

Summary
The recent clinical successes of inhibitors of the proteasome for the treatment of cancer have highlighted the therapeutic potential of this protein degradation system. Proteasome inhibitors prevent the degradation of numerous proteins, so increased specificity could be achieved by inhibiting the components of the ubiquitin-proteasome system that target specific subsets of proteins for degradation. F-box proteins are the substrate-targeting subunits of SKP1-CUL1-F-box protein (SCF) ubiquitin ligase complexes. Through the degradation of a plethora of diverse substrates, SCF ubiquitin ligases control a large number of processes at the cellular and organismal levels, and their misregulation is implicated in many pathologies. SCF ligases are characterized by a high specificity for their substrates, so they represent promising drug targets. However, the potential for therapeutic manipulation of SCF complexes remains an underdeveloped area. This review will explore and discuss potential strategies to target SCF-mediated biology to treat human diseases.
Introduction
Ubiquitin-mediated proteasomal degradation is an irreversible mechanism employed by numerous processes that feature regulation through the selective turnover of proteins, and it is used extensively by processes that proceed unidirectionally, such as the cell cycle or circadian oscillations. Ubiquitylation occurs via a sequence of enzymatic events, in which the small protein ubiquitin is activated by linkage to an E1 (ubiquitin-activating) enzyme, transferred to an E2 (ubiquitin-conjugating) enzyme, and then transferred to a free amine group in either the N-terminus or an internal lysine of a substrate that is dictated by an E3 ubiquitin ligase (Figure 1) 1. Repeated iterations of this process result in long chains of ubiquitin (polyubiquitin) on the substrate, and these chains can feature different topologies depending on the lysine residue within ubiquitin that is used for chain extension (i.e. K6, K11, K27, K29, K33, K48, or K63) or the use of the ubiquitin N-terminus 2. Substrates can also be mono-ubiquitylated (on one or more sites). These different ubiquitin configurations can result in a variety of biological outcomes 3, with K48- and, less commonly, K11-linked ubiquitin chains committing proteins to the proteasome for degradation. E3 ubiquitin ligases are functionally opposed by de-ubiquitylating (DUB) enzymes that are able to remove ubiquitin from proteins to prevent proteolysis or alter signaling 4.
Figure 1. Ubiquitin-mediated degradation.

Ubiquitin is attached to substrates by the consecutive activities of three enzymes. An E1 enzyme activates ubiquitin in an ATP-dependent reaction. An E2 enzyme subsequently transfers the activated ubiquitin to the substrate that is specifically bound to the E3 substrate selection factor. Polyubiquitylated substrates are targeted to the proteasome, a multisubunit protease, to undergo degradation. Although protein degradation is irreversible, the ubiquitylation signal can be attenuated through the action of de-ubiquitylating enzymes (DUBs). Multiple enzymatic steps within the ubiquitylation process are potentially druggable. As the selectivity factors, E3 ubiquitin ligases represent the most specific point of intervention. In contrast, proteasome inhibitors block the degradation of a large number of substrates.
The ubiquitin proteasome system (UPS) has links to an expanding array of diseases, including cancer, immunological disorders, and neurological disorders, and the validity of the UPS as a target has been confirmed by the clinical success of the proteasome inhibitor bortezomib in the treatment of multiple myeloma 5–7. The success of bortezomib has driven the production of additional proteasome inhibitors for use as cancer therapeutics (Box 1), and proteasome inhibition has been investigated in several clinical trials for several additional diseases (mostly immune in nature, including graft versus host and autoimmune disease). Despite their clinical successes and specificity for the proteasome, proteasome inhibitors remain fairly general treatments, affecting all processes that utilize ubiquitin-mediated degradation for regulation. Although bortezomib is clinically effective, some side-effects, such as neuropathy, have been reported, and multiple myelomas can evolve bortezomib resistance 8, 9. Additionally, there remains debate surrounding which molecular targets are key to growth inhibition 10, 11. However, the UPS is composed of over 1,000 proteins, and the potential exists to develop more specific drugs that inhibit distinct biological processes with greater efficacy by choosing targets other than the proteasome itself (Figure 1).
Box 1. Drugging the ubiquitin proteasome system.
The UPS can be targeted with therapeutics at multiple levels, resulting in varying degrees of specificity (Figure 1). For example, proteasome inhibitors globally inhibit the degradation of all proteins, while E3 ubiquitin ligase inhibitors will block the degradation of a small subset of proteins. Compounds affecting each point in the UPS have been explored. Although some of these compounds remain experimental, others have advanced into pre-clinical and clinical trials.
Proteasome Inhibition: Broad inhibition of all ubiquitin-dependent degradation by the proteasome inhibitor bortezomib is clinically effective in the treatment of multiple myeloma and relapsed mantle cell lymphoma. The proteasome features three dominant proteolytic activities (chymotrypsin-like, trypsin-like, and peptidyl-glutamyl peptide hydrolyzing), as well as two less characterized proteolytic activities (branched chain amino acid-preferring and small neutral amino acid-preferring) 138. The majority of proteasome inhibitors in clinical trials inhibit the chymotrypsin-like and/or trypsin-like activities. Although proteasome inhibition stabilizes anti-proliferative proteins, part of the effectiveness of proteasome inhibitors is thought to be due to the general exacerbation of the proteotoxic stress found in tumour cells, particularly antibody-secreting plasma cells, which produce large levels of immunoglobulins 10. Following the success of bortezomib, several other proteasome inhibitors, including Ixazomib, Delanzomib, Marizomib, and Oprozomib, are presently in various stages of clinical trials, and Carfilzomib has been approved for the treatment of multiple myeloma 9, 139.
p97 AAA+ ATPase inhibitors: The p97 AAA+ ATPase facilitates the degradation of chromatin, endoplasmic reticulum, and mitochondria-associated through the extraction and/or unfolding of ubiquitylated proteins, regulating the degradation of multiple proteins involved in a large variety of disease-associated processes 140, 141. p97 also plays a key role in the regulation of CRL assembly 142, 143. A number of p97 inhibitors are in pre-clinical studies and/or poised to enter phase I trials 144, 145.
Ubiquitin E1 inhibitors: There are only two human E1 enzymes, Uba1 and Uba6, and the success of proteasome inhibitors suggested that broad inhibition of protein ubiquitylation by these enzymes would have anti-proliferative effects 11, 146. The PYR-41 inhibitor, which covalently binds and irreversibly inhibits Uba1, confirmed the anti-proliferative effects of broad ubiquitylation inhibition, and several additional E1 inhibitors are in preclinical testing 147, 148. Crossreactivity of Uba1 inhibitors with Uba6 has not been reported. Inhibiting global ubiquitylation might be expected to produce very broad biological impacts because it inhibits both ubiquitin-dependent degradation and ubiquitin-mediated signaling.
Nedd8 E1 inhibitors: Like ubiquitylation, neddylation, the covalent attachment of Nedd8 to other proteins utilizes a cascade composed of an E1, E2, and E3 39. As its name implies, ubiquitin modification is nearly ubiquitous, but Nedd8 modification is much more selective. The best known Nedd8 targets are the cullins, and neddylation is required for their ubiquitin ligase activity. Because the cullins regulate many proteins involved in cell proliferation, inhibition of neddylation is an attractive target for cancer chemotherapy. MLN4924 is the first neddylation inhibitor, and it inhibits neddylation at the level of the E1 35, 39. In preclinical testing, MLN4924 was a potent anti-proliferative, and it is now entering phase I trials. Although the effects of MLN4924 are reported to be mediated through inhibition of Cdt1 degradation, it is likely that disruption of the degradation of other CRL substrates contributes to the effectiveness.
E2 inhibitors: There are nearly 40 E2 enzymes, suggesting that inhibition of E2 enzymes could provide inhibition of ubiquitylation that is slightly more specific than the global inhibition of an E1 inhibitor. However, E2s can still control the degradation of many substrates. Thus far, one specific allosteric inhibitor of Cdc34 has been reported, and this inhibitor, which binds a pocket between the E2 and the covalently-linked ubiquitin, does not interfere with either E1 or E3 binding 149, 150. For the purposes of this review, inhibitors that block E2–E3 binding are considered E3 inhibitors.
DUB Inhibitors: Because they directly regulate the stability of substrates by counteracting ubiquitylation, de-ubiquitylating (DUB) enzymes are prime targets for the development of inhibitors. There are nearly 100 different DUBs, offering the potential to inhibit a narrow range of biological functions, and as proteases, the active sites of DUBs are amenable to traditional small molecule, competitive inhibitors 4. In particular, USP7, which regulates Mdm2 and p53, is under investigation, and USP7 inhibitors increase p53 levels 151, 152. A selective USP14 inhibitor also increases proteasome activity and may be useful in pathologies characterized by proteotoxic stress and accumulation of ubiquitylated proteins 153–155. However, the development of DUB inhibitors has been slowed by several issues, including a general lack of knowledge of DUB functions and a lack of specificity in competitive inhibitors. For example, one of the most promising DUB inhibitors derives its anti-proliferative activity from inhibition of both USP7 and USP47 156, 157. Another DUB inhibitor non-selectively inhibits USP9x, USP5, USP14, and UCH37 158. Noncompetitive inhibitors of USP1, the deubiquitinase for PCNA and FANCD2/FANCI, have been characterized 159. However, the development of noncompetitive inhibitors is complicated by the lack of understanding of the biochemical mechanisms of DUBs, particularly the involvement of co-factors and other regulatory proteins 160–163.
In humans, there are two E1 ubiquitin-activating enzymes, multiple E2 enzymes, and hundreds of E3 enzymes, which together control the degradation of thousands of substrates 12, 13 (Figure1). Small molecule inhibitors of E1 and E2 enzymes have been developed (Box 1), and like proteasome inhibitors, these drugs affect a very large range of substrates, which may reduce their therapeutic value 11 (Figure 1). The UPS components that are viewed as the most promising drug targets are the deubiquitylating enzymes and E3 ubiquitin ligases, which have fewer targets and provide the specificity of the system 4, 6. While targeted inhibition of the 95 deubiquitylating enzymes is complicated by shared active sites (among only five classes of proteases) and a general lack of knowledge of the biological roles and regulation of these proteins [Box 1 and reviewed elsewhere 4], several compounds that target E3 ligases with varying degrees of specificity have been developed and hint that effective drugs targeting E3 ubiquitin ligases may be developed.
E3 enzymes can be divided into two main classes (RING or HECT E3s), of which those with RING domains are the most common 14, 15. While RING-finger E3s mediate substrate ubiquitylation by bringing the substrate in contact with the E2 enzyme, HECT domain E3 enzymes both recruit the substrate and directly participate in the ubiquitin transfer reaction. RING domain ligases can function as single subunit enzymes or as multisubunit enzymes. Multisubunit RING E3 enzymes, such as the Cullin-Ring Ligases (CRLs), allow one core scaffold to facilitate the ubiquitylation of numerous substrates via variable/exchangeable substrate recognition modules 16. Notably, the large number of ubiquitin ligases and ubiquitin ligase substrates links the UPS to numerous different biological pathways and functions, many of which are dysregulated in a wide range of diseases. Therefore, the abundance and diversity of ubiquitin ligase-substrate pairs offers the potential for the development of drugs that are highly specific at both the pathway and protein scales. This review will discuss the rationale and approaches to targeting one of the largest subfamilies of ubiquitin ligases, the SKP1-CUL1-F-box protein (SCF; also called CRL1) complexes, with novel therapeutics, using established model systems to highlight key concepts and principles.
SCF ubiquitin ligases
Cullins
In mammals, there are eight cullin proteins which form the backbone of around 200 CRLs, of which SCF complexes are the best characterized. Each CRL complex contains a different cullin subunit, which acts as an extended scaffold that simultaneously binds to the catalytic machinery (E2 ubiquitin-conjugating enzymes) at the C-terminus and substrate recognition factors at the N-terminus. The C-termini of cullins 1, 2, 3, 4A, 4B, 7, and 9 bind to the E2 enzyme via the small RING protein RBX1, while CUL5 uses the related protein RBX2 for E2 recruitment 16. In addition to recruiting the E2 for substrate ubiquitylation, the RBX proteins recruit the E2 required for the covalent attachment of NEDD8, a small ubiquitin-like protein, to the cullin backbone (Figure 2). Neddylation induces a structural rearrangement in the cullin backbone that facilitates ubiquitin transfer from the E2 to the substrate and is required for CRL function. Like ubiquitylation, this modification results from the sequential action of NEDD8-specific E1, E2, and E3 enzymes (the RBX/cullin complexes themselves) 12, 16, and it can be reversed via the actions of the COP9 signalosome (reviewed elsewhere) 17.
Figure 2. The modular structure of SCF ubiquitin ligases and points of potential therapeutic intervention.

CUL1 acts as a scaffold that brings the E2 enzyme in proximity to the substrate. It binds to SKP1 at its N-terminus, and the E2 enzyme via RBX1 at its C-terminus. The F-box protein functions as the interchangeable component (69 F-box proteins in humans) that interacts with SKP1 via its F-box domain and with its cognate substrate through its specific substrate recognition domain. CUL1 is activated by covalent conjugation with Nedd8. This modification induces a conformational change in CUL1 that facilitates the transfer of ubiquitin from the E2 to the substrate and is required for CRL function. MLN4924, a small molecule inhibitor of the Nedd8-activating enzyme, shows therapeutic potential and has progressed to phase I/II clinical trials. Because it inhibits the activity of all CRLs, it affects the degradation of a large number substrates. Strategies to inhibit SCF ligase function with more selectivity include blocking SCF complex assembly, blocking the interaction between substrate and F-box protein, and inhibiting E2 enzyme binding and/or function.
The N-terminus of each cullin binds and utilizes a unique family of proteins for substrate specification, and the large number of adaptor proteins allows the assembly of over 200 CRLs from the eight cullin scaffolds. The N-terminus of CUL1 interacts with SKP1, which binds to the F-box domain of an interchangeable F-box protein 6 (Figure 2). The F-box domain is an approximately 40 amino acid domain, which was originally defined in cyclin F (FBXO1). There are 69 F-box proteins in humans, classified into three families based on their protein interaction domains: WD40 domains (10 FBXW proteins), leucine-rich repeats (21 FBXL proteins), and those with other, diverse domains (38 FBXO proteins) 18. These protein-protein interaction domains interact with specific substrates. Most of the well-studied F-box proteins have multiple substrates 19. Therefore, it is thought that each F-box protein will have multiple substrates, making the full complement of SCF substrates very large.
F-box protein-substrate recognition
The F-box proteins typically recognize unique, short degradation motifs (termed degrons) in their substrates. In many of the well-characterized F-box protein-substrate interactions, post-translational modifications (often phosphorylation) of the substrate’s degron direct F-box protein binding, and although unmodified degrons exist, access to these degrons is usually controlled by posttranslational modifications or other regulatory mechanisms 6. The protein interaction domains in F-box proteins are highly specific degron recognition domains, often recognizing short peptide sequences. For example, SKP2, together with its cofactor CKS1, recognizes p27 only after it is phosphorylated on Thr187 by a cyclin-dependent kinase (CDK) 20–23. However, it is becoming increasingly clear that F-box proteins can recognize a wide variety of degron, allowing multiple alternative mechanisms dictate substrate selection. For example, in additional to phosphorylations, F-box proteins can recognize alternative modifications, such as glycosylations, or non-modified degrons 6. Some degrons are even protected by phosphorylations that block F-box protein binding, such as the degrons within CDT2 (degraded via SCFFBXO11) and p85β (degraded via SCFFBXL2) 24–26. In addition, F-box proteins themselves are tightly regulated by post-translational mechanisms, including by phosphorylation and proteolytic turnover. The distinct modes of substrate recognition by F-box proteins have been thoroughly discussed previously 6.
Targeting E3s
In the past 20 years, the pharmaceutical industry has focused a large amount of effort on the discovery and development of protein kinase inhibitors 27–29, and this effort to develop kinase inhibitors offers a framework for the development of ubiquitin ligase-based therapies (Box 2). However, ubiquitin ligase complexes have biochemically distinct active sites, in addition to other potential drug binding pockets, that offer multiple targets for small molecules, suggesting that ubiquitin ligase-focused therapies could achieve greater specificity than competitive kinase inhibitors. Notably, the protein-protein interaction surfaces required for both SCF assembly and substrate recognition are of sufficient size to accommodate many potential compounds (and are not too large for a small molecule approach to therapies) 31.
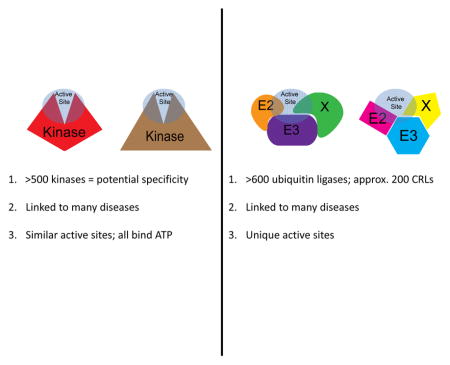
Box 2. Lessons from Kinase Inhibitor Development and Application.
In terms of rationale, an extensive literature links both the deregulation of kinases and ubiquitin ligases to many diseases, including cancer and immunological disorders, and this literature has established key mechanisms, regulators, and pathways. Additionally, both protein families are large and have many substrates: there are over 500 kinases and over 700 ubiquitin ligases encoded in the human genome, offering the promise of specific action through the inhibition of individual proteins.
There are also notable differences that impact the ability to effectively target kinases and ubiquitin ligases with novel therapeutics 164. With respect to drug development, the most important is the nature of each enzyme’s active site. Kinase active sites bind ATP, so many ATP analogs have been developed as competitive inhibitors. However, because all kinases utilize ATP and kinase active sites are often structurally similar, these competitive inhibitors often inhibit multiple kinases and/or exhibit a low dose window for specific inhibition 28. In some cases, this lack of specificity may add to the clinical efficacy by inhibiting multiple kinases in signaling cascades, such as with the successful use of Sorafenib 30, but in other cases, greater specificity is required in subsequent generations of compounds. The ubiquitylation reactions catalyzed by ubiquitin ligases do not share any small metabolites, such as ATP, and each active site for these enzymes is uniquely defined by the protein-protein interactions of the ubiquitin ligase and the substrate, avoiding the specificity issues of ATP analogs used in competitive kinase inhibitors 28. Notably, while non-competitive inhibitors of kinases can be developed, including antibody-based therapies for receptor tyrosine kinases, allosteric inhibitors, or protein-protein interaction inhibitors, a primary focus has been on small molecule, competitive inhibitors, based on ATP analogs that bind kinase active sites, such as Imatinib, Dasatinib, Sorafenib, Palbociclib, and Idelalisib, all of which are clinically effective or look extremely promising in late stage clinical trials 30, 164–170.
The development of cyclin-dependent kinase (CDK) inhibitors illustrates both the successes and failures of developing competitive kinase inhibitors. Because CDKs are key components of the machinery driving the cell cycle, inhibition of CDK1, CDK2, CDK4, or CDK6 should block cell proliferation, and indeed, the first generation inhibitors of CDKs, such as flavopiridol and roscovitine, are effective inhibitors of kinase activity and have a clear anti-proliferative effect in vitro. However, the anti-proliferative activity of these CDK inhibitors may actually stem from their inhibition of transcription and induction of apoptosis, through CDK7, CDK8, and CDK9 171. Significantly, the first generation inhibitors have met limited success in the clinic and only in the context of multi-agent therapy. More selective, next generation CDK inhibitors, such as Palbociclib, which specifically inhibits CDK4 and CDK6, may prove much more efficacious, especially when coupled with a more advanced molecular genetic understanding of individual tumours. For example, a primary target of investigation for Palbociclib is breast cancer, a disease with clear links to the activation of cyclin D/CDK4 and possibly cyclin D/CDK6.
Although the development of CDK inhibitors may suggest that specific kinases inhibitors are best, current treatments for chronic myelogenous leukemia (CML) demonstrate that both highly specific and less specific kinase inhibitors can be clinically effective. Imatinib, the frontline therapy for CML, was developed as a specific inhibitor of the BCR-ABL fusion that drives CML. When CML becomes resistant to Imatinib, Dasatinib can be an effective therapy, and unlike Imatinib, which is highly specific for only BCR-ABL, c-KIT, and PDGFR, Dasatinib also inhibits all nine Src family kinases, as well as several other tyrosine kinases 28. The utilization of these kinase inhibitors in CML demonstrates that both specificity and context are important in the application of kinase inhibitor therapies, and broad inhibition of multiple pathways can be an effective strategy.
Conversely, this distinctiveness of ubiquitin ligase active sites also creates significant hurdles to drug development as the unique features of each ligase must be known and compounds cannot be simply generated from a common molecular backbone for all ubiquitin ligases. These limitations are similar to those observed in the development of non-competitive kinase inhibitors and protein-protein-interaction inhibitors for kinases and have slowed the development of these drug classes considerably.
Non-SCF ubiquitin ligases as drug targets
The promise of specific E3 ubiquitin ligase inhibitors has been confirmed by the nutlins, a class of small molecule inhibitors of the Mdm2 ubiquitin ligase, which are currently in phase I clinical trials (Box 2) 32. The activation of the tumour suppressor p53 to block cell proliferation or induce apoptosis of tumour cells is a primary goal in the treatment of p53 positive tumours, and nutlins inhibit the interaction of Mdm2 with p53, resulting in the stabilization of p53. However, because p53 is the primary anti-tumour effector, nutlins should only be effective in the context of wild type p53, requiring knowledge of the p53 status of the tumour. Nutlins, a general name given to a series of similar compounds based on a cis-imidazoline backbone, are competitive inhibitors of p53 binding to Mdm2, and these compounds structurally mimic interactions of p53 (through Phe19, Trp23, and Leu26) with the N-terminus of Mdm2 33. In general, these compounds are active in both cell culture and xenograft model systems, resulting in p53 accumulation, apoptosis, and tumour regression. Phase I trials of RG7112 were recently completed, and this compound demonstrated activation of the p53 pathway, including the induction of apoptosis, suggesting its potential as a single agent therapy 34. Several additional Nutlins/Nutlin-like compounds are being examined in ongoing phase I studies.
While the inhibition of a single ubiquitin ligase by nutlins makes an argument for specificity, it is also possible to generate effective chemotherapies targeting multiple E3 ubiquitin ligases. The CRL family of ubiquitin ligases has multiple links to cancer, with SCFs/CRL1s and CRL4s having particularly strong links due to their control of the cell cycle, DNA replication, and DNA repair. As discussed above, the activity of CRLs is dependent upon the modification of their cullin backbones by the small ubiquitin-like protein NEDD8 (Figure 2) 16. MLN4924, which inhibits cullin neddylation, was recently found to be an effective antiproliferative agent, and thus far, the results from this drug in pre-clinical studies have been promising in a variety of cancers 35. MLN4924 inhibits the growth of lung, ovarian, breast, leukemia, lymphoma, myeloma, melanoma, and Ewing sarcoma cells in vitro, and this broad spectrum of effectiveness is also supported by experiments in xenograft and transgenic rodent models 36–45. Phase I trials for MLN4924 in lymphoma, myeloma, and melanoma have been successfully completed, and multiple other phase I trials are ongoing. The anti-tumour effect of MLN4924 was initially ascribed to the inhibition of SCF and/or CRL4-dependent degradation of Cdt1, but like proteasome inhibition, MLN4924 must affect a broad range of substrates that are targeted for degradation through the more than 200 CRL complexes 35, 46–48. (The potential impact of MLN4924 through other reported, non-cullin NEDD8 substrates is unclear 49) Notably, the evolution of resistance to MLN4924 has been observed in vitro 50, 51.
SCF ubiquitin ligases as drug targets
The success of MLN4924 and our extensive knowledge of cancer-relevant CRL targets – particularly for SCFs – suggest that, at least in some cases, the specific inhibition of individual CRLs may prove more effective and provide a better therapeutic ratio than global inhibition via MLN4924. Additionally, while initial efforts targeting the UPS have focused on the development of anti-cancer therapeutics, our expanding knowledge of the SCF family of ubiquitin ligases indicates that the targeting of specific SCF complexes may also result in effective therapies for a variety of disorders, including sleep disorders, mood disorders, inflammation, and acquired infections 52–59. Although many F-box protein-substrate pairings have been described and linked to potential biological functions, the first hurdle to generating SCF-directed therapies remains the thorough biological characterization of the family. Less than 20 of the 69 F-box proteins have established substrates, and even fewer F-box proteins have multiple confirmed substrates from which key biochemical principles for substrate targeting and ubiquitylation can be extrapolated 6, 19 (Table 2). Additionally, while some F-box proteins appear to have a universal function (i.e. either driving or stopping cell proliferation) by targeting substrates with common biological functions, other F-box proteins control substrates with disparate – or even opposing – biological functions, so a thorough understanding of each F-box protein’s role is essential for determining targets and/or targeting strategies. Finally, the great majority of studies of SCF function have focused on roles in cancer 5, 60, 61, and more research is required to define the roles of these proteins in other diseases.
Table 2. Attributes of potential SCF drug targets.
Squares are color coded to represent a scale of favorability for rational drug development. Green= favorable; Orange=potentially favorable; Pink= underdeveloped.
| SCF | Substrate | Disease links | Crystal structures | Biochemical Mechanism of substrate recognition |
|---|---|---|---|---|
| SCFs with favourable characteristics for rational drug development | ||||
| SKP2/FBXL1 | > 5 (mainly tumour suppressors) | Established oncogene |
|
Well-understood. (Cks1-dependent (cofactor dependent) and phosphodegron (S/TPxR/K)) |
| FBXW7 | >5 (mainly oncogenes) | Established tumour suppressor. Survival factor B-cells. | Fbw7-Skp1-cyclin E peptide | Well understood. Phosphodegron (TPPXS) |
| SCFs with potentially favourable characteristics for rational drug development | ||||
| FBXL3 | 1 substrate | Potential link to sleep/metabolic disorders |
|
Well understood. |
| βTrCP | Multiple substrates | Pleiotropic- regulates multiple disease pathways (oncogenes, tumour suppressors, circadian regulators, etc.) | βTrCP1-Skp1 complex bound to a βcatenin phosphopeptide | Well Understood Phosphodegron (DSGxxS) |
| FBXO4 | <5 substrates | Potential tumour suppressor |
|
No established consensus mechanism |
| SCFs whose characteristics are not sufficiently explored | ||||
| FBXO2 | <5 substrates | None known |
|
Unknown |
| FBXO3 | <5 substrates | Inflammation | Predicted structure of ApaG domain of FBXO2 | Unknown |
| FBXO31 | <5 substrates | Tumour suppressor, intellectual disability | None | No established consensus mechanism |
| FBXL4 | 1 substrate | Early onset mitochondrial encephalomyopathy | None | Unknown |
| FBXL2 | <5 substrates | HCV replication, inflammation | None | No established consensus mechanism |
| FBXO11 | <5 substrates | Tumour suppressor | None | No established consensus mechanism |
Despite these caveats, there are multiple SCF complexes with defined biology and biochemistry and clear links to human disease, and these SCF complexes illustrate the key concepts of targeting SCF complexes with novel therapies (Table 2). Direct alteration of SCF activity in diseases typically results from the overexpression or mutations (deletions or point mutations) of an F-box protein, and indirect alterations typically affect pathways controlling degron modifications or substrate degrons themselves. Each mechanism requires a different conceptual approach for the development of therapeutics.
Inhibition of SCF-mediated degradation
SKP2-mediated p27 degradation
SKP2/FBXL1 is one of the best-characterized F-box proteins and a bona fide oncoprotein in multiple tumours and animal models 62–64. Together with the CKS1 cofactor, SKP2 promotes S-phase entry by mediating the ubiquitylation and degradation of the CDK inhibitor p27, an important tumour suppressor 20, 21, 65. Notably, p27 is seldom mutated in cancer, and its tumour suppressive activity is typically removed by SKP2 overexpression. Accordingly, SKP2 overexpression correlates with high tumour grade and poor prognosis in multiple cancers 5. Other SKP2 targets, including the p57 and p21 CDK inhibitors, may also be relevant to SKP2 overexpression in cancer 61. However, in mouse models, SKP2 overexpression is phenocopied by p27 deletion, and Skp2 knockout phenotypes are rescued by p27 deletion, suggesting that the oncogenic functions of SKP2 are largely dependent on p27 degradation 62, 64. Thus, SKP2 presents an attractive target for the generation of inhibitors of p27 degradation.
The biochemistry of the SKP2-CKS1-p27 interaction is very well established. Binding of SKP2 and CKS1 to p27 is controlled by phosphorylation of the degron by a CDK, and the crystal structure of SKP2, CKS1, and the p27 phosphodegron clearly shows a pocket between SKP2 and CKS1 that is flanked by residues essential for p27 binding 20–23, 66. This thorough understanding of SKP2-dependent p27 degradation could facilitate the development of inhibitors of p27 degradation, which could target the SKP2-CKS1 degron interface or structural features of the SCFSKP2 complex (Figures 2 and 4) (Table 1). Indeed, both types of inhibitors were recently reported from in silico screens using the structure of SCFSKP2. In each of these cases, in silico screening was able to identify compounds with activity either in vitro or in tissue culture systems. One group identified small compounds that could bind the interface between SKP2, CKS1, and p27 and disrupt complex formation 67, 68. Another in silico screen identified a SKP2 inhibitor that interferes with the binding between SKP2 and SKP1, preventing the formation of an active SCF complex 69. A mechanistically similar inhibitor of the yeast F-box protein Met30 was previously identified in a screen for rapamycin sensitivity, and although both compounds might be expected to inhibit multiple F-box proteins by binding the F-box domain and blocking SKP1 binding, each compound appears specific for only one F-box protein 69, 70.
Table 1.
Drugs that target E3 ubiquitin ligases.
| Target | Disease | Compound | Mechanism of action | Stage of clinical development | Company/Reference |
|---|---|---|---|---|---|
| SCF ligase inhibitors | |||||
| Skp2 | Breast, colon, prostate, others. | C1, C2, C25 | Binds to pocket formed by Skp2 and Cks1 to prevent substrate binding | Preclinical | 67 |
| Skp2 | Breast, colon, prostate, others. | Compound A | Blocks the binding between SKP2 and SKP1 | Preclinical | 72 |
| Skp2 | Breast, colon, prostate, others. | SMIP004 | Down-regulates SKP2 | Preclinical | 74 |
| Skp2 | Breast, colon, prostate, others. | Compound #25 | Blocks binding between SKP2 and SKP1 | Preclinical | 69 |
| Fbxl3 | Gluconeogenesis, sleep disorders | KL001 | Binds to pocket in CRY2 substrate and prevents FBXL3 binding | Preclinical | 80, 82 |
| Fbxo3 | Inflammation | BC-1215 | Competitive inhibition of substrate binding to FBXO3 | Preclinical | 87, 172 |
| βTrCP | Inflammation | GS143 | Blocks the interaction between p-IκBα and βTrCP1 | Preclinical | 173 |
| Met30 (yeast) | N/A | SMER3 | Blocks Met30 binding to Skp1 | N/A | 70 |
| Cdc4 (yeast) | N/A | SCF-I2 | Allosteric inhibitor | N/A | 71 |
| βTrCP | Tumour suppression | Erioflorin | Inhibits interaction between βTRCP and PDCD4 tumour suppressor | Preclinical | 174 |
| βTrCP | Tumour suppression | UBP-036 | Competitive inhibition of substrate binding to βTRCP | Preclinical | UBPharma |
| NON SCF E3 ligase inhibitors | |||||
| Mdm2 | solid and hematological tumours | Nutlin | Blocks the interaction between Mdm2 and p53 | Phase I/II | Roche |
| CRBN | Multiple Myeloma, relapsed mantle cell lymphoma | Lenalidomi de/Pomalid omide | Retargets CrBN to Ikaros | Approved | Celgene |
The existing compounds, whether targeting the F-box protein-substrate interface or the F-box-protein-SKP1 interface, are currently effective at relatively high concentrations, but one key advantage of the in silico approaches to generating SKP2 inhibitors is that the potency of lead compounds identified by virtual screens can be optimized via traditional medicinal chemistry with a priori knowledge of the important molecular contacts and pharmacophores. Thus far, the in silico approaches to inhibiting SKP2 have focused on directly disrupting either p27 or SKP1 binding, but it may also be possible to generate allosteric inhibitors of SKP2 (Figure 3). For example, an allosteric inhibitor that binds the WD40 repeats of the yeast F-box protein Cdc4 and causes structural rearrangements that block substrate binding was identified through a more traditional in vitro screen 71.
Figure 3. Strategies to manipulate SCF ubiquitin ligase activity.

A) Unperturbed substrate recognition by an F-box protein. B) PROTACs can act to restore the function of a deleted or mutated F-box protein. They are hetero-bivalent molecules that simultaneously bind to the substrate and the SCF ligase, thereby bringing the ligase into proximity with the substrate. C) Small molecules can restore the function of mutated F-box proteins by acting as molecular glue, providing stable interaction surfaces. D) Small molecules can also function to re-purpose substrate receptors, targeting them to other substrates. E) Inhibition of SCF ligases can be accomplished by competitive inhibition of substrate binding or G) SCF complex formation. F) Allosteric inhibitors can bind to the F-box protein at a region remote from the degron site, distorting the substrate recognition site.
Traditional screening approaches for the development of inhibitors of p27 degradation have failed to identify compounds that directly affect SCFSKP2 72–74. It is unclear why traditional screens, such as those dependent on p27-degron-reporter fusions, fail to detect compounds that directly disrupt SKP2 interactions, but multiple reasons, including the limited number of compound libraries screened and cell permeability issues, may reduce the identification rate for lead compounds of this class. In other cases, cell-based screens have detected compounds that reduce SKP2 mRNA levels and do not directly affect the activity of SCFSKP2 towards p27 74. However, traditional screens do efficiently identify small molecule inhibitors of CDKs as inhibitors of the SKP2-p27 interaction, highlighting another key concept for the inhibition of SCF ubiquitin ligases 75. Because the binding of many F-box proteins to their consensus degrons requires phosphorylation of the degron, inhibition of kinases or other modifying proteins represents a viable option for the inhibition of substrate degradation, and this indirect method of inhibition can make use of many pre-existing compounds, such as kinase inhibitors (see below) 75, 76. Overall, it is likely that a combination of in silico and traditional screening approaches will prove more successful than the application of either individually.
SKP2-mediated p27 degradation is the paradigm for the development of SCF-targeted inhibitors because of our extensive understanding of the biochemistry, a high quality crystal structure, and an established link to disease. However, several other F-box proteins represent intriguing targets for the development of inhibitors (Table 2). Although the biochemistry of substrate degradation by these F-box proteins is incompletely understood and firm links to disease may not yet be established, several features of these F-box proteins underscore their potential as drug targets.
FBXL3
FBXL3 controls circadian rhythm oscillations through the degradation of the Cryptochrome proteins (CRY1 and CRY2) 77–79. Abnormalities in the circadian machinery are linked to sleep disorders, mood disorders, and a variety of metabolic disorders, suggesting that pharmacologic manipulation of circadian rhythms could be of therapeutic use. Our rapidly evolving understanding of the biochemistry of FBXL3-mediated CRY degradation presents several opportunities for drug development. The X-ray crystal structure of the FBXL3-CRY2 complex was recently solved, revealing several key features of this interaction 80. FBXL3 binds CRY2 across an extended region through its Leucine-rich repeats (LRRs). Additionally, the conserved tail of FBXL3 sticks out abruptly from the arch-like LRR domain and inserts into a deep pocket in CRY2. Intriguingly, this conserved pocket binds the redox factor FAD, raising the possibility that FAD or FAD analogs may function as competitive inhibitors of FBXL3 binding to CRY proteins. In fact, a small, FAD-like molecule (KL001) that lengthens the circadian cycle was previously identified in a screen 81, and this drug has now been shown to occupy the CRY2 cofactor pocket, likely blocking insertion of the tail of FBXL3 82, impairing the ability of FBXL3 to bind to CRY2 and mediate its ubiquitylation.
This detailed understanding of FBXL3-mediated CRY degradation – and the means to inhibit this degradation via FAD analogs - has outpaced our current understanding of the potential roles of CRY in disease. This discrepancy illustrates a key issue for the SCF field in the future. Historically, SCF research has focused on cancer and cell proliferation, and although it is clear that F-box proteins will regulate many other processes, research into these areas has lagged. Additionally, while inhibition of FBXL3 may be advantageous in terms of circadian regulation and disorders associated with CRY dysregulation, there could be other FBXL3 substrates, affecting unknown processes, so the overall biological effects of some classes of FBXL3 inhibitors may be more pleiotropic than others. Also, KL001, as an analog of a common metabolite, may influence other pathways. However, it is intriguing that KL001, through its stabilization of CRY, is able to inhibit glucagon-induced gluconeogenesis in hepatocytes in cell culture 81, hinting that inhibition of CRY degradation might be utilized beyond the regulation of sleep-wake cycles in the treatment of diabetes and metabolic disorders.
FBXL2
While the FBXL3-CRY interaction has firm biochemistry and weaker links to disease, there is a strong foundation to support the development of FBXL2 inhibitors, despite a lack of complete biochemical information. FBXL2 is one of two F-box proteins featuring CaaX motifs, which direct its geranylgeranylation and localization to cell membranes, and this membrane localization appears to be required for PI3K activation through FBXL2-mediated degradation of p85β 26. FBXL2 also binds to the Hepatitis C Virus (HCV) NS5A protein, and an undefined, SCF- and localization-dependent activity of FBXL2 is required for the replication of HCV 52. Notably, HCV was previously observed to require geranylgeranylated host proteins, which may be a reflection of this requirement for FBXL2 function 83. However, this idea requires clear demonstration, and although NS5A is not a target for FBXL2, the relevant targets of FBXL2 in the context of HCV infection remain unclear. Despite the recent development of effective HCV therapies, FBXL2 offers a distinct new target to counter the evolution of resistance to current drugs, and novel HCV therapies may prove more economical 84, 85.
The development of FBXL2 inhibitors is challenging because the biochemistry of FBXL2-mediated degradation remains unclear irrespective of HCV infection. Several potential FBXL2 substrates have been identified, but the degradation of these substrates has not been linked to HCV replication 26, 86–89. It is also unknown whether NS5A enhances ubiquitylation of normal FBXL2 substrates or retargets FBXL2 to novel substrates. In the context of uninfected cells, the limited number of reported FBXL2 substrates does not support a consensus mechanism for understanding the biochemistry of FBXL2-mediated degradation, and there is no crystal structure available. Therefore, although there is a strong rationale for developing FBXL2 inhibitors for the treatment of HCV, insufficient biochemical detail is available to support the development of effective inhibitors. As with other F-box proteins, FBXL2 has multiple substrates, and FBXL2-targeted therapies might have effects beyond limiting HCV infection. Therefore, the best FBXL2-targeted therapies may focus on the binding between FBXL2 and NS5A instead of interactions with the substrate or SCF scaffold, although further investigation of FBXL2 functions in the absence of HCV infection may suggest the application of additional FBXL2 inhibitors in settings beyond HCV infection 26, 86, 88, 90.
Retargeting the F-box protein-substrate interface
Inhibitors of SCF ligase function are needed for pathologies resulting from F-box overactivity/overexpression, such as SKP2 in tumours. However, it is often the loss of F-box protein activity that leads to disease, and in these situations, the function of a defective SCF ligase must be restored, presenting a different set of problems for generating effective therapies (Figure 3). FBXW7 is a well-established tumour suppressor in a variety of tissues 60, 91–94. The majority of FBXW7 substrates are oncoproteins, including cyclin E, c-Myc, Notch, and c-JUN, (reviewed in 60). FBXW7 is located at a genomic region that is frequently deleted in tumours, and it is estimated that 6% of all cancers have mutations in the FBXW7 gene 95–97. It is mutated most frequently in T-cell acute lymphoblastic leukemia (31%) and cholangiocarcinomas (35%), colorectal cancer (17%), endometrial cancers (16%), bladder cancers (10%), lung squamous cell carcinomas (6%) although mutations are also observed in gastrointestinal and prostate cancers. FBXW7 can be deleted, but the most common form of inactivation is through point mutations, with the most common mutations occurring in the substrate recognition domain of FBXW7 95. The substrate binding domain is composed of a series of WD-40 repeats that form an eight-bladed β-propeller structure 98, and nearly half of the tumour-associated missense mutations result in amino acid substitutions at three key arginine residues within the WD40 domain (Arg465Cys/His, Arg479Leu, or Arg505Cys) that form the main substrate recognition contacts. The F-box protein-substrate interface is also subject to mutations in its oncogenic substrates. For example, the phosphodegron of c-Myc is frequently mutated in Burkitt’s lymphoma, resulting in the stabilization of c-Myc 99, 100, and mutations that disrupt Notch binding to FBXW7 are found in T-ALL 96, 101.
Like SKP2, FBXW7 represents a model for the development of SCF-targeted drugs, but restoring the function of mutated FBXW7 requires different approaches. In the straightforward case of simple deletion, where FBXW7 is completely absent, the degradation of FBXW7 substrates must be reconstituted, creating a large challenge. However, evidence suggests that other ubiquitin ligases could be re-targeted to replace the functionality of FBXW7. Proteolysis Targeting Chimeras (PROTACS) were originally developed as hetero-bivalent, chimeric molecules that recruit an E3 ligase with a degron mimicking compound at one end and recruit a specific target substrate with a compound on the other side, effectively tethering the ligase to the substrate, resulting in target degradation (Figure 3). The first PROTAC linked the IκBα degron, which is recognized by βTrCP (βTrCP1/FBXW1 and βTrCP2/FBXW11, which share biochemical function) to ovalicin, a drug that covalently binds to MetAP-2, so this drug targeted MetAP-2 for SCFβTrCP-mediated degradation 102, 103. Variations on this theme have been shown to be effective, at least in cell culture model systems, and in addition to peptide-degron fusions, PROTACS that are entirely based on small-molecules have been developed 104. For example, the androgen receptor ligand was fused to a nutlin, to recruit the androgen receptor to the Mdm2 ubiquitin ligase 105.
The principle of ubiquitin ligase re-targeting received recent validation with the understanding of the biochemical basis for the effectiveness of lenalidomide (a thalidomide derivative) in multiple myeloma treatment. Lenalidomide has been successfully used in the clinic for many years, but its mechanism of action was entirely unknown. Strikingly, lenalidomide binds to Cereblon (CRBN), a substrate recognition subunit for a CRL4 ubiquitin ligase complex, forcing CRBN to target the Ikaros transcription factors for ubiquitylation and degradation 106, 107. Ikaros proteins are essential for the survival of multiple myeloma cells, accounting for the effects of lenalidomide. The recently solved crystal structure of lenalidomide in complex with CRBN shows that lenalidomide binds the substrate binding domain of CRBN and blocks ubiquitination of an endogenous CRBN substrate, suggesting that lenalidomide-associated phenotypes may be associated with both inhibition of endogenous substrate degradation and de novo substrate targeting 108. Notably, while lenalidomide and the other thalidomide derivatives all inhibit ubiquitination of MEIS2, Ikaros protein recruitment varies among these compounds. Crystal structures of the CRBN-lenalidomide-Ikaros and CRBN-MEIS2 complexes are likely required to fully understand the biochemical mechanisms of lenalidomide. However, our current understanding of the interaction of lenalidomide with CRBN supports the idea that similar strategies can be used to target SCF ubiquitin ligases. Due to the nature of degron recognition by F-box proteins, which often recognize small chemical modifications, SCF ubiquitin ligases may be particularly amenable to regulation through bivalent small molecules for the restoration of a lost function and/or de novo substrate targeting.
However, in addition to requiring extensive knowledge of both the ubiquitin ligase and the proposed substrate, the retargeting approach to restoring function faces other barriers. For example, a PROTAC that binds and re-targets an F-box protein may disrupt the regulation of that F-box protein’s normal substrates or target more than one new substrate, creating off target effects (although existing PROTACs have not demonstrated this problem). Indeed, the side effects of thalidomide are well documented in multiple animal systems 109. The effects of thalidomide on animal development are mainly due to the binding of CRBN, but the precise biochemical nature of thalidomide’s effect during development (i.e. whether thalidomide inhibits the degradation of a normal CRBN substrate or re-targets CRBN to an alternative substrate) is unclear 110.
An intermediate approach to restoring the function of mutated F-box proteins has been suggested by the characterization of the Arabidopsis auxin receptor TIR1, which is an F-box protein. TIR1 mediates the degradation of a family of transcription factors via a unique degron composed of the target protein and the hormone auxin. Auxin fills a cavity in the substrate-binding site of TIR1, establishing an additional molecular surface to stabilize the interaction between the substrate and TIR1111, and this auxin based system can be reconstituted in mammalian cells, demonstrating proof of concept for compounds that function in a manner similar to auxin112. By analogy to auxin, when the binding between an F-box protein and its substrate is disrupted by mutation of the substrate binding domain of the F-box protein, a small molecule may be capable of acting as a molecular glue between the proteins and restoring proper degradation.
The development of auxin-like molecular glue compounds are restricted to substrate binding surfaces and may not be feasible for all disease-associated mutations of F-box proteins, which could affect the global protein structure. However, small molecules have been effectively used to bind and stabilize the conformation of mutated p53 (specifically the Y220C mutant) 113, 114. These ‘reactivators’ act as molecular chaperones by preferentially interacting with the correctly folded conformation of p53, stabilizing its functional structure. Reactivators that bind F-box proteins outside of the substrate binding domains could stabilize or shift structural elements to restore proper substrate recognition and ubiquitylation.
Because many proteins feature mutation hotspots, a small array of compounds (either auxin- or reactivator-like) could allow the treatment of a high percentage of patients. For example, a large percentage of FBXW7 mutations occur at three arginines in the substrate binding region, making FBXW7 a prime target for auxin-like therapies that stabilize the binding interface with substrates 95. Additionally, allosteric inhibitors of yeast Cdc4, the yeast homolog of FBXW7, suggest that compounds that bind outside of the substrate binding interface can positively or negatively affect substrate recruitment, supporting the idea that reactivators can be developed for FBXW7 mutants 71.
Emerging targets for restorative therapy
More recently, another F-box protein has been reported to be widely mutated or deleted in cancers, suggesting it acts as a tumour suppressor. FBXO11 deletions or inactivating mutations are observed in ~15% of patients with Diffuse Large B Cell Lymphoma (DLBCL) and DLBCL cell lines. FBXO11 targets the BCL6 oncoprotein for degradation, and in B-cells, FBXO11 mutation directly corresponds with stabilization of BCL6 115. Accordingly, FBXO11 mutations occur predominantly in the CASH repeat domains, the presumptive substrate-binding domain of FBXO11, and block BCL6 binding. Reconstitution of FBXO11 in DLBCL cell lines induces BCL6 degradation and cell death. Although FBXO11’s potential as a tumour suppressor is highlighted by BCL6 degradation in B-cells, FBXO11 mutations are also observed in cancers of the colon, lung, ovary, head and neck, and other lymphomas, implying that FBXO11 functions as a tumour suppressor in other tissues, likely through additional substrates, as BCL6 is not normally expressed outside of B-cells 116–120. CDT2 is also targeted for degradation by FBXO11, and it is overexpressed and/or amplified in Ewing sarcomas, breast, liver, and gastrointestinal cancers, suggesting that CDT2 is oncogenic 24, 25, 121–125. Other, unidentified substrates may also contribute to the tumour suppressive activity of FBXO11, and remarkably little is known about the biochemical basis of substrate binding by FBXO11 (Table 2). The CDT2 degron is inhibited by phosphorylation, but a BCL6 degron has not been mapped 24, 25. Furthermore, no structural information is available for FBXO11. Despite this lack of information, FBXO11 remains an attractive potential target for restorative therapies.
Evidence also exists to support a role for FBXO31 and FBXL4 mutation in human disease, suggesting that these F-box proteins will be appropriate for “restoration of function” therapies. FBXO31 is linked to tumour suppression and to an inherited intellectual disability, while FBXL4 is mutated in mitochondrial encephalomyopathy 126–129. However, both FBXO31 and FBXL4 are emerging targets. Only a few targets have been proposed for each, and the relationships of these substrates to the human disorders are unclear. Additionally, little is known about the biochemistry of substrate targeting by either FBXO31 or FBXL4, preventing the rational design of targeted therapies (Table 2).
Alternative Models for Inhibition of F-box protein function
Notably, although much of this review has focused on the efforts to develop therapies based on F-box proteins with singular biological functions, the effectiveness of bortezomib and MLN4924 also support that idea that broad inhibition of degradation can be an effective strategy for cancer chemotherapy, especially when applied in a targeted manner to specific diseases and/or patients. Therefore, F-box proteins with a large number of substrates in biologically diverse pathways or F-box proteins with key functions in particular tissues may be suitable targets, although the therapeutic window may be small. These concepts are illustrated by two of the best-characterized F-box proteins: βTrCP and FBXW7.
βTrCP targets an extremely broad array of substrates in multiple biological pathways, including β-catenin, IκBα (and other IκB proteins), CDC25A, Emi1, PDCD4, BimEL, Claspin, and Period 5. The diversity of βTrCP substrates is supported, in part, by the use of a canonical phosphodegron, the motif DSGxxS, with both Serines phosphorylated. The regulation of βTrCP binding by phosphorylation of the degron allows stimulus specific degradation. When GSK3 is active, β-catenin is degraded and IκBα remains stable. Conversely, when the IKK complex is active, IκBα is degraded and β-catenin remains stable. βTrCP itself appears to be constitutively expressed and active.
Many βTrCP substrates are important in human pathologies. β-catenin, CDC25A, and others are oncogenes, but IκBα, PDCD4, BimEL, and others are tumour suppressors 5. Additionally, Period regulates circadian rhythms. All of these proteins are deregulated in various disorders, highlighting the difficulty of generating a single outcome via inhibition of βTrCP. However, there are several justifications to explore inhibition of βTrCP function as a therapy. With specific molecular genetic knowledge of a disease, βTrCP inhibitors could be applied in a targeted manner. For example, many B-cell malignancies are dependent upon active NF-κB signaling for their survival 130, and inhibition of βTrCP would increase IκB levels, decreasing this survival signaling 131. βTrCP inhibition could also induce apoptosis via BimEL stabilization in cancers with activated MAPK pathways 132. Alternatively, the pure breadth and importance of βTrCP substrates in regulating the core cell cycle could be exploited. Silencing βTrCP also sensitizes cells to genotoxic stress 133 and inhibition of βTRCP has the potential to arrest the cell cycle at multiple points, which could lead to cell death for proliferating tumour cells. Indeed, inhibitors of βTrCP that function at nanomolar concentrations have been developed, and they are effective at blocking the proliferation of breast and prostate cancer cells (personal communication N Wilkie and http://ub-pharma.com).
Beyond generating direct inhibitors of βTRCP, it may also be possible to exploit the regulation of the βTRCP phosphodegron by kinases. The inhibition of a specific kinase could block one substrate’s degradation while leaving another substrate’s degradation unaffected. Notably, this approach has been proposed for manipulation of one FBXW7 substrate. Although FBXW7 is known primarily as a tumour suppressor 60, in the B-cell lineage, it serves a pro-survival factor through the degradation of p100/NF-κB2 76. Stabilization of p100 in B-cells leads to decreased cell survival and proliferation. Like βTrCP, FBXW7 targets substrates using a phosphodegron, and the p100 degron is phosphorylated by GSK3. Blocking this phosphorylation of the p100 degron by GSK3 stabilizes p100 and blocks B-cell proliferation, highlighting the potential to indirectly inhibit F-box dependent degradation by preventing substrate modification.
FBXW7 also highlights another context specific use of F-box protein inhibitors. Although inhibition of a tumour suppressor seems an unusual treatment for cancer, the inhibition of FBXW7 has been proposed as a way to sensitize cancer stem cells to chemotherapies 91, 134–136. For example, in CML, Imatinib is a very effective therapy that can kill most of the cancerous cells, but it fails to kill the cancer initiating cells in the bone marrow, which are often quiescent 137. Upon discontinuation of therapy, the cancer returns, and this initiating cell population is a reservoir that allows the acquisition of inhibitor resistance mutations. For example, forcing these cancer stem cells out of quiescence and into the cell cycle should make them more susceptible to chemotherapy, which differentially affects proliferating cells 134. Indeed, in experimental genetic systems, deletion of Fbxw7 in a mouse CML model facilitates the eradication of these leukemia initiating cells by tyrosine kinase inhibitor therapies. This effect appears to be largely dependent on increases in c-Myc, suggesting that inhibition of c-Myc degradation by FBXW7 or universal FBXW7 inhibition might synergize with existing chemotherapies 135, 137. Whether this approach can be adapted to patients remains unclear, as are the long term effects of FBXW7 inhibition.
Future Perspectives
There has been a disproportionately large effort into the research of a small number of well-characterized F-box proteins. However, even these few F-box proteins have demonstrated that the number of diverse pathways that exploit SCF-mediated proteolysis is large. In contrast, the majority of F-box proteins remain functionally mysterious or “orphans,” with no established substrates, and our lack of understanding of these orphans is a primary impediment to the development of novel F-box protein-targeted therapies. Ideal F-box proteins for targeted therapies have defined biological roles in disease, multiple substrates, defined biochemical mechanisms, and a crystal structure, and although our knowledge of F-box proteins is expanding rapidly, research into many of these proteins is just starting. Undoubtedly, as the substrates and functions of these orphan F-box proteins are elucidated, additional drug targets will become apparent. Importantly, even for those F-box proteins with well defined biochemistry and known structures, more functional and structural data is required to facilitate the development of effective therapies. Finally, as we enter an era when it is possible to know the unique molecular genetic signature of an individual’s disease, there is significant promise in the situational/context-dependent application of inhibitors of SCF complexes.
Historically, the SCF field has focused on the role of F-box proteins in cancer, but it is evident that F-box proteins will influence many aspects of biology beyond cell proliferation and survival. Moving forward, research into non-cancer disorders, such as sleep, mood, metabolic, and intellectual disability disorders, will be required to fully define the role of the F-box family in disease and uncover potential drug targets. However, as exemplified by the established F-box proteins discussed in this review, with the proper information in hand, many different approaches are available to manipulate substrate degradation for therapeutic applications.
Acknowledgments
The authors thank T Cardozo, C Crewes, R Deshaies, R Kumar, R Mallampalli, K Nakayama, N Wilkie, and N Zheng for critical reading of the manuscript and apologize for the omission of many colleagues’ work owing to space constraints. J.R.S. is a Special Fellow of The Leukemia & Lymphoma Society. J.K.P. is supported by a fellowship from the Lymphoma Research Foundation. Work in the Pagano laboratory is supported by grants from the NIH (R37CA076584 and R01GM057587). M.P. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Rieser E, Cordier SM, Walczak H. Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem Sci. 2013;38:94–102. doi: 10.1016/j.tibs.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–29. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 4.Nijman SM, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–86. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–49. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14:369–81. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843:13–25. doi: 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki E, et al. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS One. 2011;6:e27996. doi: 10.1371/journal.pone.0027996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruschak AM, Slassi M, Kay LE, Schimmer AD. Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst. 2011;103:1007–17. doi: 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- 10.Kubiczkova L, Pour L, Sedlarikova L, Hajek R, Sevcikova S. Proteasome inhibitors - molecular basis and current perspectives in multiple myeloma. J Cell Mol Med. 2014 doi: 10.1111/jcmm.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mattern MR, Wu J, Nicholson B. Ubiquitin-based anticancer therapy: carpet bombing with proteasome inhibitors vs surgical strikes with E1, E2, E3, or DUB inhibitors. Biochim Biophys Acta. 2012;1823:2014–21. doi: 10.1016/j.bbamcr.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10:319–31. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21:301–7. doi: 10.1038/nsmb.2780. [DOI] [PubMed] [Google Scholar]
- 14.Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci. 2012;125:531–7. doi: 10.1242/jcs.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 16.Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–61. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei N, Serino G, Deng XW. The COP9 signalosome: more than a protease. Trends Biochem Sci. 2008;33:592–600. doi: 10.1016/j.tibs.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Jin J, et al. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–80. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skaar JR, D’Angiolella V, Pagan JK, Pagano M. SnapShot: F Box Proteins II. Cell. 2009;137:1358, 1358 e1. doi: 10.1016/j.cell.2009.05.040. [DOI] [PubMed] [Google Scholar]
- 20.Sutterluty H, et al. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1:207–14. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 21.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–9. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 22.Ganoth D, et al. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;3:321–4. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 23.Spruck C, et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell. 2001;7:639–50. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- 24.Abbas T, et al. CRL1-FBXO11 promotes Cdt2 ubiquitylation and degradation and regulates Pr-Set7/Set8-mediated cellular migration. Mol Cell. 2013;49:1147–58. doi: 10.1016/j.molcel.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi M, et al. Regulation of the CRL4(Cdt2) ubiquitin ligase and cell-cycle exit by the SCF(Fbxo11) ubiquitin ligase. Mol Cell. 2013;49:1159–66. doi: 10.1016/j.molcel.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuchay S, et al. FBXL2- and PTPL1-mediated degradation of p110-free p85beta regulatory subunit controls the PI(3)K signalling cascade. Nat Cell Biol. 2013;15:472–80. doi: 10.1038/ncb2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pitts TM, Davis SL, Eckhardt SG, Bradshaw-Pierce EL. Targeting nuclear kinases in cancer: Development of cell cycle kinase inhibitors. Pharmacol Ther. 2014;142:258–269. doi: 10.1016/j.pharmthera.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 29.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilhelm S, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–44. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 31.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–51. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 32.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 34.Van Maerken T, et al. Pharmacologic activation of wild-type p53 by nutlin therapy in childhood cancer. Cancer Lett. 2014;344:157–65. doi: 10.1016/j.canlet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 36.Li L, et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju083. [DOI] [PubMed] [Google Scholar]
- 37.Li H, et al. Inactivation of SAG/RBX2 E3 ubiquitin ligase suppresses KrasG12D-driven lung tumorigenesis. J Clin Invest. 2014;124:835–46. doi: 10.1172/JCI70297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu Y, et al. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood. 2014;123:3269–76. doi: 10.1182/blood-2013-08-521914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milhollen MA, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116:1515–23. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 40.Pan WW, et al. Ubiquitin E3 ligase CRL4(CDT2/DCAF2) as a potential chemotherapeutic target for ovarian surface epithelial cancer. J Biol Chem. 2013;288:29680–91. doi: 10.1074/jbc.M113.495069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackintosh C, et al. WEE1 accumulation and deregulation of S-phase proteins mediate MLN4924 potent inhibitory effect on Ewing sarcoma cells. Oncogene. 2013;32:1441–51. doi: 10.1038/onc.2012.153. [DOI] [PubMed] [Google Scholar]
- 42.Wei D, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72:282–93. doi: 10.1158/0008-5472.CAN-11-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith MA, et al. Initial testing of the investigational NEDD8-activating enzyme inhibitor MLN4924 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;59:246–53. doi: 10.1002/pbc.23357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu GW, et al. Mutations in UBA3 confer resistance to the NEDD8-activating enzyme inhibitor MLN4924 in human leukemic cells. PLoS One. 2014;9:e93530. doi: 10.1371/journal.pone.0093530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blank JL, et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013;73:225–34. doi: 10.1158/0008-5472.CAN-12-1729. [DOI] [PubMed] [Google Scholar]
- 46.Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70:10310–20. doi: 10.1158/0008-5472.CAN-10-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer. 2010;1:708–16. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiley DJ, et al. Yeast Augmented Network Analysis (YANA): a new systems approach to identify therapeutic targets for human genetic diseases. F1000Res. 2014;3:121. doi: 10.12688/f1000research.4188.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell. 2011;19:168–76. doi: 10.1016/j.ccr.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Toth JI, Yang L, Dahl R, Petroski MD. A gatekeeper residue for NEDD8-activating enzyme inhibition by MLN4924. Cell Rep. 2012;1:309–16. doi: 10.1016/j.celrep.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Milhollen MA, et al. Treatment-emergent mutations in NAEbeta confer resistance to the NEDD8-activating enzyme inhibitor MLN4924. Cancer Cell. 2012;21:388–401. doi: 10.1016/j.ccr.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Wang C, et al. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol Cell. 2005;18:425–34. doi: 10.1016/j.molcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 53.Sandberg JK, Andersson SK, Bachle SM, Nixon DF, Moll M. HIV-1 Vpu interference with innate cell-mediated immune mechanisms. Curr HIV Res. 2012;10:327–33. doi: 10.2174/157016212800792513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang EE, et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat Med. 2010;16:1152–6. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Price CT, et al. Molecular mimicry by an F-box effector of Legionella pneumophila hijacks a conserved polyubiquitination machinery within macrophages and protozoa. PLoS Pathog. 2009;5:e1000704. doi: 10.1371/journal.ppat.1000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L, Villa NY, McFadden G. Interplay between poxviruses and the cellular ubiquitin/ubiquitin-like pathways. FEBS Lett. 2009;583:607–14. doi: 10.1016/j.febslet.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 57.Dupuis J, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–16. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Fonzo A, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. 2009;72:240–5. doi: 10.1212/01.wnl.0000338144.10967.2b. [DOI] [PubMed] [Google Scholar]
- 59.Toh KL, et al. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291:1040–3. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- 60.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 61.Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14:233–47. doi: 10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kossatz U, et al. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004;18:2602–7. doi: 10.1101/gad.321004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakayama K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000;19:2069–81. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakayama K, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6:661–72. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- 65.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9:661–4. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 66.Hao B, et al. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 67.Wu L, et al. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol. 2012;19:1515–24. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pavlides SC, et al. Inhibitors of SCF-Skp2/Cks1 E3 ligase block estrogen-induced growth stimulation and degradation of nuclear p27kip1: therapeutic potential for endometrial cancer. Endocrinology. 2013;154:4030–45. doi: 10.1210/en.2013-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chan CH, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–68. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aghajan M, et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat Biotechnol. 2010;28:738–42. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Orlicky S, et al. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat Biotechnol. 2010;28:733–7. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen Q, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood. 2008;111:4690–9. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang KS, Vassilev LT. High-throughput screening for inhibitors of the Cks1-Skp2 interaction. Methods Enzymol. 2005;399:717–28. doi: 10.1016/S0076-6879(05)99047-2. [DOI] [PubMed] [Google Scholar]
- 74.Rico-Bautista E, Yang CC, Lu L, Roth GP, Wolf DA. Chemical genetics approach to restoring p27Kip1 reveals novel compounds with antiproliferative activity in prostate cancer cells. BMC Biol. 2010;8:153. doi: 10.1186/1741-7007-8-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang GJ, et al. Bioluminescent imaging of Cdk2 inhibition in vivo. Nat Med. 2004;10:643–8. doi: 10.1038/nm1047. [DOI] [PubMed] [Google Scholar]
- 76.Busino L, et al. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol. 2012;14:375–85. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Busino L, et al. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316:900–4. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- 78.Godinho SI, et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- 79.Siepka SM, et al. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–23. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xing W, et al. SCF(FBXL3) ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature. 2013;496:64–8. doi: 10.1038/nature11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hirota T, et al. Identification of small molecule activators of cryptochrome. Science. 2012;337:1094–7. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nangle S, Xing W, Zheng N. Crystal structure of mammalian cryptochrome in complex with a small molecule competitor of its ubiquitin ligase. Cell Res. 2013;23:1417–9. doi: 10.1038/cr.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ye J, et al. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci U S A. 2003;100:15865–70. doi: 10.1073/pnas.2237238100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feeney ER, Chung RT. Antiviral treatment of hepatitis C. BMJ. 2014;349:g3308. doi: 10.1136/bmj.g3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pawlotsky JM. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology. 2014;146:1176–92. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 86.Chen BB, Coon TA, Glasser JR, Mallampalli RK. Calmodulin antagonizes a calcium-activated SCF ubiquitin E3 ligase subunit, FBXL2, to regulate surfactant homeostasis. Mol Cell Biol. 2011;31:1905–20. doi: 10.1128/MCB.00723-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen BB, et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol. 2013;14:470–9. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen BB, Glasser JR, Coon TA, Mallampalli RK. Skp-cullin-F box E3 ligase component FBXL2 ubiquitinates Aurora B to inhibit tumorigenesis. Cell Death Dis. 2013;4:e759. doi: 10.1038/cddis.2013.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen BB, Glasser JR, Coon TA, Mallampalli RK. F-box protein FBXL2 exerts human lung tumor suppressor-like activity by ubiquitin-mediated degradation of cyclin D3 resulting in cell cycle arrest. Oncogene. 2012;31:2566–79. doi: 10.1038/onc.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen BB, et al. F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood. 2012;119:3132–41. doi: 10.1182/blood-2011-06-358911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matsuoka S, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008;22:986–91. doi: 10.1101/gad.1621808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Onoyama I, et al. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med. 2007;204:2875–88. doi: 10.1084/jem.20062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thompson BJ, et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. 2008;205:1395–408. doi: 10.1084/jem.20080277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crusio KM, King B, Reavie LB, Aifantis I. The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene. 2010;29:4865–73. doi: 10.1038/onc.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Akhoondi S, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–12. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 96.Maser RS, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–71. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Annunziata CM, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol. 2000;20:2423–35. doi: 10.1128/mcb.20.7.2423-2435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bahram F, von der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95:2104–10. [PubMed] [Google Scholar]
- 101.Weng AP, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 102.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–9. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sakamoto KM, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2:1350–8. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 104.Bouchie A, Allison M, Webb S, Defrancesco L. Nature Biotechnology’s academic spinouts of 2013. Nat Biotechnol. 2014;32:229–38. doi: 10.1038/nbt.2846. [DOI] [PubMed] [Google Scholar]
- 105.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904–8. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kronke J, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–5. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu G, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–9. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fischer ES, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014 doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ito T, Ando H, Handa H. Teratogenic effects of thalidomide: molecular mechanisms. Cell Mol Life Sci. 2011;68:1569–79. doi: 10.1007/s00018-010-0619-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ito T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–50. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 111.Tan X, et al. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature. 2007;446:640–5. doi: 10.1038/nature05731. [DOI] [PubMed] [Google Scholar]
- 112.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–22. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- 113.Basse N, et al. Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant. Chem Biol. 2010;17:46–56. doi: 10.1016/j.chembiol.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 114.Boeckler FM, et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A. 2008;105:10360–5. doi: 10.1073/pnas.0805326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duan S, et al. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012;481:90–3. doi: 10.1038/nature10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lohr JG, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–84. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 118.Stransky N, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 120.Richter J, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012;44:1316–1320. doi: 10.1038/ng.2469. [DOI] [PubMed] [Google Scholar]
- 121.Baraniskin A, et al. MiR-30a-5p suppresses tumor growth in colon carcinoma by targeting DTL. Carcinogenesis. 2012;33:732–9. doi: 10.1093/carcin/bgs020. [DOI] [PubMed] [Google Scholar]
- 122.Pan HW, et al. Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell Cycle. 2006;5:2676–87. doi: 10.4161/cc.5.22.3500. [DOI] [PubMed] [Google Scholar]
- 123.Ueki T, et al. Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene. 2008;27:5672–83. doi: 10.1038/onc.2008.186. [DOI] [PubMed] [Google Scholar]
- 124.Mackintosh C, et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene. 2012;31:1287–98. doi: 10.1038/onc.2011.317. [DOI] [PubMed] [Google Scholar]
- 125.Li J, et al. Identification of retinoic acid-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. Br J Cancer. 2009;101:691–8. doi: 10.1038/sj.bjc.6605202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mir A, et al. Truncation of the E3 ubiquitin ligase component FBXO31 causes non-syndromic autosomal recessive intellectual disability in a Pakistani family. Hum Genet. 2014 doi: 10.1007/s00439-014-1438-0. [DOI] [PubMed] [Google Scholar]
- 127.Seeley AH, et al. Macrocerebellum, epilepsy, intellectual disability, and gut malrotation in a child with a 16q24.1-q24.2 contiguous gene deletion. Am J Med Genet A. 2014 doi: 10.1002/ajmg.a.36569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bonnen PE, et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet. 2013;93:471–81. doi: 10.1016/j.ajhg.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gai X, et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet. 2013;93:482–95. doi: 10.1016/j.ajhg.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yaron A, et al. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396:590–4. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 132.Dehan E, et al. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell. 2009;33:109–16. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Soldatenkov VA, Dritschilo A, Ronai Z, Fuchs SY. Inhibition of homologue of Slimb (HOS) function sensitizes human melanoma cells for apoptosis. Cancer Res. 1999;59:5085–8. [PubMed] [Google Scholar]
- 134.Takeishi S, Nakayama KI. Role of Fbxw7 in the maintenance of normal stem cells and cancer-initiating cells. Br J Cancer. 2014 doi: 10.1038/bjc.2014.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Takeishi S, et al. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23:347–61. doi: 10.1016/j.ccr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 136.King B, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552–66. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reavie L, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23:362–75. doi: 10.1016/j.ccr.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Navon A, Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J Biol Chem. 2009;284:33713–8. doi: 10.1074/jbc.R109.018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Buckley DL, Crews CM. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew Chem Int Ed Engl. 2014;53:2312–30. doi: 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ye Y. Diverse functions with a common regulator: ubiquitin takes command of an AAA ATPase. J Struct Biol. 2006;156:29–40. doi: 10.1016/j.jsb.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 141.Fessart D, Marza E, Taouji S, Delom F, Chevet E. P97/CDC-48: proteostasis control in tumor cell biology. Cancer Lett. 2013;337:26–34. doi: 10.1016/j.canlet.2013.05.030. [DOI] [PubMed] [Google Scholar]
- 142.Yen JL, et al. Signal-induced disassembly of the SCF ubiquitin ligase complex by Cdc48/p97. Mol Cell. 2012;48:288–97. doi: 10.1016/j.molcel.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.den Besten W, Verma R, Kleiger G, Oania RS, Deshaies RJ. NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat Struct Mol Biol. 2012;19:511–6. S1. doi: 10.1038/nsmb.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chou TF, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem. 2013;8:297–312. doi: 10.1002/cmdc.201200520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Magnaghi P, et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol. 2013;9:548–56. doi: 10.1038/nchembio.1313. [DOI] [PubMed] [Google Scholar]
- 146.Groettrup M, Pelzer C, Schmidtke G, Hofmann K. Activating the ubiquitin family: UBA6 challenges the field. Trends Biochem Sci. 2008;33:230–7. doi: 10.1016/j.tibs.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 147.Yang Y, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67:9472–81. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- 148.Chen JJ, et al. Mechanistic studies of substrate-assisted inhibition of ubiquitin-activating enzyme by adenosine sulfamate analogues. J Biol Chem. 2011;286:40867–77. doi: 10.1074/jbc.M111.279984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ceccarelli DF, et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145:1075–87. doi: 10.1016/j.cell.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 150.Huang H, et al. E2 enzyme inhibition by stabilization of a low-affinity interface with ubiquitin. Nat Chem Biol. 2014;10:156–63. doi: 10.1038/nchembio.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60:61–8. doi: 10.1007/s12013-011-9185-5. [DOI] [PubMed] [Google Scholar]
- 152.Colland F, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther. 2009;8:2286–95. doi: 10.1158/1535-7163.MCT-09-0097. [DOI] [PubMed] [Google Scholar]
- 153.Lee BH, Finley D, King RW. A High-Throughput Screening Method for Identification of Inhibitors of the Deubiquitinating Enzyme USP14. Curr Protoc Chem Biol. 2012;4:311–30. doi: 10.1002/9780470559277.ch120078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee BH, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–84. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tian Z, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood. 2014;123:706–16. doi: 10.1182/blood-2013-05-500033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chauhan D, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345–58. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Weinstock J, et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP47. ACS Medicinal Chemistry Letters. 2012;3:789–792. doi: 10.1021/ml200276j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kapuria V, et al. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70:9265–76. doi: 10.1158/0008-5472.CAN-10-1530. [DOI] [PubMed] [Google Scholar]
- 159.Chen J, et al. Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18:1390–400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Faesen AC, et al. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol Cell. 2011;44:147–59. doi: 10.1016/j.molcel.2011.06.034. [DOI] [PubMed] [Google Scholar]
- 161.Cohn MA, et al. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell. 2007;28:786–97. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 162.Villamil MA, et al. Serine phosphorylation is critical for the activation of ubiquitin-specific protease 1 and its interaction with WD40-repeat protein UAF1. Biochemistry. 2012;51:9112–23. doi: 10.1021/bi300845s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Villamil MA, Chen J, Liang Q, Zhuang Z. A noncanonical cysteine protease USP1 is activated through active site modulation by USP1-associated factor 1. Biochemistry. 2012;51:2829–39. doi: 10.1021/bi3000512. [DOI] [PubMed] [Google Scholar]
- 164.Cohen P, Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell. 2010;143:686–93. doi: 10.1016/j.cell.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 165.Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10:130–7. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schwartz GK, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1) Br J Cancer. 2011;104:1862–8. doi: 10.1038/bjc.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rocca A, Farolfi A, Bravaccini S, Schirone A, Amadori D. Palbociclib (PD 0332991) : targeting the cell cycle machinery in breast cancer. Expert Opin Pharmacother. 2014;15:407–20. doi: 10.1517/14656566.2014.870555. [DOI] [PubMed] [Google Scholar]
- 168.Fry DW, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–38. [PubMed] [Google Scholar]
- 169.Macias-Perez IM, Flinn IW. GS-1101: a delta-specific PI3K inhibitor in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2013;8:22–7. doi: 10.1007/s11899-012-0142-1. [DOI] [PubMed] [Google Scholar]
- 170.Fruman DA, Rommel C. PI3Kdelta inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Discov. 2011;1:562–72. doi: 10.1158/2159-8290.CD-11-0249. [DOI] [PubMed] [Google Scholar]
- 171.Blagosklonny MV. Flavopiridol, an inhibitor of transcription: implications, problems and solutions. Cell Cycle. 2004;3:1537–42. doi: 10.4161/cc.3.12.1278. [DOI] [PubMed] [Google Scholar]
- 172.Mallampalli RK, et al. Targeting F box protein Fbxo3 to control cytokine-driven inflammation. J Immunol. 2013;191:5247–55. doi: 10.4049/jimmunol.1300456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nakajima H, Fujiwara H, Furuichi Y, Tanaka K, Shimbara N. A novel small-molecule inhibitor of NF-kappaB signaling. Biochem Biophys Res Commun. 2008;368:1007–13. doi: 10.1016/j.bbrc.2008.01.166. [DOI] [PubMed] [Google Scholar]
- 174.Blees JS, et al. Erioflorin Stabilizes the Tumor Suppressor Pdcd4 by Inhibiting Its Interaction with the E3-ligase beta-TrCP1. PLoS One. 2012;7:e46567. doi: 10.1371/journal.pone.0046567. [DOI] [PMC free article] [PubMed] [Google Scholar]