

Abstract
Background
The tumor suppressors p14ARF (ARF) and p16INK4A (p16) are encoded by overlapping reading frames at the CDKN2A/INK4A locus on chromosome 9p21. In human melanoma, the accumulated evidence has suggested that the predominant tumor suppressor at 9p21 is p16, not ARF. However, recent observations from melanoma-prone families and murine melanoma models suggest a p16-independent tumor suppressor role for ARF. We analyzed a group of melanoma metastases and cell lines to investigate directly whether somatic alterations to the ARF gene support its role as a p16-independent tumor suppressor in human melanoma, assuming that two alterations (genetic and/or epigenetic) would be required to inactivate a gene.
Methods
We examined the p16/ARF locus in 60 melanoma metastases from 58 patients and in 9 human melanoma cell lines using multiplex ligation-dependent probe amplification and multiplex polymerase chain reaction (PCR) to detect deletions, methylation-specific PCR to detect promoter methylation, direct sequencing to detect mutations affecting ARF and p16, and, in a subset of 20 tumors, immunohistochemistry to determine the effect of these alterations on p16 protein expression. All statistical tests were two-sided.
Results
We observed two or more alterations to the ARF gene in 26/60 (43%) metastases. The p16 gene sustained two or more alterations in 13/60 (22%) metastases (P =.03). Inactivation of ARF in the presence of wild-type p16 was seen in 18/60 (30%) metastases.
Conclusion
Genetic and epigenetic analyses of the human 9p21 locus indicate that modifications of ARF occur independently of p16 inactivation in human melanoma and suggest that ARF is more frequently inactivated than p16.
The CDKN2A/INK4A locus at chromosome 9p21, which includes two tumor suppressor genes that share a common second exon, is a critical target of inactivation in cancer biology. p16INK4A (p16) is transcribed from exons 1α, 2, and 3, with exon 3 encoding only four amino acids. p14ARF (ARF) is encoded by an alternative exon 1 (1β) and the shared exon 2. Exon 1β is located approximately 20 kb upstream of p16 exon 1α. The p16 and ARF transcripts are translated in different reading frames; thus the two proteins have no physical homology. Both proteins function as tumor suppressors, acting through different pathways: ARF via the p53 pathway (1, 2) and p16 via the retinoblastoma (RB) pathway (3). Evidence of a role for p16 in human melanoma includes frequent genetic and epigenetic alterations in human melanoma specimens (4–6) and cell lines (7), the presence of germline mutations in this gene in 10%–50% of familial melanoma cases (8–11), and data from murine models in which the specific deletion of exon 1α combined with melanocyte-specific expression of a mutant Ras transgene produced melanomas (12). In vitro studies of cells from a patient with biallelic germline mutations at 9p21 suggested that in humans it is p16, rather than ARF, that is critical in Ras-induced arrest of fibroblasts (13). The role of ARF in susceptibility to melanoma has therefore been questioned.
More recent evidence, however, challenges the central role of p16 in melanomagenesis. Members of several melanoma families have been reported to share an exon 1β germline deletion (14–16) or mutation in either the coding region or splice donor site of this exon (17–19), suggesting an important role for the ARF gene in the human disease. In murine melanoma models using either a Ras transgene driven by the melanocyte-specific tyrosinase promoter or the Met ligand hepatocyte growth factor/scatter factor driven by the metallothonein promoter, it has been observed that targeted deletion of exon 1β alone leads to spontaneous melanoma. Moreover, melanomas develop in these mice with a substantially shorter latency period than similarly constructed mice with a deletion of only exon 1α (12, 20). Most recently, ARF has been demonstrated to be a regulator of melanocyte senescence, independent of p53 activity (20). These data suggest a p16-independent role for ARF in melanomagenesis.
Somatic ARF inactivation has also been observed in a variety of human tumors (21–23), but no in-depth studies have, to our knowledge, directly assessed the status of the ARF gene in human melanoma tumors. Therefore, we performed a comprehensive analysis of the pattern of genetic and epigenetic alterations to the p16 and ARF tumor suppressor loci in melanoma.
Methods
Human Tumor Specimens and Cell Lines
Sixty metastatic melanoma tumor samples and corresponding normal tissues removed at the time of metastasectomy were obtained from 58 patients. Specimens were embedded in a cryopreservative solution, ornithine carbonyl transferase compound (Miles Laboratories, Elkhart, IN); snap frozen in isopentane; and stored at −70°C in the Memorial Sloan-Kettering Cancer Center Tumor Bank. Cut sections were stained with hematoxylin and eosin and examined to verify the presence of at least 70% metastatic melanoma tissue. Thirty-eight of sixty lesions (63%) were from lymph nodes or soft tissue sites; 22 of 60 (36.7%) were from visceral or other sites. Informed consent was obtained from all patients, and the institutional review boards of both Memorial Sloan-Kettering Cancer Center and New York University approved the conduct of this study.
Nine human metastatic melanoma cell lines (SK-MEL 19, 94, 100, 103, 147, 173, 187, 192, and 197) derived in one of our laboratories (A. Houghton) were maintained in culture using DMEM with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin (BioWhittaker, Walkerville, MD). LoVo colon cancer cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained in FK-12 medium with 10% FCS and 1% penicillin/streptomycin (BioWhittaker).
Immunoblotting
Cell lysates were prepared by collecting cells using trypsinization and centrifugation and by incubation of the cell pellets in 2× Laemmli buffer for 20 minutes at 95°C. Fifty micrograms of protein from cell line lysates were loaded onto 4%–20% SDS–PAGE gels (Bio Rad, Hercules, CA), electrophoresed, and transferred to a nitrocellulose membrane. The membrane was blocked at room temperature for 20 minutes with a solution of 5% nonfat dry milk and 0.02% sodium azide in TBST (10 mM Tris-HCl, pH 7.4, 0.9% NaCl, and 0.05% v/v Tween-20). The blocking solution was changed and the membrane incubated with rocking at 4°C overnight with primary antibodies to ARF (Ab-2 from Oncogene, Cambridge, MA) and p16 (Ab-1, Oncogene), each at 1:100 dilution. The membrane was washed three times with TBST and incubated for 1 hour at room temperature with rocking with a 1:2000 dilution of mouse IgG (Oncogene). Equal loading of lanes was verified using the anti-Ran clone C-20 (Santa Cruz Biotechnology, Santa Cruz, CA) as a primary antibody at a 1:100 dilution with anti-goat secondary antibody (Santa Cruz) at a 1:2000 dilution. Proteins were visualized on autoradiography film (Labscientific, Livingston, NJ) using the SuperSignal West Pico chemiluminescent system (Pierce, Rockford, IL).
DNA Isolation and Polymerase Chain Reaction
Eight to ten 30-μm sections were cut from each specimen, and genomic DNA was isolated using the QIAamp DNA mini kit (Qiagen, Valencia, CA). DNA concentrations were standardized in 1.5% agarose gels in reference to the Low DNA Mass Ladder (Invitrogen, Carlsbad, CA). Amplifications were carried out in 20 μL using 100 ng DNA of genomic DNA and a mastermix consisting of purified, autoclaved water; MgCl2 buffer (Qiagen); DMSO (Sigma-Aldrich, St Louis, MO); mixed dinucleotide triphosphates (dNTPs) (Roche, Indianapolis, IN); forward and reverse primers (Applied Biosystems, Foster City, CA); and Hot Start Taq (Qiagen) according to the manufacturer's instructions. Bovine serum albumin at a final concentration of 1 μg/μL (Sigma-Aldrich) was added to buffer excess melanin. Polymerase chain reaction (PCR) conditions were optimized using an Eppendorf Mastercycler Gradient thermal cycler, and PCRs were performed on that machine or the GeneAmp PCR System 9700 (Applied Biosystems). The laboratory was divided into pre- and post-PCR rooms to avoid contamination of genomic material with aerosolized amplification products.
Sequencing
Fifty to one hundred nanograms of genomic DNA were used in direct sequencing, which was begun with a 40-cycle, primer-exhaustive PCR. Primer sequences for p14ARF exon 1β and p16INK4a exon 1α were from Fitzgerald et al. (24) and Kamb et al. (25), respectively, and the ARF and p16–shared exon 2 was from Hussussian et al. (26) Ten nanograms of PCR product was used in a cycle-sequencing reaction, and sequencing was performed on the ABI PRISM 310 Genetic Analyzer (Applied Biosystems). Sequence quality was screened using SeqScape 1.1 software (Applied Biosystems). All mutations were confirmed by bidirectional sequencing.
Deletion Analysis
Three methods were used to assess deletions at the 9p21 locus: multiplex ligation-dependent probe amplification (MLPA); semiquantitative, fluorescence-based multiplex PCR; and microsatellite analysis. In MLPA, hybridization, ligation, and PCR reactions were performed with the P024 kit (MRC-Holland, Amsterdam, The Netherlands) according to the manufacturer's specifications with a mix of 17 uniquely sized control probes and 21 target probes that spanned 9p21. DNA samples were used at a final concentration of approximately 25 ng/μL, and amplification products were visualized on an ABI 3100 DNA sequencer (Applied Biosystems). Tumor amplification products were normalized to amplification products from their normal tissue counterpart control probes. Normal samples showing a standard deviation of more than 0.29% among the WT control probes were considered to have failed the analysis. In these instances, the results of the two other deletion detection methods (described below) were used to make the final deletion determinations. Final calculation of gene dosage quotients was based on the method proposed by Schouten et al. (27) but using peak height rather than peak area as the indicator of the amount of template DNA present (28). The dosage quotient score, that is, peak height relative to control, was adjusted from a theoretical model of 0.0 for homozygous loss, 0.5 for hemizygous loss, and 1.0 for wild type (WT) to the following: 0.0–0.19 (peak height relative to control) = homozygous loss, 0.20–0.42 = borderline between homozygous and hemizygous losses, 0.43–0.69 = hemizygous loss, 0.70–0.75 = borderline between hemizygous loss and WT, and 0.76 or above = WT. These adjustments were based on the results using a panel of 100 known Taqman-validated WT blood samples as references to model dosage quotients.
In the multiplex PCR analysis, p16INK4a or p14ARF exons were simultaneously amplified along with a control gene, p53 exon 4. The sizes of the amplicons were 346 and 410 bp for p16 exons 1α and 2, respectively, and 416 and 262 bp for ARF exon 1β and p53 exon 4, respectively. p53 was used as the control because previous studies from our group and others found few gene losses and mutations at this location (29–31). The sense primer for each amplicon was labeled with a 5′-amidite for detection using an ABI 310 Genetic Analyzer. Primer concentrations were adjusted to obtain comparably strong products over a range of DNA template amounts. For each reaction, 50–100 ng of genomic DNA was amplified for 30 cycles at an annealing temperature of 58.0°C Peak heights corresponding to particular amplicons were analyzed as a ratio using the p53 peak height as the denominator. Samples were scored as WT (>75% of signal strength), deleted (<50%), or borderline (50%–75%) based on a standard curve generated by mixing DNA from cell lines known to have homozygous deletions in the region with normal DNA in known proportions. An example of the standard curve for exon 2 is shown in Supplementary Figure 1 (available online).
For microsatellite analysis, the three nearest markers were used to verify exonic deletion in cases that had failed MLPA analysis and were determined to be borderline between WT and deleted by multiplex PCR. Thirty-cycle reactions were run for the markers D9S1752, D9S974, and D9S2136 at annealing temperatures of 54.0°C, 55.0°C, and 59.0°C, respectively. Primer sequences were obtained from the GDB Human Genome Database (www.gdb.org), and the forward oligonucleotide was labeled with a 5′-amidite for detection using an ABI 310 Genetic Analyzer. Samples were considered informative if two peaks, corresponding to the two parental alleles, were observed in the normal specimen. R values were calculated by first determining the ratio between the heights of the two peaks of the normal and tumor specimens, respectively. The ratio of the tumor peak heights was divided by the ratio of the normal peak heights to yield the R value. An R value of either less than 0.5 or greater than 1.5 was considered to represent loss of one allele.
Gene loss determinations were made by comparing the results of MLPA, multiplex PCR, and microsatellite analyses. Within each method, the most conservative result (ie, the one least consistent with gene loss) of several runs was taken as the overall result. In five cases, the MLPA analysis failed. In the remaining 55 cases, gene loss determinations for each of the three ARF and p16 exons (totaling 165 determinations) were based primarily on the MLPA results. These results were confirmed by multiplex PCR in 130/165 (79%) exons. Of the 35 results that were not concordant for these methods, differences between WT and hemizgous deletions were observed in 30/35 exons, differences between hemizygous deletions and homozygous deletions in 2/35 exons, differences between WT by MLPA and gene loss by multiplex PCR in 2/35 exons, and differences between a homozygous deletion (MLPA) and WT gene dosage by multiplex PCR in 1 exon. For these 35 exons, calls were finalized based on analysis of flanking MLPA probes. Specifically, deletions were confirmed if probes on both sides of the exon under study showed losses. For the five cases lacking MLPA data, results were based on multiplex PCR and microsatellite analysis. Cases were considered to have gene losses if both assays confirmed the loss, or in one case, if the multiplex PCR showed a loss and the microsatellite analysis was noninformative. Because tissue specimens had varying degrees of normal tissue contamination, determinations of hemizygosity were accepted if scored as a deletion by MLPA and borderline by multiplex PCR. For a subset of nine cases with sufficient DNA, array comparative genomic hybridization (CGH) was also performed and the results interpreted as previously described (32).
Methylation
CpG island promoter hypermethylation was determined by methylation-specific PCR following bisulfite modification, as described previously by Herman et al. (33). Genomic DNA (300–500 μg) was modified with the CpGenome DNA Modification Kit (Chemicon International, Temecula, CA). p16 was amplified with the CpG Wiz p16 Amplification Kit (Serologicals Corporation, Norcross, GA), according to the manufacturer's instructions. For p14ARF, primers used were those previously described by Esteller et al. (22). Amplification of methylated DNA and unmethylated DNA was carried out for 45 and 40 cycles, respectively, at an annealing temperature of 60°C. All PCR products were separated on 2.5% agarose gels and stained with ethidium bromide. Positive samples were those in which two separate reactions (out of a maximum of three attempts) produced a result consistent with promoter hypermethylation. All 60 cases were analyzed for hypermethylation of both promoters.
Immunohistochemistry
Tissue sections (4-μm thick) were deparaffinized in xylene, rehydrated using a graded series of ethanols, and rinsed in distilled water. Heat-induced epitope retrieval was performed in 10 mM citrate buffer at pH 6.0 for 20 minutes in a 1200-W microwave oven at 90% power. Sections were allowed to cool for 30 minutes and then rinsed in distilled water. Antibody incubations and detection were carried out on a NEXes instrument (Ventana Medical Systems Tucson, AZ) using Ventana buffer and detection kit. In brief, endogenous peroxidase activity was blocked with hydrogen peroxide. The primary antibody (mouse anti-human p16, clone G175-405 [BD Pharmingen, San Diego, CA]) was diluted 1:40 and incubated overnight at room temperature. Detected was accomplished using a biotinylated secondary goat anti-mouse antibody, followed by application of streptavidin–peroxidase conjugate. The immune complex was visualized with 3,3-diaminobenzidene and the signal enhanced by copper sulfate addition. Slides were washed in distilled water, counterstained with hematoxylin, dehydrated, and mounted with permanent media. Cell blocks of positive (SK-MEL 94 and 147) and negative (SK-MEL 173) control cell lines were also included.
To determine the specificity of ARF antibodies in sections, we induced ARF expression in cell lines SK-MEL 192 and LoVo with methyltransferase inhibitors (described below) before placing them in cell blocks. Immunohistochemistry was performed as described above. The primary antibody was either rabbit anti-p14ARF NB200-111 or NB100-57549 (Novus Biologicals, Littleton, CO), and the secondary antibody was a goat anti-rabbit antibody. To reactivate ARF expression in SK-MEL 192 cells, zebularine (Sigma, St Louis, MO) was added to the culture medium at a final concentration of either 0.1 or 0.4 mM and cells were incubated for either 24 or 96 hours before collecting lysates. To reactivate ARF expression in the colon cancer cell line LoVo (American Type Culture Collection), 5-aza-2′-deoxycytidine (Sigma) was added to the culture medium at a final concentration of 10 μM and the cells harvested after 72 hours of treatment.
Statistical Methods
In the immunohistochemical analysis, we used a two-sided Fisher exact test to determine if there was an association between the number of “hits” to the p16 gene and expression of its protein. To determine if there was a statistically significant difference in the proportion of cases in which p16 and ARF were inactivated, we made the assumption that two “hits” were required to inactivate a gene. We compared the proportions of cases with inactivation of p16 and ARF by Fisher exact test. Confidence intervals for each proportion were calculated using the Wald method and Graphpad Software (La Jolla, CA) to assess the precision of the obtained estimates.
Results
Alterations of ARF and p16 in Human Melanoma Cell Lines
To test the feasibility of our approach, we screened nine human melanoma cell lines for expression of ARF and p16. p16 was detected by western blotting in two of nine lines, but ARF was undetectable in all nine (Figure 1). To characterize the mechanisms responsible for lack of expression of both ARF and p16, we performed assays to identify gene mutations and deletions and promoter methylation. Deletion was the most common mechanism inactivating both loci with seven of nine lines affected by either hemizygous (n = 1) or homozygous (n = 6) losses. In the six lines with homozygous losses, the most common area of deletion localized to the region between ARF exon 1β and p16 exon 1α (MLPA probes 8 and 9), and in each case the losses also encompassed either ARF (3/6), p16 (2/6), or both (1/6) (Supplementary Table 1). In the two lines (94 and 147) in which p16 could be detected by western blotting, hemizygous losses of p16 were observed, suggesting that a second mutation may not have occurred to inactivate the gene. Point mutations were observed in three lines, and methylation of the p16 promoter was noted in one (Table 1). In three of nine lines, the ARF promoter was methylated (173, 192, and 197). Because ARF methylation had not to our knowledge been previously reported in melanoma cell lines, we verified the promoter methylation results by treating SK-MEL 192 with zebularine, an inhibitor of DNA methylation (34). We detected markedly increased expression of ARF in response to a low dose of zebularine (0.1 mM) for 24 hours. When a higher dose was used (0.4 mM), the effect lasted for several days (Figure 1, B).
Figure 1.
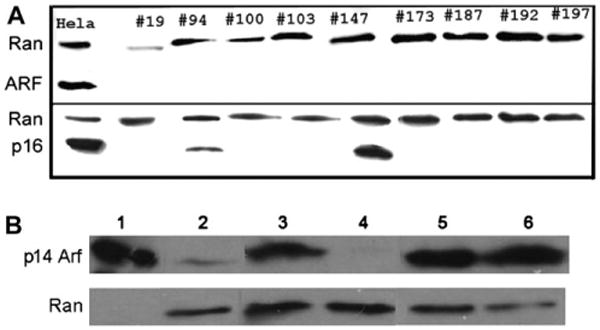
p16 and ARF expression in melanoma cell lines. A) Lysates from melanoma cell lines were subject to electrophoresis, and separated proteins were transferred to nitrocellulose membranes and probed with antibodies to p14ARF (ARF) (top) and p16INK4a (bottom). Blots were also probed with an antibody to the Ran protein as a control for equal loading and transfer. HeLa cells were used as a positive control for the expression of both ARF and p16. B) Treatment of SK-MEL 192 cells with the demethylating agent zebularine (Z). HeLa cells (underloaded due to the high expression of p14 ARF, lane 1) were used as a positive control. Lanes 2–6 were loaded with lysates prepared from SK-MEL 192 cells. Lane 2, no Z; lanes 3 and 4, after treatment with 0.1 mM Z for 24 hours (lane 3) or 96 hours (lane 4); lanes 5 and 6, after treatment with 0.4 mM Z for 24 hours (lane 5) or 96 hours (lane 6). Note the absence of p14ARF expression after 96 hours of treatment with a low dose of zebularine, compared with the sustained expression seen using the higher dose.
Table 1. Genetic and epigenetic modifications to the ARF and p16 genes in human melanoma cell lines *.
| Cell line | ARF | p16 | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Protein expression† | Promoter methylation‡ | Deletion status | Gene seqǁ | Protein expression† | Promoter methylation‡ | Deletion status | Gene seqǁ | ||
|
|
|
||||||||
| Ex 1β§ | Ex 2/3§ | Ex 1α/2/3§ | |||||||
| 19 | Absent | No | WT | WT | P72L | Absent | No | WT | R58Stop |
| 94 | Absent | No | Hemi | Hemi | WT | Present | No | Hemi | WT |
| 100 | Absent | HD | HD | HD | HD | Absent | HD | HD | HD |
| 103 | Absent | HD | HD | Hemi | WT | Absent | No | Hemi | P114L |
| 147 | Absent | HD | HD | Hemi | WT | Present | No | Hemi | P114L |
| 173 | Absent | Yes | WT | HD | WT | Absent | HD | HD | HD |
| 187 | Absent | HD | HD | WT | WT | Absent | No | WT | WT |
| 192 | Absent | Yes | WT | WT | WT | Absent | Yes | WT | WT |
| 197 | Absent | Yes | WT | HD | WT | Absent | HD | HD | HD |
HD = homozygous deletion; Hemi = hemizygous deletion; Ex = exon; WT = wild type; P = proline; L = leucine; R = arginine; Seq = sequence change.
Western blot analysis.
Promoter CpG island hypermethylation assay.
Combined deletion analysis (see text for details).
Gene sequencing was used to predict amino acid sequences.
Deletions of ARF and p16 in Metastatic Melanoma Tumors
To further assess the frequency of ARF and p16 alterations in human melanoma, we used a combination of MLPA, multiplex PCR, and microsatellite analysis to determine the presence of deletions in 60 human melanoma metastases from 58 patients (Figures 2, 3, and 4). This combination of techniques provided high resolution over the 9p21 region, including direct assessment of exons 1β, 1α, and 2 using two methodologies. The MLPA analysis, using seven probes to cover approximately 26 kb, had particularly high resolution for the region spanning exon 1β to exon 3 (Figure 2). Overall, 41/60 (68%) tumors showed deletions that included exons 1α, 1β, or 2, and 35/60 (58%) tumors sustained losses of exon 1β. In 11 of these 35 cases (31%), the p16 coding regions were completely intact. In contrast, deletions of sequences encoding p16 without concomitant loss of exon 1β were only observed in 6/60 (10%) tumors. The most common area of deletion defined by MLPA analysis was ARF exon 1β. Overall, these results suggest that ARF deletion can occur independently of p16 deletion in melanoma.
Figure 2.
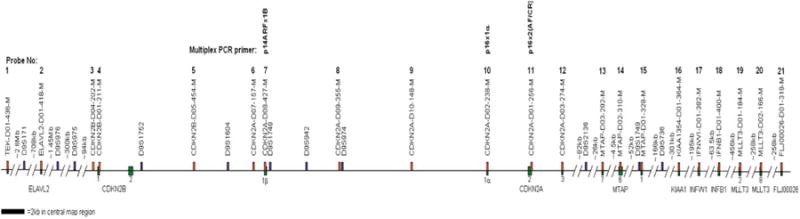
Chromosome 9p21 physical map. The locations of all primers used in deletion screening are shown. Map is to scale at the CDKN2 locus. Multiplex ligation-dependent probe amplification (MLPA) probes are indicated with orange-filled rectangles; microsatellite markers with blue-filled rectangles and multiplex PCR primers are located over MLPA probes 7, 10, and 11. Green-filled rectangles refer to exons of specific genes.
Figure 3.
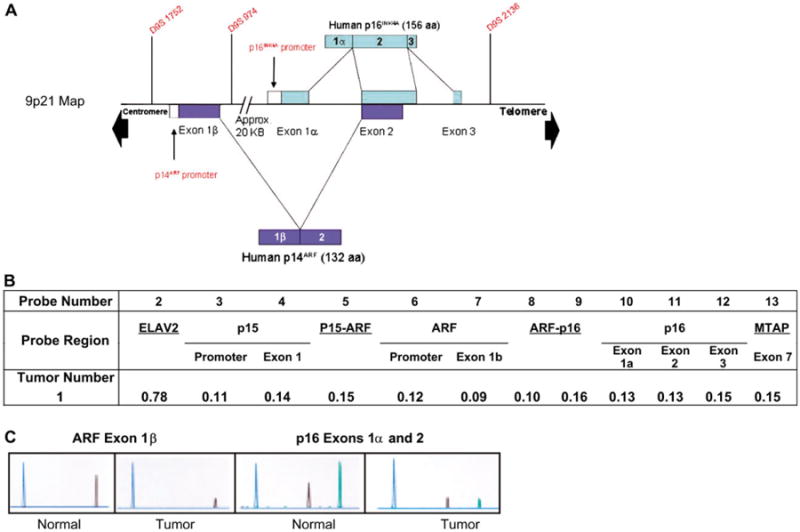
Combined deletion analysis techniques. A) Map of the 9p21 region depicting relative locations of exons and probes. B) Multiplex ligation-dependent probe amplification (MLPA) of the 9p21 locus in tumor sample 1 in which there was complete loss of p14ARF exon 1β, p16INK4a exon 1α, and the ARF/p16-shared exon 2. Of the 21 probes spanning the region, only the CDKN2 locus and flanking probes are shown. Shown are the results of calculating the gene dosage quotients for each probe. Final calculation of these quotients was based on the method proposed by Schouten et al. (27), but using peak height rather than peak area as the indicator of the amount of template DNA present (28). The scoring used was as follows: 0.0–0.19 = homozygous loss, 0.20–0.42 = borderline between homozygous and hemizygous losses, 0.43–0.69 = hemizygous loss, 0.70–0.75 = borderline between hemizygous loss and wild type (WT), and 0.76 or above = WT. See “Methods” for more details. (C) Exonic deletion analysis via semiquantitative fluorescence-based multiplex PCR of cells from tumor 1. The blue peak represents the control amplicon. For ARF, the black peak corresponds to the amplicon from exon 1β. For p16, the black peak corresponds to the amplicon from exon 1α and the green peak to the shared exon 2.
Figure 4.

Schematic diagram of alterations in 60 metastatic melanoma tissue specimens. The genomic region is shown at the top of the diagram. Multiplex ligation-dependent probe analysis (MLPA) probes are indicated with orange-filled rectangles, microsatellite markers with blue-filled rectangles, and exons with green-filled rectangles. Circles are located at each site that was tested. A black circle corresponds to a homozygous deletion, gray circle to a hemizygous loss, and an open circle to no loss. Samples in which the ARF or p16 promoter sequences were methylated are shown with a blue box around the circle corresponding to the relevant exon. Exons with mutations are shown as a red box around the relevant circle. Noninformative microsatellite marker results are shown as a circle with a central black square. Samples that failed MLPA analysis were those in which the corresponding normal samples showed a standard deviation of > 0.29 % among the wild-type control probes. These samples are shown as a circle with an “X”.
The results of the MLPA and multiplex PCR approaches were compared with data from array CGH on a subset of nine cases from which enough DNA was available for analysis. Of the five cases in which genomic losses were detected by MLPA and multiplex PCR, array CGH detected genomic losses in two. A lower detection rate by CGH is expected because the array platform used has a substantially lower resolution than more focused approaches such as MLPA and multiplex PCR, which probe smaller regions of DNA. In no case did CGH detect a loss that was not also detected using MLPA and multiplex PCR.
Mutations in ARF and/or p16 in Metastatic Melanoma Tumors
As in the melanoma cell lines, small sequence alterations such as point mutation or microdeletions and insertions in the metastatic tumors were uncommon, with only 5/60 (8%) tumors showing these types of mutations (Table 2). Only three mutations affected ARF, and all occurred in exon 2, including a novel mutation (c.242C>A) that produced amino acid substitutions that affected both ARF and p16. No mutations were found in ARF exon 1β. In one tumor, there was a 4-bp deletion in the p16 exon 1α, (c.43_46del4). Although this mutation has not, to our knowledge, been described in melanoma, it has been detected in pancreatic adenocarcinoma (36) and esophageal carcinoma (40). Corresponding normal tissues were sequenced for all patients in which a mutation was identified but no germline mutations were found.
Table 2. Mutations in p16 and ARF detected in a set of 60 human melanomas*,†.
| Nucleotide | Amino acid change(s) in p16 | Amino acid change in ARF | Previous report in somatic cells | Previous report in germline cells |
|---|---|---|---|---|
| c.43_46del4‡ | All downstream of Asp14 | None | Caldas, 1994 (36) | None |
| c.143C>T | Pro48Leu | None | Gretarsdottir, 1998 (37) | Platz, 1997 (8) |
| c.172C>T | Arg58Stop | Pro72Leu | Cairns, 1994 (38) | Hussussian, 1994 (26) |
| c.238C>T | Arg80Stop | Pro94Leu | Gruis, 1995 (39) | None |
| c.242C>A | Pro81His | Arg96Ser | None | None |
C = cytosine; T = thymine; A = adenine; Pro = proline; Leu = leucine; Arg = arginine; His = histidine; Ser = serine
Also identified but not included were a silent mutation, c.387C>T, in p16 and not affecting ARF (n = 1), and the c.442G>A (Ala148Thr) polymorphism (n = 10). ARF amino acids are from Stott et al. (35).
Sequence for the c.43_46del4 microdeletion identified in p16 exon 1α reads 5′-TGGCTGGC-3′. It can alternately be described as c.47_50del4. Sequence alteration results in a frameshift.
Methylation of the ARF and p16 Promoters
To further evaluate alterations of ARF and p16, all samples were tested for promoter hypermethylation using methylation-specific PCR assays. The ARF promoter was methylated in 34/60 (57%) tumors; the p16 promoter was methylated in 16/60 (27%) tumors (Figures 4 and 5). Seven of 60 (12%) tumors were methylated for both genes. In two patients, two metastatic tumor samples were available from each patient. The patterns of methylation were the same for p16 (all four samples methylated); however, in both patients, ARF was methylated in only one of the two metastases. This rate of ARF promoter methylation is relatively high compared with the rates of 20%–55% (21–23) reported in other tumor types and has not to our knowledge been observed previously in melanoma.
Figure 5.
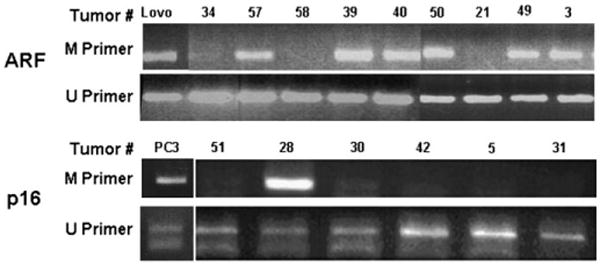
Methylation-specific polymerase chain reaction (PCR). ARF (upper panel) and p16 (lower panel) promoters were amplified. Methylation-specific primers (M) amplify a band of 122 bp (ARF promoter) or 145 bp (p16 promoter) from samples that are methylated in the promoter region. Primers specific for the unmethylated promoter sequences (U) always show a band because samples contain some infiltrating normal tissue. PCR products using the “U” primers are 132 and 154 bp for ARF and p16, respectively. Numbers above each lane correspond to tumor specimen numbers. DNA from the colon carcinoma cell line LoVo was used as a positive control for ARF promoter methylation; DNA from the prostate cancer cell line PC3 was used as a positive control for p16 promoter methylation.
Analysis of p16 Protein Expression in the Context of Genetic and Epigenetic Alterations to the Gene
To confirm that the genetic and epigenetic alterations we observed in the melanoma metastases resulted in functional inactivation of p16, we studied p16 protein expression in these specimens using immunohistochemistry. We first validated the specificity of the antibody using the melanoma cell lines we had characterized by western blotting (Figure 1); p16 expression was readily detected in SK-MEL 94 and 147 cells but not in SK-MEL 173 cells, which sustained a homozygous deletion of p16 but retained sequences encoding the adjacent p15INK4B, a protein of very similar structure and function (Figure 6, A). The expression was primarily cytoplasmic, with some nuclear reactivity also observed. We attempted to use a validated immunohistochemical approach for ARF that was similar to the one we used to detect p16 expression. To induce ARF expression, we treated the SK-MEL 192 and LoVo cell lines with methyltransferase inhibitors (zebularine for SK-MEL 192, 5-azacytidine for LoVo) and confirmed reexpression of ARF by immunoblotting (data not shown). We then tested the anti-ARF antibodies on cell blocks made with treated or control untreated SK-MEL 192 or LoVo cells. Unfortunately, the immunohistochemistry results with two different antibodies did not differ between the cells expressing or not expressing ARF. Therefore, we were unable to analyze ARF protein expression using immunohistochemistry on formalin-fixed material.
Figure 6.

Immunohistochemistry of p16. A) Melanoma cell lines tested for p16 expression. In SK-MEL 94 and 147, expression is mostly cytoplasmic, with some nuclei showing reactivity. SK-MEL 173 is negative. Each panel is shown at ×200 magnification. B) Expression of p16 in melanoma tumor samples. Alterations to the p16 and ARF loci are listed for each tumor. HD = homozygous deletion; Hemi = hemizygous; Meth = promoter methylation; Hemi–Meth = hemizygous and promoter methylation; WT = wild type. Expression is seen in tumors in which either no alteration to the locus was observed (tumors 4 and 31) or only one alteration was observed (tumor 13). No expression was seen in tumor 34, in which two alterations to the locus were observed (hemizygous loss and promoter methylation). The ARF locus sustained two alterations in tumors 4, 13, and 31, which were positive for p16 expression.
Detection of p16 in the melanoma metastases was performed on a subset of 20 tumors for which paraffin-embedded material was available from the same fresh tumor specimen analyzed for DNA alterations. As in the cell lines, tumors with positive expression exhibited a predominantly homogeneous pattern of cytoplasmic reactivity, with occasional nuclear reactivity observed (Figure 6, B). In this subset of tumors, 12/20 (60%) exhibited p16 protein expression. In 11/12 (92%) cases, the positive result was in concordance with the genetic and epigenetic data, which showed the status of the p16 locus to be either WT (n = 6) or to have sustained only one alteration, such as hemizygous loss (n = 2) or promoter methylation alone (n = 3) (Table 3). In one instance, protein expression was observed in a case that sustained a hemizygous loss and promoter methylation. Among the eight tumors lacking p16 protein expression, five had sustained two alterations to the locus. The lack of protein expression in the other three cases may be due to factors besides genetic or epigenetic changes [eg, altered expression of transcription factors (41)]. Under the assumption that two alterations (epigenetic or genetic) would be required to abolish protein expression (ie, inactivate the gene), we observed a concordance between protein expression and the number of alterations in 16/20 (80%) cases (P =.0181, Fisher exact test) (Table 3).
Table 3. p16 protein expression according to genetic alterations observed in the p16 gene*.
| Genetic and epigenetic alterations to the p16 gene | p16 Expression | |
|---|---|---|
|
| ||
| Positive† | Negative† | |
| None | 6‡ | 1§ |
| One hit | ||
| Hemizygous deletion | 2‡ | 2§ |
| Promoter methylation | 3‡ | 0§ |
| Two hits | ||
| Homozygous deletion | 0‡ | 1‡ |
| Hemizygous loss plus promoter methylation | 1§ | 3‡ |
| Hemizygous loss plus mutation | 0‡ | 1‡ |
| Total | 12 | 8 |
Twenty tumors were analyzed for p16 expression using immunohistochemistry. Images of tumors 4, 13, 31, and 34 are provided in Figure 6, B.
Positive and negative scoring as described in the text.
Concordant results between protein expression and genetic/epigenetic alterations (n = 16).
Discordant results between protein expression and genetic/epigenetic alterations (n = 4).
Comparison of ARF and p16 Inactivation
We compared our data on genetic and epigenetic changes in tumor DNA sequences with protein expression data to determine whether ARF inactivation occurs independently of p16 inactivation in human melanoma. Among the subset of 20 tumors in which p16 expression was determined, we found that 5/20 (25%) tumors had sustained two alterations to the ARF locus (either hemizygous deletions combined with promoter hypermethylation [n = 4] or homozygous deletion of exon 1β [n = 1]) in the context of intact p16 expression. In the entire cohort, alterations involving both genes were frequent, with 55/60 (92%) tumors showing an alteration of either ARF or p16. However, ARF sustained two alterations in 26/60 cases (0.43; 95% confidence interval [CI] = 0.32 to 0.56) compared with 13/60 cases (0.22; 95% CI = 0.13 to 0.34) for p16 (Table 4). This difference was statistically significant (P =.03, Fisher exact test). Among the 47 tumors in which p16 was not inactivated, 14 (30%) sustained two alterations to the ARF locus. Overall, inactivation of p16 alone was only observed in 3/60 (5%) cases, whereas inactivation of ARF alone was observed in 15/60 (25%) cases.
Table 4. Genetic and epigenetic alterations to the ARF and p16 loci in the entire cohort (n = 60).
| Genetic and epigenetic alterations | ARF locus | p16 locus |
|---|---|---|
| None | 10 | 16 |
| One hit | ||
| Hemizygous deletion | 14 | 17 |
| Promoter methylation | 10 | 14 |
| Total one hit | 24 | 31 |
| Two hits | ||
| Homozygous deletion | 7 | 5 |
| Hemizygous loss plus promoter methylation | 19 | 3 |
| Hemizygous loss plus mutation | 0 | 5 |
| Total two hits | 26 | 13 |
Discussion
The CDKN2A locus on chromosome 9p21 encodes the tumor suppressors p16INK4A and p14ARF, which function in the RB and p53 pathways, respectively. The role of p16 in melanoma tumorigenesis is well established, but the contribution of ARF to this process has been controversial. To address this question, we performed an analysis of genetic and epigenetic alterations to the 9p21 locus. We used several techniques including high-resolution MLPA to map deletions, the most common alteration to this locus. The deletion and mutation analyses were combined with a detailed analysis of the methylation status of the p16 and ARF promoters. We also analyzed the effect of these alterations on p16 protein expression using immunohistochemistry. To insure the specificity of this assay for p16, we first tested the antibody on cell blocks from the cell lines we had characterized using the other techniques. Using the validated assay, we verified that two alterations to the p16 gene (genetic or a combination of genetic and epigenetic) were required for its inactivation. We subsequently used this “two-hit” requirement to assess the inactivation of each gene separately. The combined analyses revealed that not only is ARF often inactivated in the context of an intact p16 gene but also it is a more frequent target of inactivation than p16 itself.
Previous studies of p16 and p14ARF inactivation in human melanoma specimens have been limited by the lack of a combined genetic and epigenetic analysis of both p16 and ARF in the same specimens. In one of the most comprehensive studies, Flores et al. (6) examined several microsatellite markers spanning the 9p21 region and analyzed the status of the p16 gene by Southern blotting with a 960-bp complementary DNA probe to exons 1α and 2. They reported p16 inactivation in 28% of melanoma specimens, with a rate of loss of heterozygosity (LOH) of 55%. No exon 1β–specific assays were described in that paper. Other reports have either used a small number of probes (42) or did not examine epigenetic modification of ARF (4).
In this study, the ARF promoter was methylated in 57% of metastatic melanoma tumors, a higher rate than has previously been reported in melanoma tumors, and a high rate relative to other tumor types, where the rate of ARF promoter methylation has been reported to range between 20% and 55% (22, 23, 43). We verified the promoter methylation assay result by treating one of the cell lines with the DNA methylation inhibitor zebularine. This treatment led to the reexpression of ARF, thus providing additional support for the reliability of the promoter methylation assay. We did observe promoter methylation in three tumors that sustained homozygous deletions comprising exon 1β and its upstream sequences. It is possible that the PCR-based methylation assay detected a subclone of tumor cells in which the promoter region was hypermethlyated but not deleted. Several published reports document similar intratumoral heterogeneity with respect to genetic alterations such as deletions. For example Fujii et al. (44) demonstrated heterogeneity with respect to allelic losses among different neoplastic foci in 8/20 (40%) breast cancer cases. Nakayama et al. (45) found intratumor heterogeneity in 6/26 (23%) melanoma satellite metastases that were studied using laser-capture microdissection and LOH analysis. Although they state in their abstract that this level of intratumor heterogeneity was not statistically significant, their data indicate that it does occur, albeit infrequently, as evaluated by their methodology. Finally, Baisse et al. (46) using microdissection of sporadic colorectal carcinomas found that 10/15 (67%) tumors were heterogeneous for at least one genetic alteration and that between two and six genetically distinct clones could be identified in each of these tumor specimens. They found that heterogeneity was more frequent for allelic losses than for point mutations.
The rate of p16 promoter methylation (27%) in metastatic tumor samples in this study was slightly higher than the 10%–19% reported previously in studies of earlier stage tumors (4, 47). In one of these studies, p16 promoter hypermethylation was associated with a worse clinical outcome (4, 48). The current observations are consistent with those of Straume et al. (4) who studied p16 protein expression, promoter hypermethylation, mutation, 3′ untranslated region polymorphisms, and LOH using four microsatellite markers in a large number of primary melanoma specimens. They observed as we did that p16 promoter methylation alone did not correlate with absence of p16 protein expression; however in the 25% of tumors in which one or more genetic or epigenetic alterations to the gene were present, there was an associated absence or reduction of p16 protein expression (4). It is possible that deletions of exon 1β could produce subtle effects on p16 expression. Such deletions would presumably include the loss of distant upstream transcriptional regulatory sites for p16. Although we cannot rule out an effect of alterations to exon 1β on p16 expression, ARF was inactivated primarily by the combination of hemizygous loss and methylation of its promoter. Therefore, at least one ARF allele was retained in the majority of cases. There were seven cases in which ARF was inactivated by homozygous deletion; however, p16 was similarly inactivated in five of these seven cases.
The observation that the most common inactivating alteration sustained to the ARF locus was hemizygous deletion combined with methylation of the ARF promoter raises interesting therapeutic possibilities. In a collaborative study, our group previously reported that more than 40% of metastatic melanomas suffered LOH of the apoptosis regulator Apoptotic Protease Activating Factor 1 (APAF-1) (30). Expression of APAF-1 could be restored by treatment with the demethylating agent, 5-aza-2′-deoxycytidine. Taken together, these observations support the hypothesis that investigations utilizing demethylating agents may lead to the restoration of cell growth control by reactivating several genes acting through different pathways. For these strategies to be effective, however, they would need to be applied to tumors in which the relevant genes were not substantially compromised by homozygous gene losses.
Previous observations in murine models of melanoma demonstrated that mice null for both p19Arf and p16Ink4a have shorter latencies of melanoma onset than mice null for either p16 exon 1α or p19Arf exon 1β. However, loss of either p19Arf or p16 was sufficient to drive melanomagenesis in the context of an oncogenic Ras transgene (12, 20). Furthermore, mice hemizygous for p19Arf and lacking p16Ink4a were more likely to develop tumors (including melanomas) and to develop them more rapidly than those that only lacked p16Ink4a (49, 50). Although the study by Krimpenfort et al. (51) did not find p19Arf promoter methylation, their analysis was limited by the use of a methylation-specific restriction enzyme assay, which may lack the necessary sensitivity to detect methylation outside the recognition sites of the enzymes used in the study. Our results also differ from these engineered models in that inactivation of ARF was often accomplished in the mice by deletion of exons 2 and 3 of p16Ink4a, whereas we observed that among ARF-altered specimens, only 3/50 (6%) were affected solely at the shared exon 2. Despite these differences in the mechanisms affecting ARF and p16, human and mouse data provide strong evidence that ARF plays an important independent role in the pathogenesis of human melanoma.
These combined human and murine observations suggest that alterations in either ARF or p16 (or both) could translate into different biological behaviors of the respective tumors. Several studies using melanoma tissue specimens have demonstrated that reduced expression of p16 correlated with histopathologic tumor progression and decreased patient survival (52). The role of ARF loss in melanoma prognosis is currently unknown, in part due to the lack of ARF-specific antibodies that could be applied to archival pathology specimens linked to clinical follow-up data.
In conclusion, with highly detailed mapping afforded by the MLPA and exon-specific multiplex PCR analyses combined with the study of promoter methylation for both p16 and ARF, we were able to identify ARF as the 9p21 gene most commonly inactivated by combined genetic and epigenetic alterations and a gene frequently targeted independently of p16.
Supplementary Material
Context and Caveats.
Prior knowledge
The role of p14ARF, whose reading frame overlaps that of the tumor suppressor p16INK4A, in human melanoma was unclear.
Study design
Using a variety of molecular assays, the p16/ARF locus was analyzed in melanoma metastases for mutations and promoter methylation.
Contribution
The study provided a detailed description of genetic and epigenetic changes at the p16/ARF locus in melanoma metastases. Inactivation of the gene encoding p14ARF occurred frequently, sometimes in the absence of p16 inactivation, suggesting that alterations in p14ARF function may play a role in melanoma pathogenesis.
Implications
Additional work is needed to define the relative contributions of alterations in p14ARF and p16 function to melanoma pathogenesis.
Limitations
Lack of an adequate antibody to p14ARF prevented the authors from measuring the changes in this protein's expression that corresponded to the genetic changes observed.
Acknowledgments
Funding: National Institutes of Health (K08 AR02129 to D.P., R33 CA95300 and P01 CA025874-25-A1 to B.C.B.); Cancer Research UK (to S.H.R., J.A.N.B.). Also supported in part by the use of facilities at the Manhattan Veterans Affairs Medical Center, New York, NY (to D.P.).
The authors thank Paul Christos for statistical advice, Eva Hernando and Molly Yancovitz for careful review of the manuscript, and Maryann Mikhail and Jessie Yu for their assistance with the preparation of the figures. The funding agencies had no role in the the design, conduct, and analysis of the work and the writing of the manuscript.
References
- 1.Pomerantz J, Schreiber-Agus N, Liegeois NJ, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92(6):713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92(6):725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60(14):3689–3695. [PubMed] [Google Scholar]
- 4.Straume O, Smeds J, Kumar R, Hemminki K, Akslen LA. Significant impact of promoter hypermethylation and the 540 C>T polymorphism of CDKN2A in cutaneous melanoma of the vertical growth phase. Am J Pathol. 2002;161(1):229–237. doi: 10.1016/S0002-9440(10)64174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bastian BC, LeBoit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58(10):2170–2175. [PubMed] [Google Scholar]
- 6.Flores JF, Walker GJ, Glendening JM, et al. Loss of the p16INK4a and p15INK4b genes, as well as neighboring 9p21 markers, in sporadic melanoma. Cancer Res. 1996;56(21):5023–5032. [PubMed] [Google Scholar]
- 7.Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264(5157):436–440. doi: 10.1126/science.8153634. see comments. [DOI] [PubMed] [Google Scholar]
- 8.Platz A, Hansson J, Mansson-Brahme E, et al. Screening of germline mutations in the CDKN2A and CDKN2B genes in Swedish families with hereditary cutaneous melanoma. J Natl Cancer Inst. 1997;89(10):697–702. doi: 10.1093/jnci/89.10.697. [DOI] [PubMed] [Google Scholar]
- 9.Aitken J, Welch J, Duffy D, et al. CDKN2A variants in a population-based sample of Queensland families with melanoma. J Natl Cancer Inst. 1999;91(5):446–452. doi: 10.1093/jnci/91.5.446. [DOI] [PubMed] [Google Scholar]
- 10.Harland M, Meloni R, Gruis N, et al. Germline mutations of the CDKN2 gene in UK melanoma families. Hum Mol Genet. 1997;6(12):2061–2067. doi: 10.1093/hmg/6.12.2061. [DOI] [PubMed] [Google Scholar]
- 11.Della Torre G, Pasini B, Frigerio S, et al. CDKN2A and CDK4 mutation analysis in Italian melanoma-prone families: functional characterization of a novel CDKN2A germ line mutation. Br J Cancer. 2001;85(6):836–844. doi: 10.1054/bjoc.2001.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharpless E, Chin L. The INK4a/ARF locus and melanoma. Oncogene. 2003;22(20):3092–3098. doi: 10.1038/sj.onc.1206461. [DOI] [PubMed] [Google Scholar]
- 13.Huot TJ, Rowe J, Harland M, et al. Biallelic mutations in p16(INK4a) confer resistance to Ras- and Ets-induced senescence in human diploid fibroblasts. Mol Cell Biol. 2002;22(23):8135–8143. doi: 10.1128/MCB.22.23.8135-8143.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laud K, Marian C, Avril MF, et al. Comprehensive analysis of CDKN2A (p16INK4A/p14ARF) and CDKN2B genes in 53 melanoma index cases considered to be at heightened risk of melanoma. J Med Genet. 2006;43(1):39–47. doi: 10.1136/jmg.2005.033498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mistry SH, Taylor C, Randerson-Moor JA, et al. Prevalence of 9p21 deletions in UK melanoma families. Genes Chromosomes Cancer. 2005;44(3):292–300. doi: 10.1002/gcc.20238. [DOI] [PubMed] [Google Scholar]
- 16.Randerson-Moor JA, Harland M, Williams S, et al. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet. 2001;10(1):55–62. doi: 10.1093/hmg/10.1.55. [DOI] [PubMed] [Google Scholar]
- 17.Hewitt C, Lee Wu C, Evans G, et al. Germline mutation of ARF in a melanoma kindred. Hum Mol Genet. 2002;11(11):1273–1279. doi: 10.1093/hmg/11.11.1273. [DOI] [PubMed] [Google Scholar]
- 18.Rizos H, Puig S, Badenas C, et al. A melanoma-associated germline mutation in exon 1beta inactivates p14ARF. Oncogene. 2001;20(39):5543–5547. doi: 10.1038/sj.onc.1204728. [DOI] [PubMed] [Google Scholar]
- 19.Harland M, Taylor CF, Chambers PA, et al. A mutation hotspot at the p14ARF splice site. Oncogene. 2005;24(28):4604–4608. doi: 10.1038/sj.onc.1208678. [DOI] [PubMed] [Google Scholar]
- 20.Ha L, Ichikawa T, Anver M, et al. ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc Natl Acad Sci USA. 2007;104(26):10968–10973. doi: 10.1073/pnas.0611638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krassenstein R, Sauter E, Dulaimi E, et al. Detection of breast cancer in nipple aspirate fluid by CpG island hypermethylation. Clin Cancer Res. 2004;10(1 pt 1):28–32. doi: 10.1158/1078-0432.ccr-0410-3. [DOI] [PubMed] [Google Scholar]
- 22.Esteller M, Tortola S, Toyota M, et al. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000;60(1):129–133. [PubMed] [Google Scholar]
- 23.Rousseau E, Ruchoux MM, Scaravilli F, et al. CDKN2A, CDKN2B and p14ARF are frequently and differentially methylated in ependymal tumours. Neuropathol Appl Neurobiol. 2003;29(6):574–583. doi: 10.1046/j.0305-1846.2003.00505.x. [DOI] [PubMed] [Google Scholar]
- 24.FitzGerald MG, Harkin DP, Silva-Arrieta S, et al. Prevalence of germline mutations in p16, p19ARF, and CDK4 in familial melanoma: analysis of a clinic-based population. Proc Natl Acad Sci USA. 1996;93(16):8541–8545. doi: 10.1073/pnas.93.16.8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamb A, Shattuck-Eidens D, Eeles R, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8(1):23–26. doi: 10.1038/ng0994-22. [DOI] [PubMed] [Google Scholar]
- 26.Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8(1):15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 27.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hogervorst FB, Nederlof PM, Gille JJ, et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res. 2003;63(7):1449–1453. [PubMed] [Google Scholar]
- 29.Albino AP, Vidal MJ, McNutt NS, et al. Mutation and expression of the p53 gene in human malignant melanoma. Melanoma Res. 1994;4(1):35–45. doi: 10.1097/00008390-199402000-00006. [DOI] [PubMed] [Google Scholar]
- 30.Soengas MS, Capodieci P, Polsky D, et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409(6817):207–211. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]
- 31.Castresana JS, Rubio MP, Vazquez JJ, et al. Lack of allelic deletion and point mutation as mechanisms of p53 activation in human malignant melanoma. Int J Cancer. 1993;55(4):562–565. doi: 10.1002/ijc.2910550407. [DOI] [PubMed] [Google Scholar]
- 32.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 33.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93(18):9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoo CB, Cheng JC, Jones PA. Zebularine: a new drug for epigenetic therapy. Biochem Soc Trans. 2004;32(pt 6):910–912. doi: 10.1042/BST0320910. [DOI] [PubMed] [Google Scholar]
- 35.Stott FJ, Bates S, James MC, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. Embo J. 1998;17(17):5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caldas C, Hahn SA, da Costa LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8(1):27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 37.Gretarsdottir S, Olafsdottir GH, Borg A. Five novel somatic CDKN2/p16 mutations identified in melanoma, glioma and carcinoma of the pancreas. Mutations in brief no. 170. Online. Hum Mutat. 1998;12(3):212. [PubMed] [Google Scholar]
- 38.Cairns P, Mao L, Merlo A, et al. Rates of p16 (MTS1) mutations in primary tumors with 9p loss. Science. 1994;265(5170):415–7. doi: 10.1126/science.8023167. [DOI] [PubMed] [Google Scholar]
- 39.Gruis NA, Weaver-Feldhaus J, Liu Q, et al. Genetic evidence in melanoma and bladder cancers that p16 and p53 function in separate pathways of tumor suppression. Am J Pathol. 1995;146(5):1199–206. [PMC free article] [PubMed] [Google Scholar]
- 40.Esteve A, Martel-Planche G, Sylla BS, Hollstein M, Hainaut P, Montesano R. Low frequency of p16/CDKN2 gene mutations in esophageal carcinomas. Int J Cancer. 1996;66(3):301–304. doi: 10.1002/(SICI)1097-0215(19960503)66:3<301::AID-IJC5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 41.Polsky D, Young AZ, Busam KJ, Alani RM. The transcriptional repressor of p16/Ink4a, Id1, is up-regulated in early melanomas. Cancer Res. 2001;61(16):6008–6011. [PubMed] [Google Scholar]
- 42.Grafstrom E, Egyhazi S, Ringborg U, Hansson J, Platz A. Biallelic deletions in INK4 in cutaneous melanoma are common and associated with decreased survival. Clin Cancer Res. 2005;11(8):2991–2997. doi: 10.1158/1078-0432.CCR-04-1731. [DOI] [PubMed] [Google Scholar]
- 43.Dominguez G, Carballido J, Silva J, et al. p14ARF promoter hypermethylation in plasma DNA as an indicator of disease recurrence in bladder cancer patients. Clin Cancer Res. 2002;8(4):980–985. [PubMed] [Google Scholar]
- 44.Fujii H, Marsh C, Cairns P, Sidransky D, Gabrielson E. Genetic divergence in the clonal evolution of breast cancer. Cancer Res. 1996;56(7):1493–1497. [PubMed] [Google Scholar]
- 45.Nakayama T, Taback B, Turner R, Morton DL, Hoon DS. Molecular clonality of in-transit melanoma metastasis. Am J Pathol. 2001;158(4):1371–1378. doi: 10.1016/S0002-9440(10)64088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baisse B, Bouzourene H, Saraga EP, Bosman FT, Benhattar J. Intratumor genetic heterogeneity in advanced human colorectal adenocarcinoma. Int J Cancer. 2001;93(3):346–352. doi: 10.1002/ijc.1343. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalgo ML, Bender CM, You EH, et al. Low frequency of p16/CDKN2A methylation in sporadic melanoma: comparative approaches for methylation analysis of primary tumors. Cancer Res. 1997;57(23):5336–5347. [PubMed] [Google Scholar]
- 48.van der Velden PA, Metzelaar-Blok JA, Bergman W, et al. Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001;61(13):5303–5306. [PubMed] [Google Scholar]
- 49.Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413(6851):83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- 50.Sharpless NE, Bardeesy N, Lee KH, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413(6851):86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 51.Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413(6851):83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- 52.Li W, Sanki A, Karim RZ, et al. The role of cell cycle regulatory proteins in the pathogenesis of melanoma. Pathology. 2006;38(4):287–301. doi: 10.1080/00313020600817951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.