

Serum provides a relatively inexpensive source of biomolecules, including proteins and proteoglycans, that can support cell growth on various substrates.[1] For example, serum-borne biomolecules can adsorb onto a culture substrate, while also binding to a cell-surface receptor to mediate cell adhesion onto the underlying material. However, biomolecule adsorption onto culture materials is random and non-specific, which introduces difficulties when trying to immobilize specific subsets of biomolecules.[2] In addition, a single serum-borne bio-molecule may interact with different cell surface receptors and elicit different cell responses. For example, fibronectin includes domains that bind to the integrin family of cell adhesion receptors,[3] as well as a heparin-binding domain,[4] and a variety of growth factor binding sites.[5] However, the interaction of an adsorbed protein’s functional domain with its cognate receptor may be sterically hindered, either due to the proximity of the substrate or neighboring adsorbed biomolecules, and domain activity may be altered by substrate-induced changes in protein conformation or orientation.[6] To circumvent these limitations, numerous groups have developed chemically well-defined cell culture materials with covalently immobilized cell adhesion or heparin-binding motifs.[7] These materials are commonly used to characterize cell response resulting from the direct interaction of immobilized heparin-binding or cell adhesion motifs with cell surface proteoglycans or receptors, respectively.[8–11] However, less attention has been paid to materials that can non-covalently sequester soluble molecules (e.g., heparin or growth factors) at the cell-material interface.
Here we explored endothelial cell responses to oligo(ethylene glycol) (OEG) self-assembled monolayers (SAMs) presenting a heparin-binding peptide derived from fibroblast growth factor-2 (FGF-2),[12,13] Lys-Arg-Thr-Gly-Gln-Tyr-Lys-Leu (KRTGQYKL) (Figure 1A). We hypothesized that this peptide would sequester heparin from standard fetal bovine serum, while the underlying OEG groups would prevent non-specific protein adsorption. In turn, we reasoned that sequestered, serum-borne heparin would mediate binding of a heparin-binding growth factor, fibroblast growth factor-2 (FGF-2), onto the SAM (Figure 1B), resulting in amplified FGF-2 bioactivity at the cell-material interface. In total, this report establishes a general concept wherein materials engineered to non-covalently sequester specific soluble biomolecules at the cell-material interface can enable detailed studies on the influence of common cell culture supplements, such as serum and growth factors, on cell behavior. Importantly, the materials introduced in this report are likely to provide useful basic cell biology tools that can probe the role of diverse biomolecules on cell behavior, as well as novel biomaterials that can modulate cell behavior for tissue engineering applications in vitro and in vivo.
Figure 1.
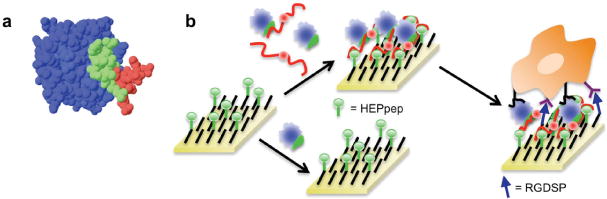
Cell culture substrates presenting a heparin-binding peptide sequester heparin from serum and, in turn, amplify recombinant growth factor activity. A) 3-D rendition of a heparin tetrasaccharide (red) binding to HEPpep, which is derived from FGF-2 (HEPpep is green, the rest of the FGF-2 protein is blue). PDB accession number 1BFB. B) A SAM presenting a heparin-binding peptide (green) sequesters a growth factor (blue) in the presence of serum-borne heparin (red), and presents the growth factor to cell surface receptors, resulting in amplified growth factor activity.
To characterize sequestering of soluble, serum-borne heparin, we exposed SAMs presenting a heparin-binding peptide (“HEPpep SAMs”) to fetal bovine serum (FBS), a standard cell culture component known to contain heparin both as a glycosaminoglycan (GAG) and a component of proteoglycans (PGs).[14] We used polarization-modulation infrared reflectance-absorbance spectroscopy (PM-IRRAS) to characterize the composition of molecules sequestered on the surface. 1% HEPpep SAMs incubated in a 50% FBS solution showed increased IR absorbance at wavenumbers corresponding to amide I, amide II, and sulfate functionalities (λ = 1666 cm−1, 1550 cm−1, 1230 cm−1, 1030 cm−1, respectively) (Figure 2A). These wavenumbers were similar to previously published IR data of proteins[15] and heparin GAGs[16] on substrates, suggesting that 1% HEPpep SAMs sequester heparin, either as a PG or GAG, as well as heparin-binding proteins from FBS. In contrast, SAMs presenting 1% of a scrambled, non-functional version of HEPpep with identical amino acid composition and net charge (“scramble”) demonstrated no change from baseline when incubated in a 50% FBS solution (Figure 2A), indicating that heparin sequestering was specific. To gain further insight into the composition of biomolecules sequestered from serum, we pre-treated FBS with heparin lyase I (HLyI), an enzyme that selectively cleaves heparin GAGs.[17] IR absorbance of 1% HEPpep SAMs incubated with HLyI-treated FBS did not change compared to baseline (Figure 2A), indicating that no serum-derived molecules bind to HEPpep SAMs after heparin digestion. Taken together, these binding results indicate that HEPpep SAMs sequester heparin from serum.
Figure 2.
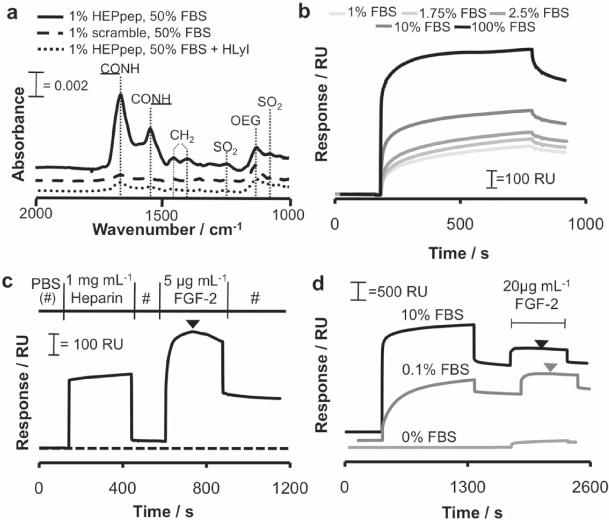
Analysis of heparin and growth factor binding to HEPpep SAMs. (A) PM-IRRAS spectra of a 1% HEPpep SAM exposed to 50% FBS (solid line), a 1% ‘scramble’ SAM exposed to 50% FBS (dashed line), or a 1% HEPpep SAM exposed to 50% FBS treated with 10 units of heparin lyase I (dotted line). B) SPR sensorgrams of 1% HEPpep SAMs exposed to 1–100% FBS. C) SPR sensorgram of 1 mg mL−1 heparin followed by 5 μg mL−1 FGF-2 passed over a 1% HEPpep SAM. D) SPR sensorgram of 0% (light gray), 0.1% (gray), or 10% (black) FBS followed by 20 μg mL−1 FGF-2 passed over a 1% HEPpep SAM. Arrows represent point of sensorgram inflection where heparin is released from HEPpep SAMs due to FGF-2 out-competing HEPpep
We then used surface plasmon resonance (SPR) to characterize the binding kinetics associated with serum-borne biomolecule binding to HEPpep SAMs. The SPR signal increased as a function of serum volume fraction over the range of 1–100% FBS (Figure 2B), and binding approached saturation when 100% FBS was passed over the SAM. Next, we calculated the observed binding rate constant (kobs) and dissociation rate constant (koff) from the SPR signals detected when 1–100% FBS was passed over a 1% HEPpep SAM. The change in kobs as a function of serum volume fraction was non-linear when 1% HEPpep SAMs were exposed to 0–100% FBS (Supporting Information, Figure 1A). Similarly, the dependence of koff on serum volume fraction was also non-linear when 1% HEPpep SAMs were exposed to 0–100% FBS (Supporting Information, Figure 1B). Such non-linear behavior is associated with equilibrium binding mechanisms involving at least two steps, where one step is concentration-dependent.[18] This observation is not surprising, as serum-borne heparin and heparin-binding proteins are likely to bind sequentially to HEPpep SAMs exposed to FBS. The rate of protein binding to HEPpep SAMs is dependent on the density of heparin bound to the surface, and the density of bound heparin is expected to increase with FBS volume fraction. In addition, koff is dependent on the release of heparin and heparin-binding proteins, either alone or in combination, from the surface (Supporting Information, Figure 1C). As the density of heparin and heparin-binding proteins bound to HEPpep SAMs increases, it is expected that koff will become increasingly dependent on the release of each species from the surface.
Next, we used SPR to characterize the sequential binding of heparin and a heparin-binding growth factor of high biological relevance, fibroblast growth factor-2 (FGF-2). Passing a 1 mg mL−1 heparin glycosaminoglycan (GAG) solution over a 1% HEPpep SAM resulted in a 44 response unit (RU) shift (Figure 2C). Subsequently, passing a 5 μg mL−1 recombinant human FGF-2 solution over this SAM resulted in a 564 RU shift (Figure 2C). During the FGF-2 binding phase, we also observed that FGF-2 in solution began out-competing immobilized HEPpep for heparin GAG binding, resulting in release of heparin GAGs from the surface as observed by an inflection point in the SPR sensorgram (indicated by black arrowhead in Figure 2C). Thus, heparin bound to HEPpep SAMs can subsequently sequester the heparin-binding growth factor FGF-2 onto the substrate.
Importantly, heparin sequestered from standard cell culture medium also led to subsequent binding of FGF-2. When 0.1% or 10% FBS was first passed over a 1% HEPpep SAM followed by 20 μg mL−1 FGF-2, there was a substantial increase in SPR response after the serum binding phase (1419 or 2109 RU, respectively), and a significant secondary increase in SPR response during the FGF-2 binding phase (582 or 491 RU, respectively) (Figure 2D). As the concentration of FGF-2 passed over the SAM was increased, the maximum shift in SPR response due to FGF-2 binding also increased, as expected (Supporting Information, Figure 2). Similar to the heparin binding competition observed between immobilized HEPpep and soluble FGF-2 (Figure 2C), we also observed an inflection point in SPR sensorgrams when FGF-2 was passed over HEPpep SAMs exposed to FBS (arrowheads in Figure 2D), which demonstrates that soluble FGF-2 can compete with immobilized HEPpep for bound, serum-borne heparin. In contrast, when 1× PBS (0% FBS) followed by 20 μg mL−1 FGF-2 was passed over a 1% HEPpep SAM, only a slight 150 RU shift was observed (Figure 2D), which is consistent with low-level non-specific protein binding.[8] Taken together, these results demonstrate that HEPpep SAMs sequester serum-borne heparin, which, in turn, can sequester FGF-2 onto the substrate.
SAMs presenting HEPpep alongside the cell adhesion peptide RGDSP were then used to characterize the influence of heparin sequestering on a key biological outcome of FGF-2 signaling -endothelial cell proliferation. Here, mixed HEPpep/RGDSP SAMs were prepared by reacting COOH-terminated SAMs with aqueous buffered solutions containing 50 mole percent RGD and 50 mole percent HEPpep + scramble, as this method provides control over the density of each peptide immobilized on a SAM (Supporting Information, Figure 3). A 2% HEPpep surface density enhanced HUVEC proliferation in medium supplemented with 10% FBS and 1 ng mL−1 FGF-2, while a HEPpep surface density ≥ 1% HEPpep enhanced HUVEC proliferation in medium supplemented with 10% FBS and 5 ng mL−1 FGF-2 over 72 hours (Figure 3A–B). Of note, HUVEC growth rate on 2% HEPpep SAMs in serum-free medium supplemented with 5 ng mL−1 FGF-2 was unchanged compared to cells on control SAMs over 72 hours (Figure 3C), which indicates that serum-borne biomolecules are required to amplify growth factor activity on HEPpep SAMs. When taken together with the observation that serum-borne heparin binds to HEPpep SAMs (Figure 2), these results suggest that sequestered serum-borne heparin can concentrate a recombinant FGF-2 supplement at the cell-material interface, thereby amplifying HUVEC proliferation. It is worth noting, however, that additional heparin-binding, serum-borne biomolecules may also be concentrated at the cell-material interface, where they can work in concert with FGF-2 and heparin to amplify HUVEC proliferation.
Figure 3.
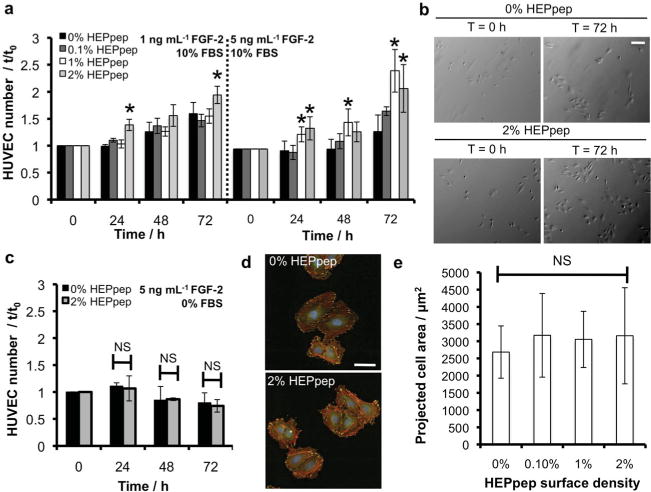
Heparin sequestered from serum amplifies FGF-2-mediated HUVEC proliferation. HUVEC number on SAMs presenting 2% RGDSP and various densities of HEPpep/scramble (for all conditions mol% HEPpep + mol% scramble = 2%) in medium with 10% FBS and 1 ng mL−1 FGF-2 or 10% FBS and 5 ng mL−1 FGF-2. B) Bright-field photomicrographs of HUVECs on SAMs presenting 2% RGDSP and 0% or 2% HEPpep at t = 0 and 72 h in medium supplemented with 10% FBS and 5 ng mL−1 FGF-2 (scalebar = 100 μ m). C) HUVEC number on SAMs presenting 2% RGDSP and 2% HEPpep or 2% scramble (0% HEPpep) in serum-free medium supplemented with 5 ng mL−1 FGF-2. D) Fluorescence photomicroscopy images of HUVECs in 10% FBS on HEPpep/RGDSP SAMs stained with Phalloidin (red), anti-vinculin (green) and dapi (blue) at t = 0 h (scalebar = 200 μ m). E) HUVEC projected cell area on SAMs presenting 2% RGDSP and 0%, 0.1%, or 1% HEPpep at t = 72 h. NS represents no significant difference, * represents significant difference compared to ‘0%’ condition, ANOVA with Dunnett’s post-hoc comparison (N = 3 for proliferation, N = 10 for spreading; P < 0.05).
In view of previous demonstrations that integrin- and heparin-binding peptides can work synergistically to influence cell adhesion,[9] and that heparin PGs can mediate cell adhesion to immobilized FGF-2,[19] we performed additional analyses to characterize the influence of HEPpep on HUVEC adhesion. Specifically, an increase in cell spreading would be indicative of a direct interaction between cell surface receptors and sequestered serum-borne heparin. There was no difference in HUVEC spreading on control or RGDSP/HEPpep SAMs at t = 0 or 72 h (Figure 3D–E), which suggests that there is no direct interaction between cell surface receptors and sequestered serum-borne heparin. These data are consistent with the assertion that enhanced HUVEC proliferation on HEPpep SAMs is due to amplified growth factor signaling resulting from indirect sequestering rather than direct cell-material adhesion.
Materials that can sequester a particular type of serum-borne biomolecule address key challenges in biomaterials design: nonspecific adsorption and heterogeneous display of serum-borne biomolecules at the cell-material interface. Here we demonstrated that serum-borne heparin can be sequestered by a material presenting a heparin-binding ligand, and sequestered heparin amplifies the activity of FGF-2 by non-covalently localizing the growth factor at the cell-material interface. The heparin-binding materials described in this report may enable further efforts to characterize the influence of sequestered heparin on the activity of the broad array of other known heparin-binding proteins, as well as the influence of heparin on additional cell types, such as multipotent stem cells. In addition, this general approach may be broadly applicable to other serum-borne biomolecules by changing the identity of the sequestering ligand immobilized on the substrate.
Experimental Section
Polarization-Modulation Infrared Reflectance-Absorbance Spectroscopy
1% HEPpep or 1% scramble peptide SAMs were incubated in 1× PBS solutions containing 50% FBS or 50% FBS and 10 units heparin lyase I for 20 minutes. A Nicolet Magna-IR 860 FT-IR spectrometer with photoelastic modulator (PEM-90, Hinds Instruments, Hillsboro, OR), synchronous sampling demodulator (SSD-100, GWC Technologies, Madison, WI), and a liquid nitrogen-cooled mercury cadmium telluride detector was used (incident angle = 83°, modulation center = 1500 cm−1, 500 scans per modulation center, resolution = 4 cm−1). Data were collected as differential reflectance versus wavenumber, and then normalized and converted to absorbance units versus wavenumber using the method outlined by Frey and co-workers.[20]
Surface Plasmon Resonance
Biomolecule binding onto mixed SAMs consisting of 99 mol% HS-EG3 and 1 mol% HEPpep or 1 mol% scramble was analyzed using a BIAcore 2000 system (Piscataway, NJ). Typical binding experiments were performed as follows: 1) phosphate buffered saline (pH 7.4) (PBS) was passed over the SAM to establish the plasmon resonance baseline of the surface, 2) 100 μL of 1× PBS solutions containing various concentrations of heparin, FBS or FGF-2 were passed over the SAM, and 3) PBS was passed over the SAM to establish the change in surface plasmon resonance signal due to the presence of adsorbed protein. kobs and koff were calculated using nonlinear regression analysis in GraphPad Prism software.
FGF-2 Mediated HUVEC Proliferation
HUVECs were cultured according the protocol supplied by the manufacturer. Sub-confluent cells were harvested from the plate, suspended in M199 supplemented with 10% FBS, and counted using a hemacytometer. Cells were collected as a pellet by centrifugation at 1100 rpm for 5 minutes and resuspended in fresh 10% FBS-supplemented M199 at a density of 80 000 cells mL−1. HUVECs were then seeded onto SAM substrates at a density of 10,000 cells cm−2. After allowing the HUVECs to attach to the SAMs overnight, the substrates were washed with 1× PBS to remove any loosely bound cells and placed in M199 supplemented with either 10% FBS and 1 ng mL−1 FGF-2, 10% FBS and 5 ng mL−1 FGF-2, or 0% FBS and 5 ng mL−1 FGF-2. T = 0 h for each experiment was designated as the point immediately after placing the SAMs in fresh culture medium supplemented with FGF-2. Brightfield photomicroscopy images (40× mag) of each substrate were collected on an Olympus IX51 inverted epifluorescent microscope at t = 0, 24, 48, and 72 h, and the total number of cells per viewing area was counted manually.
Analysis of Projected Cell Area
Brightfield photomicroscopy images (100× mag) of HUVECs cultured on HEPpep/RGDSP SAMs or control SAMs were collected using an Olympus IX51 inverted microscope at t = 0 h. Individual cells within each viewing area were outlined manually and the area of the outlined region was measured using ImageJ software.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01HL093282) and the University of Wisconsin (UW) Stem Cell and Regenerative Medicine Center. PM-IRRAS was performed in the National Science Foundation-funded UW-MRSEC facility and SPR was performed in the UW Biophysics Instrumentation Facility.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Gregory A. Hudalla, Department of Biomedical Engineering, University of Wisconsin-Madison, 5009 Wisconsin Institutes for Medical Research, 1111 Highland Ave., Madison, WI 53705, USA.
Justin T. Koepsel, Department of Biomedical Engineering, University of Wisconsin-Madison, 5009 Wisconsin Institutes for Medical Research, 1111 Highland Ave., Madison, WI 53705, USA
Prof. William L. Murphy, Email: wlmurphy@wisc.edu, Department of Biomedical Engineering, University of Wisconsin-Madison, 5009 Wisconsin Institutes for Medical Research, 1111 Highland Ave., Madison, WI 53705, USA Department of Biomedical Orthopedics and Rehabilitation, Department of Pharmacology, University of Wisconsin-Madison, 5009 Wisconsin Institutes for Medical Research, 1111 Highland Ave., Madison, WI 53705, USA.
References
- 1.Horbett TA. Colloids Surf B. 1994;2:225. [Google Scholar]
- 2.Horbett TA. Thromb Haemost. 1984;51:174. [PubMed] [Google Scholar]
- 3.Pierschbacher MD, Ruoslahti E. Proc Natl Acad Sci USA. 1984;81:5985. doi: 10.1073/pnas.81.19.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingham KC, Brew SA, Atha DH. Biochem J. 1990;272:605. doi: 10.1042/bj2720605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martino MM, Hubbell JA. FASEB J. 2010;24:4711. doi: 10.1096/fj.09-151282. [DOI] [PubMed] [Google Scholar]
- 6.Lhoest JB, Detrait E, van den Bosch de Aguilar P, Bertrand P. J Biomed Mater Res. 1998;41:95. doi: 10.1002/(sici)1097-4636(199807)41:1<95::aid-jbm12>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 7.Hudalla GA, Murphy WL. Soft Matter. 2011;7:9561. doi: 10.1039/C1SM05596H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hudalla GA, Murphy WL. Langmuir. 2009;25:5737. doi: 10.1021/la804077t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudalla GA, Murphy WL. Langmuir. 2010;26:6449. doi: 10.1021/la1008208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klim JR, Li L, Wrighton PJ, Piekarczyk MS, Kiessling LL. Nat Methods. 2010;7:989. doi: 10.1038/nmeth.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rezania A, Healy KE. Biotechnol Prog. 1999;15:19. doi: 10.1021/bp980083b. [DOI] [PubMed] [Google Scholar]
- 12.Thompson LD, Pantoliano MW, Springer BA. Biochemistry. 1994;33:3831. doi: 10.1021/bi00179a006. [DOI] [PubMed] [Google Scholar]
- 13.Li LY, Safran M, Aviezer D, Bohlen P, Seddon AP, Yayon A. Biochemistry. 1994;33:10999. doi: 10.1021/bi00202a020. [DOI] [PubMed] [Google Scholar]
- 14.Lu H, McDowell LM, Studelska DR, Zhang L. Glycobiol Insights. 2010;2010:13. [PMC free article] [PubMed] [Google Scholar]
- 15.Boucher J, Trudel E, Methot M, Desmeules P, Salesse C. Colloids Surf B. 2007;58:73. doi: 10.1016/j.colsurfb.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 16.Martins MC, Curtin SA, Freitas SC, Salgueiro P, Ratner BD, Barbosa MA. J Biomed Mater Res A. 2009;88:162. doi: 10.1002/jbm.a.31849. [DOI] [PubMed] [Google Scholar]
- 17.Desai UR, Wang HM, Linhardt RJ. Arch Biochem Biophys. 1993;306:461. doi: 10.1006/abbi.1993.1538. [DOI] [PubMed] [Google Scholar]
- 18.Strickland S, Palmer G, Massey V. J Biol Chem. 1975;250:4048. [PubMed] [Google Scholar]
- 19.Levenstein ME, Berggren WT, Lee JE, Conard KR, Llanas RA, Wagner RJ, Smith LM, Thomson JA. Stem Cells. 2008;26:3099. doi: 10.1634/stemcells.2007-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey BL, Corn RM, Weibel SC. Handbook of Vibrational Spectroscopy. Vol. 2. Wiley & Sons; New York: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.