

Abstract
Replication protein A (RPA) is the main eukaryotic single-stranded DNA (ssDNA) binding protein, having essential roles in all DNA metabolic reactions involving ssDNA. RPA binds ssDNA with high affinity, thereby preventing the formation of secondary structures and protecting ssDNA from the action of nucleases, and directly interacts with other DNA processing proteins. Here we discuss recent results supporting the idea that one function of RPA is to prevent annealing between short repeats that can lead to chromosome rearrangements by microhomology-mediated end joining or the formation of hairpin structures that are substrates for structure-selective nucleases. We suggest that replication fork catastrophe caused by depletion of RPA could result from cleavage of secondary structures by nucleases, and that failure to cleave hairpin structures formed at DNA ends could lead to gene amplification. These studies highlight the important role RPA plays in maintaining genome integrity.
Keywords: chromosome rearrangements, DNA replication, end joining, homologous recombination, Mre11, RPA, Rad51, Sae2
Introduction
RPA is a heterotrimeric complex consisting of 70, 32 and 14 kDa subunits that binds with high affinity to single-stranded DNA (ssDNA) [1, 2]. RPA was originally identified as a factor required for in vitro reconstitution of simian virus 40 (SV40) replication [3]. These studies demonstrated an important role for RPA in stabilizing the unwound ssDNA generated by SV40 large T antigen at the replication origin, and in stimulating the elongation phase of DNA synthesis [2]. RPA is highly conserved among eukaryotes and the genes encoding each subunit, RFA1, RFA2 and RFA3 in budding yeast (RPA1, RPA2 and RPA3 in vertebrates), are essential for viability [4]. In addition to its essential role during DNA synthesis, RPA functions in most DNA repair reactions that involve ssDNA intermediates, including nucleotide excision repair, mismatch repair and homologous recombination (HR), and plays critical roles in DNA damage signaling [2, 5]. The role of RPA in these diverse DNA metabolic processes is to remove secondary structures from ssDNA that would interfere with DNA processing, to shield ssDNA from nucleases, and to interact physically and functionally with other DNA processing proteins. Here, we will discuss how RPA bound to ssDNA stabilizes fragile intermediates during DNA double-strand break (DSB) repair and thereby prevents genome rearrangements.
RPA: A modular DNA binding protein
RPA interacts with ssDNA with a defined 5′-3′ polarity using four OB (oligonucleotide/oligosaccharide binding) folds, also referred to as DNA binding domains DBD-A, -B –C and –D located in the RPA70 and RPA32 subunits (Fig. 1) [1, 6–11]. Two additional OB domains are present in RPA70 and RPA14, but do not contribute to DNA binding [1, 7]. RPA binds to ssDNA in two modes: the low-affinity mode has an occluded binding site of ~8 nucleotides (nt) and involves DBD-A and DBD-B; the high-affinity mode engages all four DBDs and has an occluded binding site of ~30 nt [6]. As an individual domain, DBD-A has the highest affinity for ssDNA and the affinity increases by ~100-fold for the DBD-A-DBD-B peptide [1, 6]. A stepwise transition to the high-affinity mode has been suggested: this involves sequential binding of the DBD-A and DBD-B domain, DBD-C and then DBD-D [1]. The DBD-C and DBD-D domains of RPA70 and RPA32, respectively, are also required for inter-subunit interaction, and the OB fold of RPA14 interacts with DBD-D connecting the three subunits and stabilizing the complex (Fig. 1).
Figure 1.
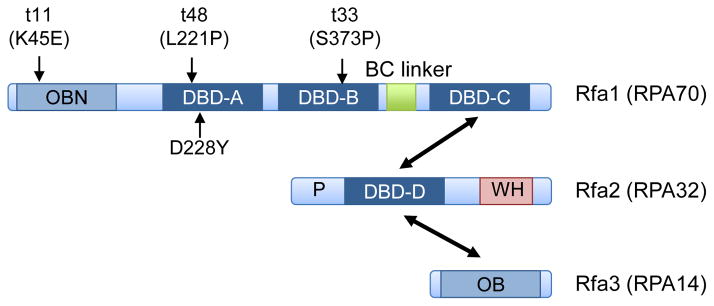
Schematic showing the domains present in the RPA subunits. The names shown are for the budding yeast proteins with the human protein names in parentheses. The approximate locations of the rfa1 mutations described in the text are shown. The DNA binding domains (DBD) are indicated by the dark blue boxes and the OB folds that do not interact with DNA are shown in light blue. WH refers to the winged helix domain; double-headed arrows show the domains involved in subunit interactions.
Binding and extension of ssDNA through the removal of secondary structures has been directly visualized by single molecule imaging using a fluorescently tagged variant of yeast RPA (RPA-eGFP) [12]. RPA rapidly extended ssDNA and remained bound to the substrate with little or no dissociation for more than 60 min when free protein was absent from the solution [12, 13]. However, the bound RPA-eGFP could be rapidly replaced with free RPA-mCherry, or with another ssDNA binding protein, when added to the flow cell [13]. The slow dissociation of RPA, but ability to be replaced, suggests RPA undergoes constant local dissociation of the DBDs, and that complete dissociation of the complex only occurs when free protein present in solution competes with the bound form as a result of mass action. This mode of binding is consistent with the modular organization of the DBDs, and ensures that ssDNA remains protected by RPA, but allows RPA to be displaced by other ssDNA binding proteins when necessary.
In addition to DNA binding, RPA interacts directly with a large number of DNA replication and repair proteins via the N-terminal OB fold of RPA70 and the winged helix domain in the C-terminus of RPA32. The N-terminal region of RPA32 undergoes extensive phosphorylation during the cell cycle and in response to DNA damage [14]: indeed, antibodies directed against specific RPA32 phosphorylation sites can be used to monitor activation of the ATR and ATM kinases in human cells by immunoblotting [15]. RPA1 is also SUMOylated after treatment of human cells with agents that generate DSBs, and this modification increases the interaction between RPA and RAD51 to promote homology-directed repair [16].
Many ways to repair a broken chromosome
Chromosomal DSBs are considered one of the most toxic lesions to the cell, and their accurate repair is essential to maintain genome integrity. Failure to repair DSBs or inaccurate repair can result in loss of genetic information, gross chromosome rearrangements (GCRs) or even cell death. DSBs are repaired by one of two main mechanisms: homologous recombination (HR) or non-homologous end joining (NHEJ) (Fig. 2). HR requires a homologous DNA duplex to template repair of both broken strands, and is generally restricted to the S and G2 phases of the cell cycle when a sister a chromatid is available as a donor duplex [17]. NHEJ involves the direct ligation of the DNA ends and is considered to be more error-prone than HR. NHEJ is the primary mechanism to repair DSBs in human cells, whereas HR is the predominant mechanism in bacteria and yeast. The biased use of HR in organisms with compact genomes may be to minimize mutations caused by erroneous NHEJ in coding DNA. On the other hand, for organisms with many repeated sequences HR needs to be tightly regulated because associated crossovers can generate chromosome rearrangements.
Figure 2.

Models for DSB repair. The DNA ends can be ligated by NHEJ or undergo resection to form 3′ ssDNA tails. The initial processing of the DNA ends has the potential to reveal short homologies internal to the ends that can be used for MMEJ. The long ssDNA tails are initially bound by RPA, which is then replaced by Rad51 with the assistance of Rad52 (or BRCA2). Rad51 promotes invasion of a donor duplex and the 3′ end is extended by DNA synthesis. The extended invading strand can be displaced by a helicase and can pair with the other resected end; alternatively, the displaced strand from the donor duplex pairs with the other break end and after gap filling and ligation a double Holliday junction (dHJ) intermediate is formed. The dHJ is acted on by a helicase and topoisomerase to dissolve the dHJ forming non-crossover products or cleaved by nucleases to generate crossover or non-crossover products.
An essential first step for HR is the 5′-3′ nucleolytic degradation of the DNA ends to produce 3′ ssDNA tails, a process referred to as end resection [18]. The Mre11-Rad50-Xrs2/NBS1 (MRX/N) complex and Sae2 (CtIP in mammalian cells) initiate resection by endonucleolytic cleavage of the 5′-terminated strand ~50–100 nt from the end, creating a short overhang sufficient for high affinity RPA binding [19]. More extensive resection to generate long 3′ ssDNA tails requires the Exo1 nuclease or Sgs1 helicase in conjunction with Dna2 endonuclease [20]. RPA binds to the ssDNA generated by end resection and is frequently used as a cytological marker for resection of DSBs [21, 22]. Because efficient recombination requires >200 nt homology [23], RPA stabilization of ssDNA intermediates is particularly important for this cellular process. In order for HR to proceed, Rad51 has to displace RPA from the ssDNA, a reaction that requires the Rad52 or BRCA2 mediator proteins [24–28]. Once Rad51 is bound to the 3′ ssDNA tail it catalyzes pairing with a homologous duplex, and exchanges one strand of the duplex with the incoming ssDNA tail to form a displacement loop (D-loop) [18]. The 3′ end of the invading strand is then used to prime DNA synthesis, templated by the donor duplex. In the simplest version of recombination models, the extended invading strand is displaced by the action of a helicase and can then pair with the resected end at the other side of the DSB [29]. Alternatively, the displaced strand of the D-loop pairs with the other DSB end and after gap filling and ligation a double Holliday junction intermediate is formed, which is subsequently processed to form crossover or non-crossover products [30].
NHEJ involves direct joining of the fragmented chromosome through end protection by the Ku complex and ligation via DNA ligase IV [31]. NHEJ can join blunt or complementary cohesive ends (such as those produced by restriction endonucleases), or more complex ends with the assistance of end processing factors. The initiation of end resection is inhibitory to end joining because Ku has reduced affinity for long ssDNA overhangs relative to blunt ends [32]. RPA binding to ssDNA-tailed duplexes is likely to create an additional impediment to Ku binding.
In the absence of Ku and Ligase IV, end joining can still occur at a significant frequency in mammalian cells and at a much lower frequency in budding yeast [33–36]. The more efficient use of MMEJ in mammalian cells is likely due to proteins that can synapse and ligate ends, such as PARP-1 and DNA ligase III, respectively [37–40], that are absent from budding yeast. The alternative end joining mechanism in yeast utilizes microhomologies (MH) in the range of 5–20 nucleotides internal to the ends to align them for repair, and consequently has been named microhomology-mediated end joining (MMEJ) [35, 41]. Alternative end joining in mammalian cells also entails use of MH; however, these tracts tend to be shorter than those observed in yeast cells [34, 42, 43]. Interest in the mechanism of MMEJ has grown with the recognition that many chromosome rearrangements associated with human genetic disease, including translocations, copy number variants and chromothripsis, have MH at the breakpoints [44–46]. End resection is needed to expose MH internal to the ends; thus, MMEJ has some of the same requirements as those for HR (Fig. 2). MMEJ is similar to another DSB repair mechanism called single-strand annealing (SSA) that operates between long direct repeats flanking a DSB [18]. SSA requires sufficient resection to expose complementary strands of each repeat, the homologous sequences are annealed and, if necessary, 3′ heterologous flaps are removed by nucleases to allow DNA synthesis and ligation. In yeast, SSA is highly dependent on RAD52 and is independent of RAD51 [18].
RPA plays a critical role in homologous recombination
Much of our understanding of how RPA functions during HR derives from reconstitution of early steps of DSB repair with purified components. In vitro reconstitution of the extensive resection mechanisms revealed an essential role for RPA in Sgs1-Dna2 catalyzed resection, and a stimulatory role with Exo1. RPA stabilizes DNA unwound by the Sgs1 helicase and specifically targets the 5′-terminated strand for degradation by Dna2 [47, 48]. RPA acts indirectly to promote Exo1-mediated resection by preventing non-productive association of Exo1 with ssDNA [49]. Biochemical reconstitution of Rad51-catalyzed strand exchange demonstrated a crucial role for RPA to remove secondary structure from the ssDNA; thus facilitating polymerization of Rad51 to form the Rad51-ssDNA nucleoprotein filament (Fig. 2) [50, 51]. For RPA to promote Rad51 binding to ssDNA, Rad51 must be added to the reaction before RPA. If the proteins are present at the same time, resembling physiological conditions, then Rad52 or BRCA2 is required to facilitate RPA displacement by Rad51 [24–28]. Rad52 binds directly to both Rad51 and RPA, providing a high local concentration of Rad51 to compete with RPA for DNA binding [52–54].
Rad52 also promotes annealing of ssDNA in vitro and this activity is likely important for capture of the second end in HR and for the SSA mechanism of recombination [55–58], RPA is inhibitory to ssDNA annealing in vitro; however, Rad52 is able to overcome this inhibition [59]. Short complementary oligonucleotides (~50 nucleotides in length) undergo efficient spontaneous annealing and the rate is greatly accelerated by Rad52, but for longer ssDNA with the potential to form extensive secondary structure, Rad52-catalyzed annealing requires RPA [55, 56, 59].
Because all of the subunits of RPA are essential for cell viability, studies to elucidate RPA function during HR in vivo have relied on use of conditional or hypomorphic alleles to determine the cellular consequence of RPA loss or malfunction. The budding yeast, Saccharomyces cerevisiae, has been particularly informative for the genetic studies. Random mutational analysis of the RFA1 and RFA2 genes identified alleles with a temperature sensitive growth defect and/or sensitivity to DNA damaging agents [60–62]. The rfa1-t11 mutant is extremely defective for both RAD51 dependent strand invasion and RAD51-independent SSA [62, 63]. Biochemical analysis revealed much slower displacement of RPAt11 by Rad51, indicative of higher binding to ssDNA [64]. In contrast, the RPAt33 and RPAt48 mutant complexes show less stable binding to ssDNA [65], but are also defective for DSB-induced HR [62, 66]. Hypomorphic alleles of RFA1 have also been recovered from genetic screens for altered frequencies of HR. The rfa1-44 mutant is defective for DSB-induced recombination and exhibits genetic interaction with RAD52 [67], whereas the rfa1-D228Y mutation was recovered as a suppressor of the SSA defect of the rad52 mutant [68]. RAD52 is required to promote annealing between the resected repeats during SSA; however, when Rfa1 is impaired by the D228Y substitution, SSA can occur in the absence of RAD52. Presumably the RPAD228Y complex is still able to remove most of the secondary structures from ssDNA, but is less efficient in preventing spontaneous annealing [69]. It is interesting to note that the lower limit for Rad52 to promote annealing in vivo is ~14 nt [70], the transition point at which most sequences are unique in yeast. Thus, there is a balance in which RPA prevents strand annealing and Rad52 promotes annealing when the homologous stretches are sufficiently long that the sequences are likely to be unique. This minimizes illegitimate recombination events.
GCRs are elevated when RPA is dysfunctional
The yeast CAN1 gene is frequently used to measure forward mutation rates because loss of function results in resistance to the drug canavanine (CanR). RPA was first recognized as a suppressor of GCRs by the unusual spectrum of spontaneous CanR mutations recovered from rfa1 mutants [71]. Although the mutation rate in rfa1 mutants was only moderately elevated compared to wild type, several of the CanR events analyzed from rfa1 mutants were generated by large deletions or translocations, in contrast to CAN1 inactivation by point mutations in wild-type cells. The assay was refined to specifically detect GCRs by inserting a second counter-selectable marker (URA3) adjacent to CAN1 and then measuring simultaneous loss of both markers [72]. Because CAN1 is within a non-essential region of chromosome V, complete loss of the terminal fragment encompassing the CAN1 locus is compatible with viability in haploid cells. The frequency of GCRs was extremely low in wild-type cells, and most of the events recovered resulted from loss of the terminal chromosome fragment containing the URA3 and CAN1 genes followed by de novo telomere addition. Several rfa1 and rfa2 mutants were tested in this assay and showed a 50–5,000-fold elevated rate of GCRs relative to wild type [72]. Furthermore, many of the GCR events recovered from rfa1 mutants were due to translocations, deletions or inversions that had MH at the junctions. The rfa1 mutational signature is unique; GCRs also occur at greatly elevated frequencies in the mre11Δ mutant, but are not mediated by MH [72].
The role of RPA in suppressing GCRs is not restricted to yeast. The rfa1-t48 mutation, which confers a 5,000-fold increase in the frequency of GCRs, has been modeled in mouse cells [73]. Although yeast cells harboring the rfa1-t48 mutation are viable, mice homozygous for the Rpa1t48 allele display embryonic lethality. The Rpa1t48/+ heterozygous animals were viable, but they had a reduced lifespan and developed lymphomas [73]. Tumors analyzed from the heterozygous animals exhibited multiple genome alterations, including chromosome aneuploidy and segmental gains or losses. The breakpoints of rearrangements were not sequenced, and it would be of interest to determine whether they exhibit increased use of MH.
The increased frequency of chromosome translocations with MH at the junctions in rfa1 mutants could be due to a defect during DNA synthesis resulting in more DSBs and subsequent channeling of DSBs to a repair mechanism rarely used in wild-type cells. A recent study by Deng et al. supports the notion that RPA suppresses mutagenic repair of DSBs by MMEJ [65]. As noted above, MMEJ requires resection to reveal MH internal to the break ends and is driven by the annealing between short MH (>5 nt) [35]. The length, percent homology and G-C content of the MH are determinants of the frequency of MMEJ in yeast, suggesting that the process is spontaneous and thermodynamically driven [70, 74, 75]. Using a genetic assay to detect MMEJ between 12 bp direct repeats flanking a site-specific DSB, Deng et al. showed that MMEJ occurs at very low frequencies in wild-type cells, whereas several of the rfa1 hypomorphic mutants tested showed greatly elevated frequencies of MMEJ (Fig. 3) [65]. The rfa1-t48 mutant displayed the greatest increase in MMEJ (350-fold higher than wild type), consistent with the high frequency of GCRs reported by Chen and Kolodner [72]. The rfa1-t48 mutation results from substitution of a highly conserved leucine at position 221 of Rfa1 with proline. L221 is within DBD-A and a site of direct contact with ssDNA [6]. The purified yeast RPAt48 mutant complex exhibited reduced DNA binding in vitro and was unable to fully extend ssDNA, indicating that it is impaired in removal of secondary structures [65]. The human RPAL221P mutant complex showed a greater defect in DNA binding than observed for the yeast RPAt48 complex, and was unable to support SV40 DNA replication in vitro, in agreement with the lethal phenotype conferred by this mutation in mouse cells [73,76]. The rfa1-D228Y and rfa1-t33 alleles, which encode proteins with amino acid substitutions in DBD-A and DBD-B, respectively, that do not make direct contacts with DNA [6], also increased MMEJ, but not to the same extent as rfa1-t48 [6, 65]. MMEJ was not increased in the rfa1-t11 mutant, consistent with the RPAt11 complex retaining high affinity DNA binding. These studies highlight an important role for RPA in preventing promiscuous annealing between short repeats during DSB repair in yeast.
Figure 3.

RPA prevents inter- and intra-molecular annealing between short repeats to prevent MMEJ or formation of hairpin-capped DNA ends. RPA binding to the long ssDNA tracts formed by end resection is required to prevent pairing between short repeats flanking the DSB or potentially with another DSB end elsewhere in the genome. After pairing heterologous flaps are removed, gaps filled by DNA synthesis and the ends are ligated yielding deletions, translocations or inversions. Pairing between short inverted repeats within the ssDNA tail can lead to the formation of hairpin-capped ends. MRX acts with Sae2 to cleave hairpins creating another substrate for end resection and repair. Failure to cleave the hairpin can lead to palindromic gene duplication and possibly more complex rearrangements if the resulting chromosome fragment has two centromeres.
RPA facilitates extensive resection and shields 3′ ends from degradation
The role of DNA end resection is to create a ssDNA substrate for Rad51 polymerization and a 3′ end that can be extended by DNA synthesis following Rad51-catalyzed strand invasion. Most studies of end resection in budding yeast indicate that the 3′ ends at a DSB remains stable for several hours [77, 78]. To test the hypothesis that RPA protects the ssDNA formed by end resection against degradation, Chen et al. [79] used a heat-inducible rfa1 degron allele (td-RFA1) to rapidly deplete RPA from cells at the same time as the induction of a site-specific DSB by the HO endonuclease. Resection initiation was unaltered by depletion of RPA, but extensive resection was almost completely abolished. Furthermore, the short 3′ ssDNA tails generated by MRX and Sae2 were unstable, indicating that RPA is indeed directly protecting ssDNA from improper processing.
The block to extensive resection caused by RPA depletion is in part due to failure of Dna2 to localize to DSBs by its direct interaction with RPA [79, 80]. However, the Exo1-dependent extensive resection pathway was also blocked by RPA depletion in vivo, and Exo1 has not been found to physically interact with RPA and can localize to break sites independently of RPA [79]. One possible explanation is that naked ssDNA with secondary structure is a poor substrate for Exo1 nuclease. In support of this view, Cannavo et al. [49] showed that Exo1 acts preferentially on a dsDNA end with a 3′-ssDNA overhang and unprotected ssDNA can sequester free Exo1.
Both Rad52 and Rad51 failed to localize to DSB sites upon loss of RPA [79, 81]; thus, preventing the repair of DSBs by HR. The dependence of Rad52 and Rad51 recruitment on RPA can be partially explained by their direct interactions, but could also be due to the lack of a suitable substrate [53, 82]. Similarly, ATRIP/Ddc2 localization to damage sites is reduced when RPA is depleted from cells [5], likely due to loss of both long tracts of ssDNA and the direct association of the ATRIP/Ddc2 with RPA.
DNA secondary structures are potential targets for nucleases
In addition to 3′ strand loss, RPA depletion also resulted in the formation of fold-back structures by intramolecular pairing between short inverted repeats (5–9 bp) in the 3′ ssDNA overhang formed by end resection, followed by DNA synthesis and ligation to form hairpin-capped ends (Fig. 3) [79]. These DNA secondary structures could be dangerous for cells for several reasons. First, they are likely an unfavorable conformation to be recognized by DNA repair factors that require ends or ssDNA for binding; thus, perturbing end resection and subsequent HR repair.
Second, the hairpin structures at break ends resulting from RPA depletion are potential substrates for structure-selective endonucleases. Eukaryotic cells encode several nucleases that cleave at ssDNA-dsDNA transitions and that could potentially target secondary structures in ssDNA [83]. The Mre11-Rad50 complex is able to cleave hairpins in vitro and genetic studies support a role for MRX in resolving hairpin-capped ends [84, 85]. Sae2 is also required for resolving hairpin structures formed at long inverted repeats in vivo [84], even though MR can cleave model hairpins in vitro in the absence of Sae2 [86]. The hairpin-capped DNA ends formed after RPA depletion were stabilized in mre11Δ and sae2Δ mutants [79]. Indeed, it is possible that one of the many functions of MRX and Sae2 at DSBs is to remove fold-back structures formed inadvertently at short inverted repeats within the ssDNA tails the result from resection. Although hairpin-capped ends were not detected at the site of a single DSB unless RPA was depleted, fold-back structures have been identified at meiotic DSBs in recombination-defective rad52Δ cells [57]. This suggests that hyper-resection at multiple DSBs (in budding yeast, ~150 DSBs are generated during meiosis) due to deficient HR might exhaust the nuclear pool of free RPA.
Pathological RPA depletion can be caused by ATR deficiency in human cells under replication stress, where unscheduled origin firing generates excessive amounts of ssDNA [15]. This global exhaustion of RPA leads to the simultaneous breakage of all active forks, known as replication catastrophe. How depletion of RPA causes DNA breakage at replication forks is not known, but one attractive hypothesis is for MRX/N-Sae2/CtIP to cleave secondary DNA structures formed within unprotected ssDNA. Alternatively, the stalled replication forks resulting from replication stress, or forks remodeled by DNA translocases to form structures resembling Holliday junctions, could be acted on by the Mus81-Mms4/EME1 and/or Slx1-Slx4 endonucleases resulting in DSBs [87–89]. The pool of free RPA also becomes limiting when mouse embryonic fibroblasts deficient for DNA polymerase ζ are treated with UV light due to sequestration of RPA at damaged replication forks, and this interferes with global nucleotide excision repair [90, 91].
Third, it is also possible that certain types of DNA secondary structures formed when RPA is dysfunctional escape cleavage or cannot be processed by nucleases. Replication of a chromosome fragment with a hairpin-capped end would result in the formation of a giant palindromic chromosome (Fig. 3). If the chromosome fragment contained a centromere it would become dicentric and could then be broken at mitosis and undergo further rearrangements. In support of this idea, palindromic gene amplification at telomeres has been reported in telomerase- and recombination-defective yeast as a means to escape senescence [92]. DNA sequence analysis provided evidence for pairing between short (5–9 bp) inverted repeats within the ssDNA formed by resection from the telomeres. We speculate that RPA is depleted as a result of extensive degradation of many chromosome ends, and that the ssDNA forms fold-back structures. Similar types of event have been reported at a site-specific DSB adjacent to a large inverted repeat, but their recovery was only seen in the absence of Sae2 or MRX, presumably because these factors normally process hairpin-capped ends [93, 94].
Conclusions and outlook
Here, we have focused on the important role that RPA plays in preventing inter- and intra-molecular annealing between short repeats during the repair of DSBs. It would be of interest to determine whether this function of RPA is conserved in mammalian cells and acts to prevent MMEJ, as demonstrated in yeast. RPA inhibition of strand annealing likely plays an important role during DNA replication to prevent aberrant processing of secondary structures within ssDNA formed during lagging strand synthesis, or when the replicative helicase and leading strand polymerase become uncoupled due to replication stress. In addition to the threat posed by short inverted repeats, eukaryotic genomes contain many repetitive sequences or motifs that can form stable secondary structures, including trinucleotide repeats, long inverted repeats, AT- and GC-rich mini (10–60 bp repeat) and microsatellites (2–5 bp repeats), and G-rich sequences with the potential to form G-quadruplexes. These sequence motifs are known to stall replication, and induce high levels of chromosome fragility, recombination and chromosome rearrangements [84, 95–99]. Decreased expression of RFA2, achieved by using a doxycycline-repressible promoter, resulted in a dramatic increase in GCRs at long (GAA)n tracts and at palindromic sequences, suggesting that RPA is indeed a critical safeguard for genomic integrity at trinucleotide and inverted repeats, and possibly at other “at-risk motifs” as well [100, 101].
Acknowledgments
We thank W. K. Holloman for comments on the manuscript. Research in our laboratory cited in this review was supported by a grant from National Institutes of Health (GM041784).
Abbreviations
- DSB
double-stranded break
- dsDNA
double-stranded DNA
- GCR
gross chromosome rearrangement
- HR
homologous recombination
- MH
microhomology
- MMEJ
microhomology-mediated end joining
- MRX/N
Mre11-Rad50-Xrs2/NBS1 complex
- NHEJ
non-homologous end joining
- nt
nucleotide
- OB
oligonucleotide/oligosaccharide binding
- RPA
replication protein A
- ssDNA
single-stranded DNA
- SV40
simian virus 40
Footnotes
The authors have no conflict of interest to declare.
References
- 1.Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–37. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 3.Wold MS, Kelly T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc Natl Acad Sci USA. 1988;85:2523–7. doi: 10.1073/pnas.85.8.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brill SJ, Stillman B. Replication factor-A from Saccharomyces cerevisiae is encoded by three essential genes coordinately expressed at S phase. Genes Dev. 1991;5:1589–600. doi: 10.1101/gad.5.9.1589. [DOI] [PubMed] [Google Scholar]
- 5.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 6.Fan J, Pavletich NP. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012;26:2337–47. doi: 10.1101/gad.194787.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bochkarev A, Bochkareva E, Frappier L, Edwards AM. The crystal structure of the complex of replication protein A subunits RPA32 and RPA14 reveals a mechanism for single-stranded DNA binding. EMBO J. 1999;18:4498–504. doi: 10.1093/emboj/18.16.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997;385:176–81. doi: 10.1038/385176a0. [DOI] [PubMed] [Google Scholar]
- 9.Bochkareva E, Frappier L, Edwards AM, Bochkarev A. The RPA32 subunit of human replication protein A contains a single-stranded DNA-binding domain. J Biol Chem. 1998;273:3932–6. doi: 10.1074/jbc.273.7.3932. [DOI] [PubMed] [Google Scholar]
- 10.Bastin-Shanower SA, Brill SJ. Functional analysis of the four DNA binding domains of replication protein A. The role of RPA2 in ssDNA binding. J Biol Chem. 2001;276:36446–53. doi: 10.1074/jbc.M104386200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brill SJ, Bastin-Shanower S. Identification and characterization of the fourth single-stranded-DNA binding domain of replication protein A. Mol Cell Biol. 1998;18:7225–34. doi: 10.1128/mcb.18.12.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibb B, Silverstein TD, Finkelstein IJ, Greene EC. Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Anal Chem. 2012;84:7607–12. doi: 10.1021/ac302117z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibb B, Ye LF, Gergoudis SC, Kwon Y, et al. Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014;9:e87922. doi: 10.1371/journal.pone.0087922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Binz SK, Sheehan AM, Wold MS. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair. 2004;3:1015–24. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 15.Toledo LI, Altmeyer M, Rask MB, Lukas C, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 16.Dou H, Huang C, Singh M, Carpenter PB, et al. Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol Cell. 2010;39:333–45. doi: 10.1016/j.molcel.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ira G, Pellicioli A, Balijja A, Wang X, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–7. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–71. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 19.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–4. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 21.Barlow JH, Lisby M, Rothstein R. Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol Cell. 2008;30:73–85. doi: 10.1016/j.molcel.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jazayeri A, Falck J, Lukas C, Bartek J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 23.Jinks-Robertson S, Michelitch M, Ramcharan S. Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3937–50. doi: 10.1128/mcb.13.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–83. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature. 1998;391:407–10. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- 26.Shinohara A, Ogawa T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature. 1998;391:404–7. doi: 10.1038/34943. [DOI] [PubMed] [Google Scholar]
- 27.Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem. 1997;272:28194–7. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- 28.Yang H, Li Q, Fan J, Holloman WK, et al. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005;433:653–7. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 29.Ferguson DO, Holloman WK. Recombinational repair of gaps in DNA is asymmetric in Ustilago maydis and can be explained by a migrating D-loop model. Proc Natl Acad Sci USA. 1996;93:5419–24. doi: 10.1073/pnas.93.11.5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci. 2014;39:409–19. doi: 10.1016/j.tibs.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foster SS, Balestrini A, Petrini JH. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol. 2011;31:4379–89. doi: 10.1128/MCB.05854-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4 :e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 35.Ma JL, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol. 2003;23:8820–8. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truong LN, Li Y, Shi LZ, Hwang PY, et al. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci USA. 2013;110:7720–5. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279:55117–26. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 38.Liang L, Deng L, Nguyen SC, Zhao X, et al. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008;36:3297–310. doi: 10.1093/nar/gkn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simsek D, Brunet E, Wong SY, Katyal S, et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011;7:e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang M, Wu W, Wu W, Rosidi B, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee K, Lee SE. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics. 2007;176:2003–14. doi: 10.1534/genetics.107.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brunet E, Simsek D, Tomishima M, DeKelver R, et al. Chromosomal translocations induced at specified loci in human stem cells. Proc Natl Acad Sci USA. 2009;106:10620–5. doi: 10.1073/pnas.0902076106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–6. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;22:211–20. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stephens PJ, McBride DJ, Lin ML, Varela I, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–10. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stephens PJ, Greenman CD, Fu B, Yang F, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, et al. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010;467:112–6. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niu H, Chung WH, Zhu Z, Kwon Y, et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature. 2010;467:108–11. doi: 10.1038/nature09318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cannavo E, Cejka P, Kowalczykowski SC. Relationship of DNA degradation by Saccharomyces cerevisiae exonuclease 1 and its stimulation by RPA and Mre11-Rad50-Xrs2 to DNA end resection. Proc Natl Acad Sci USA. 2013;110:E1661–8. doi: 10.1073/pnas.1305166110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–3. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 51.Sugiyama T, Kowalczykowski SC. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J Biol Chem. 2002;277:31663–72. doi: 10.1074/jbc.M203494200. [DOI] [PubMed] [Google Scholar]
- 52.Gibb B, Ye LF, Kwon Y, Niu H, et al. Protein dynamics during presynaptic-complex assembly on individual single-stranded DNA molecules. Nat Struct Mol Biol. 2014;21:893–900. doi: 10.1038/nsmb.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hays SL, Firmenich AA, Massey P, Banerjee R, et al. Studies of the interaction between Rad52 protein and the yeast single-stranded DNA binding protein RPA. Mol Cell Biol. 1998;18:4400–6. doi: 10.1128/mcb.18.7.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Milne GT, Weaver DT. Dominant negative alleles of RAD52 reveal a DNA repair/recombination complex including Rad51 and Rad52. Genes Dev. 1993;7:1755–65. doi: 10.1101/gad.7.9.1755. [DOI] [PubMed] [Google Scholar]
- 55.Mortensen UH, Bendixen C, Sunjevaric I, Rothstein R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci USA. 1996;93:10729–34. doi: 10.1073/pnas.93.20.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shinohara A, Shinohara M, Ohta T, Matsuda S, et al. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. GenesCells. 1998;3:145–56. doi: 10.1046/j.1365-2443.1998.00176.x. [DOI] [PubMed] [Google Scholar]
- 57.Lao JP, Oh SD, Shinohara M, Shinohara A, et al. Rad52 promotes postinvasion steps of meiotic double-strand-break repair. Mol Cell. 2008;29:517–24. doi: 10.1016/j.molcel.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugawara N, Haber JE. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol. 1992;12:563–75. doi: 10.1128/mcb.12.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci USA. 1998;95:6049–54. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Longhese MP, Plevani P, Lucchini G. Replication factor A is required in vivo for DNA replication, repair, and recombination. Mol Cell Biol. 1994;14:7884–90. doi: 10.1128/mcb.14.12.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santocanale C, Neecke H, Longhese MP, Lucchini G, et al. Mutations in the gene encoding the 34 kDa subunit of yeast replication protein A cause defective S phase progression. J Mol Biol. 1995;254:595–607. doi: 10.1006/jmbi.1995.0641. [DOI] [PubMed] [Google Scholar]
- 62.Umezu K, Sugawara N, Chen C, Haber JE, et al. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics. 1998;148:989–1005. doi: 10.1093/genetics/148.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X, Haber JE. Role of Saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. PLoS Biol. 2004;2:E21. doi: 10.1371/journal.pbio.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kantake N, Sugiyama T, Kolodner RD, Kowalczykowski SC. The recombination-deficient mutant RPA (rfa1-t11) is displaced slowly from single-stranded DNA by Rad51 protein. J Biol Chem. 2003;278:23410–7. doi: 10.1074/jbc.M302995200. [DOI] [PubMed] [Google Scholar]
- 65.Deng SK, Gibb B, de Almeida MJ, Greene EC, et al. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:405–12. doi: 10.1038/nsmb.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soustelle C, Vedel M, Kolodner R, Nicolas A. Replication protein A is required for meiotic recombination in Saccharomyces cerevisiae. Genetics. 2002;161:535–47. doi: 10.1093/genetics/161.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Firmenich AA, Elias-Arnanz M, Berg P. A novel allele of Saccharomyces cerevisiae RFA1 that is deficient in recombination and repair and suppressible by RAD52. Mol Cell Biol. 1995;15:1620–31. doi: 10.1128/mcb.15.3.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith J, Rothstein R. A mutation in the gene encoding the Saccharomyces cerevisiae single-stranded DNA-binding protein Rfa1 stimulates a RAD52-independent pathway for direct-repeat recombination. Mol Cell Biol. 1995;15:1632–41. doi: 10.1128/mcb.15.3.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith J, Rothstein R. An allele of RFA1 suppresses RAD52-dependent double-strand break repair in Saccharomyces cerevisiae. Genetics. 1999;151:447–58. doi: 10.1093/genetics/151.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Villarreal DD, Lee K, Deem A, Shim EY, et al. Microhomology directs diverse DNA break repair pathways and chromosomal translocations. PLoS Genet. 2012;8:e1003026. doi: 10.1371/journal.pgen.1003026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen C, Umezu K, Kolodner RD. Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell. 1998;2:9–22. doi: 10.1016/s1097-2765(00)80109-4. [DOI] [PubMed] [Google Scholar]
- 72.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 73.Wang Y, Putnam CD, Kane MF, Zhang W, et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat Genet. 2005;37:750–5. doi: 10.1038/ng1587. [DOI] [PubMed] [Google Scholar]
- 74.Daley JM, Wilson TE. Rejoining of DNA double-strand breaks as a function of overhang length. Mol Cell Bbiol. 2005;25:896–906. doi: 10.1128/MCB.25.3.896-906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Decottignies A. Microhomology-mediated end joining in fission yeast is repressed by pku70 and relies on genes involved in homologous recombination. Genetics. 2007;176:1403–15. doi: 10.1534/genetics.107.071621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hass CS, Gakhar L, Wold MS. Functional characterization of a cancer causing mutation in human replication protein A. Mol Cancer Res. 2010;8:1017–26. doi: 10.1158/1541-7786.MCR-10-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ray BL, White CI, Haber JE. Heteroduplex formation and mismatch repair of the “stuck” mutation during mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:5372–80. doi: 10.1128/mcb.11.10.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zierhut C, Diffley JF. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008;27:1875–85. doi: 10.1038/emboj.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen H, Lisby M, Symington LS. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell. 2013;50:589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bae KH, Kim HS, Bae SH, Kang HY, et al. Bimodal interaction between replication-protein A and Dna2 is critical for Dna2 function both in vivo and in vitro. Nucleic Acids Res. 2003;31:3006–15. doi: 10.1093/nar/gkg422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 82.Golub EI, Gupta RC, Haaf T, Wold MS, et al. Interaction of human rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res. 1998;26 :5388–93. doi: 10.1093/nar/26.23.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwartz EK, Heyer WD. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma. 2011;120:109–27. doi: 10.1007/s00412-010-0304-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–93. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 85.Trujillo KM, Sung P. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50*Mre11 complex. J Biol Chem. 2001;276:35458–64. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- 86.Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–5. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- 87.Hanada K, Budzowska M, Davies SL, van Drunen E, et al. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol. 2007;14:1096–104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
- 88.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–23. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ragland RL, Patel S, Rivard RS, Smith K, et al. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev. 2013;27:2259–73. doi: 10.1101/gad.223180.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Overmeer RM, Moser J, Volker M, Kool H, et al. Replication protein A safeguards genome integrity by controlling NER incision events. J Cell Biol. 2011;192:401–15. doi: 10.1083/jcb.201006011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsaalbi-Shtylik A, Moser J, Mullenders LH, Jansen JG, et al. Persistently stalled replication forks inhibit nucleotide excision repair in trans by sequestering Replication protein A. Nucleic Acids Res. 2014;42:4406–13. doi: 10.1093/nar/gkt1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maringele L, Lydall D. Telomerase- and recombination-independent immortalization of budding yeast. Genes Dev. 2004;18:2663–75. doi: 10.1101/gad.316504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rattray AJ, Shafer BK, Neelam B, Strathern JN. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005;19:1390–9. doi: 10.1101/gad.1315805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rattray AJ, McGill CB, Shafer BK, Strathern JN. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics. 2001;158:109–22. doi: 10.1093/genetics/158.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA. 2008;105:9936–41. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Freudenreich CH, Kantrow SM, Zakian VA. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279:853–6. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- 97.Paeschke K, Bochman ML, Garcia PD, Cejka P, et al. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013;497:458–62. doi: 10.1038/nature12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Piazza A, Serero A, Boule JB, Legoix-Ne P, et al. Stimulation of gross chromosomal rearrangements by the human CEB1 and CEB25 minisatellites in Saccharomyces cerevisiae depends on G-quadruplexes or Cdc13. PLoS Genet. 2012;8:e1003033. doi: 10.1371/journal.pgen.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ribeyre C, Lopes J, Boule JB, Piazza A, et al. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 2009;5:e1000475. doi: 10.1371/journal.pgen.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang Y, Saini N, Sheng Z, Lobachev KS. Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLoS Genet. 2013;9:e1003979. doi: 10.1371/journal.pgen.1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, et al. Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol Cell. 2012;48:254–65. doi: 10.1016/j.molcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]