pool in the slow neuromodulatory pathway (Figure 1); this is thought to drive the sustained increase in sympathetic outflow. When ouabain administration is discontinued, plasma EO normalizes within in hours in the normal rodent119 while it takes ~3–7 days before BP normalizes78,79. Thus, the drop in BP is delayed relative to plasma ouabain and, again, the absence of a steady state obscures the relationship between ouabain (EO) and BP. But how is the delay in the normalization of BP explained, and does it have relevance to the “lag hypothesis” in the setting of the hemodialysis patient?
It is apparent that the slow reversal of ouabain-dependent hypertension is not necessarily explained by the clearance of ouabain from the circulation per se. In normal rats, the half time for the clearance of ouabain (EO) is < 5 hours120, whereas in normal humans it is ~23 hours and ~50 hours in patients with renal failure120,121. As dialysis per se is unable to effectively remove EO from the circulation, we may ask whether dialysis can remove the volume (or other) stimulus to EO secretion and reduce BP by that mechanism? There are numerous considerations: 1). With short-term rapid dialysis, the answer is probably no. Even if we assume that the chronic volume stimulus to EO secretion could be effectively switched off by a single dry weight dialysis, then in patients with renal failure, the decline in circulating EO would be simply too slow to allow for a significant decline in BP through an EO-dependent pressor mechanism. 2). Even presuming hemodialysis as an effective way to remove EO, it is clear based upon the above half times for clearance, that no significant BP drop would be expected for several days and, by then, the patient’s (re)expanded volume and other stimuli to EO secretion would have returned. In other words, in the CKD setting where EO is chronically elevated, probing of its role in the lag phenomenon does not appear to be easily amenable to investigation by manipulation of dialysis protocols per se. 3). In the above context, the ability to specifically and chronically suppress EO secretion and/or physically remove it from the circulation is needed. Much work suggests that the adrenal cortex provides the majority of the circulating EO49 but currently there are no specific ways to block adrenal EO production that do not affect classical corticosteroid pathways. In experimental studies, MR blockers given into the CNS reduce circulating EO66 but this is not feasible in patients. However, another approach that may be practical involves the capture of EO by immobilized Fab fragments (Digifab™) during dialysis122. A well designed cartridge with Fab fragments inserted into the dialysis chain might also be amenable to regeneration and reusability over several weeks in the same patient. 4). The peripheral, and perhaps CNS, actions of EO on Na+ pumps might be blocked by rostafuroxin, but this has yet to be tested in CKD. While canrenone, a metabolite of spironolactone, may already perform a similar function, rostafuroxin may have the advantage in that it does not provoke hyperkalemia. 5). Another more radical approach is to actively immunize patients against EO. As EO is produced and secreted slowly (production is <10,000 times lower than angiotensin II), the production of endogenous antibodies would be more than fast enough to neutralize circulating EO. Indeed, in Dahl salt-sensitive rats where there is a renal abnormality driving chronic sodium retention and volume excess, the slow neuromodulatory pathway is overactive123–127, and active immunity against EO lowers BP significantly128.
Does circulating EO explain the slow decline in BP that accompanies prolonged dry weight dialysis? Figure 7 of Twardowski115 indicates that dry weight dialysis requires ~9 months to normalize BP and that the apparent half-time for the decline in BP is approximately one month. For EO to be the mediator of this prolonged BP lag, we suggest there are two potential mechanisms that may be relevant. In the first mechanism, the chronic elevation of plasma EO (ouabain) in CKD, and by analogy with ouabain administration in experimental studies, is expected to lead to prominent accumulation of this steroid in the hypothalamus, anterior pituitary, and especially the kidney78. Muscle and many another tissues (with the exception of the adrenal glands) also take up this CTS. Thus, a significant body burden of EO is likely in CKD. Once inside cells, the half times for the turnover of EO (ouabain) are slow, ranging from ~20 hr ex vivo to 4 weeks in vivo129,130. Turnover of body EO “stores” is likely to be especially slow in humans because the volume of distribution for EO (ouabain) is larger than rats. Thus, the BP lag might readily be explained by the slow loss of accumulated intracellular EO to the circulation and its subsequent clearance. As noted in point 3 above, the impact of increasing EO clearance on the lag time could be tested clinically. A second distinct mechanism for the lag phenomenon may involve the plasticity of synaptic connections within the CNS. In response to CKD, the prolonged activation of the electrical elements in the slow neuromodulatory pathway (Figure 1) will likely promote formation of new synaptic connections consistent with Hebbian theory131. Thus, in addition to pathway activation by signals from the renal failure, new synaptic connections will likely reinforce downstream signaling events (i.e., increased sympathetic outflow and plasma EO). When the stimulus to the slow neuromodulatory pathway is removed by sustained dry weight dialysis, electrical activity in the pathway will decline but may not normalize until such time as synaptic pruning has reversed the new connections. Changes in synaptic plasticity occur in key slow pathway nuclei during hypertension132–134 as well as in the peripheral sympathetic ganglia86,135. The full reversal of these changes, and the impact of their electrical consequences on BP, may take several months.
But which of these two possibilities is more likely? Both appear to have general kinetic behavior that is quantitatively relevant to the lag phenomenon. However, there are some paradoxes. In normal humans, reducing total body sodium with diuretics raises EO92. This physiological increase in RAAS, EO, and activation of the slow neurohumoral pathway may help to support BP when total body sodium and/or sodium intake is low. Thus, the initial impact of dry weight dialysis seems more likely to us to stimulate the slow pathway, adding to the ongoing chronic CNS activation (stress), synaptic rewiring136, sympathetic activity, and circulating EO. The predicted time course of these factors would seem to fit the initial changes in total peripheral resistance according to Figure 3 of Twardowski115.
During chronic dry weight dialysis, removal of the volume stimulus to the slow CNS pathway would permit sympathetic activity and EO to decline secondary to changes in synaptic activity and wiring. If the above-noted ideas are correct, circulating EO and BP (i.e., indices of slow pathway activity) should decline more-or-less in parallel over many months of sustained dry weight dialysis. The importance of the slow neurohumoral pathway and synaptic plasticity is shown by the dramatic impact of hypothalamic lesions. Lesions that impact slow pathway nuclei prevent or reverse renal137–140, salt and volume-dependent141–143, and ouabain144 forms of experimental hypertension. Thus, it is difficult to avoid the conclusion that the CNS exerts a dominant role in determining whether major deficits in renal function, sodium handling and volume excess can raise BP and sustain hypertension. Furthermore, it seems likely that altered synaptic activity and wiring have a fundamental influence on the lag phenomenon in CKD.
ESRD, Resistant Hypertension (rHT), Mineralocorticoid Receptor (MR) Antagonists and Renal Denervation
As defined by the eighth Joint National Committee (JNC 8), rHT is the inability to achieve a BP lower than 140/90 mm Hg despite optimal doses of 3 or more antihypertensive drugs, including a diuretic109. The prevalence of rHT varies from 10% to 15% among treated hypertensive patients145 after exclusion of pseudo-resistant hypertension from non-adherence to medications or from the white coat syndrome. The etiology of rHT typically involves multiple factors, including obesity, older age, high dietary salt, renal artery stenosis, chronic kidney disease, and aldosterone excess146. Classical primary aldosteronism and lesser degrees of aldosterone excess, possibly originating from visceral adipocytes, contribute to rHT. Furthermore, many hypertensives with hyperaldosteronism have elevated circulating levels of EO.104
MR blockers decrease proteinuria in CKD subjects147 but their use in patients with low or minimal filtration is limited by the risk of life-threatening hyperkalemia. Novel non-steroidal antagonists with high affinity and selectivity for the MR receptor (BR4628 and SM368229) are being developed 148,149 but it is not yet clear if these drugs will avoid the negative impact on plasma potassium. MR antagonists are particularly effective for treatment of rHT regardless of circulating aldosterone levels. This raises the obvious question: what is their mechanism of action? Spironolactone and its major bioactive metabolite, canrenone, block aldosterone and cortisol binding to MR yet it is unlikely that their antihypertensive effect is mediated by renal MR in dialysis patients with rHT simply because tubular reabsorption becomes increasingly less relevant in the face of ever diminishing filtration. Therefore, powerful extrarenal sites of action of MR blockers are required. Instillation of MR blockers into the CNS, in amounts too small to have any peripheral effect, lowers sympathetic activity, plasma renin activity, BP, and reduces end organ damage in many experimental models of cardiovascular disease150–151. Further, the block of brain ouabain has correspondingly large effects in heart failure154–156 and appear to be mediated via slow pathway effects that are also amenable to central MR blockers125. The slow pathway is chronically activated by a number of signals (plasma angiotensin II, plasma sodium, volume), one or more of which are abnormal in CKD. It is important to note that the slow neuromodulatory pathway differs from the rapidly acting neural networks that govern the sympathetic response to flight or fight in that it is much slower to activate and deactivate, and that it involves a number of neuroendocrine elements (Ang II receptors, aldosterone generation, MR, epithelial sodium channels and EO) that are likely arrayed in a functional sequence in the CNS. Activation of the slow pathway by Ang II leads to sustained increases in circulating EO and BP, and all effects are blocked by central aldosterone antagonists66. Thus, the beneficial effect of MR antagonists in many patients with rHT is most likely related to block of slow CNS pathway effects (Figure 1).
In addition, to targeting MR, spironolactone and canrenone compete with EO (ouabain) and eCTS steroids for binding to Na+ pumps. One of the first clues that canrenone was a CTS receptor antagonist was the ability of therapeutic doses to reduce the effect of digitalis and ouabain on the heart157–162. Subsequent work showed canrenone suppressed ouabain binding to the Na+ pump163,164 and was antihypertensive in some experimental models of hypertension where ouabain-like factors were implicated165–168. In therapeutically relevant concentrations, canrenone blocked the effect of ouabain on basal aortic tone77 and myogenic tone in pressured mesenteric arteries169.
MR blockers also reduce the impact of EO in heart failure and severe hypertension.67–69 and reduce the fibrotic effect of MBG70. In other words, the clinical efficacy of MR blockers likely benefits from their polyspecific actions. Based on available dose-response relationships, and the therapeutic circulating levels of MR blockers (1–5 μM), their BP lowering effects in rHT are likely mediated by block of MR and/or competition with central and/or peripheral eCTS for binding to Na+ pumps. The negative inotropic effects of canrenone on cardiac and vascular contractility are due to block of L-type Ca2+ channels170,171. These latter effects occur at concentrations ~50–200 fold higher than those achieved in routine therapy and seem unlikely to be of clinical relevance.
There are no reports on the impact of renal denervation on EO or MBG in CKD. Only a minority of patients with apparent rHT may be eligible; the most frequent cause of ineligibility (46.9%) is BP normalization following optimization of therapy that included low-dose spironolactone172.
Muscle fatigue in CKF and ESRD a role for eCTS?
In both heart failure and CRD, the fatigability of skeletal muscle is increased173,174. The underlying mechanisms are unclear but may be common to both conditions. Elevated EO is present in both CKD and heart failure and it is of interest that ouabain induces muscle fatigue175,176. In skeletal muscle, the highly ouabain (and EO) sensitive α-2 isoform of the Na+ pump is expressed in the t-tubular system where it influences Ca2+ handling. Reduced expression (activity) of this isoform in the heart or skeletal muscle (i.e., mimicking the functional effects of the high EO state in CKD) leads to hypocontractility and the early onset of fatigue. Although the nephrologist is not ordinarily concerned with skeletal muscle performance in CKD, MR blockers and other agents that block eCTS may have the added benefit of improving exercise performance/tolerance in the kidney disease setting.
Summary
The brain, heart, circulation, and kidneys of patients with CKD are exposed to a potentially devastating mixture of neural and humoral agents, many of which appear to be activated by volume and other signals generated by, or in response to, poor kidney function. In the circulation alone, there is a toxic cocktail comprised of elevated levels of RAAS components, EO, MBG, endothelin, inhibitors of nitric oxide generation, and others. Carefully crafted, as well as bold and imaginative clinical and experimental studies are needed to test fundamental ideas and concepts in CKD, including the roles of the CNS and eCTS, and the potential impact of anti eCTS therapies.
Clinical Summary.
The failing kidney enhances signals which activate a slow neurohumoral pathway in the brain that, in turn, controls the long term level (i.e., non-reflexive) of sympathetic nerve activity and the circulating level of endogenous ouabain.
Inappropriate activation of the brain pathway raises sympathetic activity, circulating endogenous ouabain and blood pressure, and all these effects are prevented by agents that block the binding of angiotensin II, mineralocorticoids, and endogenous ouabain with their respective receptors in the brain and periphery.
In patients with end stage renal failure, two endogenous cardiotonic steroids (endogenous ouabain and marinobufagenin) circulate in elevated and clinically significant amounts and exacerbate cardiac hypertrophy and renal fibrosis. It may be possible to specifically remove these materials during dialysis.
The “lag phenomenon”, i.e., the delayed return of blood pressure following dialysis to dry weight, may be related to plasticity changes in the brain and periphery, i.e., the slow rewiring of synaptic connections in the CNS and/or sympathetic ganglia that reduce sympathetic outflow to the vasculature.
Research directed to improving therapy for chronic renal failure should consider the role of the CNS, humoral cardiotonic steroid mediators, and test methods that chronically reduce or neutralize these steroids.
Acknowledgments
Supported in part by USPHS Grant HL107555.
Footnotes
Conflict of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Wardener HE, Mills IH, Clapham WF, Hayter CJ. Studies on the efferent mechanism of the sodium diuresis which follows the administration of intravenous saline in the dog. Clin Sci. 1961;21:249–58. [PubMed] [Google Scholar]
- 2.Cort JH, Lichardus B. The natriuretic activity of jugular vein blood during carotid occlusion. Physiol Bohemoslov. 1963;12:497–501. [PubMed] [Google Scholar]
- 3.Hamlyn JM. Natriuretic hormones, endogenous ouabain and related sodium transport inhibitors. Frontiers in Endocrinology. 2014 Dec 03; doi: 10.3389/fendo.2014.00199. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kariya K, Sano H, Yamanishi J, Saito K, Furuta Y, Fukuzaki H. A circulating Na+-K+ATPase inhibitor, erythrocyte sodium transport and hypertension in patients with chronic renal failure. Clin Exp Hypertens A. 1986;8(2):167–83. doi: 10.3109/10641968609074770. [DOI] [PubMed] [Google Scholar]
- 5.Gambhir KK, Parui R, Agarwal V, Cruz I. The effect of hemodialysis on the transport of sodium in erythrocytes from chronic renal failure patients maintained on hemodialysis. Life Sci. 2002;71(14):1615–21. doi: 10.1016/s0024-3205(02)01855-6. [DOI] [PubMed] [Google Scholar]
- 6.Prasad R, Mond R, Jain S, Kaur G, Chugh KS. Modulation of ouabain sensitive sodium potassium pump of erythrocytes from patients with chronic renal failure: role of acute hemodialysis. Biochem Mol Biol Int. 1996;40(6):1087–94. doi: 10.1080/15216549600201723. [DOI] [PubMed] [Google Scholar]
- 7.Fervenza FC, Hendry BM, Ellory JC. Effects of dialysis and transplantation on red cell Na pump function in renal failure. Nephron. 1989;53(2):121–8. doi: 10.1159/000185723. [DOI] [PubMed] [Google Scholar]
- 8.Czarkowski M, Ignatowska-Switalska H, Jabłońska-Skwiecińska E, Drygieniec D, Wardyn K, Rojek-Trebicka J, Staniszewska K, Chodakowska J. Effect of hemodialysis on the level of serum digoxin-like substance and sodium-potassium pump activity in erythrocytes from patients with chronic renal failure. Pol Arch Med Wewn. 1992;87(6):325–31. [PubMed] [Google Scholar]
- 9.Cumberbatch M, Morgan DB. Relations between sodium transport and sodium concentration in human erythrocytes in health and disease. Clin Sci (Lond) 1981;60(5):555–64. doi: 10.1042/cs0600555. [DOI] [PubMed] [Google Scholar]
- 10.Thomas TH, Mansy H, Wilkinson R. Erythrocyte sodium and sodium flux in relation to hypertension in chronic renal failure. Nephrol Dial Transplant. 1989;4(1):21–6. [PubMed] [Google Scholar]
- 11.Greiber S, O’Neill WC, Mitch WE. Impaired cation transport in thymocytes of rats with chronic uremia includes the Na+/H+ antiporter. J Am Soc Nephrol. 1995;5(9):1689–96. doi: 10.1681/ASN.V591689. [DOI] [PubMed] [Google Scholar]
- 12.Sohn HJ, Stokes GS, Johnston H. An Na,K-ATPase inhibitor from ultrafiltrate obtained by hemodialysis of patients with uremia. J Lab Clin Med. 1992;120(2):264–71. [PubMed] [Google Scholar]
- 13.Tao QF, Soszynski PA, Hollenberg NK, Graves SW. Specificity of the volume-sensitive sodium pump inhibitor isolated from human peritoneal dialysate in chronic renal failure. Kidney Int. 1996;49(2):420–9. doi: 10.1038/ki.1996.61. [DOI] [PubMed] [Google Scholar]
- 14.Okamoto S. Endogenous digitalis-like substance and Na-K-ATPase inhibitor in cardiovascular and renal disease. Nihon Naibunpi Gakkai Zasshi. 1988;64(1):39–50. doi: 10.1507/endocrine1927.64.1_39. [DOI] [PubMed] [Google Scholar]
- 15.Kolmakova EV, Haller ST, Kennedy DJ, Isachkina AN, Budny GV, Frolova EV, Piecha G, Nikitina ER, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY. Endogenous cardiotonic steroids in chronic renal failure. Nephrol Dial Transplant. 2011;26(9):2912–9. doi: 10.1093/ndt/gfq772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Druml W, Kelly RA, May RC, Mitch WE. Abnormal cation transport in uremia. Mechanisms in adipocytes and skeletal muscle from uremic rats. J Clin Invest. 1988;81(4):1197–203. doi: 10.1172/JCI113435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deray G, Pernollet MG, Devynck MA, Zingraff J, Touam A, Rosenfeld J, Meyer P. Plasma digitalislike activity in essential hypertension or end-stage renal disease. Hypertension. 1986;8(7):632–8. doi: 10.1161/01.hyp.8.7.632. [DOI] [PubMed] [Google Scholar]
- 18.Graves SW, Glatter KA, Lazarus JM, Williams GH, Hollenberg NK. Volume expansion in renal failure patients: a paradigm for a clinically relevant [Na,K]ATPase inhibitor. J Cardiovasc Pharmacol. 1993;22 (Suppl 2):S54–7. [PubMed] [Google Scholar]
- 19.Weinberg U, Dolev S, Werber MM, Shapiro MS, Shilo L, Shenkman L. Identification and preliminary characterization of two human digitalis-like substances that are structurally related to digoxin and ouabain. Biochem Biophys Res Commun. 1992;188(3):1024–9. doi: 10.1016/0006-291x(92)91334-m. [DOI] [PubMed] [Google Scholar]
- 20.Bisordi JE, Holt S. Digitalislike immunoreactive substances and extracellular fluid volume status in chronic hemodialysis patients. Am J Kidney Dis. 1989;13(5):396–403. doi: 10.1016/s0272-6386(89)80023-x. [DOI] [PubMed] [Google Scholar]
- 21.Vasdev S, Johnson E, Longerich L, Prabhakaran VM, Gault MH. Plasma endogenous digitalis-like factors in healthy individuals and in dialysis-dependent and kidney transplant patients. Clin Nephrol. 1987;27(4):169–74. [PubMed] [Google Scholar]
- 22.Graves SW, Brown B, Valdes R., Jr An endogenous digoxin-like substance in patients with renal impairment. Ann Intern Med. 1983;99(5):604–8. doi: 10.7326/0003-4819-99-5-604. [DOI] [PubMed] [Google Scholar]
- 23.Stella P, Manunta P, Mallamaci F, Melandri M, Spotti D, Tripepi G, Hamlyn JM, Malatino LS, Bianchi G, Zoccali C. Endogenous ouabain and cardiomyopathy in dialysis patients. J Intern Med. 2008;263(3):274–80. doi: 10.1111/j.1365-2796.2007.01883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, Takahashi H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem. 2005;38(1):36–45. doi: 10.1016/j.clinbiochem.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Valdes R., Jr Endogenous digoxin-like immunoreactive factors: impact on digoxin measurements and potential physiological implications. Clin Chem. 1985;31(9):1525–32. [PubMed] [Google Scholar]
- 26.Witherspoon L, Shuler S, Alyea K, Figueroa J, Neely H. Digoxin-like substance in term pregnancy, newborns, and renal failure. J Nucl Med. 1986;27(9):1418–22. [PubMed] [Google Scholar]
- 27.Witherspoon L, Shuler S, Neely H, Sonnemaker R, Alyea K, Gilbert S. Digoxin radioimmunoassay that does not detect digoxin-like substance in serum of newborns, infants, or patients with renal failure. Clin Chem. 1987;33(3):420. [PubMed] [Google Scholar]
- 28.Naomi S, Graves S, Lazarus M, Williams GH, Hollenberg NK. Variation in apparent serum digitalis-like factor levels with different digoxin antibodies. The “immunochemical fingerprint”. Am J Hypertension. 1991;4(10 Pt 1):795–801. doi: 10.1093/ajh/4.10.795. [DOI] [PubMed] [Google Scholar]
- 29.Kramer HJ, Pennig J, Klingmüller D, Kipnowski J, Glänzer K, Düsing R. Digoxin-like immunoreacting substance(s) in the serum of patients with chronic uremia. Nephron. 1985;40(3):297–302. doi: 10.1159/000183482. [DOI] [PubMed] [Google Scholar]
- 30.Bourgoignie JJ, Weisser F, Rolf D, Klahr S, Bricker NS. Demonstration of a low molecular weight natriuretic factor in uremic serum. Trans Assoc Am Physicians. 1970;83:277–287. [PubMed] [Google Scholar]
- 31.Kaplan MA, Bourgoignie JJ, Rosecan J, Bricker NS. The effects of the natriuretic factor from uremic urine on sodium transport, water and electrolyte content, and pyruvate oxidation by the isolated toad bladder. J Clin Invest. 1974;53(6):1568–77. doi: 10.1172/JCI107707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt RW, Bourgoignie JJ, Bricker NS. On the adaptation in sodium excretion in chronic uremia. The effects of “proportional reduction” of sodium intake. J Clin Invest. 1974;53(6):1736–41. doi: 10.1172/JCI107725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bricker NS. On the pathogenesis of the uremic state. An exposition of the “trade-off hypothesis”. N Engl J Med. 1972;286(20):1093–9. doi: 10.1056/NEJM197205182862009. [DOI] [PubMed] [Google Scholar]
- 34.Haddy FJ, Overbeck HW. The role of humoral agents in volume expanded hypertension. Life Sci. 1976;19(7):935–47. doi: 10.1016/0024-3205(76)90284-8. [DOI] [PubMed] [Google Scholar]
- 35.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci U S A. 1991;88(14):6259–63. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagrov AY, Fedorova OV, Dmitrieva RI, Howald WN, Hunter AP, Kuznetsova EA, Shpen VM. Characterization of a urinary bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension. 1998;31(5):1097–103. doi: 10.1161/01.hyp.31.5.1097. [DOI] [PubMed] [Google Scholar]
- 37.Harris DW, Clark MA, Fisher JF, Hamlyn JM, Kolbasa KP, Ludens JH, DuCharme DW. Development of an immunoassay for endogenous digitalislike factor. Hypertension. 1991;17(6 Pt 2):936–43. doi: 10.1161/01.hyp.17.6.936. [DOI] [PubMed] [Google Scholar]
- 38.Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev. 2009;61(1):9–38. doi: 10.1124/pr.108.000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zulian A, Linde CI, Pulina MV, Baryshnikov SG, Papparella I, Hamlyn JM, Golovina VA. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca2+ signaling in arterial smooth muscle cells. Am J Physiol Cell Physiol. 2013;304(4):C324–33. doi: 10.1152/ajpcell.00337.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldstein I, Lerer E, Laiba E, Mallet J, Mujaheed M, Laurent C, Rosen H, Ebstein RP, Lichtstein D. Association between sodium- and potassium-activated adenosine triphosphatase alpha isoforms and bipolar disorders. Biol Psychiatry. 2009;65(11):985–91. doi: 10.1016/j.biopsych.2008.10.033. [DOI] [PubMed] [Google Scholar]
- 41.Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S, Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D, Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension. 2007;49(1):215–24. doi: 10.1161/01.HYP.0000252409.36927.05. [DOI] [PubMed] [Google Scholar]
- 42.Dvela-Levitt M, Cohen-Ben Ami H, Rosen H, Ornoy A, Hochner-Celnikier D, Granat M, Lichtstein D. Reduction in Maternal Circulating Ouabain Impairs Offspring Growth and Kidney Development. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2014020130. ASN.2014020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lichtstein D, Gati I, Samuelov S, Berson D, Rozenman Y, Landau L, Deutsch J. Identification of digitalis-like compounds in human cataractous lenses. Eur J Biochem. 1993;216(1):261–8. doi: 10.1111/j.1432-1033.1993.tb18141.x. [DOI] [PubMed] [Google Scholar]
- 44.Puschett JB, Agunanne E, Uddin MN. Emerging role of the bufadienolides in cardiovascular and kidney diseases. Am J Kidney Dis. 2010;56(2):359–70. doi: 10.1053/j.ajkd.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 45.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol. 2007;293(2):C509–36. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]
- 46.Boulanger BR, Lilly MP, Hamlyn JM, Laredo J, Shurtleff D, Gann DS. Ouabain is secreted by the adrenal gland in awake dogs. Am J Physiol. 1993;264(3 Pt 1):E413–9. doi: 10.1152/ajpendo.1993.264.3.E413. [DOI] [PubMed] [Google Scholar]
- 47.Laredo J, Hamilton BP, Hamlyn JM. Ouabain is secreted by bovine adrenocortical cells. Endocrinology. 1994;135(2):794–7. doi: 10.1210/endo.135.2.8033829. [DOI] [PubMed] [Google Scholar]
- 48.Hamlyn JM, Laredo J, Shah JR, Lu ZR, Hamilton BP. 11-hydroxylation in the biosynthesis of endogenous ouabain: multiple implications. Ann N Y Acad Sci. 2003;986:685–93. doi: 10.1111/j.1749-6632.2003.tb07283.x. [DOI] [PubMed] [Google Scholar]
- 49.Manunta P, Ferrandi M, Bianchi G, Hamlyn JM. Endogenous ouabain in cardiovascular function and disease. J Hypertension. 2009;27(1):9–18. doi: 10.1097/HJH.0b013e32831cf2c6. [DOI] [PubMed] [Google Scholar]
- 50.Lichtstein D, Steinitz M, Gati I, Samuelov S, Deutsch J, Orly J. Biosynthesis of digitalis-like compounds in rat adrenal cells: hydroxycholesterol as possible precursor. Life Sci. 1998;62(23):2109–26. doi: 10.1016/s0024-3205(98)00186-6. [DOI] [PubMed] [Google Scholar]
- 51.Qazzaz HM, Cao Z, Bolanowski DD, Clark BJ, Valdes R., Jr De novo biosynthesis and radiolabeling of mammalian digitalis-like factors. Clin Chem. 2004;50(3):612–20. doi: 10.1373/clinchem.2003.022715. [DOI] [PubMed] [Google Scholar]
- 52.Gonick HC, Ding Y, Vaziri ND, Bagrov AY, Fedorova OV. Simultaneous measurement of marinobufagenin, ouabain, and hypertension-associated protein in various disease states. Clin Exp Hypertension. 1998;20(5–6):617–27. doi: 10.3109/10641969809053240. [DOI] [PubMed] [Google Scholar]
- 53.Harwood S, Mullen AM, McMahon AC, Dawnay A. Plasma OLC is elevated in mild experimental uremia but is not associated with hypertension. Am J Hypertens. 2001;14(11 Pt 1):1112–5. doi: 10.1016/s0895-7061(01)02219-1. [DOI] [PubMed] [Google Scholar]
- 54.Fedorova LV, Raju V, El-Okdi N, Shidyak A, Kennedy DJ, Vetteth S, Giovannucci DR, Bagrov AY, Fedorova OV, Shapiro JI, Malhotra D. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: implication of epithelial-to-mesenchymal transition. Am J Physiol Renal Physiol. 2009;296(4):F922–34. doi: 10.1152/ajprenal.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bignami E, Casamassima N, Frati E, Lanzani C, Corno L, Alfieri O, Gottlieb S, Simonini M, Shah KB, Mizzi A, Messaggio E, Zangrillo A, Ferrandi M, Ferrari P, Bianchi G, Hamlyn JM, Manunta P. Preoperative endogenous ouabain predicts acute kidney injury in cardiac surgery patients. Crit Care Med. 2013;41(3):744–55. doi: 10.1097/CCM.0b013e3182741599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferrandi M, Molinari I, Rastaldi MP, Ferrari P, Bianchi G, Manunta P. Rostafuroxin protects from podocyte injury and proteinuria induced by adducin genetic variants and ouabain. J Pharmacol Exp Ther. 2014;351(2):278–87. doi: 10.1124/jpet.114.217133. [DOI] [PubMed] [Google Scholar]
- 57.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341(10):709–17. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 58.Epstein M. Aldosterone blockade: an emerging strategy for abrogating progressive renal disease. Am J Med. 2006;119(11):912–9. doi: 10.1016/j.amjmed.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 59.Namsolleck P, Unger T. Aldosterone synthase inhibitors in cardiovascular and renal diseases. Nephrol Dial Transplant. 2014;29 (Suppl 1):i62–i68. doi: 10.1093/ndt/gft402. [DOI] [PubMed] [Google Scholar]
- 60.Tian J, Shidyak A, Periyasamy SM, Haller S, Taleb M, El-Okdi N, Elkareh J, Gupta S, Gohara S, Fedorova OV, Cooper CJ, Xie Z, Malhotra D, Bagrov AY, Shapiro JI. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension. 2009;54(6):1313–20. doi: 10.1161/HYPERTENSIONAHA.109.140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Finotti P, Palatini P. Canrenone as a partial agonist at the digitalis receptor site of sodium-potassium-activated adenosine triphosphatase. J Pharmacol Exp Ther. 1981;217(3):784–90. [PubMed] [Google Scholar]
- 62.Sorrentino R, Cirino G, Calignano A, Mancuso F, Sorrentino L, Andriuoli G, Pinto A. Increase in the basal tone of guinea pig thoracic aorta induced by ouabain is inhibited by spironolactone canrenone and potassium canrenoate. J Cardiovasc Pharmacol. 1996;28(4):519–25. doi: 10.1097/00005344-199610000-00007. [DOI] [PubMed] [Google Scholar]
- 63.Oka M, Manku MS. Spironolactone inhibits vascular reactivity by a prostaglandin related mechanism unconnected with aldosterone. Prostaglandins Med. 1981;7(4):305–19. doi: 10.1016/0161-4630(81)90135-x. [DOI] [PubMed] [Google Scholar]
- 64.Leenen FH. Actions of circulating angiotensin II and aldosterone in the brain contributing to hypertension. Am J Hypertension. 2014;27(8):1024–32. doi: 10.1093/ajh/hpu066. [DOI] [PubMed] [Google Scholar]
- 65.Huang BS, Leenen FH. The brain renin-angiotensin-aldosterone system: a major mechanism for sympathetic hyperactivity and left ventricular remodeling and dysfunction after myocardial infarction. Curr Heart Fail Rep. 2009;6(2):81–8. doi: 10.1007/s11897-009-0013-9. [DOI] [PubMed] [Google Scholar]
- 66.Hamlyn JM, Linde CI, Gao J, Huang BS, Golovina VA, Blaustein MP, Leenen FH. Neuroendocrine Humoral and Vascular Components in the Pressor Pathway for Brain Angiotensin II: A New Axis in Long Term Blood Pressure Control. PLOS ONE. 2014;9(9):e108916. doi: 10.1371/journal.pone.0108916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pisoni R, Acelajado MC, Cartmill FR, Dudenbostel T, Dell’Italia LJ, Cofield SS, Oparil S, Calhoun DA. Long-term effects of aldosterone blockade in resistant hypertension associated with chronic kidney disease. J Hum Hypertension. 2012;26(8):502–6. doi: 10.1038/jhh.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006;70(12):2116–23. doi: 10.1038/sj.ki.5001854. [DOI] [PubMed] [Google Scholar]
- 69.Gottlieb SS, Rogowski AC, Weinberg M, Krichten CM, Hamilton BP, Hamlyn JM. Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation. 1992;86(2):420–5. doi: 10.1161/01.cir.86.2.420. [DOI] [PubMed] [Google Scholar]
- 70.Pitzalis MV, Hamlyn JM, Messaggio E, Iacoviello M, Forleo C, Romito R, de Tommasi E, Rizzon P, Bianchi G, Manunta P. Independent and incremental prognostic value of endogenous ouabain in idiopathic dilated cardiomyopathy. Eur J Heart Fail. 2006;8(2):179–86. doi: 10.1016/j.ejheart.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 71.Bagrov AY, Kuznetsova EA, Fedorova OV. Endogenous digoxin-like factor in acute myocardial infarction. J Intern Med. 1994;235(1):63–7. doi: 10.1111/j.1365-2796.1994.tb01033.x. [DOI] [PubMed] [Google Scholar]
- 72.Mohmand B, Malhotra DK, Shapiro JI. Uremic cardiomyopathy: role of circulating digitalis-like substances. Front Biosci. 2005;10:2036–2044. doi: 10.2741/1679. [DOI] [PubMed] [Google Scholar]
- 73.Middleton RJ, Parfrey PS, Foley RN. Left ventricular hypertrophy in the renal patient. J Am Soc Nephrol. 2001;12(5):1079–84. doi: 10.1681/ASN.V1251079. [DOI] [PubMed] [Google Scholar]
- 74.Kennedy D, Omran E, Periyasamy SM, Nadoor J, Priyadarshi A, Willey JC, Malhotra D, Xie Z, Shapiro JI. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J Am Soc Nephrol. 2003;14(1):90–7. doi: 10.1097/01.asn.0000037403.95126.03. [DOI] [PubMed] [Google Scholar]
- 75.Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY, Shapiro JI. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47(3):488–95. doi: 10.1161/01.HYP.0000202594.82271.92. [DOI] [PubMed] [Google Scholar]
- 76.Haller ST, Kennedy DJ, Shidyak A, Budny GV, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am J Hypertension. 2012;25(6):690–6. doi: 10.1038/ajh.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hamlyn JM, Manunta P. Endogenous ouabain: a link between sodium intake and hypertension. Curr Hypertens Rep. 2011;13(1):14–20. doi: 10.1007/s11906-010-0161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Manunta P, Rogowski AC, Hamilton BP, Hamlyn JM. Ouabain induced hypertension in the rat: relationships among circulating and tissue ouabain and blood pressure. J Hypertension. 1994;12:549–560. [PubMed] [Google Scholar]
- 79.Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res. 2000;23(Suppl):S77–85. doi: 10.1291/hypres.23.supplement_s77. [DOI] [PubMed] [Google Scholar]
- 80.Hamlyn JM, Hamilton BP, Manunta P. Endogenous ouabain, sodium balance and blood pressure: a review and a hypothesis. J Hypertension. 1996;14(2):151–67. doi: 10.1097/00004872-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 81.Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J, Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol. 2012;302(5):H1031–49. doi: 10.1152/ajpheart.00899.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat Med. 2004;10(11):1193–9. doi: 10.1038/nm1118. [DOI] [PubMed] [Google Scholar]
- 83.Zhang J, Hamlyn JM, Karashima E, Raina H, Mauban JR, Izuka M, Berra-Romani R, Zulian A, Wier WG, Blaustein MP. Low-dose ouabain constricts small arteries from ouabain-hypertensive rats: implications for sustained elevation of vascular resistance. Am J Physiol Heart Circ Physiol. 2009;297(3):H1140–50. doi: 10.1152/ajpheart.00436.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leenen FH. The central role of the brain aldosterone-“ouabain” pathway in salt-sensitive hypertension. Biochim Biophys Acta. 2010;1802(12):1132–9. doi: 10.1016/j.bbadis.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 85.Huang BS, Ahmadi S, Ahmad M, White RA, Leenen FH. Central neuronal activation and pressor responses induced by circulating ANG II: role of the brain aldosterone-“ouabain” pathway. Am J Physiol Heart Circ Physiol. 2010;299(2):H422–30. doi: 10.1152/ajpheart.00256.2010. [DOI] [PubMed] [Google Scholar]
- 86.Aileru AA, De Albuquerque A, Hamlyn JM, Manunta P, Shah JR, Hamilton MJ, Weinreich D. Synaptic plasticity in sympathetic ganglia from acquired and inherited forms of ouabain-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2001;281(2):R635–44. doi: 10.1152/ajpregu.2001.281.2.R635. [DOI] [PubMed] [Google Scholar]
- 87.Raina H, Zhang Q, Rhee AY, Pallone TL, Wier WG. Sympathetic nerves and the endothelium influence the vasoconstrictor effect of low concentrations of ouabain in pressurized small arteries. Am J Physiol Heart Circ Physiol. 2010;298(6):H2093–101. doi: 10.1152/ajpheart.01045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;298(1):H263–74. doi: 10.1152/ajpheart.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Linde CI, Antos LK, Golovina VA, Blaustein MP. Nanomolar ouabain increases NCX1 expression and enhances Ca2+ signaling in human arterial myocytes: a mechanism that links salt to increased vascular resistance? Am J Physiol Heart Circ Physiol. 2012;303(7):H784–94. doi: 10.1152/ajpheart.00399.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tian J, Haller S, Periyasamy S, Brewster P, Zhang H, Adlakha S, Fedorova OV, Xie ZJ, Bagrov AY, Shapiro JI, Cooper CJ. Renal ischemia regulates marinobufagenin release in humans. Hypertension. 2010;56(5):914–9. doi: 10.1161/HYPERTENSIONAHA.110.155564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xie JX, Shapiro AP, Shapiro JI. The Trade-Off between Dietary Salt and Cardiovascular Disease; A Role for Na/K-ATPase Signaling? Front Endocrinol (Lausanne) 2014;5:97. doi: 10.3389/fendo.2014.00097. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manunta P, Hamilton BP, Hamlyn JM. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am J Physiol Regul Integr Comp Physiol. 2006;290(3):R553–9. doi: 10.1152/ajpregu.00648.2005. [DOI] [PubMed] [Google Scholar]
- 93.Vöhringer HF, Rietbrock N. Digitalis therapy in renal failure with special regard to digitoxin. Int J Clin Pharmacol Ther Toxicol. 1981;19(4):175–84. [PubMed] [Google Scholar]
- 94.Mikkaichi T, Suzuki T, Onogawa T, Tanemoto M, Mizutamari H, Okada M, Chaki T, Masuda S, Tokui T, Eto N, Abe M, Satoh F, Unno M, Hishinuma T, Inui K, Ito S, Goto J, Abe T. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci USA. 2004;101:3569–3574. doi: 10.1073/pnas.0304987101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Inui KI, Masuda S, Saito H. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 2000;58:944–958. doi: 10.1046/j.1523-1755.2000.00251.x. [DOI] [PubMed] [Google Scholar]
- 96.Tripodi G, Citterio L, Kouznetsova T, Lanzani C, Florio M, Modica R, Messaggio E, Hamlyn JM, Zagato L, Bianchi G, Staessen JA, Manunta P. Steroid biosynthesis and renal excretion in human essential hypertension: association with blood pressure and endogenous ouabain. Am J Hypertension. 2009;22(4):357–63. doi: 10.1038/ajh.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manunta P, Hamlyn JM, Simonini M, Messaggio E, Lanzani C, Bracale M, Argiolas G, Casamassima N, Brioni E, Glorioso N, Bianchi G. Endogenous ouabain and the renin-angiotensin-aldosterone system: distinct effects on Na handling and blood pressure in human hypertension. J Hypertension. 2011;29(2):349–56. doi: 10.1097/HJH.0b013e32833ea821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bignami E, Casamassima N, Frati E, Messaggio E, Corno L, Zangrillo A, Manunta P. Endogenous Ouabain Changes Rapidly During Cardiac Pulmonary by Pass. Journal of Steroids & Hormonal Science. 2011 http://dx.doi.org/10.4172/2157-7536.S3-002.
- 99.Simonini M, Lanzani C, Bignami E, Casamassima N, Frati E, Meroni R, Messaggio E, Alfieri O, Hamlyn J, Body SC, Collard CD, Zangrillo A, Manunta P CABG Genomics Investigators. A new clinical multivariable model that predicts postoperative acute kidney injury: impact of endogenous ouabain. Nephrol Dial Transplant. 2014;29(9):1696–701. doi: 10.1093/ndt/gfu200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ferrari P, Torielli L, Ferrandi M, Padoani G, Duzzi L, Florio M, Conti F, Melloni P, Vesci L, Corsico N, Bianchi G. PST2238: a new antihypertensive compound that antagonizes the long-term pressor effect of ouabain. J Pharmacol Exp Ther. 1998;285(1):83–94. [PubMed] [Google Scholar]
- 101.Quadri L, Bianchi G, Cerri A, Fedrizzi G, Ferrari P, Gobbini M, Melloni P, Sputore S, Torri M. 17 beta-(3-furyl)-5 beta-androstane-3 beta, 14 beta, 17 alpha-triol (PST 2238). A very potent antihypertensive agent with a novel mechanism of action. J Med Chem. 1997;40(11):1561–4. doi: 10.1021/jm970162e. [DOI] [PubMed] [Google Scholar]
- 102.Wang H, Haas M, Liang M, Cai T, Tian J, Li S, Xie Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem. 2004;279:17250–9. doi: 10.1074/jbc.M313239200. [DOI] [PubMed] [Google Scholar]
- 103.Ferrandi M, Molinari I, Barassi P, Minotti E, Bianchi G, Ferrari P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J Biol Chem. 2004;279(32):33306–14. doi: 10.1074/jbc.M402187200. [DOI] [PubMed] [Google Scholar]
- 104.Song H, Karashima E, Hamlyn JM, Blaustein MP. Ouabain-digoxin antagonism in rat arteries and neurones. J Physiol. 2014;592(Pt 5):941–69. doi: 10.1113/jphysiol.2013.266866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wenceslau CF, Rossoni LV. Rostafuroxin ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats: the role of Na+K+-ATPase/cSRC pathway. J Hypertension. 2014;32(3):542–54. doi: 10.1097/HJH.0000000000000059. [DOI] [PubMed] [Google Scholar]
- 106.Rossi G, Manunta P, Hamlyn JM, Pavan E, De Toni R, Semplicini A, Pessina AC. Immunoreactive endogenous ouabain in primary aldosteronism and essential hypertension: relationship with plasma renin, aldosterone and blood pressure levels. J Hypertension. 1995;13(10):1181–91. doi: 10.1097/00004872-199510000-00013. [DOI] [PubMed] [Google Scholar]
- 107.Lanzani C, Citterio L, Glorioso N, Manunta P, Tripodi G, Salvi E, Carpini SD, Ferrandi M, Messaggio E, Staessen JA, Cusi D, Macciardi F, Argiolas G, Valentini G, Ferrari P, Bianchi G. Adducin- and ouabain-related gene variants predict the antihypertensive activity of rostafuroxin, part 2: clinical studies. Science Transl Med. 2010;2(59):59ra87. doi: 10.1126/scitranslmed.3001814. [DOI] [PubMed] [Google Scholar]
- 108.Staessen JA, Thijs L, Stolarz-Skrzypek K, Bacchieri A, Barton J, Espositi ED, de Leeuw PW, Dłużniewski M, Glorioso N, Januszewicz A, Manunta P, Milyagin V, Nikitin Y, Souček M, Lanzani C, Citterio L, Timio M, Tykarski A, Ferrari P, Valentini G, Kawecka-Jaszcz K, Bianchi G. Main results of the Ouabain and Adducin for Specific Intervention on Sodium in Hypertension Trial (OASIS-HT): a randomized placebo-controlled phase-2 dose-finding study of rostafuroxin. Trials. 2011;12:13. doi: 10.1186/1745-6215-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Swart S, Bing RF, Swales JD, Thurston H. Plasma renin in long-term diuretic treatment of hypertension: Effect of discontinuation and restarting therapy. Clin Sci. 1982;63:121–5. doi: 10.1042/cs0630121. [DOI] [PubMed] [Google Scholar]
- 110.Paull JR, Widdop RE. Persistent cardiovascular effects of chronic renin-angiotensin system inhibition following withdrawal in adult spontaneously hypertensive rats. J Hypertens. 2001;19:1393–1402. doi: 10.1097/00004872-200108000-00007. [DOI] [PubMed] [Google Scholar]
- 111.Bangalore S, Gong Y, Cooper-DeHoff RM, Pepine CJ, Messerli FH. 2014 Eighth Joint National Committee panel recommendation for blood pressure targets revisited: results from the INVEST study. J Am Coll Cardiol. 2014;64(8):784–93. doi: 10.1016/j.jacc.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chan KE, Lazarus JM, Hakim RM. Digoxin associates with mortality in ESRD. J Am Soc Nephrol. 2010;21(9):1550–9. doi: 10.1681/ASN.2009101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Testani JM, Brisco MA, Tang WH, Kimmel SE, Tiku-Owens A, Forfia PR, Coca SG. Potential effects of digoxin on long-term renal and clinical outcomes in chronic heart failure. J Card Fail. 2013;19(5):295–302. doi: 10.1016/j.cardfail.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Khosla UM, Johnson RJ. Hypertension in the hemodialysis patient and the “lag phenomenon”: insights into pathophysiology and clinical management. Am J Kidney Dis. 2004;43(4):739–51. doi: 10.1053/j.ajkd.2003.12.036. [DOI] [PubMed] [Google Scholar]
- 115.Twardowski ZJ. Sodium, hypertension, and an explanation of the “lag phenomenon” in hemodialysis patients. Hemodialysis Int. 2008;12:412–425. doi: 10.1111/j.1542-4758.2008.00304.x. [DOI] [PubMed] [Google Scholar]
- 116.Shaldon S. Beneficial effect of strict volume control on blood pressure and mortality in patients on hemodialysis. Nature Reviews Nephrology. 2007;3:130–131. doi: 10.1038/ncpneph0403. [DOI] [PubMed] [Google Scholar]
- 117.Chazot C, Jean G. The advantages and challenges of increasing the duration and frequency of maintenance dialysis sessions. Nat Clin Pract Nephrol. 2009;5(1):34–44. doi: 10.1038/ncpneph0979. [DOI] [PubMed] [Google Scholar]
- 118.Charra B. From adequate to optimal dialysis long 3 × 8 hr dialysis: a reasonable compromise. Nefrologia. 2005;25 (Suppl 2):19–24. [PubMed] [Google Scholar]
- 119.Yuan CM, Manunta P, Hamlyn JM, Chen S, Bohen E, Yeun J, Haddy FJ, Pamnani MB. Long-term ouabain administration produces hypertension in rats. Hypertension. 1993;22(2):178–87. doi: 10.1161/01.hyp.22.2.178. [DOI] [PubMed] [Google Scholar]
- 120.Selden R, Margolies MN, Smith TW. Renal and gastrointestinal excretion of ouabain in dog and man. J Pharmacol Exp Ther. 1974;188(3):615–23. [PubMed] [Google Scholar]
- 121.Selden R, Haynie G. Ouabain plasma level kinetics and removal by dialysis in chronic renal failure. A study in fourteen patients. Ann Intern Med. 1975 Jul;83(1):15–9. doi: 10.7326/0003-4819-83-1-15. [DOI] [PubMed] [Google Scholar]
- 122.Pullen MA, Harpel MR, Danoff TM, Brooks DP. Comparison of non-digitalis binding properties of digoxin-specific Fabs using direct binding methods. J Immunol Methods. 2008;336(2):235–41. doi: 10.1016/j.jim.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 123.Gomez-Sanchez EP, Gomez-Sanchez CM, Plonczynski M, Gomez-Sanchez CE. Aldosterone synthesis in the brain contributes to Dahl salt-sensitive rat hypertension. Exp Physiol. 2010;95(1):120–30. doi: 10.1113/expphysiol.2009.048900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol. 1992;262(1 Pt 1):E96–9. doi: 10.1152/ajpendo.1992.262.1.E96. [DOI] [PubMed] [Google Scholar]
- 125.Leenen FH. The central role of the brain aldosterone-“ouabain” pathway in salt-sensitive hypertension. Biochim Biophys Acta. 2010;1802(12):1132–9. doi: 10.1016/j.bbadis.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 126.Budzikowski AS, Huang BS, Leenen FH. Brain “ouabain”, a neurosteroid, mediates sympathetic hyperactivity in salt-sensitive hypertension. Clin Exp Hypertens. 1998;20(2):119–40. doi: 10.3109/10641969809053211. [DOI] [PubMed] [Google Scholar]
- 127.Huang BS, White RA, Jeng AY, Leenen FH. Role of central nervous system aldosterone synthase and mineralocorticoid receptors in salt-induced hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2009;296(4):R994–R1000. doi: 10.1152/ajpregu.90903.2008. [DOI] [PubMed] [Google Scholar]
- 128.Gomez-Sanchez EP, Gomez-Sanchez CE, Fort C. Immunization of Dahl SS/jr rats with an ouabain conjugate mitigates hypertension. Am J Hypertens. 1994;7(7 Pt 1):591–6. doi: 10.1093/ajh/7.7.591. [DOI] [PubMed] [Google Scholar]
- 129.Lamb JF, Ogden P. Internalization of ouabain and replacement of sodium pumps in the plasma membranes of HeLa cells following block with cardiac glycosides. Q J Exp Physiol. 1982;67(1):105–19. doi: 10.1113/expphysiol.1982.sp002605. [DOI] [PubMed] [Google Scholar]
- 130.Kitano S, Morimoto S, Nishibe A, Fukuo K, Hirotani A, Nakahashi T, Yasuda O, Ogihara T. Exogenous ouabain is accumulated in the adrenals and mimics the kinetics of endogenous digitalis-like factor in rats. Hypertens Res. 1998;21(1):47–56. doi: 10.1291/hypres.21.47. [DOI] [PubMed] [Google Scholar]
- 131.Hebb DO. The Organization of Behavior. New York: Wiley & Sons; 1949. p. 335. [Google Scholar]
- 132.Li DP, Yang Q, Pan HM, Pan HL. Plasticity of pre- and postsynaptic GABAB receptor function in the paraventricular nucleus in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295(2):H807–15. doi: 10.1152/ajpheart.00259.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brooks VL, Haywood JR, Johnson AK. Translation of salt retention to central activation of the sympathetic nervous system in hypertension. Clin Exp Pharmacol Physiol. 2005;32(5–6):426–32. doi: 10.1111/j.1440-1681.2005.04206.x. [DOI] [PubMed] [Google Scholar]
- 134.Theodosis DT, Poulain DA. Neuronal-glial and synaptic plasticity in the adult rat paraventricular nucleus. Brain Res. 1989;484(1–2):361–6. doi: 10.1016/0006-8993(89)90382-x. [DOI] [PubMed] [Google Scholar]
- 135.Alkadhi KA, Otoom SA, Tanner FL, Sockwell D, Hogan YH. Inhibition of ganglionic long-term potentiation decreases blood pressure in spontaneously hypertensive rats. Exp Biol Med (Maywood) 2001;226(11):1024–30. doi: 10.1177/153537020122601109. [DOI] [PubMed] [Google Scholar]
- 136.Nathan PJ, Cobb SR, Lu B, Bullmore ET, Davies CH. Studying synaptic plasticity in the human brain and opportunities for drug discovery. Curr Opin Pharmacol. 2011;11(5):540–8. doi: 10.1016/j.coph.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 137.Songu-Mize E, Bealer SL, Caldwell RW. Effect of anteroventral third ventricle lesions on vascular sodium-pump activity in two-kidney Goldblatt hypertension. Hypertension. 1983;5(2 Pt 2):I89–93. doi: 10.1161/01.hyp.5.2_pt_2.i89. [DOI] [PubMed] [Google Scholar]
- 138.Johnson AK, Buggy J, Fink GD, Brody MJ. Prevention of renal hypertension and of the central pressor effect of angiotensin by ventromedial hypothalamic ablation. Brain Res. 1981;205(2):255–64. doi: 10.1016/0006-8993(81)90337-1. [DOI] [PubMed] [Google Scholar]
- 139.Marson O, Saragoça MA, Ribeiro AB, Bossolan D, Tufik S, Ramos OL. Anteroventral third ventricle and renin-angiotensin system interaction in the two-kidney, one clip hypertensive rat. Hypertension. 1983;5(6 Pt 3):V90–3. doi: 10.1161/01.hyp.5.6_pt_3.v90. [DOI] [PubMed] [Google Scholar]
- 140.Buggy J, Huot S, Pamnani M, Haddy F. Periventricular forebrain mechanisms for blood pressure regulation. Fed Proc. 1984;43(1):25–31. [PubMed] [Google Scholar]
- 141.Azar S, Ernsberger P, Livingston S, Azar P, Iwai J. Paraventricular--suprachiasmatic lesions prevent salt-induced hypertension in Dahl rats. Clin Sci (Lond) 1981;61 (Suppl 7):49s–51s. doi: 10.1042/cs061049s. [DOI] [PubMed] [Google Scholar]
- 142.Ernsberger P, Azar S, Azar P. The role of the anteromedial hypothalamus in Dahl hypertension. Brain Res Bull. 1985;15(6):651–6. doi: 10.1016/0361-9230(85)90214-x. [DOI] [PubMed] [Google Scholar]
- 143.Songu-Mize E, Bealer SL, Caldwell RW. Effect of AV3V lesions on development of DOCA-salt hypertension and vascular Na+-pump activity. Hypertension. 1982;4(5):575–80. doi: 10.1161/01.hyp.4.5.575. [DOI] [PubMed] [Google Scholar]
- 144.Veerasingham SJ, Leenen FH. Ouabain- and central sodium-induced hypertension depend on the ventral anteroventral third ventricle region. Am J Physiol. 1999;276(1 Pt 2):H63–70. doi: 10.1152/ajpheart.1999.276.1.H63. [DOI] [PubMed] [Google Scholar]
- 145.Persell Stephen D. Prevalence of Resistant Hypertension in the United States, 2003–2008. Hypertension. 2011;57(6):1076–80. doi: 10.1161/HypertensionAHA.111.170308.. [DOI] [PubMed] [Google Scholar]
- 146.Calhoun David A. Hyperaldosteronism as a Common Cause of Resistant Hypertension. Annual Review of Medicine. 2013;64(1):233–47. doi: 10.1146/annurev-med-042711-135929.. [DOI] [PubMed] [Google Scholar]
- 147.Chrysostomou A, Becker G. Spironolactone in addition to ACE inhibition to reduce proteinuria in patients with chronic renal disease. N Engl J Med. 2001;345:925–6. doi: 10.1056/NEJM200109203451215. [DOI] [PubMed] [Google Scholar]
- 148.Fagart J, Hillisch A, Huyet J, Barfacker L, Fay M, Pleiss U, Pook E, Schafer S, Rafestin-Oblin ME, Kolkhof P. A new mode of mineralocorticoid receptor antagonism by a potent and selective nonsteroidal molecule. J Biol Chem. 2010;285:29932–40. doi: 10.1074/jbc.M110.131342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nariai T, Fujita K, Mori M, Katayama S, Hori S, Matsui K. SM-368229, a novel selective and potent non-steroidal mineralocorticoid receptor antagonist with strong urinary Na+ excretion activity. J Pharmacol Sci. 2011;115:346–53. doi: 10.1254/jphs.10285fp. [DOI] [PubMed] [Google Scholar]
- 150.Gabor A, Leenen FH. Mechanisms in the PVN mediating local and central sodium-induced hypertension in Wistar rats. Am J Physiol Regul Integr Comp Physiol. 2009;296(3):R618–30. doi: 10.1152/ajpregu.90417.2008. [DOI] [PubMed] [Google Scholar]
- 151.Huang BS, White RA, Ahmad M, Leenen FH. Role of brain corticosterone and aldosterone in central angiotensin II-induced hypertension. Hypertension. 2013;62(3):564–71. doi: 10.1161/HYPERTENSIONAHA.113.01557. [DOI] [PubMed] [Google Scholar]
- 152.Gabor A, Leenen FH. Central mineralocorticoid receptors and the role of angiotensin II and glutamate in the paraventricular nucleus of rats with angiotensin II-induced hypertension. Hypertension. 2013;61(5):1083–90. doi: 10.1161/HYPERTENSIONAHA.111.00797. [DOI] [PubMed] [Google Scholar]
- 153.Kang YM, Zhang ZH, Johnson RF, Yu Y, Beltz T, Johnson AK, Weiss RM, Felder RB. Novel effect of mineralocorticoid receptor antagonism to reduce proinflammatory cytokines and hypothalamic activation in rats with ischemia-induced heart failure. Circ Res. 2006;99(7):758–66. doi: 10.1161/01.RES.0000244092.95152.86. [DOI] [PubMed] [Google Scholar]
- 154.Huang BS, Yuan B, Leenen FH. Chronic blockade of brain “ouabain” prevents sympathetic hyper-reactivity and impairment of acute baroreflex resetting in rats with congestive heart failure. Can J Physiol Pharmacol. 2000;78(1):45–53. doi: 10.1139/cjpp-78-1-45. [DOI] [PubMed] [Google Scholar]
- 155.Leenen FH, Yuan B, Huang BS. Brain “ouabain” and angiotensin II contribute to cardiac dysfunction after myocardial infarction. Am J Physiol. 1999;277(5 Pt 2):H1786–92. doi: 10.1152/ajpheart.1999.277.5.H1786. [DOI] [PubMed] [Google Scholar]
- 156.Leenen FH, Huang BS, Yu H, Yuan B. Brain ‘ouabain’ mediates sympathetic hyperactivity in congestive heart failure. Circ Res. 1995;77(5):993–1000. doi: 10.1161/01.res.77.5.993. [DOI] [PubMed] [Google Scholar]
- 157.Selye H, Krajny M, Savoie L. Digitoxin poisoning: prevention by spironolactone. Science. 1969;164(3881):842–3. doi: 10.1126/science.164.3881.842. [DOI] [PubMed] [Google Scholar]
- 158.Schaumann W, Koch K, Mattern HR. Extrarenal actions of aldosterone antagonists in guinea pigs. II. Reduction of the toxicity of beta-methyl-digoxin. Naunyn Schmiedebergs Arch Pharmacol. 1973;279(1):9–18. doi: 10.1007/BF00502063. [DOI] [PubMed] [Google Scholar]
- 159.Yeh BK, Sung PK. Prevention and abolition of ouabain-induced ventricular tachycardia by potassium canrenoate in dogs. Eur J Pharmacol. 1972;19(2):151–5. doi: 10.1016/0014-2999(72)90002-7. [DOI] [PubMed] [Google Scholar]
- 160.deGuzman NT, Yeh BK. Potassium canrenoate in the treatment of long-term digoxin-induced arrhythmias in conscious dogs. Am J Cardiol. 1975;35(3):413–20. doi: 10.1016/0002-9149(75)90035-1. [DOI] [PubMed] [Google Scholar]
- 161.Yeh BK, Chiang BN, Sung PK. Antiarrhythmic activity of potassium canrenoate in man. Am Heart J. 1976;92(3):308–14. doi: 10.1016/s0002-8703(76)80112-3. [DOI] [PubMed] [Google Scholar]
- 162.Vassallo PF, Stefanon I, Rossoni LV, França A, Vassallo DV. Small doses of canrenone block the effects of ouabain on the mechanical activity of the heart and vessels of the rat. J Cardiovasc Pharmacol. 1998;32(5):679–85. doi: 10.1097/00005344-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 163.Finotti P, Palatini P. Canrenone as a partial agonist at the digitalis receptor site of sodium-potassium-activated adenosine triphosphatase. J Pharmacol Exp Ther. 1981;217(3):784–90. [PubMed] [Google Scholar]
- 164.Balzan S, Nicolini G, Bellitto L, Ghione S, Biver P, Montali U. Effect of canrenone on the digitalis site of Na+/K(+)-ATPase in human placental membranes and in erythrocytes. J Cardiovasc Pharmacol. 2003;42(1):32–6. doi: 10.1097/00005344-200307000-00005. [DOI] [PubMed] [Google Scholar]
- 165.de Mendonça M, Grichois ML, Pernollet MG, Wauquier I, Trouillet-Thormann B, Meyer P, Devynck MA, Garay R. Antihypertensive effect of canrenone in a model where endogenous ouabain-like factors are present. J Cardiovasc Pharmacol. 1988;11(1):75–83. doi: 10.1097/00005344-198801000-00012. [DOI] [PubMed] [Google Scholar]
- 166.Preuss HG, al Karadaghi P, Yousufi A, MacArthy P. Effects of canrenone on RRM-sucrose hypertension in WKY. Clin Exp Hypertens A. 1991;13(5):917–23. doi: 10.3109/10641969109042097. [DOI] [PubMed] [Google Scholar]
- 167.Semplicini A, Serena L, Valle R, Ceolotto G, Felice M, Fontebasso A, Pessina AC. Ouabain-inhibiting activity of aldosterone antagonists. Steroids. 1995;60(1):110–3. doi: 10.1016/0039-128x(94)00005-w. [DOI] [PubMed] [Google Scholar]
- 168.Pamnani MB, Whitehorn WV, Clough DL, Haddy FJ. Effects of canrenone on blood pressure in rats with reduced renal mass. Am J Hypertens. 1990;3(3):188–95. doi: 10.1093/ajh/3.3.188. [DOI] [PubMed] [Google Scholar]
- 169.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP. Sodium pump alpha2 subunits control myogenic tone and blood pressure in mice. J Physiol. 2005;569(Pt 1):243–56. doi: 10.1113/jphysiol.2005.091801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cargnelli G, Trevisi L, Debetto P, Luciani S, Bova S. Effects of canrenone on aorta and right ventricle of the rat. J Cardiovasc Pharmacol. 2001;37(5):540–7. doi: 10.1097/00005344-200105000-00006. [DOI] [PubMed] [Google Scholar]
- 171.Costa AR, Torres LB, Medei E, Ricardo RA, França JP, Smaili S, Nascimento JH, Oshiro ME, Bassani JW, Ferreira AT, Tucci PJ. The negative inotropic action of canrenone is mediated by L-type calcium current blockade and reduced intracellular calcium transients. Br J Pharmacol. 2009;158(2):580–7. doi: 10.1111/j.1476-5381.2009.00329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Persu A, Jin Y, Azizi M, Baelen M, Völz S, Elvan A, Severino F, Rosa J, Adiyaman A, Fadl Elmula FE, Taylor A, Pechère-Bertschi A, Wuerzner G, Jokhaji F, Kahan T, Renkin J, Monge M, Widimský P, Jacobs L, Burnier M, Mark PB, Kjeldsen SE, Andersson B, Sapoval M, Staessen JA. European Network COordinating research on Renal Denervation (ENCOReD). Blood Pressure Changes After Renal Denervation at 10 European Expert Centers. Journal of Human Hypertension. 2013;26:150–6. doi: 10.1038/jhh.2013.88.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Diesel W, Noakes TD, Swanepoel C, Lambert M. Isokinetic muscle strength predicts maximum exercise tolerance in renal patients on chronic hemodialysis. Am J Kidney Dis. 1990;16(2):109–14. doi: 10.1016/s0272-6386(12)80563-4. [DOI] [PubMed] [Google Scholar]
- 174.Lunde PK, Verburg E, Vøllestad NK, Sejersted OM. Skeletal muscle fatigue in normal subjects and heart failure patients. Is there a common mechanism? Acta Physiol Scand. 1998;162(3):215–28. doi: 10.1046/j.1365-201X.1998.0343f.x. [DOI] [PubMed] [Google Scholar]
- 175.Müller-Ehmsen J, McDonough AA, Farley RA, Schwinger RH. Sodium pump isoform expression in heart failure: implication for treatment. Basic Res Cardiol. 2002;97 (Suppl 1):I25–30. doi: 10.1007/s003950200025. [DOI] [PubMed] [Google Scholar]
- 176.Radzyukevich TL, Neumann JC, Rindler TN, Oshiro N, Goldhamer DJ, Lingrel JB, Heiny JA. Tissue-specific role of the Na,K-ATPase α2 isozyme in skeletal muscle. J Biol Chem. 2013;288(2):1226–37. doi: 10.1074/jbc.M112.424663. [DOI] [PMC free article] [PubMed] [Google Scholar]