

Abstract
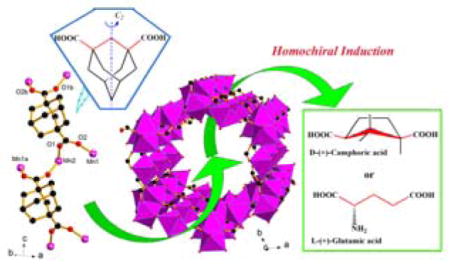
Chirality Induction in Porous MOF The chiral induction reagent (e.g., D-, L-camphoric acid) exhibits two distinct roles: (1) enable and catalyze growth of chiral crystals, and (2) control bulk chirality of Mn3(HCOO)4(adc) crystals (H2adc = adamantane-1,3-dicarboxylic acid. Time-dependent experiments show the initial crystallization of an achiral phase Mn(adc), which persists in the absence of chiral induction agent, but is slowly converted into enantioenriched Mn3(HCOO)4(adc) in the presence of chiral induction agent such as D-camphoric acid, operating in synergy with in situ generated formate.
Keywords: asymmetric crystallization, microporous, crystal structure, Homochirality, catalysis
There is currently an enormous demand for practical synthetic methods to allow the preparation of chiral compounds as single enantiomers.[1,2] While homochirality is a ubiquitous feature in living systems, the generation of homochirality using chemical methods remains highly challenging.[3,4,5] For the synthesis of enantiopure or enantioenriched molecules, a great progress has been achieved in the past several decades through the use of asymmetric homogeneous catalysts.[6] In comparison, there have been very little progresses in the area of asymmetric crystallization from achiral precursors to form enantiopure or enantioenriched crystalline nanoporous materials. The latter is very important for the development of heterogeneous porous asymmetric catalysts based on crystalline solids.
In the past decade, there has been an increasing interest in creating crystalline homochiral porous materials that may be utilized for enantioselective catalysis, separation etc.[1–3] The main objective is to devise homochiral 3-D frameworks with tunable pore geometry and catalytic properties for enantioselective progresses. With few exceptions, homochiral porous solids prepared so far acquire homochirality through the incorporation of enantiopure organic ligands that are bonded to the crystalline framework either as a crosslinking ligand or pendant ligand.[1–3, 7–9]
In the absence of enantiopure building blocks, the chirality can be generated from achiral precursors through crystallization, as evidenced by many crystals (such as quartz) reported in enantiomorphous space groups.[4] In these crystals, chirality comes from the spatial organization of achiral building blocks. While individual crystals can be homochiral through a process called spontaneous resolution, the bulk sample tends to be a conglomerate, an equal mixture of crystals with opposite handedness.[10]
It is worth emphasizing that the generation of chirality itself is usually not an asymmetric process (because of the formation of racemates) and is not uncommon either. In the area of homochiral crystalline porous materials, it is the generation of bulk homochirality from achiral building blocks that is still rare and highly challenging. [11] Several well-known examples of homochiral crystallization (sometimes also called total spontaneous resolution or symmetry breaking) from achiral precursors (e.g., NaClO3) are known.[11a] However, these examples are based on statistical fluctuation of initial nucleation events (e.g., single-colony growth induced by secondary nucleation) and the particular handedness of crystals is not controllable. Such experiments, when repeated multiple times, would normally lead to an overall racemic product. In addition, crystals in these examples are not 3-D porous materials.
One example in the control of the absolute chirality of porous materials built from achiral units was provided by the work of Rosseinsky and co-workers, in which they show that the absolute chirality of an intrinsically chiral 3-connected net can be induced by the coordination of enantiopure solvent molecules to the framework.[12] More recently, controllable homochiral crystallization in 3-D open-framework materials was demonstrated by Morris et al. through the use of the chiral ionic liquid solvent (1-butyl 3-methylimidazolium L-aspartate).[13] One unique and unprecedented aspect of Morris’s work is that the handedness of 3-D framework materials is controlled without having any enantiopure ligand being incorporated into the framework (either through bridging or pendant mode). Complementary to the use of chiral ionic liquid solvents, naturally occurring enantiopure alkaloids such as cinchonidine and cinchonine were recently found to induce the homochiral crystallization of a metal-organic framework.[14]
Despite of these progresses, the controllable asymmetric crystallization of 3-D open-framework materials from achiral precursors remains little explored. It is unlikely that a particular chiral induction reagent can induce absolute chirality in different types of chiral crystals. It is conceivable that the controlled asymmetric crystallization of a porous material with a particular composition and topology, if possible, would likely require a unique chiral induction agent. It is therefore essential to study what types of open-framework structures can be crystallized asymmetrically and with what kinds of chiral induction agents.
In this work, we explore the enantioselective effect of enantiopure organic acids and naturally occurring amino acids. The ultimate goal is to create a library of inexpensive chiral chemicals that can effect asymmetric crystallization of 3-D crystalline porous materials and to use these inexpensive asymmetric molecular catalysts such as organic acids and amino acids to create new crystalline heterogeneous asymmetric catalysts. Furthermore, by exploring different types of chiral induction agents, it is possible to develop a better understanding about chemical or structural factors that contribute to the asymmetric crystallization.
Crystals of (+)-Mn3(HCOO)4(adc) (denoted 1D-Mn, H2adc = adamantane-1,3-dicarboxylic acid) and (−)-Mn3(HCOO)4(adc) (1L-Mn) were solvothermally synthesized (Scheme 1). They consist of 3-D Mn-O-Mn framework of [Mn3(HCOO)4]n2n+ with honeycomb channels along the c axis. Decorative achiral organic chains of adc ligands line up the honeycomb channels by attaching to the wall of [Mn3(HCOO)4]n2n+ through Mn-adc bonds (Figure 1a–b, 1d). It is worth noting that [Mn3(HCOO)4]n2n+ is chiral and forms 31 or 32 helical structures along the c axis. The most interesting aspect of its synthesis is the asymmetric crystallization catalyzed by chiral additives (Figure 1c). In addition, through a comparative study of four different crystallization processes with D-, L-, DL-camphoric acid (H2cam) and without them (Scheme 1), we are able to gain a much better understanding into the chirality induction mechanism. The permanent microporosity of 1D-Mn was characterized by the CO2 adsorption isotherm which shows a significant adsorption of 25.1 cm3·g−1 at ~1 atm and 273 K (Figure S5 in the supporting information).
Scheme 1.
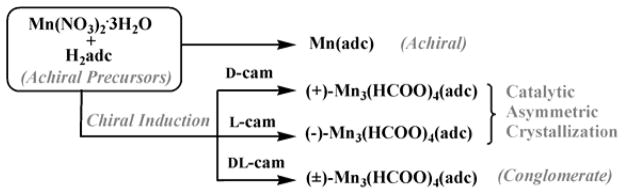
Illustration of four crystallization processes showing that the camphorate ligand not only controls the absolute chirality of crystals, but also enable and catalyze the growth of chiral crystals. The synthesis and crystallization were performed in mixed DMF/EtOH solvents at 120°C. The formate ligand was generated in situ from the solvent DMF.
Figure 1.

(a) The [Mn(adc)]n chain based on achiral adc ligand with μ4-coordination; (b) the porous [Mn3(HCOO)4]n2+ channel based on inorganic Mn-O-Mn connectivity; (c) two types of enantiopure catalysts used for synthesis and chiral induction of 1D-Mn; The directions of arrows show the possible mechanism of chiral induction. D-camphoric acids initially control the absolute chirality of [Mn3(HCOO)4]n2+ frameworks, but are later displaced by adc. (d) the 3-D hybrid framework of 1D-Mn, showing the achiral [Mn(adc)]n chains attached to the wall of the nano-sized channels; (e) the solid-state CD spectra of 1D-Mn (red solid line) and 1L-Mn (blue dashed line).
Crystals of 1D-Mn were found to be predominant in the batch synthesized by using a small amount of enantiopure D-(+)-H2cam as chiral catalyst, while crystals of 1L-Mn were found to be predominant in the batch synthesized by using a small amount of enantiopure L-(−)-H2cam. Note that in both cases, enantiopure camphoric acid is not incorporated into the crystal structure. However, without camphoric acid, crystals of 1D-Mn and 1L-Mn can not even be synthesized, not to mention the control of the absolute chirality.
To confirm the asymmetric crystallization, crystal structures of twenty crystals were randomly picked from the batch catalyzed with D-H2cam and were refined using single-crystal X-ray diffraction data. The Flack parameter of each refinement indicates 18 crystals belong to the P3221 space group while 2 crystals adopt the opposite P3121 space group (Table S1 in the supporting information). This suggests that when D-H2cam is used as the additive, the product is enantioenriched with Mn-1D.
For the batch catalyzed with L-H2cam, another set of twenty crystals were also randomly picked and refined using single-crystal X-ray diffraction data. The Flack parameter of each refinement indicates 18 crystals belong to the P3121 space group while 2 crystals adopt the opposite P3221 space group (Table S2 in the supporting information). This suggests that when L-H2cam is used as the additive, the product is enantioenriched with Mn-1L.
Additional evidence for the asymmetric crystallization comes from the solid-state circular dichroism (CD) spectroscopy. The CD spectra for the five batches catalyzed with D-H2cam show that the bulk sample exhibits a positive CD signal at 218 nm (Figure 1e). In comparison, the CD spectra for the three batches catalyzed with L-H2cam show that the bulk sample exhibits a negative CD signal at a similar wavelength (Figure 1e).
A comparative study of the four different crystallization processes (performed with D-, L- or DL-H2cam and without any H2cam) not only further confirms the observed asymmetric crystallization, but also offers an insight into the mechanism of asymmetric crystallization (Scheme 1). When racemic DL-camphoric was used in place of enantiopure D- or L-H2cam, the resulting bulk sample is a racemic conglomerate. With four randomly picked crystals, two belong to P3221 and the other two belong to P3121. Furthermore, no CD signal was detected for the bulk sample (Table S3 and Figure S3 in the supporting information).
Interestingly, without the addition of H2cam in any chiral form (D-, L- or DL-), a new achiral layered compound Mn(adc) (2) was formed under otherwise identical reaction conditions. This demonstrates that the role of chiral H2cam goes beyond just the control of the absolute chirality in the crystallization of 1D-Mn or 1L-Mn. Camphoric acid is in fact essential for growth of Mn3(HCOO)4(adc) crystals.
The asymmetric crystallization of 1D-Mn and 1L-Mn is believed to result from cooperative interactions between chiral H2cam and achiral adc ligand. Because H2cam has similar coordination geometry with adc (in both, two -COO− groups are separated by three C atoms), camphorate ligand is capable of directly participating in the nucleation and crystallization process, which allows the control of the absolute helicity of [Mn3(HCOO)4]n2n+ frameworks. However, adc ligands eventually replace the camphorate ligands in the final crystals.
The fact that adc and camphorate ligands have similar binding features can be experimentally demonstrated. Indeed, without the competing adc ligand, camphoric acid can itself bond to the [Mn3(HCOO)4]n2n+ framework to directly control the absolute helicity of the [Mn3(HCOO)4]n2n+ as evidenced by isostructural crystal structures of homochiral [Mn3(HCOO)4](D-cam) or [Mn3(HCOO)4](L-cam) reported previously.[16] When competing adc ligands are introduced, Mn3(HCOO)4(adc) crystals are formed and camphorate ligands can no longer be retained in the final crystals. Interestingly, despite being excluded from the final crystals, the camphorate ligand exhibits a chirality induction effect by controlling the absolute chirality of Mn3(HCOO)4(adc) crystals. Such two different ways (i.e., direct binding to the framework or catalytic chiral induction, depending on whether the competing adc ligand is present or not) for the camphorate ligand to control the absolute chirality of 3-D porous [Mn3(HCOO)4]2+ framework is truly unprecedented and extraordinary.
To gain further insight into the crystallization process, time-dependent experiments were performed. After the clear solution of H2adc, D-H2cam and Mn(NO3)2·3H2O in the mixed DMF/ethanol solvent was heated at 120 °C for 3 hours, a large amount of long needle-like crystals of achiral Mn(adc) 2 (Yield: 89 %) were formed. In comparison, for the reaction examined after 12 hours of heating, a small amount of hexagonal-prism-shaped crystals (1D-Mn) was also present together with achiral Mn(adc) 2 crystals, as confirmed by single-crystal X-ray diffraction. After four days, hexagonal prismatic 1D-Mn crystals were obtained in the high yield (80%). Because the needle-like crystals of achiral Mn(adc) 2 can be washed away by ethanol, hexagonal prismatic 1D-Mn crystals can be easily separated out. The chiral property of the bulk sample was subsequently verified by CD spectra.
A four-step mechanism was shown in Scheme 2. The first step is the fast crystallization of achiral Mn(adc) 2. If the chiral induction agent such as D-H2cam is not present, the process is over and only achiral 2 could be obtained, even after four days. The second step is the slow release of HCOO− from the decomposition of DMF molecules, which is common in solvothermal synthesis. With the slow release of HCOO− from DMF, the homochiral nuclei of [Mn3(HCOO)4](D-cam) (3) might be formed (the third step). While the direct experimental observation of intermediate nuclei of 3 has not been made, there are other experimental data to support the existence of such intermediate nuclei 3. If the achiral adc ligand is not added, large crystals of 3 would form.[16] Finally, chiral D-cam induction agent is replaced by achiral adc ligand, which is the fourth step in the proposed mechanism.
Scheme 2.
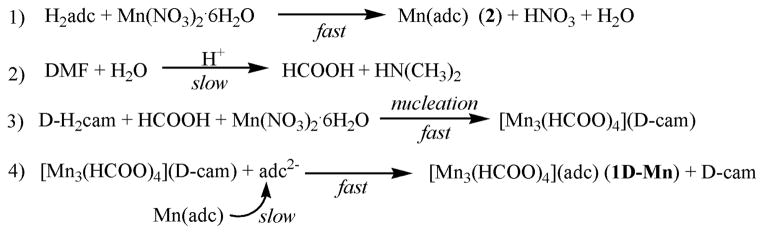
The four-step mechanism for the crystallization of homochiral 1D-Mn. The achiral Mn(adc) (2) crystals are formed first in the initial hours and can persist for days in the absence of camphoric acid. However, in the presence of D-camphoric acid, the achiral Mn(adc) is converted into enantioenriched chiral [Mn3(HCOO)4](adc).
The gradual release of HCOO− anions from DMF is worth noting. The formate not only provides the necessary building units for 1D-Mn, but is closely associated with the kinetics of the crystallization process. However, the presence of too much formate anions all at once through the direct addition of HCOOH or HCOO− at the beginning of the reaction led to the formation of manganese formate.
Recognizing the structural similarity between adc and camphorate ligands, we envisaged that other ligands with the coordination geometry similar to H2cam might also be able to function as the catalyst for the asymmetric crystallization of [Mn3(HCOO)4](adc). Indeed, the use of glutamic acid also leads to the asymmetric crystallization of Mn3(HCOO)4](adc) as evidenced by the CD spectra (Fig. S3). However, different from D- and L-camphoric acids which give 1D-Mn and 1L-Mn, respectively, the L-glutamic acid catalyzes the formation of 1D-Mn while the D-glutamic acid catalyzes the formation of 1L-Mn.
The above study with glutamic acid further suggests that in order to induce asymmetric crystallization, the chiral induction agent may need to possess proper coordination chemistry that matches well with the achiral framework building block. A similar example was recently found in the crystallization of a chiral zincophosphate.[17] To further investigate this idea, we have also studied the effect of other amino acids (L-alanine, L-histidine, and L-aspartic acid) in the crystallization process of Mn3(HCOO)4](adc). For L-alanine, the achiral compound 2, Mn(adc), was obtained, similar to the reaction performed without the use of any induction agent. For L-histidine, the resulting product is an unknown polycrystalline phase. For aspartic acid, neither achiral Mn(adc) nor chiral Mn3(HCOO)4(adc) crystallized and only aspartic acid crystals were re-crystallized.
In conclusion, we demonstrate here an unusual asymmetric crystallization of a new 3-D porous material constructed entirely from achiral building units by using enantiopure organic acids or amino acids as the chirality induction agents. In addition to control the absolute chirality, the chiral induction agent is also essential to initiate the nucleation process of the chiral crystals. It is suggested here that the chirality control is achieved through cooperative binding between enantiopure chiral reagents and achiral structural building units and that enantiopure chiral reagents control the absolute chirality of crystals by participating in the nucleation and crystallization processes, but are later replaced with achiral ligands in the resulting crystals. The discovery that camphoric acid can control the absolute chirality of 3-D porous [Mn3(HCOO)4]2+ framework by either direct binding to the framework or by catalytic chiral induction (depending on whether the competing adc ligand is present or not) is truly unprecedented and extraordinary.
Experimental Section
Typical synthesis of (+)-Mn3(HCOO)4(adc) (1D-Mn)
Adamantane-1,3-dicarboxylic acid (H2adc, 0.0798 g, 0.48 mmol), D-(+)-camphoric acid (0.0280 g, 0.13 mmol) and Mn(NO3)2·3H2O (0.1390 g, 0.90 mmol) in mixed dimethylformamide (DMF, 4.0415 g, ethanol 0.8812 g) solvent were placed in a 20 ml vial. The sample was heated at 120 °C for 4 days, and then cooled to room-temperature. After washing by ethanol, the colorless crystals were obtained (yield: 80%).
Crystal data for 1D-Mn
C16H18Mn3O12, Mr = 567.12, Trigonal, space group P3221, a = b = 15.1841(9) Å, c = 7.8474(9) Å, V = 1566.9(2) Å3, Z = 3, Dc = 1.803 g/cm3, Flack parameter = 0.03(5), R1(wR2) = 0.0510 (0.1366) and S = 1.099 for 1602 reflections with I > 2σ(I).
Crystal data for 1L-Mn
C16H18Mn3O12, Mr = 567.12, Trigonal, space group P3121, a = b = 15.2162(10) Å, c = 7.9063(11) Å, V = 1585.3(3) Å3, Z = 3, Dc = 1.782 g/cm3, Flack parameter = 0.00(4), R1(wR2) = 0.0445 (0.0944) and S = 0.980 for 1424 reflections with I > 2σ(I).
Crystal data for layered compound Mn(adc) (2)
C12H14MnO4, Mr = 277.17, Monoclinic, space group Cc, a = 7.7609(3) Å, b = 20.5827(9) Å, c = 7.0717(3) Å, β = 93.411(3)°, V = 1127.63(8) Å3, Z = 4, Dc = 1.633 g/cm3, Flack parameter = 0.59(3), R1(wR2) = 0.0393 (0.0759) and S = 0.956 for 1843 reflections with I > 2σ(I).
CCDC-736430 (1D-Mn), 736431 (1L-Mn), 736432 (2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html.
Measurement of solid CD spectra
The mixture of about 3-mg sample and 40 mg dried KBr powder was well grounded and then pressed into a disk for use in the CD measurement, using a J-810 spectropolarimeter.
Supplementary Material
Acknowledgments
We thank the support of this work by NIH (X. B. 2 S06 GM063119-05), NSF (X. B. DMR-0846958), and DOE (P. F. 004407-002). X. B is a Henry Dreyfus Teacher Scholar.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Prof. Dr. Jian Zhang, Department of Chemistry and Biochemistry, California State University, Long Beach, 1250 Bellflower Boulevard, Long Beach, CA 90840, Fax: (+) 562-985-8557. State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, CAS Fuzhou, Fujian, China 350002.
Dr. Shumei Chen, Department of Chemistry and Biochemistry, California State University, Long Beach, 1250 Bellflower Boulevard, Long Beach, CA 90840, Fax: (+) 562-985-8557.
Ruben A. Nieto, Department of Chemistry and Biochemistry, California State University, Long Beach, 1250 Bellflower Boulevard, Long Beach, CA 90840, Fax: (+) 562-985-8557
Tao Wu, Department of Chemistry, University of California, Riverside, CA 92521.
Prof. Dr. Pingyun Feng, Department of Chemistry, University of California, Riverside, CA 92521
Prof. Dr. Xianhui Bu, Email: xbu@csulb.edu, Department of Chemistry and Biochemistry, California State University, Long Beach, 1250 Bellflower Boulevard, Long Beach, CA 90840, Fax: (+) 562-985-8557
References
- 1.a) Kesanli B, Lin W. Coord Chem Rev. 2003;246:305. [Google Scholar]; b) Ma L, Abney C, Lin W. Chem Soc Rev. 2009;38:1248–1256. doi: 10.1039/b807083k. [DOI] [PubMed] [Google Scholar]
- 2.Seo JS, Whang D, Lee H, Jun SI, Oh J, Jeon YJ, Kim K. Nature. 2000;404:982. doi: 10.1038/35010088. [DOI] [PubMed] [Google Scholar]
- 3.a) Férey G. Chem Soc Rev. 2008;37:191. doi: 10.1039/b618320b. [DOI] [PubMed] [Google Scholar]; b) Sun J, Bonneau C, Cantín Á, Corma A, Díaz-Cabañas MJ, Moliner M, Zhang D, Li M, Zou X. Nature. 2009;458:1154–1157. doi: 10.1038/nature07957. [DOI] [PubMed] [Google Scholar]; c) Han Y, Zhang DL, Chng LL, Sun JL, Zhao L, Zou XD, Ying JY. Nature Chemistry. 2009;1:123–127. doi: 10.1038/nchem.166. [DOI] [PubMed] [Google Scholar]
- 4.Alexander AJ. Cryst Growth Des. 2008;8:2630. [Google Scholar]
- 5.Lahav M, Leiserowitz L. Cryst Growth & Des. 2006;6:619. [Google Scholar]
- 6.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito, California: 2009. [Google Scholar]
- 7.Vaidhyanathan R, Bradshaw D, Rebilly JN, Barrio JP, Gould JA, Berry NG, Rosseinsky MJ. Angew Chem. 2006;118:6645. doi: 10.1002/anie.200602242. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:6495. [Google Scholar]
- 8.Dybtsev DN, Nuzhdin AL, Chun H, Bryliakov KP, Talsi EP, Fedin VP, Kim K. Angew Chem Int Ed. 2006;45:916. doi: 10.1002/anie.200503023. [DOI] [PubMed] [Google Scholar]
- 9.Anokhina EV, Go YB, Lee Y, Vogt T, Jacobson AJ. J Am Chem Soc. 2006;128:9957. doi: 10.1021/ja062743b. [DOI] [PubMed] [Google Scholar]
- 10.Pérez-García L, Amabilino DB. Chem Soc Rev. 2002;31:342. doi: 10.1039/b201099m. [DOI] [PubMed] [Google Scholar]
- 11.(a) Kondepudi DK, Kaufman RJ, Singh N. Science. 1990;250:975. doi: 10.1126/science.250.4983.975. [DOI] [PubMed] [Google Scholar]; (b) Ezuhara T, Endo K, Aoyama Y. J Am Chem Soc. 1999;121:3279. [Google Scholar]; (c) Wu S, Wu Y, Kang Q, Zhang H, Long L, Zheng Z, Huang R, Zheng L. Angew Chem Int Ed. 2007;46:8475. doi: 10.1002/anie.200703443. [DOI] [PubMed] [Google Scholar]
- 12.Bradshaw D, Prior TJ, Cussen EJ, Claridge JB, Rosseinsky MJ. J Am Chem Soc. 2004;126:6106. doi: 10.1021/ja0316420. [DOI] [PubMed] [Google Scholar]
- 13.Lin Z, Slawin AMZ, Morris RE. J Am Chem Soc. 2007;129:4880. doi: 10.1021/ja070671y. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Chen S, Wu T, Feng P, Bu X. J Am Chem Soc. 2008;130:12882. doi: 10.1021/ja805272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tulashie SK, Lorenz H, Seidel-Morgenstern A. Cryst Growth Des. 2009;9:2387. [Google Scholar]
- 16.Zhang J, Chen S, Valle H, Wong M, Austria C, Cruz M, Bu X. J Am Chem Soc. 2007;129:14168. doi: 10.1021/ja076532y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Chen S, Bu X. Angew Chem Int Ed. 2009;48:6049. doi: 10.1002/anie.200903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.