

Abstract
Accumulation and cytotoxicity of amyloid beta (Aβ) are understood as the major cause of Alzheimer's disease (AD). There is evidence that naturally occurring antibodies against amyloid beta (Aβ) protein play a role in Aβ-clearance, and such a mechanism appears to be impaired in AD. In the present study, the anti-Aβ antibodies in the serum from individuals with and without late onset AD were measured using ELISA and dot-blot methods. Aβ auto-antibodies in serum were mainly targeted to Aβ1-15 epitope and its titer was significantly lower in AD patients than elderly non-AD controls (NC). The dot-blot analysis further demonstrated that auto-antibodies against fibrillar Aβ42, Aβ1-15 and Aβ16-30 epitopes were all in a lower level in AD than in NC. The isotypes of the auto-antibodies were mainly non-inflammatory IgG2 type. We also analyzed the relationship of auto-Aβ antibodies levels with the genotypes of Apolipoprotein E (ApoE) and ANKK1/DRD2 gene.
Keywords: Alzheimer's disease, Amyloid beta antibodies, Human serum, ELISA, Dot blot, Apolipoprotein E (ApoE), ANKK1/DRD2
1. Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disease associated with disruption of neuronal function in the hippocampus and cerebral cortex, which gradually deteriorates the cognition, function, and behavior of the patients [1]. Increased accumulation and deposition of amyloid β (Aβ) protein in the form of plaques is thought to be the leading cause, and clearance of Aβ from the brain has been a major focus for the prevention and treatment of AD. Active immunization with Aβ peptide increased blood anti-Aβ antibodies and thus decreased brain Aβ plaque burden in AD mouse models [1, 2] as well as in AD patients [3, 4]. Administration of N-terminal Aβ antibodies was also effective in reducing plaques in AD mouse models and in AD patients [5, 6]. These data indicate that pre-existing auto-antibodies against Aβ in the blood may play an important role in the plaque formation and such an immune mechanism may have been impaired in AD.
Many studies have been conducted to compare serum levels of Aβ auto-antibodies in AD and in age-matched non-AD control (NC) subjects. The results of these studies have been inconsistent: some reported that Aβ auto-antibodies in AD were lower than in normal subjects [7, 8, 9, 10], some unaltered [11, 12, 13], and some increased [14, 15, 16]. The inconsistent results may have been caused by several factors including nonspecific bindings [17], serum Aβ interference [18], incorrect diagnosis [19, 20], structural conformation of Aβ1-42, and/or small sample size. Despite the relatively large number of studies being conducted, the epitope-specific binding and isotyping of the auto-antibodies against Aβ1-42 have not been reported. The present study therefore has sought to measure epitope-specific auto-antibodies against Aβ1-42 peptide in AD patients by comparison with the normal age-matched control subjects (NC). Our results indicate that naturally occurred Aβ antibodies mainly target Aβ1-15 epitope in both AD and NC subjects, as evidenced by the measurements of various Aβ epitopes with ELISA method, and in addition, its levels are significantly reduced in AD, especially in patients over 65 years of age, in comparison with those of NC subjects. Dot-blot analysis further demonstrated that antibody levels against fibrillar Aβ1-42, Aβ1-15 and Aβ16-30 were all significantly lower in AD than in NC subjects. The low level of Ab1-15 auto-antibodies is also in a trend of association with ApoE 4/4 alleles and with ANKK-CC alleles.
2. Patients and Methods
2.1. Patients and control subjects
We studied 113 subjects who were recruited from the Alzheimer's disease Center at the University of Texas Southwestern Medical Center. The AD group was consisted of 53 subjects, and the control group was consisted of 60 non-cognitively impaired patients (see Table 1 for breakdown by age, race and gender). All subjects with AD had physical and neurological examinations, neuropsychological testing, laboratory studies, and brain imaging to exclude reversible causes of dementia. The average score of mini–mental state examination (MMSE) for the AD group was 19.8 ± 6.3 and for controls 28.9 ± 2.6 (mean ± SD). All patients met International Classification of Diseases (ICD)-10 criteria for dementia as well as National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria for probable AD. For some patients with AD, informants reported a family history of cognitive impairment, whereas normal controls had no reported family history. Control subjects had no significant decline or impairment in cognition on clinical examination. They had no history or evidence of neurologic disease with potential to affect cognition. All individuals or their legally authorized representatives have signed/supplied written informed consent. The blood was drawn and the serum was prepared, coded, and frozen at -80°C within one hour of collection.
Table 1. Subject Summary.
| Diagnosis | Gender | Age | Race | Vitamin | MMSE |
|---|---|---|---|---|---|
| AD (53) | 33 male | 55-65 (8) | 2 African | 22 No | 19.8 ± 6.3 |
| 20 female | 66-75 (27) | 3 Hispanic | 31 yes | ||
| 76-89 (18) | 48 Caucasian | ||||
| 72.3 ± 7.5 | |||||
|
| |||||
| NC (60) | 21 male | 55-65 (19) | 2 African | 24 No | 28.9 ± 2.6 |
| 39 female | 66-75 (30) | 3 Hispanic | 36 Yes | ||
| 76-82 (11) | 55 Caucasian | ||||
| 67.6 ± 5.8 | |||||
Ages are reported as in 10 ears separate with Means ± SD. AD, Alzheimer's Disease, NC, non-cognitive impaired control. MMSE, mini–mental state examination (Means ± SD).
2.2. Amyloid beta peptides
Aβ1-42, Aβ1-15, Aβ16-30 and Aβ31-42 peptides were synthesized by BioBasic Inc., Canada. The peptides were first dissolved in water and then by adding 10× PBS to make stock solution (2 mg/ml) in 1× PBS. Fibrillar Aβ1-42 was prepared by incubation of Aβ1-42 solution in 37°C overnight [1].
2.3. ELISA for measuring Aβ auto-antibodies in serum and Aβ1-42 peptide in plasma
The levels of anti-Aβ antibodies in sera of all subjects were measured using an enzyme-linked immunosorbent assay (ELISA). The wells of the ELISA plate (Immulon 2 HB U, Thermo Scientific) were coated with 2 μg/ml of human Aβ peptide in a 0.1 M bicarbonate-carbonate buffer (pH 9.0) at 4°C overnight (50 μl/well). The wells were then blocked with 100 μl 1% bovine serum albumin (BSA) in PBS containing 0.05% (v/v) Tween-20 (PBST) for 1 h and then washed three times with PBST. The human serum (50 μl) was added to each well by a 1:100 dilution with 1% BSA in PBST, left for overnight at 4° C to produce an Aβ-antibody complex. The plates were then washed three times with PBST and incubated for 2 h at room temperature with the secondary antibody (horseradish peroxidase labeled anti-human antibody, Bio-Rad) by 1:2000 dilution with 1% BSA-PBST. The plates were then washed three times with PBST. Finally, a 50 μl solution of 3,3,5,5-tetramethylbenzidine (TMB) was added to the wells, to form a colored reaction product indicating the presence of anti-Aβ antibodies, and absorbance was measured at a wavelength of 405 nm with a plate reader (ThermoMax). A mouse anti-human Aβ monoclonal antibody (4G8) was used as the standard.
For the isotyping of Aβ1-15 antibodies in the serum, highly positive samples from both AD (n=15) and NC (n=15) subjects were selected to add to the wells coated with Aβ1-15 peptide. The binding antibodies were detected with rabbit anti-human IgG1, IgG2 and IgG3 (Sigma-Aldrich, St. Louis, MO) followed by anti-rabbit IgG antibodies conjugated with HRP. The OD405 values were recorded.
2.4. Dot blot assay for detection of Aβ auto-antibodies in the serum
Aβ peptide (2 μg/2 μl) is spotted onto nitrocellulose membrane, dried in room temperature, washed with TBST once (20 mM Tris-HCl, 150 mM NaCl, pH 7.5, 0.05% Tween20), and blocked in 10% milk TBST at 4° C overnight. The membrane is then incubated with human serum (1:1000) in 5% milk in TBST for 2 h, washed with TBST (3 × 5 min), incubated with secondary rabbit anti-human IgG antibody labeled with HRP in 1:2000 dilution (Biomada, Foster city, CA) for 2 h at room temperature, washed three times with TBST (5 min each), one time with TBS (5 min), and one time with water (5 min). It was further incubated with ECL reagent for 1 minute, covered with Saran-wrap (remove excessive solution from the surface), and exposed to X-ray film in the dark room. The spot area and intensity were quantified using Image J image processing and analysis software (NIH).
2.5. APOE and ANKK1/DRD2 gene genotype
Genomic DNA was extracted from white blood cells using Qiagen DNA Blood kits (#51162; Qiagen Inc., Valencia, CA). Two TaqMan assays (Rs429358 and Rs7412, Applied Biosystems) were used for ApoE genotyping, and RS 1800497 (Applied Biosystems) for DRD2/ANKK1 Taq1A genotype.
2.6. Statistical analysis
Each blood sample was counted as an individual value for statistical analysis. The significance of difference was calculated by Student's t test (tails = 2, type = 2) expressed as the p value.
3. Results
We have established a sensitive, reproducible ELISA method to quantify the concentration of anti-Aβ immunoglobulins in human serum. The assay values obtained from this method are consistent with less than 10% variation between inter- and intra-assays. We first tested the binding capacity of the serum to various epitopes of the Aβ42 peptide, including Aβ1-42, Aβ1-42 fibrils (Aβ42F), Aβ1-15, Aβ16-30, and Aβ31-42 with this ELISA method. The anti-Aβ1-15 epitope antibodies were demonstrated to be in the highest reading values followed sequentially by Aβ16-30, Aβ1-42F, Aβ31-42 and Aβ1-42 in both AD and NC patients (Fig 1A). When the antibody levels between AD and NC subjects were compared, Aβ1-15 targeted antibodies were the only one showing a significant reduction in AD compared with those in NC subjects. The average level is 0.59 ± 0.05 μg/ml serum (mean ± SEM; n=53) in AD and 0.82 ± 0.08 μg/ml serum in NC (n=60; p = 0.02, Fig. 1B). The anti-Aβ16-30 and Aβ42F antibodies also showed trend of reduction in AD compared with NC subjects, but statistical significance cannot be reached in the current sample size.
Fig 1.
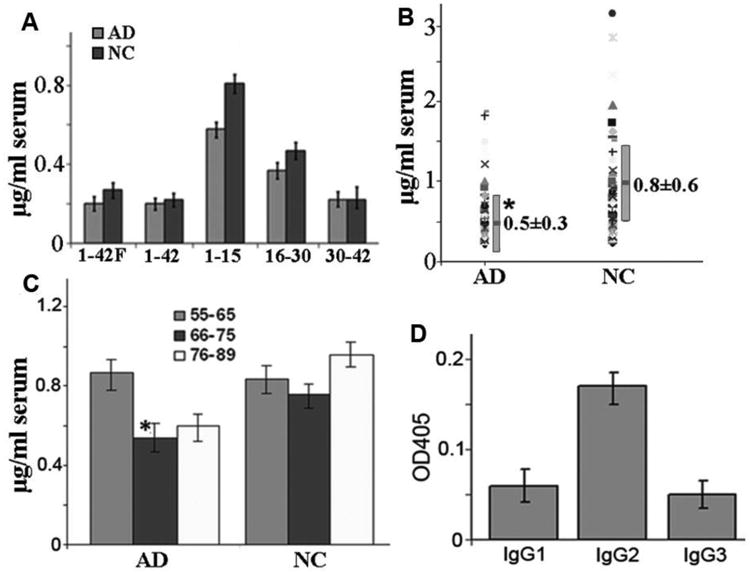
Anti-Aβ antibodies in serum detected with ELISA. A. Anti-Aβ1-15 antibodies were in highest levels among the epitopes in both AD (n=53) and NC (n=60) groups. B. Distribution patterns of Aβ1-15 antibodies in AD (n=53) and NC (n=60) groups and its levels are significantly lower in AD than NC groups (p < 0.02). C. Age-associated Aβ1-15 antibody reduction in AD patients in 66-75 years of age (n = 27) compared with those of 55-65 (n = 8) or 76 - 89 (n = 19) years of age (p = 0.03 and 0.08 respectively). No age-related differences in NC among 66-75 (n = 30) than 55-65 (n = 19) and 76-89 (n = 11) years old subjects (p = 0.9). D. Isotyping ELISA showed that IgG2 was the major isotype of Aβ1-15 antibodies among IgG1-3. Shown are Mean ± SE.
We also found age-associated reduction in Aβ1-15 antibody levels in AD group as measured by ELISA. Aβ1-15 antibodies were decreased significantly in AD patients of over 65 years of age (0.53 ± 0.26) compared with AD patients younger than 65 years of age (0.78 ± 0.6, p = 0.03). No such a difference was observed in NC (0.75 ± 0.4 for over 65 years of age and 0.83 ± 0.8 for younger than 65 years old subjects, p = 0.9). It is more interesting that even higher levels of the antibodies were found in NC in the older than 75 years old group (0.97 ± 0.58). These data indicate that increased levels of Aβ antibodies may be a protective factor against AD (Fig. 1C). We also demonstrated that the Aβ1-15 antibodies in human serum were distributed to different isotypes from IgG1 to IgG3. (Fig. 1D). There is no difference in Aβ1-15 auto-antibodies between a low (below 20) and high (above 20) score of MMSE in AD, and between male and female in both AD and NC subjects.
The data were also collected using ImageJ quantization of dot-blot. As shown in Fig. 2A, dot-blot scores were generally higher in NC samples compared with those in AD. In 1:1000 diluted sera the most frequently detected epitope was the Aβ42F, followed by Aβ1-15, and then Aβ16-30. The Aβ42 and Aβ31-42 epitope were rarely detectable with the dot-blot method, which is consistent with the ELISA result. We also apply ImageJ quantization method to quantify the density of stained spot that is clearly discernible (n=24 for AD, and n=34 for NC). The average density of a stained spot was calculated by multiplication of the mean intensity with the staining area and the results were shown in Fig. 1B. Three epitope antibodies, Aβ42F, Aβ1-15 and Aβ16-31 detected with this dot blot method, were significantly lower in AD subjects compared with the NC group. The most significant differences were Aβ1-15 antibodies with the density values of 3490 ± 64 in the AD group, vs. 6190 ± 94 in the NC group (mean ± SEM; p = 0.0008). The lesser difference was the Aβ16-31 with the density of 3770 ± 57 in NC vs. 1250 ± 46 in AD (p = 0.004), and Aβ42F (2520 ± 50 in NC vs. 595 ± 35 in AD, p = 0.018). The anti-Aβ42 and Aβ31-42 antibodies were not detectable by dot blot method, which is consistent with the ELISA results shown above.
Fig. 2.
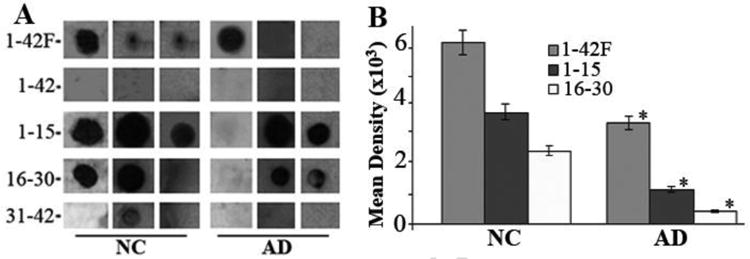
A. Dot blot assay of Aβ peptide detected by AD and NC sera demonstrating binding variability of the sera samples. B. Mean density of the dot-blot detected with the serum of AD (n = 24) and NC (n = 44) subjects for Aβ1-42F, Aβ1-15, and Aβ16-30, as measured by ImageJ software. All these epitope antibodies are significantly decreased in AD than NC groups. Shown are Mean ± SE. *P < 0.01. Aβ1-42 and Aβ31-42 peptide were not detectable with dot-blot assay.
The distribution pattern of ApoE alleles in the AD and the NC group is shown in Table 2. None of the AD patients carried E2 alleles (0%, 0/50) with statistically significant difference from that of the NC group (11.0%, 8/56, p < 0.02). Double E4 alleles were 20.0% (10/50) in the AD group and only 0.04% (2/56) in the NC group (p < 0.002 in comparison with AD). The level of Aβ1-15 antibodies was lower in E4/E4 patients in both AD and NC groups (p =0.09) in comparison with other alleles (Fig. 3 Top).
Table 2. Distribution of Apo-E and ANKK1 gene alleles in percentage.
| ApoE | AD (n=50) | NC% (n=56) | ANNK1 | AD% (n=50) | NC% (n=57) |
|---|---|---|---|---|---|
| 2/2,2/3,2/4 | 0/0/0 | 11 (1,6,1)* | TT | 2.0 (1) | 3.5 (2) |
| 3/3 | 36.0 (18) | 51.8 (29) | CT | 28.0 (14) | 40.4 (23) |
| 3/4 | 44.0 (22) | 30.4 (17) | CC | 70.0 (35) | 56.1 (32) |
| 4/4 | 20.0 (10)* | 0.04 (2) |
AD, Alzheimer's Disease, NC, non-cognitive impaired control. There is statistically significant differences in ApoE2 allele (p = 0.003) and ApoE4 (p = 0.03) between AD and NC. There is a trend of higher number of CC (A2/A2) allele in AD than NC (p = 0.33) for ANNK1 gene.
Fig. 3.
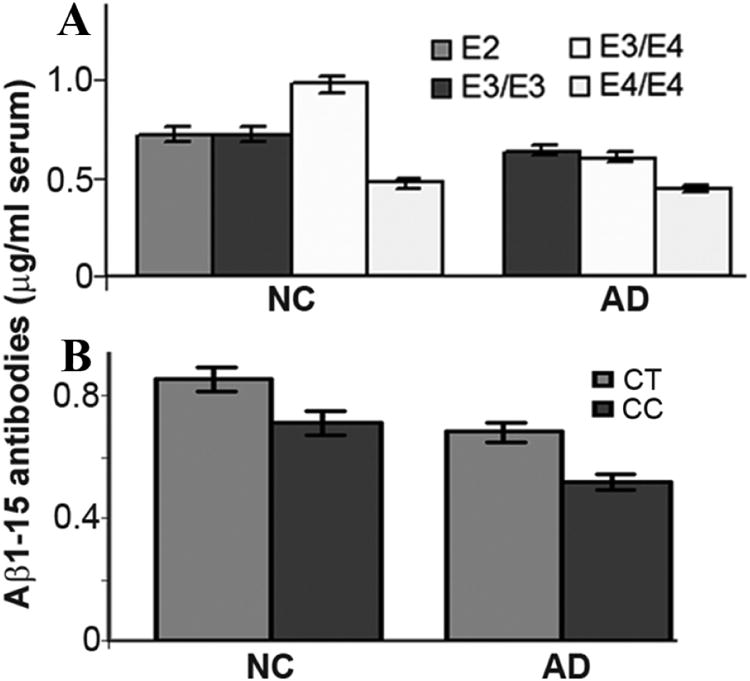
Variations of the serum Aβ1-15 antibody levels in patients carrying different ApoE gene alleles (A) and ANKK1/DRD2 gene alleles (B). A. The serum Aβ1-15 antibodies were tend to be lower in E4/E4 carrying patients in both AD and NC groups although statistically significant could not be reached in the present number of patients compared with the other alleles (p =0.09). B. The levels of Ap1-15 antibodies were tend to be lower in ANKK1 gene CC allele than A1 (CT or TT) allele patietennts (p = 0.11 and 0.39 in AD and NC group).
The genotype distribution pattern of ANKK1 gene was also shown in Table 2. A2/A2 (CC alleles) genotype was 70% (35/50) in the AD, which is slightly higher than that of the NC (56%, 32/57, p = 0.33). The Ab1-15 antibodies in the AD group were in a tendency of lower level in CC alleles than the A1+ (CT + TT) alleles, although it is insignificant statistically (p = 0.11 for the AD and 0.39 for the NC group in comparison with the non-CC alleles).
4. Discussion
This study demonstrates that naturally generated Aβ42 antibodies mainly target the N-terminal Aβ1-15 epitope, as measured by ELISA. The N-terminal antibody against Aβ42 peptide is the major immunoglobulin induced by active Aβ42 peptide immunization and in clinic has been applied for passive immunization to treat the patients with AD [21, 22]. The anti-Aβ16-30 and anti-Aβ1-42F antibodies were less detectable with the ELISA method but significantly different in AD from those in NC as measured by the dot-blot assay. The C-terminal Aβ30-42 antibodies were undetectable in both ELISA and dot-blot approaches, indicating this part of the peptide may not be targeted by the humoral immune system, or in an undetectable under the current method although the c-terminal antibodies have been identified using phage display method (36). The Aβ30-42 epitope was reported to be more hydrophobic and less immunogenic than the other Aβ epitopes [23, 24].
The autoantibodies against full-length Aβ1-42 peptide were relatively low as measured by both ELISA and dot blot in spite of Aβ1-15 epitope is present within it. This phenomenon may be partly due to the conformational interferences of the c-terminal part to make the N-terminal epitope unreachable.
While only Aβ1-15 antibodies were significantly different in AD from NC subjects with ELISA approach, the dot-blot assay additionally demonstrated significant reduction of the fibrillar Aβ1-42 and Aβ16-30 epitope autoantibodies in AD serum compared with NC. It has been reported that fibril Aβ1-42 peptide is elevated in AD CSF [25] and its autoantibodies were reduced in AD [8]. Our dot-blot results of reduced antibodies against fibrillar Aβ1-42 in AD serum are consistent with the above report.
We have noticed that the serum autoantibodies binding to fibril Aβ1-42 exceed Aβ1-15 in dot blot while in ELISA the result was just opposite. The reasons behind this phenomenon are unclear. In general, the sensitivity of dot blot is better than that of ELISA. In ELISA method, the antigens bind to the polystyrene surface with intermolecular attraction forces, thereby the most part of the peptide are attainable even with a short peptide i.e. Aβ1-15. However, in dot-blot, the proteins bound to nitrocellulose with variety forces including hydrophobic interactions, electrostatic interactions and hydrogen bonding. These tightly binding forces may impede the further antibody binding capacity especially with the short peptide while larger fibril Aβ1-42 molecules take advantage of these factors in dot blot with more epitope exposure and less spatial hinder effects. Further studies may reveal more on it.
Another interesting phenomenon is the age-related reduction of Aβ1-15 antibodies in AD but not in NC group, indicating a diagnostic value in AD over 65 years of age. Higher levels of Aβ1-15 antibodies in NC subjects, especially in those after 75 years of age, might be an indication of preventive factor of the auto-antibodies on AD, or a necessary factor for a longer life span.
It must be indicated that studies on Aβ autoantibody levels in AD were inconsistent as measured by ELISA. Many factors can impede the ELISA results including nonspecific bindings, incorrect diagnosis, Aβ interference, and small sample size. We considered that low binding efficiency of full-length Aβ1-42 peptide to the autoantibodies in coating plate could be one of the important interfering factors. Our study demonstrated that Aβ1-15 peptide exhibited best binding and its autoantibody levels were lower in AD than in control. These results were consistent with a few studies (7, 8, 9, 10). Additionally, there were reports to indicate that autoantibodies-Aβ complex levels increased in AD (38, 39). As indicated in their discussion, the complex increase could be associated with defective immune system in AD. In general, the macrophages in healthy individuals capture the antigen-antibodies complex and transferred to liver or spleen for degradation [40, 41]. However, in AD patients, such a clearance mechanism is defected due to low avidity of the autoantibodies [42]. In this aspect, lower non-bound autoantibodies in AD measured in our results can draw the same conclusion as increase of bounded autoantibodies in AD. Treatment with Aβ antibodies were beneficial for AD patient by correction of defective immune system [43]. The Aβ antibodies could prevent A β neurotoxicity [26, 27], inhibit Aβ oligomer formation [17, 28, 37] and increase phagocytic clearance of Aβ [29]. However, Aβ autoantibodies have also been associated with angiopathy in brain in some AD patients who has been treated with the IV autoantidodies (44). Therefor it is still not clear what roles it plays for naturally occurring Aβ antibodies in circulation in both AD. With the result of our present study, it is desirable to further study the possibility of Aβ 1-15 autoantibodies as a diagnostic marker or as a marker to monitor therapeutic effects on AD. Additionally, fibril Aβ42 antibodies could also be a valuable marker when dot blot method is used.
Isotype antibodies such as IgG1, IgG2 and IgG3 to Aβ1-15 were all detectable in the serum. Although levels of IgG2 showed slightly higher in this test, more studies are needed to clarify the significances of these phenomena. IgG1 is the major IgG isotype in human serum and IgG2 is the non-inflammatory antibodies. It is desirable to do more studies on comparing IgG isotype antibodies in AD with non-AD control.
We further evaluated the relationship of the Aβ1-15 antibodies with the genotype of ApoE and ANKK1/DRD2 genes. The distribution of ApoE alleles in this set of patients is consistent with the literatures reported that higher number of E4/E4 alleles exhibited in AD patients than those of the none-AD subjects [31,32]. It has been known that E4 allele of ApoE protein is less efficient in proteolysis of beta amyloid, thus increases the brain amyloid deposition in individuals with this gene variant [33]. Interestingly, the patients with double E4 alleles in both AD and NC groups exhibited low levels of amyloid antibodies in the serum in this study. Although only a limited number of sample were tested, it is convincible since all the double E4 patients consistently exhibited low levels of antibodies in both AD and NC groups. A statistically significant result could be reached if more numbers of E4/E4 subjects were tested. The significance of this phenomenon remains to be further explored.
A1 mutation of ANKK1 gene has been associated with alcoholism, smoking and drug addiction [34,35]. The relationship of this mutation with AD has not been reported. In this study, AD patients exhibited less A1 mutation in comparison with the NC group. In consideration of variable factors in pathogenesis of AD, ANKK1 gene may deserve further investigation to clarify its association with AD. The A1 mutation also resulted in a trend of higher levels of Ab1-15 antibodies than non A1 carriers, indicating that A1 mutation might be a potential factor in prevention of AD through increase of anti-Aβ antibody production.
Acknowledgments
This work was supported by P30 AG12300 (Cor B), R01 HD048179, and NIDRR H133A020526 (to RDA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 2.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–5. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 3.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM AN1792(QS-21)-201 Study Team. Clinical effects of A- beta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 4.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JAR. Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. The Lancet. 2008;372:216–233. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 5.DeMattos RB, Bales Kelly R, Cummins David J, Cosme Dodart Jean, Paul Steven M, Holtzman David M. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- 7.Weksler ME, Relkin N, Turkenich R, LaRusse S, Zhou L, Szabo P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp Gerontol. 2002;37:943–948. doi: 10.1016/s0531-5565(02)00029-3. [DOI] [PubMed] [Google Scholar]
- 8.Moir RD, Tseitlin KA, Soscia S, Hyman BT, Irizarry MC, Tanzi RE. Autoantibodies to redox-modified oligomeric Aβ are attenuated in the plasma of Alzheimer's disease patients. J Biol Chem. 2005;280:17458–17463. doi: 10.1074/jbc.M414176200. [DOI] [PubMed] [Google Scholar]
- 9.Brettschneider S, Morgenthaler NG, Teipel SJ, Fischer-Schulz C, Bürger K, Dodel R, Du Y, Möller HJ, Bergmann A, Hampel H. Decreased serum amyloid beta(1-42) autoantibody levels in Alzheimer's disease, determined by a newly developed immuno-precipitation assay with radio labeled amyloid beta(1-42) peptide. Biol Psychiatry. 2005;57:813–816. doi: 10.1016/j.biopsych.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Sohn JH, So JO, Hong HJ, Kim JW, Na DR, Kim M, Kim H, Nam E, Ha HJ, Kim YH, Mook-Jung I. Identification of autoantibody against beta-amyloid peptide in the serum of elderly. Front Biosci. 2009;14:3879–3883. doi: 10.2741/3496. [DOI] [PubMed] [Google Scholar]
- 11.Hyman BT, Smith C, Buldyrev I, Whelan C, Brown H, Tang MX, Mayeux R. Autoantibodies to amyloid-beta and Alzheimer's disease. Ann Neurol. 2001;49:808–810. doi: 10.1002/ana.1061. [DOI] [PubMed] [Google Scholar]
- 12.Baril L, Nicolas L, Croisile B, Crozier P, Hessler C, Sassolas A, McCormick JB, Trannoy E. Immune response to Aβ-peptides in peripheral blood from patients with Alzheimer's disease and control subjects. Neurosci Lett. 2004;355:226–230. doi: 10.1016/j.neulet.2003.10.071. [DOI] [PubMed] [Google Scholar]
- 13.Britschgi M, Olin CE, Johns HT, Takeda-Uchimura Y, LeMieux MC, Rufibach K, Rajadas J, Zhang H, Tomooka B, Robinson WH, Clark CM, Fagan AM, Galasko DR, Holtzman DM, Jutel M, Kaye JA, Lemere CA, Leszek J, Li G, Peskind ER, Quinn JF, Yesavage JA, Ghiso JA, Wyss Coray T. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:12145–12150. doi: 10.1073/pnas.0904866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nath A, Hall E, Tuzova M, Dobbs M, Jons M, Anderson C, Woodward J, Guo Z, Fu W, Kryscio R, Wekstein D, Smith C, Markesbery WR, Mattson MP. Autoantibodies to amyloid beta peptide (Aβ) are increased in Alzheimer's disease patients and Aβ antibodies can enhance Aβ neurotoxicity: implications for disease pathogenesis and vaccine development. Neuromolecular Med. 2003;3:29–39. doi: 10.1385/nmm:3:1:29. [DOI] [PubMed] [Google Scholar]
- 15.Gruden MA, Davidova TB, Malisauskas M, Sewell RD, Voskresenskaya NI, Wilhelm K, Elistratova EI, Sherstnev VV, Morozova-Roche LA. Differential neuroimmune markers to the onset of Alzheimer's disease neurodegeneration and dementia: autoantibodies to Aβ(25-35) oligomers, S100b and neurotransmitters. J Neuroimmunol. 2007;186:181–192. doi: 10.1016/j.jneuroim.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 16.Gustaw KA, Garrett MR, Lee HG, Castellani RJ, Zagorski MG, Prakasam A, Siedlak SL, Zhu X, Perry G, Petersen RB, Friedland RP, Smith MA. Antigen-antibody dissociation in Alzheimer disease: a novel approach to diagnosis. J Neurochem. 2008;106:1350–1356. doi: 10.1111/j.1471-4159.2008.05477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klaver AC, Patrias LM, Coffey MP, Finke JM, Loeffler DA. Measurement of anti-Aβ1- 42 antibodies in intravenous immunoglobulin with indirect ELISA: the problem of nonspecific binding. J Neurosci Methods. 2010;187:263–269. doi: 10.1016/j.jneumeth.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Li JP, Zhibing Y, Wei Q, Zhikai C, Jie X, Jinbiao L. Low avidity and level of serum anti Aβ antibodies in Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:127–132. doi: 10.1097/00002093-200607000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Klatka LA, Schiffer RB, Powers JM, Kazee AM. Incorrect diagnosis of Alzheimer's disease. A clinicopathologic study. Arch Neurol. 1996;53:35–42. doi: 10.1001/archneur.1996.00550010045015. [DOI] [PubMed] [Google Scholar]
- 20.Jellinger KA. Criteria for the neuropathological diagnosis of dementing disorders: routes out of the swamp? Acta Neuropathol. 2009;117:101–110. doi: 10.1007/s00401-008-0466-z. [DOI] [PubMed] [Google Scholar]
- 21.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Aβ species removal after Aβ42 immunization. J Neuropathol Exp Neurol. 2006 Nov;65(11):1040–8. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 22.Fu HJ, Liu B, Frost JL, Lemere CA. Amyloid-β Immunotherapy for Alzheimer's Disease. CNS Neurol Disord Drug Targets. 2010;9:197–206. doi: 10.2174/187152710791012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramos I, Fabris D, Qi W, Fernandez EJ, Good TA. Kinetic Study of β-Amyloid residue accessibility using Reductive Alkylation and Mass Spectrometry. Biotechnol Bioeng. 2009;104:181–192. doi: 10.1002/bit.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M, Allsop D, Nakagawa M. High-molecular-weight β-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. The FASEB J. 2012;24:2716–2726. doi: 10.1096/fj.09-150359. [DOI] [PubMed] [Google Scholar]
- 26.Du Y, Wei X, Dodel R, Sommer N, Hampel H, Gao F, Ma Z, Zhao L, Oertel WH, Farlow M. Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicity. Brain. 2003;126:1935–1939. doi: 10.1093/brain/awg191. [DOI] [PubMed] [Google Scholar]
- 27.Dodel R, Balakrishnan K, Keyvani K, Deuster O, Neff F, Andrei-Selmer LC, Röskam S, Stüer C, Al-Aβed Y, Noelker C, Balzer-Geldsetzer M, Oertel W, Du Y, Bacher M. Naturally occurring autoantibodies against {beta}-amyloid: investigating their role in transgenic animal and in vitro models of Alzheimer's disease. J Neurosci. 2011;31:5847–5854. doi: 10.1523/JNEUROSCI.4401-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legleiter J, Czilli DL, Gitter B, DeMattos RB, Holtzman DM, Kowalewski T. Effect of different anti-Aβ antibodies on Aβ fibrillogenesis as assessed by atomic force microscopy. J Mol Biol. 2004;335:997–1006. doi: 10.1016/j.jmb.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 29.Istrin G, Bosis E, Solomon B. Intravenous immunoglobulin enhances the clearance of fibrillar amyloid-beta peptide. J Neurosci Res. 2006;84:434–443. doi: 10.1002/jnr.20886. [DOI] [PubMed] [Google Scholar]
- 30.Bachmeier C, Paris D, Beaulieu-Abdelahad D, Mouzon B, Mullan M, Crawford F. A Multifaceted Role for apoE in the Clearance of Beta-Amyloid across the Blood-Brain Barrier. Neurodegener Dis. 2012;8 doi: 10.1159/000337231. [DOI] [PubMed] [Google Scholar]
- 31.Corder EH, Saunders AM, Strittmatter WJ, et al. Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 32.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 33.Leduc V, Domenger D, De Beaumont L, Lalonde D, Bélanger-Jasmin S, Poirier J. Function and comorbidities of apolipoprotein e in Alzheimer's disease. Int J Alzheimers Dis. 2011;2011:1–22. doi: 10.4061/2011/974361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyers JL, Nyman E, Loukola A, Rose RJ, Kaprio J, Dick DM. The association between DRD2/ANKK1 and genetically informed measures of alcohol use and problems. Addict Biol. 2013;18:523–36. doi: 10.1111/j.1369-1600.2012.00490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuo Y, Gilbert DG, Rabinovich NE, Riise H, Needham R, Huggenvik JI. DRD2-related TaqIA polymorphism modulates motivation to smoke. Nicotine Tob Res. 2009;11:1321–9. doi: 10.1093/ntr/ntp141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshihara T, Takiguchi S, Kyuno A, et al. Immunoreactivity of phage library-derived human single-chain antibodies to amyloid beta conformers in vitro. J Biochem. 2008;143:475–86. doi: 10.1093/jb/mvm239. [DOI] [PubMed] [Google Scholar]
- 37.Szabo P, Relkin N, Weksler ME. Natural human antibodies to amyloid beta peptide. Autoimmun Rev. 2008;7:415–20. doi: 10.1016/j.autrev.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Maftei M, Thrum F, Schnack C, et al. Increased levels of antigen-bound β-amyloid autoantibodies in serum and cerebrospinal fluid of alzheimer's disease patients. Plose One. 2013;8:e68996. doi: 10.1371/journal.pone.0068996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stordce D, Cammarata S, Doghi R. Elevation of β-amyloid 1-42 autoantibodies in the blood of amnestic patient with mild cognitive impairment. Arch Neurol. 2010;67:867–72. doi: 10.1001/archneurol.2010.137. [DOI] [PubMed] [Google Scholar]
- 40.Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–19. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 41.Magga J, Puli L, Pihlaja R, et al. Human intravenous immunoglobulin provides protection against Aβ toxicity by multiple mechanisms in a mouse model of Alzheimer's disease. J Neuroinflammation. 2010;7:90. doi: 10.1186/1742-2094-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jinaping L, Zhibing Y, Wei Q, et al. Low avidity and level of serum anti-abeta antibodies in Alzheimer disease. Alzheimer Drs Assoc Disord. 2006;20:127–32. doi: 10.1097/00002093-200607000-00001. [DOI] [PubMed] [Google Scholar]
- 43.Dodel R, Neff F, Noelker C, et al. Intravenous immunoglobulins as a treatment for Alzheimer's disease: rationale and current evidence. Drugs. 2010;70:513–28. doi: 10.2165/11533070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 44.Piazz F, Greenberg S, Savoiardo M, et al. Anti-amyloid b autoantibodies in cerebral amyloid angiopathy-related inflammation: Implications for amyloid-modifying therapies. Ann Neurol. 2013;73:449–58. doi: 10.1002/ana.23857. [DOI] [PubMed] [Google Scholar]