

Abstract
Mutagenesis provides insight into proteins, but only recently have assays that couple genotype to phenotype been used to assess the activities of as many as a million mutant versions of a protein in a single experiment. This approach – “deep mutational scanning” – yields large-scale datasets that can reveal intrinsic protein properties, protein behavior within cells and the consequences of human genetic variation. Deep mutational scanning is transforming the study of proteins, but many challenges must be tackled to fulfill its promise.
As the central players in the cell’s machinery, proteins have been the subject of numerous mutagenesis approaches that seek to characterize their function. Nonetheless, our ability to measure the effects of mutations in proteins has been limited to a relatively small number of mutations. But what if we knew the functional consequences of every possible single amino acid change at every position in a protein? What if we knew the biochemical activity of hundreds of thousands of different variants of a protein, each containing two, three or even more mutations? Recent technologies known collectively as “deep mutational scanning” make mutagenesis studies of this magnitude a reality.
The key problem that deep mutational scanning solves is our limited ability to predict the most informative mutations in a protein to analyze. Changes to amino acids that are distant from binding or active sites can have drastic effects on the thermodynamic stability or enzymatic activity of a protein1. Highly conservative mutations, whose consequences can be difficult to predict, may be neutral, deleterious or hyper-activating2,3. Multiple mutations can combine for unexpectedly large increases or decreases in activity4,5. By enabling the impact of mutations to be examined in an unbiased fashion, deep mutational scanning can reveal the unexpected. It can also address otherwise intractable cases in which it is necessary to measure the activity of a huge number of variants. For example, functional analyses of genomes and of protein engineering experiments increasingly demand this scale of data.
Carrying out a deep mutational scan requires an assay amenable to a coupled genotype-phenotype platform (Fig. 1). Such platforms include cell-based assays, with a protein typically expressed from a plasmid or virus, or in vitro systems, like phage or ribosome display. A library of mutated variants of the gene is synthesized, cloned into the appropriate vector and introduced, for example, into cells where the protein encoded by the gene carries out a function that can be selected for. The selection enriches cells with active protein variants and depletes those with inactive ones. The library is retrieved from both input and post-selection cells, and the frequency of each variant in the two libraries is determined by high-throughput DNA sequencing. The change in the frequency of each variant from input to selection serves as a measure of its function. Separation technologies, like cell sorting, can also be used to place variants into bins, with the variants in each bin scored by DNA read counts.
Figure 1. Deep mutational scanning generates large-scale mutational data.

Deep mutational scanning draws on high-throughput DNA sequencing to assess the functional capacity of a large number of variants of a protein simultaneously. First, a library of protein variants is created and introduced into a system where the genotype of each variant is linked to a selectable phenotype. Second, a selection for the function of the protein is imposed. Variants with high activity increase in frequency, whereas variants with low activity decrease in frequency. High-throughput DNA sequencing is used to measure the frequency of each variant before and after selection. These frequency data are analyzed to generate functional score for each of the protein variants.
The assays amenable to deep mutational scanning vary as widely as the activities that proteins can display. These include binding of a protein to a peptide, to another protein, to DNA, RNA or other ligands, and enzymatic activities such as phosphorylation or ubiquitination. Cellular assays can take advantage of a growth or drug selection, or expression of a protein that may be fluorescent or epitope-tagged. In vitro approaches can enrich active variants based on enzymatic activity, which can be combined with the use of an antibody that recognizes a post-translational modification. Because of the astronomical scale of DNA sequencing, millions of individual protein variants can be examined in a single experiment. This approach has been applied to a growing number of disparate proteins in a variety of contexts (Table 1). Nevertheless, establishing the infrastructure to carry out a deep mutational scan for the first time can be challenging, but it is becoming less so as reagents, software and methods are developed6.
Table 1.
Deep mutational scanning targets
| Scanned protein | Model | Selection |
|---|---|---|
| Fab antibody fragment36 | Ribosome display | Ligand binding |
| YAP65 WW domain13,37 | T7 bacteriophage | Ligand binding |
| E4B ubiquitin ligase14 | T7 bacteriophage | Ubiquitination activity |
| PKA regulatory subunit38 | T7 bacteriophage | Ligand binding |
| Synthetic PDZ domain39 | M13 bacteriophage | Ligand binding |
| CcdB16 | E. coli | Toxin activity |
| PSD 95 PDZ domain40 | E. coli | Ligand binding |
| G protein-coupled receptor41 | E. coli | Ligand binding |
| Designed influenza inhibitor29 | S. cerevisiae surface display | Ligand binding |
| Designed lysozyme inhibitor42 | S. cerevisiae surface display | Ligand binding |
| Designed digoxigenin binder43 | S. cerevisiae surface display | Small molecule binding |
| IgG1 CH3 domain44 | S. cerevisiae surface display | Ligand binding after thermal stress |
| Hsp9045,46 | S. cerevisiae complementation | Growth rate |
| Matα2 degron23 | S. cerevisiae fusion protein | Growth rate |
| Ubiquitin47 | S. cerevisiae complementation | Growth rate |
| Pab117 | S. cerevisiae complementation | Growth rate |
| Neuraminidase48 | Mammalian cell | Oseltamivir resistance |
| IgG CDRs49 | Mammalian cell display | Ligand binding |
| BRAF50 | Mammalian cell | Vemurafenib resistance |
On the simplest level, the large-scale mutational data that result from a deep mutational scan reveal the functional consequences of all possible single mutations. These data can be organized into a sequence–function map (Fig. 2). Such a map can be viewed as an all-residue scan, in which each position has been mutated to every other amino acid. These maps are dense with information, with each position having a unique pattern of functional effects; most substitutions are likely to be deleterious but a few may enhance activity. In addition to characterizing the effects of single mutations, deep mutational scanning can also examine the effects of multiple mutations. Collectively, these data can yield insights into protein structure and function, but gleaning these insights is a challenge that tests both experimental and computational biologists.
Figure 2. Large-scale mutational data illustrate how protein sequence impacts function.

A hypothetical sequence–function heat map is shown for a 25 amino acid long portion of a protein, illustrating the functional consequences of making every single amino acid mutation at every position. Positions are indicated numerically, and each mutation is indicated by its single letter code. The color of each element of the heat map illustrates the functional score of the indicated mutation
Inference of fundamental protein properties
A number of biochemical methods are customarily used to directly assay the fundamental properties of proteins: for example, chemical denaturation analyzes thermodynamic stability, enzyme kinetics reveal mechanism, X-ray crystallography provides structure, and light scattering measures particle size. These methods apply purpose-built instrumentation in the context of a specialized workflow, generally feasible for no more than a handful of variants.
Instead of using such methods to measure protein properties in a serial fashion, we might infer some properties from large-scale mutational data. This approach draws on our knowledge of proteins derived from more than a century of study, including principles of how proteins fold and unfold, how they act in catalysis, how they interact with solvent, and how they evolve. For a given protein property, such prior knowledge has the potential to generate a model or algorithm that relates the functional consequences of mutations to the property in question, and the model could be applied to the large-scale data obtained in a deep mutational scan (Box 1). This approach could augment and eventually supplant some traditional methods that are time-, cost- and labor-intensive. We highlight three areas in which this approach has progressed.
Box 1. Interpreting large-scale mutational data.
The initial stages of data analysis focus on producing a set of high-quality functional scores from raw sequence data51. In the simplest case, reads are aligned to a wild type template, variants are enumerated and functional scores are calculated by taking the ratio of the frequency of each variant before and after selection37. More complex cases (e.g. incorporating time-series data) can be dealt with using linear models13,14. Nevertheless, clear standards for analyzing deep mutational scanning data have yet to emerge. Enrich, an interactive software package for accomplishing the first data analysis phase, is publicly available, but requires command-line expertise to use52. Enrich guides users through the process of transforming raw high-throughput sequencing data into a set of variant functional scores. Enrich also generates a comprehensive sequence–function map from the data. However, deeper analyses of the functional scores are considerably more challenging and depend on the questions being asked. In some cases, analytical paradigms are already emerging, including those that examine how multiple mutations interact and how large-scale mutagenesis data change under different experimental conditions. Data analysis remains a significant challenge, but not an intractable one.
For example when engineering a protein or when classifying mutations in a disease-related protein, the experimenter may be interested only in how single mutations impact protein activity. In this case, data for single amino acid substitutions derived from a deep mutational scan can be displayed as a heat map relating sequence to function (Fig. 2). Further analysis can yield insights into such topics as fundamental proteins properties, the behavior of proteins inside cells and the paths of protein evolution, but is typically a slow and complex undertaking.
Successful interpretation of deep mutational scanning data starts with proper experimental design. Will the experimenter take advantage of a direct selection for a protein property of interest? Will the analysis require only single mutations, or will multiple mutations be needed? Will the analysis need large numbers of variants or will a few thousand suffice? To give an idea of how one might answer these questions, we highlight three broad experimental designs and give examples of how an experimenter might go about analyzing the resulting data sets.
Direct selection for a protein property of interest results in the most straightforward analysis of large-scale mutational data. Examples include:
thermodynamic stability of a library of IgG variants was measured using yeast display selection and thermal denaturation44
in vivo protein stability of a library of yeast degron variants was measured using a metabolic reporter protein fusion23
inhibitor resistance of a library of BRAF variants was measured using a cell-based resistance assay50
Knowledge-based inference is a more complex type of analysis, which can be applied when direct selection is not possible for the desired protein property. For example, directly selecting for mutations that change an enzyme’s mechanism would be difficult. Here, the experimenter selects for protein function without using specialized conditions (e.g. higher temperature to select for stability or the presence of an inhibitor to select for resistance) and then carries out an analysis that relates the functional scores to the property of interest. Examples include:
thermodynamically stabilizing mutations were identified because they rescue multiple destabilizing mutations13
buried positions were identified because they tolerate fewer substitutions than solvent-exposed ones16
core positions were identified because they exhibit similar patterns of preference for hydrophobic amino acids17
mechanism-altering mutations were identified because they are hyperactivating14
In even more complex cases, no analytic framework for the mutational data yet exists and will need to be developed. Examples include:
benchmarking and improving computational approaches for interpreting human genetic variation
improving the correlation of biochemical properties with disease risk
enhancing de novo protein structure/activity prediction algorithms
understanding protein evolution
First, because stabilizing mutations can rescue destabilizing mutations7–9, large-scale mutational data can be analyzed to identify thermodynamically stabilizing mutations. Stabilizing mutations are important for engineering proteins for pharmaceutical or industrial uses, and are difficult to identify; most mutations are either neutral or destabilizing. Current methods to identify stabilizing mutations have limitations, including poor performance for large or atypical proteins, extensive validation requirements, limited output and the identification of mutations that, while stabilizing, also result in an unintended loss of activity10–12. In previous work, we developed a computational model that measured the effectiveness of single mutations to rescue many other deleterious single mutations when they co-occur in a doubly mutated variant13. We applied this model to measurements of the peptide binding capacity of ~50,000 variants of a WW domain, and identified new stabilizing mutations.
Second, because mutations can perturb enzyme function, analysis of large-scale mutational data can reveal aspects of a protein’s catalytic mechanism. Rare variants can be identified that have enhanced activity or altered specificity. Such unusual variants were recently identified based on the ubiquitination activity of ~100,000 variants of an E3 ubiquitin ligase, and these hyperactive variants were used to unlock mechanistic details by further biochemical and structural approaches14. Other analyses of enzyme mechanism from large-scale mutagenesis data make use of the observation that the most mutation-intolerant positions in a protein frequently correspond to residues directly involved in contacting the substrate or performing catalysis. Another potential starting point are hyperactivating single mutations paired with deleterious single mutations that affect folding, stability, substrate interaction or other properties. For example, a mutation that enhances catalysis might be expected to rescue deleterious mutations that destabilize the protein but not those that block substrate binding.
Third, because mutations can perturb protein structure, large-scale mutational data can contribute to structural efforts. X-ray crystallography and nuclear magnetic resonance yield detailed structures, but do not work for every protein, particularly transmembrane proteins and large protein complexes15. De novo prediction of protein structure, while useful, cannot routinely provide useful structures of even average-sized proteins. Mutational data can help discriminate among predicted protein structures. In one example of such a study, the functional consequences of mutation at each position in the bacterial toxin CcdB were shown to correlate with distance to the protein surface in a known structure. Adkar et al.16 used this observation to select accurate predicted CcdB structures from among a large set of predictions based on which positions were buried. In another example from our own work, positions found to be sensitive to most substitutions except to hydrophobic amino acids constituted the core hydrophobic structure of the protein17.
In the future, large-scale mutational data could facilitate the prediction of protein secondary structure. Typically, algorithms base predictions on the amino acid preferences in each type of secondary structure (α-helix, β-sheet or loop) in a training set of proteins with known structures18,19. As an alternative, large-scale mutational data on proteins with known structures could also reveal amino acid preferences within structural elements, and the resulting preferences used to enhance structure prediction algorithms. A provocative challenge is using deep mutational scanning data to generate structural models. We suggest that these data could be analyzed to determine co-varying positions in a protein’s sequence, with the expectation that these positions will be close by in the three-dimensional folded structure. These experimentally determined distance constraints could then be combined with protein structure modeling software such as Rosetta to produce a plausible structural model20. Indeed the fact that co-variation between positions derived from the natural evolution of a protein can be used to predict structure if the multiple sequence alignment for the protein is sufficiently large21 hints that this approach is feasible.
Analysing large-scale mutational data is challenging because the principles according to which fundamental protein properties relate to mutational data are not fully understood (Box 1). In some cases, lessons learned from the study of a small number of mutations will generalize well, but in other cases refinement of our understanding will be required. Furthermore, these analyses require high quality mutational data in order to succeed. High-throughput methods are notoriously susceptible to problems with data quality. Thus, practitioners will need to develop and apply standards, especially regarding appropriate replication and models for controlling systematic and stochastic error. Nevertheless, the potential payoff is huge: a common method for understanding fundamental properties of proteins in their native environment.
Understanding how proteins behave in cells
Deep mutational scanning can be conducted in cells and thus offers the opportunity to marry protein science with cell-based approaches. Furthermore, the power of the technology is magnified by the fact that, for a particular protein, scans can be redone in a number of “sensitized” backgrounds or conditions (Fig. 3) – a veritable Hershey heaven22 where repeating the same experiment with slight alterations yields novel data. We discuss three examples of this approach.
Figure 3. Deep mutational scanning in sensitized backgrounds as a strategy for uncovering protein features.
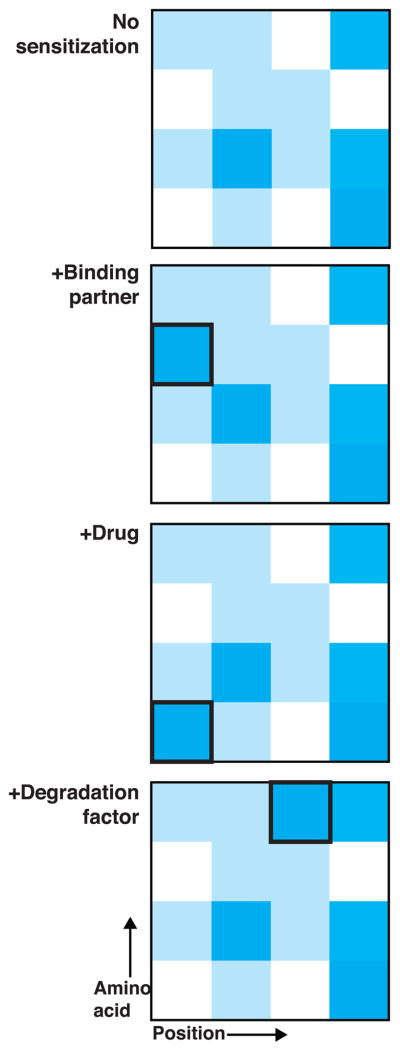
Hypothetical sequence-function heat maps collected under different conditions are shown. Once a deep mutational scan has been performed, it can be repeated in a sensitized background, which can be created by altering the cellular or chemical environment in which the scan is conducted, as indicated. The difference in functional effect for a particular mutation in a sensitized background could reveal the importance of an amino acid at a given position for the process under study.
First, deep mutational scanning can be used to probe protein-protein interactions. Structural approaches for studying protein-protein interactions, such as co-crystallization, yield high-resolution information but are inherently low throughput. High-throughput approaches, such as yeast two-hybrid or mass spectrometry, provide little, if any, structural detail. A library of variants can be screened for interaction in cells that overproduce a partner protein. The expectation is that a subset of mutations that in the initial (non-sensitized) screen were deleterious might be neutral in the presence of a binding partner, revealing positions in the protein relevant to the interaction.
Second, mutational scanning can measure the stability in cells of protein variants that are tagged with a required metabolic enzyme23. If the stability of the enzyme depends on the stability of the variant to which it is fused, then cells harboring a long-lived variant will have high levels of the enzyme and grow faster. The influence of protein degradation factors could be investigated by varying the level of these factors.
Third, mutational scanning using cell-based protein aggregation models could yield details of the biophysical processes driving aggregation in vivo. For example, variants of an aggregation-prone protein could be fused to an essential enzyme whose activity diminishes as the aggregation state of the variant increases24. Furthermore, by again varying the expression of chaperones and degradation factors, the experimenter might better understand how these factors identify and degrade aggregation-prone proteins.
Protein evolution and engineering
Experimental evolution approaches offer the opportunity to watch evolution of proteins as it occurs, but to date they have been limited either to examining a handful of variants or to making population-based measurements. Owing to the vast size of the sequence landscape, conclusions arising from these studies have been incomplete and sometimes contradictory. Protein evolution has also been treated theoretically, but many predictions remain untested. Deep mutational scanning approaches when applied to experimental evolution of proteins offer the ability to explicitly track the fate of hundreds of thousands of sequences simultaneously. They can thus begin to address fundamental questions25, such as: How many paths can evolution take? How many mutations are required to produce new function? Are there many distinct sequences that could evolve to solve the same problem? In short, these approaches offer the opportunity to experimentally explore the protein fitness landscapes that shape evolutionary trajectories. For example, large-scale mutational data on a WW domain13 and on an HIV protease and reverse transcriptase26 revealed that some combinations of mutations within variants interact to produce unexpectedly large functional effects, leading to the formation of intramolecular mutation interaction ‘hotspots’ within these proteins. High-throughput sequencing of T7 RNA polymerase evolving to bind new promoter sequences revealed distinct classes of convergently evolved solutions27.
Deep mutational scanning experiments should also be instrumental in realizing the promise of protein engineering, which improves existing proteins, and de novo design, which imagines novel ones with desired features. Currently, engineering and design efforts proceed from rule-based design28 or use blind selection to identify one or a few variants with improved functionality among a library. In both cases, deep mutational scanning approaches could be transformative, enabling the identification of large numbers of useful mutations that can be combined to refine engineered or designed proteins. For example, this approach was used to optimize a computationally-designed hemagglutinin-binding protein that inhibits influenza virus29, resulting in the identification of five mutations that combined to produce a 25-fold improvement in affinity. Traditional affinity maturation approaches would not have resulted in the final, high-affinity inhibitor because such approaches cannot effectively explore the staggeringly large number of mutant combinations required to find a variant with five mutations. Large-scale mutagenesis data offer the opportunity to improve the protein design process by enabling designers to exhaustively examine where and why their algorithms fail29,30.
Deep mutational scanning and human genetics
A large component of the genetic basis of disease lies in rare variation, with every human carrying, on average, ~300 rare, protein-coding variants31. For everyone from physicians, pharmacists and patients to casual users of personalized DNA testing, knowing the functional consequences of rare mutations in important genes is critical. Most existing experimental approaches are not practical for assessing the rapidly increasing number of these rare mutations being identified. They simply cannot achieve the scale necessary to measure the phenotypic consequences of the variation that can occur in a typical human protein, which comprises 375 amino acids subject to 7,500 possible single mutations (including to stop codons)32. The challenge is highlighted by the fact that 10% of women harboring a missense mutation in the BRCA1 gene, which may predispose them to breast cancer, are told they harbor a “variant of unknown significance”33. That BRCA1, one of the best-studied proteins, still generates such diagnoses indicates that the situation for the average protein implicated in human disease is far worse. Furthermore, we will not be able to repeat the investment of time and money spent on BRCA1 on each of these thousands of other proteins.
Currently, computational prediction of the functional consequences of mutations with programs like Condel, GERP, Polyphen-2, and SIFT is the best we can do. But these computational approaches are limited in their accuracy34. For example, when Condel, PolyPhen-2 and SIFT predicted the functional consequences of a set of known deleterious mutations, they produced correct and concordant results in fewer than half the cases35. Because these tools are based on evolutionary conservation of individual positions and/or the physicochemical properties of amino acids, they are relatively successful only on average. But they fail in an unacceptably large fraction of cases, making them far from ideal for clinical use.
Large-scale mutational data could empower these computational approaches. First, these data provide a new resource for benchmarking computational approaches. Second, analysis of a modest number of large-scale mutagenesis data sets derived from proteins with diverse structures and functions could enhance our understanding of how, in a general sense, mutations impact protein function. This information should be useful for improving the accuracy of physicochemical models of the impact of mutations. Third, large-scale mutagenesis data in model organisms that are selected for their fitness could even contribute to developing computational models that predict the effects of mutations on a more complex organism.
In principle, experimental characterization of the functional consequences of all possible single amino acid substitutions using a deep mutational scanning approach could obviate the need for computational inference in interpreting coding variation by furnishing sequence-function maps of disease-related proteins (Fig. 2). This task seems daunting, as thousands of sequence–function maps for proteins with an enormous range of functions would be required. However, the challenge may not be quite as formidable as it appears: many disease-related proteins fall into well-studied classes like transcription factors, protein kinases, surface receptors and DNA repair proteins that may allow some existing, generic assays to be used (Fig. 4). No doubt, before such data are applied in the clinic, assays used to determine protein function scores must be vetted for their capacity to adequately reflect disease risk, pathogenicity or progression. The jury is still out on which in vitro assays will do so. Furthermore, a simple functional assay that is amenable to a deep mutational scan cannot be generated for every protein. The possible rewards for such an approach are nevertheless considerable. A large, coordinated project could, for example, generate sequence-function maps for a set of cancer-related proteins, providing an invaluable clinical resource.
Figure 4. Sequence–function maps of proteins important in disease.

A hypothetical cancer cell is shown; mutations in drug transporters, drug metabolic enzymes, transcription factors, and signaling proteins all have the capacity to influence the effectiveness of treatment. Deep mutational scanning of cancer-related proteins could revolutionize our understanding of the consequences of mutations in these proteins and enable genomic medicine.
Unresolved questions
Between the promise and the reality of deep mutational scanning lie many questions. Is there as much useful protein information latent within these large datasets as we speculate that there is? We have learned already that large-scale mutational data contain a rich array of information. But developing analytic methods to reveal some of this information, such as protein structure, will likely require substantial development. Furthermore, it may be difficult to design assays that couple some cell-based properties, such as localization or post-translational modification, to the sequencing readout required for a deep mutational scan.
For an effective scan, the development of an appropriate assay for the function of interest is perhaps even more important than the methods used for mutagenesis, library construction, sequencing and computational analysis. Can the scale of assay development match the pace of progress in DNA synthesis and sequencing? Critically, the selection condition must alter or separate library members proportional to their functional capacity, ideally across a wide range of activity levels. The assay must enable the production of DNA libraries that are amenable to high-throughput sequencing, not a given for every assay. Although we can draw on decades of collective experience in crafting these functional assays, choosing and calibrating an assay that works at high throughput remains a formidable undertaking.
Will these approaches be put into place soon enough to deal with the deluge of human genetic variation, and will the mutational data generated in vitro adequately reflect the complex roles of disease proteins? The concern is that simple assays that can be scored at high throughput may not adequately reflect human disease. Undoubtedly, the limits of simple assays must be respected. For example, assays for proteins that act extracellularly or that are poorly conserved are likely not good candidates. Assays for well-conserved intracellular proteins will likely be useful, though they will need to be validated to ensure they adequately reflect disease risk. Advances in genome editing could pave the way for deep mutational scanning experiments in human cell lines, partially alleviating this concern, although even human cell-based assays are limited in their ability to model organ or whole-organism disease phenotypes. We suggest that for proteins with simple molecular functions (e.g. metabolic enzymes), large-scale mutagenesis data might have potential for direct use in the clinic. For other proteins with complex functions (e.g. signaling proteins), large-scale mutagenesis data will need to be combined with an integrative computational model. In either case, we will need extensive sets of protein variants whose activity scores can be compared to known disease risk and outcome to establish clinical utility of these data.
In summary, deep mutational scanning can be used to generate large-scale mutational data for nearly any protein. Because this approach is rooted in a rapidly developing technology – high throughput sequencing – it is likely that its power and scope will continue to grow. We have highlighted some of the ways in which we predict large-scale mutational data could transform protein science. The many challenges to this transformation also provide many opportunities to protein scientists. Understanding the vast number of protein variants within humans demands that experimental and computational methods be developed. Deep mutational scanning strategies provide one avenue to address this need.
Acknowledgments
We thank A. Merz, M. Hochstrasser, C. Queitsch, A. Gitler, J. Bloom, E. Marcotte, E. Phizicky and M. Wickens for helpful discussions and comments. This work was supported by P41 GM103533 (to S.F.) and F32 GM084699 (to D.M.F.) from the National Institute of General Medical Sciences. S.F. is supported by the Howard Hughes Medical Institute.
Footnotes
Competing Financial Interests
The authors have no competing financial interests to declare.
References
- 1.Freeman AM, Mole BM, Silversmith RE, Bourret RB. Action at a distance: amino acid substitutions that affect binding of the phosphorylated CheY response regulator and catalysis of dephosphorylation can be far from the CheZ phosphatase active site. J Bacteriol. 2011;193:4709–4718. doi: 10.1128/JB.00070-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jonson PH, Petersen SB. A critical view on conservative mutations. Protein Eng Des Sel. 2001;14:397–402. doi: 10.1093/protein/14.6.397. [DOI] [PubMed] [Google Scholar]
- 3.Gilbert GE, Novakovic VA, Kaufman RJ, Miao H, Pipe SW. Conservative mutations in the C2 domains of factor VIII and factor V alter phospholipid binding and cofactor activity. Blood. 2012;120:1923–1932. doi: 10.1182/blood-2012-01-408245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang W, Dourado DFAR, Fernandes PA, Ramos MJ, Mannervik B. Multidimensional epistasis and fitness landscapes in enzyme evolution. Biochem J. 2012;445:39–46. doi: 10.1042/BJ20120136. [DOI] [PubMed] [Google Scholar]
- 5.Natarajan C, et al. Epistasis among adaptive mutations in deer mouse hemoglobin. Science. 2013;340:1324–1327. doi: 10.1126/science.1236862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fowler DM, Stephany JJ, SS F. Measuring the Activity of Protein Variants at Large-Scale Using Deep Mutational Scanning. Nature Protocols. doi: 10.1038/nprot.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 8.Bloom JD, Arnold FH. In the light of directed evolution: pathways of adaptive protein evolution. Proc Natl Acad Sci USA. 2009;106 (Suppl 1):9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature. 2006;444:929–932. doi: 10.1038/nature05385. [DOI] [PubMed] [Google Scholar]
- 10.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 11.Magliery TJ, Lavinder JJ, Sullivan BJ. Protein stability by number: high-throughput and statistical approaches to one of protein science’s most difficult problems. Curr Opin Chem Biol. 2011;15:443–451. doi: 10.1016/j.cbpa.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foit L, et al. Optimizing protein stability in vivo. Mol Cell. 2009;36:861–871. doi: 10.1016/j.molcel.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Araya CL, et al. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc Natl Acad Sci USA. 2012;109:16858–16863. doi: 10.1073/pnas.1209751109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starita LM, et al. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc Natl Acad Sci USA. 2013;110:E1263–72. doi: 10.1073/pnas.1303309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lander GC, Saibil HR, Nogales E. Go hybrid: EM, crystallography, and beyond. Curr Opin Struct Biol. 2012;22:627–635. doi: 10.1016/j.sbi.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adkar BV, et al. Protein model discrimination using mutational sensitivity derived from deep sequencing. Structure. 2012;20:371–381. doi: 10.1016/j.str.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 17.Melamed D, Young DL, Gamble CE, Miller CR, Fields S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA. 2013;19:1537–1551. doi: 10.1261/rna.040709.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aydin Z, Singh A, Bilmes J, Noble WS. Learning sparse models for a dynamic Bayesian network classifier of protein secondary structure. BMC Bioinformatics 2011 12:242. 2011;12:154. doi: 10.1186/1471-2105-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen K, Kurgan L. Computational prediction of secondary and supersecondary structures. Methods Mol Biol. 2013;932:63–86. doi: 10.1007/978-1-62703-065-6_5. [DOI] [PubMed] [Google Scholar]
- 20.Kim DE, DiMaio F, Yu-Ruei Wang R, Song Y, Baker D. One contact for every twelve residues allows robust and accurate topology-level protein structure modeling. Proteins. 2014;82 (Suppl 2):208–218. doi: 10.1002/prot.24374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marks DS, Hopf TA, Sander C. Protein structure prediction from sequence variation. Nat Biotechnol. 2012;30:1072–1080. doi: 10.1038/nbt.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Creager ANH. Hershey heaven. Nat Struct Biol. 2001;8:18–19. doi: 10.1038/82991. [DOI] [PubMed] [Google Scholar]
- 23.Kim I, Miller CR, Young DL, Fields S. High-throughput analysis of in vivo protein stability. Mol Cell Proteomics. 2013;12:3370–3378. doi: 10.1074/mcp.O113.031708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morell M, de Groot NS, Vendrell J, Avilés FX, Ventura S. Linking amyloid protein aggregation and yeast survival. Mol Biosyst. 2011;7:1121–1128. doi: 10.1039/c0mb00297f. [DOI] [PubMed] [Google Scholar]
- 25.Dean AM, Thornton JW. Mechanistic approaches to the study of evolution: the functional synthesis. Nat Rev Genet. 2007;8:675–688. doi: 10.1038/nrg2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinkley T, et al. A systems analysis of mutational effects in HIV-1 protease and reverse transcriptase. Nat Genet. 2011;43:487–489. doi: 10.1038/ng.795. [DOI] [PubMed] [Google Scholar]
- 27.Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR. Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc Natl Acad Sci USA. 2013;110:9007–9012. doi: 10.1073/pnas.1220670110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koga N, et al. Principles for designing ideal protein structures. Nature. 2012;491:222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitehead TA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moretti R, et al. Community-wide evaluation of methods for predicting the effect of mutations on protein-protein interactions. Proteins. 2013;81:1980–1987. doi: 10.1002/prot.24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tennessen JA, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brocchieri L, Karlin S. Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 2005;33:3390–3400. doi: 10.1093/nar/gki615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Millot GA, et al. A guide for functional analysis of BRCA1 variants of uncertain significance. Human Mutation. 2012;33:1526–1537. doi: 10.1002/humu.22150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gnad F, Baucom A, Mukhyala K, Manning G, Zhang Z. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics. 2013;14 (Suppl 3):S7. doi: 10.1186/1471-2164-14-S3-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray VE, Kukurba KR, Kumar S. Performance of computational tools in evaluating the functional impact of laboratory-induced amino acid mutations. Bioinformatics. 2012;28:2093–2096. doi: 10.1093/bioinformatics/bts336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujino Y, et al. Robust in vitro affinity maturation strategy based on interface-focused high-throughput mutational scanning. Biochem Biophys Res Commun. 2012;428:395–400. doi: 10.1016/j.bbrc.2012.10.066. [DOI] [PubMed] [Google Scholar]
- 37.Fowler DM, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gold MG, et al. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell. 2006;24:383–395. doi: 10.1016/j.molcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 39.Ernst A, et al. Coevolution of PDZ domain-ligand interactions analyzed by high-throughput phage display and deep sequencing. Mol Biosyst. 2010;6:1782–1790. doi: 10.1039/c0mb00061b. [DOI] [PubMed] [Google Scholar]
- 40.McLaughlin RN, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlinkmann KM, et al. Critical features for biosynthesis, stability, and functionality of a G protein-coupled receptor uncovered by all-versus-all mutations. Proc Natl Acad Sci USA. 2012;109:9810–9815. doi: 10.1073/pnas.1202107109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Procko E, et al. Computational design of a protein-based enzyme inhibitor. J Mol Biol. 2013;425:3563–3575. doi: 10.1016/j.jmb.2013.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tinberg CE, et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501:212–216. doi: 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Traxlmayr MW, et al. Construction of a stability landscape of the CH3 domain of human IgG1 by combining directed evolution with high throughput sequencing. J Mol Biol. 2012;423:397–412. doi: 10.1016/j.jmb.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang L, Mishra P, Hietpas RT, Zeldovich KB, Bolon DNA. Latent effects of Hsp90 mutants revealed at reduced expression levels. PLoS Genet. 2013;9:e1003600. doi: 10.1371/journal.pgen.1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hietpas RT, Jensen JD, Bolon DNA. Experimental illumination of a fitness landscape. Proc Natl Acad Sci USA. 2011;108:7896–7901. doi: 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DNA. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J Mol Biol. 2013;425:1363–1377. doi: 10.1016/j.jmb.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu NC, et al. Systematic Identification of H274Y Compensatory Mutations in Influenza A Virus Neuraminidase by High-Throughput Screening. J Virol. 2013;87:1193–1199. doi: 10.1128/JVI.01658-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forsyth CM, et al. Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. MAbs. 2013;5:523–532. doi: 10.4161/mabs.24979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagenaar TR, et al. Resistance to vemurafenib resulting from a novel mutation in the BRAFV600E kinase domain. Pigment Cell Melanoma Res. 2014;27:124–133. doi: 10.1111/pcmr.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Araya CL, Fowler DM. Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol. 2011;29:435–442. doi: 10.1016/j.tibtech.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fowler DM, Araya CL, Gerard W, Fields S. Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics. 2011;27:3430–3431. doi: 10.1093/bioinformatics/btr577. [DOI] [PMC free article] [PubMed] [Google Scholar]