

Abstract
We demonstrate through PdO doping that creation of heterojunctions on Co3O4 nanoparticles can quantitatively adjust band-gap and Fermi energy levels to study the impact of metal oxide nanoparticle semiconductor properties on cellular redox homeostasis and hazard potential. Flame spray pyrolysis (FSP) was used to synthesize a nanoparticle library in which the gradual increase in the PdO content (0–8.9%) allowed electron transfer from Co3O4 to PdO to align Fermi energy levels across the heterojunctions. This alignment was accompanied by free hole accumulation at the Co3O4 interface and production of hydroxyl radicals. Interestingly, there was no concomitant superoxide generation, which could reflect the hole dominance of a p-type semiconductor. Although the electron flux across the heterojunctions induced upward band bending, the Ec levels of the doped particles showed energy overlap with the biological redox potential (BRP). This allows electron capture from the redox couples that maintain the BRP from −4.12 to −4.84 eV, causing disruption of cellular redox homeostasis and induction of oxidative stress. PdO/Co3O4 nanoparticles showed significant increases in cytotoxicity at 25, 50, 100, and 200 μg/mL, which was enhanced incrementally by PdO doping in BEAS-2B and RAW 264.7 cells. Oxidative stress presented as a tiered cellular response involving superoxide generation, glutathione depletion, cytokine production, and cytotoxicity in epithelial and macrophage cell lines. A progressive series of acute pro-inflammatory effects could also be seen in the lungs of animals exposed to incremental PdO-doped particles. All considered, generation of a combinatorial PdO/Co3O4 nanoparticle library with incremental heterojunction density allowed us to demonstrate the integrated role of Ev, Ec, and Ef levels in the generation of oxidant injury and inflammation by the p-type semiconductor, Co3O4.
Introduction
Metal oxide (MOx) nanoparticles (NPs) represent an industrially important category of nanomaterials that is produced in high volume and frequently used for their semiconductor properties.1−3 At the same time, it is important to consider that commercialization of these materials could cause adverse biological effects in humans and the environment.4−17 While a wide range of nanoparticle physicochemical properties, including particle size,12,18 shape,18,19 metal ion shedding,6,20 crystallinity,21,22 surface defects,23 and surface reactivity,24 could contribute to the generation of biological hazard, an interesting observation in a recent analysis of 24 MOx has been that MOx electronic properties and conduction band energy can be linked to the generation of oxidative stress injury and inflammation.25 Not only did we observe that the conduction band energies (Ec) of five (Co3O4, Cr2O3, Ni2O3, Mn2O3, and CoO) materials predict their ability to induce cellular redox stress and inflammation but we have demonstrated Ec overlap with the biological redox potential (BRP)26 can identify the nanoparticles that cause acute pulmonary inflammation.25 The BRP, which ranges from −4.12 to −4.84 eV, is determined by a series of cellular redox couples.25−27 While at first glance these findings may surprise biologists, it is well known in the semiconductor industry that the equivalence of band-gap and Fermi (Ef) energy levels plays a critical role in allowing electron transfer between the semiconductor and bystander materials. Thus, the awareness that the overlap of Ec with BRP plays a role in MOx hazard potential has helped to establish a quantifiable material characteristic that can be used for hazard ranking of MOx NPs.25,27,28
Extensive further work is required to understand the relationship of Ec to the valence band and Ef energy levels in activation of cellular responses. This could help us to understand why the materials with Ec overlap with the BRP are all p-type semiconductors (Co3O4, Cr2O3, Ni2O3, Mn2O3, CoO); these materials display more holes (h+) than electrons (e–) and show Ef levels that are more closely spaced to the valence band.25 Thus, in order to more completely understand the integrated relationship of Ec, Ef, and Ev to the BRP, it was necessary to use one of the implicated MOx to adjust the semiconductor status while preserving physicochemical properties such as size, surface area, crystallinity, and surface charge. One approach is to dope the crystal lattice or functionalize the particle surface to adjust band-gap and Ef levels.16,29 In light of our expertise in FSP,6,16,30 we envisaged that it should be possible to generate heterojunctions that will allow electron flow and separation of h+/e– pairs to adjust Ec, Ev, and Ef in discrete quantities, thereby yielding a library of materials to study the effects on cellular redox homeostasis. Creation of heterojunctions is frequently used by industry to generate doped nanomaterials for energy generation or catalysis.29,31−33
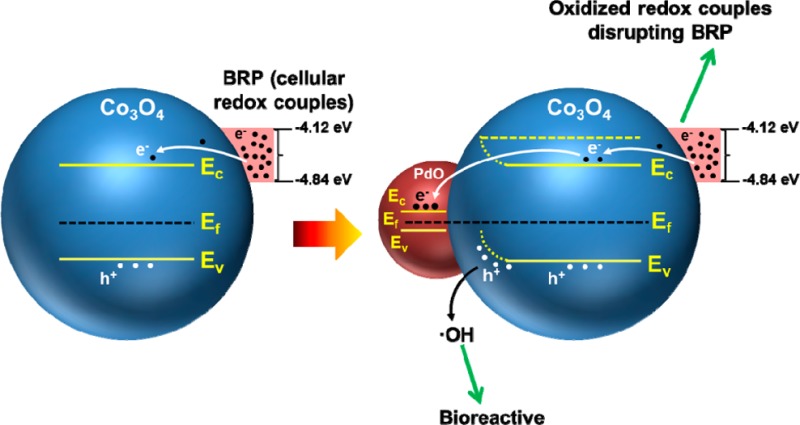
In this article, we demonstrate that through the use of incremental PdO doping, using our versatile flame spray pyrolysis (FSP) process, it is possible to generate a combinatorial Co3O4 nanoparticle library in which variance of the heterojunction density can be used to adjust the Ec, Ev, and Ef levels to study the integrated effect on biological redox regulation. Co3O4 nanoparticles have been widely investigated for electrode materials in lithium batteries, carbon monoxide oxidation, electrochromic materials, and gas sensors.34−37 In addition to clarifying the mechanism of oxidative stress injury by the semiconductor, use of a noble metal-doped Co3O4 library is relevant to nanomaterial safety because many of these materials are used in applications such as Fischer–Tropsch catalysis, biosensors, energy storage, and the solar industry where workers may come into contact with these materials.38−42 To thoroughly investigate the role of band-gap energy and the alignment of Ef energy levels cross the heterojunctions, Co3O4 nanoparticles were doped with incremental amounts (0–8.9%) of PdO. The facilitated electron transfer across the PdO/Co3O4 heterojunctions allowed upward bending of Ec and Ev energy levels and accentuation of the mismatch of holes with electrons in the Co3O4 NP (Scheme 1). The excess free h+ was accompanied by increased generation of hydroxyl radicals (•OH) (Scheme 1).16 Creation of this redox-active library allowed us to study the hazard impact from the perspective of the redox equilibrium in epithelial cells and macrophages, including extrapolating those findings to pro-inflammatory effects in the lungs of mice. Our results demonstrate that it is possible to demonstrate in coherent fashion the interplay between material band-gap and Fermi energy levels and hazard generation through disruption of cellular redox homeostasis.
Scheme 1. Heterojunction Formation by PdO Doping of Co3O4 To Yield a Combinatorial NP Library for Studying the Biological Response to a p-Type Semiconductor Material.

(a) PdO and Co3O4 have different Fermi energies (Ef). Ef of Co3O4 is higher than that of PdO. (b) Formation of PdO/Co3O4 heterojunctions allows electron transfer from Co3O4 to PdO, with the potential to increase the hole dominance of the p-type semiconductor. Electron transfer continues until the Fermi energies of Co3O4 and PdO are in equilibrium. (c) Space–charge separation across the heterojunction is accompanied by band bending (BB), with the possibility that the free holes can generate hydroxyl radicals (•OH), while the free electrons could lead to formation of superoxide radicals (O2•–).
Experimental Section
Chemicals
All chemicals were reagent grade and used without further purification or modification unless otherwise indicated. Reagent-grade water used in all experimental procedures was obtained from a Milli-Q water purification system (Millipore, Bedford, MA).
Synthesis of PdO-Doped Co3O4 Nanoparticles
PdO-doped Co3O4 nanoparticles were synthesized using a FSP reactor.6,43,44 The required amounts of metalloorganic precursors, cobalt napthenate (Strem Chemical, 6% Co) and palladium acetylacetonate (Strem Chemicals, 99.9% pure), were mixed together with xylene (Co concentration 0.5 M for all the mixtures) to provide 1–8% Pd by weight after combustion. As an example, 84 mg of Pd acetlyacetonate was mixed with 100 mL of Co-precursor solution (0.5 M) to obtain 1% Pd in Co3O4. The solution was warmed to 50 °C for 10 min prior to spraying to ensure dissolution of the palladium acetylacetonate in cobalt napthenate. For FSP, the liquid precursor was delivered at 5 mL/min using a syringe pump and atomized by a two-phase nozzle using 5 L/min O2 at a constant drop of 1.5 bar at the nozzle tip. The spray was ignited by a premixed codelivery of CH4 and O2 (1.5 L/min, 3.2 L/min) forming a spray flame. Ultrafine particles were formed by reaction, nucleation, surface growth, coagulation, and coalescence in the flame environment. Particles were collected from the 257 mm glass filter placed in the flame reactor at a distance of 60 cm from the flame.
Physicochemical Characterization of PdO-Doped Co3O4 Nanoparticles
TEM was performed on samples dispensed from aqueous suspension onto a carbon-coated TEM grid using a JEOL 2010 microscope operated at 200 keV. Pd and Co contents of the PdO-doped Co3O4 NPs were determined by energy-dispersive X-ray spectroscopy (EDX) using a FEI Titan 80/300 microscope. Specific surface area was determined by the BET method from N2 sorption isotherms, acquired using a Micrometrics ASAP 2010 sorption instrument, following gassing out under a vacuum for 12 h at 120 °C. X-ray diffraction (XRD) patterns were collected using a Panalytical X’Pert Pro diffractometer (Cu Kα radiation) with a step size of 0.02° and a counting time of 0.5 s per step over a range of 15–80° 2θ. Zeta potential and dynamic light scattering (DLS) data were obtained using a Malvern Nanosizer ZS for nanoparticles dispersed with DI water at a concentration of 50 μg/mL. X-ray photoelectron spectroscopy (XPS) and ultraviolet photoelectron spectroscopy (UPS) measurements were performed on a Kratos AXIS Ultra DLD X-ray/ultraviolet photoelectron spectrometer equipped with monochromatic Al Kα radiation (hν = 1486.6 eV) as X-ray source and a He I radiator (hν = 21.21 eV) as UV source. All spectra were collected at room temperature in an ultra-high-vacuum environment (base pressure of analysis chamber < 5 × 10–8 Torr). XPS resolution was determined by fitting the Au 4 f7/2 core-level peak from clean Au foil to the convolution of a Gaussian and a Doniach–Sunjic function of full width at half-maximum (fwhm). The UPS was carried out in normal emission with a sample bias of −10 V. All UPS binding energies are referenced to the Fermi edge of a clean Au foil. The valence band maximum (VBM) was determined by linear extrapolation of the leading edge of the UPS spectrum, and the work function was determined from one-half the height of the secondary electron onset.45 Band-gap energies were obtained from diffuse reflectance (DR) UV–vis spectroscopic analysis (Cary 5000 UV–vis–NIR spectrometer equipped with a Praying Mantis accessory). All measurements were conducted in ambient air using a bandwidth of 1.0 nm. Collected DR UV–vis spectra were converted into Kubelka–Munk function [F(R∞)] spectra using the Cary Win UV software.25
Abiotic Assessment of Total Reactive Oxygen Species (ROS), Hydroxyl Radical (•OH), and Superoxide Radical (O2•–) Generation as Well as GSH Oxidation
Total ROS and specific hydroxyl radical levels were determined by 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) fluorescence,46 3′-(p-aminophenyl) fluorescein (APF) fluorescence,47 and 2,3-bis(2-methoxy-4-nitro-5-sulfophehyl)-2H-tetrazolium-5-carboxanilide) (XTT) absorbance. The DCF working solution was prepared by mixing 50 μg of H2DCFDA with 17.3 μL of ethanol, followed by addition of 692 μL of a 0.01 mol/L sodium hydroxide solution. The resulting solution was incubated for 30 min, and 3500 μL of a sodium phosphate buffer (25 mmol/L, pH = 7.4) was added to form 29 μmol/L DCF solution. To each well of a 96 multiwell black plate (Costar, Corning, NY) we added 80 μL of one of following solutions: 29 μmol/L DCF, 10 μmol/L APF, or 100 μmol/L XTT in PBS. A 20 μL amount of 1 mg/mL nanoparticle suspension was subsequently added to each well, followed by 2 h incubation. DCF fluorescence emission spectra in the range of 500–600 nm were collected using a SpectraMax M5 microplate reader with an excitation wavelength of 490 nm. APF fluorescence emission spectra were collected at 480–600 nm with an excitation wavelength of 455 nm, while XTT absorbance spectra were recorded in the range of 410–550 nm.
To each well of a 96 multiwell white plate (Costar, Corning, NY) we added 40 μL of 50 μmol/L GSH in PBS and 10 μL of 1 mg/mL nanoparticle suspension, followed by 6 h incubation. Each well received 50 μL of 2 × GSH-Glo reagent (Promega Corp.) for 0.5 h incubation at room temperature. A 100 μL amount of Luciferin Detection Reagent was added for 15 min at room temperature, according to the manufacturer’s instructions. The luminescence intensity of the solution in each well was recorded on a SpectraMax M5 microplate reader.
Cell Culture
Human bronchial epithelial (BEAS-2B) and mouse macrophages cell lines (RAW 264.7) were cultured in vented T-75 cm2 flasks (Corning, Fisher Scientific, Pittsburgh, PA) at 37 °C in a humidified 5% CO2 atmosphere. These cultures were passaged at 70–80% confluency every 2–4 days. BEAS-2B cells were cultured in bronchial epithelial basal medium (BEBM) (Lonza, Walkersville, MD), supplemented with growth factors from the SingleQuot kit (Lonza) to reconstitute BEGM. RAW 264.7 cells were cultured in DMEM medium containing 10% fetal calf serum (FCS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine.
Nanoparticle Dispersion in Cell Culture Medium
Nanoparticle stock solutions (5 mg/mL) were prepared by dispersing the dry particles in deionized water through probe sonication (3 W).48 The stock solution was used to remove 40 μL aliquots, which were mixed with an equal volume of 4% bovine serum albumin (BSA) (Fraction-V, Gemini Bioproducts, USA) and equilibrated for 1 h at room temperature. Cell culture medium (920 μL) was added to the BSA-coated nanoparticle suspensions. Nanoparticle suspensions were sonicated (3 W) for 15 s prior to conducting cellular studies. BEGM was supplemented with 2 mg/mL BSA to prepare a dilution series of nanoparticle suspensions.
Cytotoxicity Assessment
Cell viability was determined by an MTS assay, which was carried out with CellTiter 96 AQueous (Promega Corp.) kit.25 There were 1 × 104 BEAS-2B or RAW 264.7 cells in 100 μL of culture medium plated in each well of a 96 multiwell black plate (Costar, Corning, NY) for overnight growth. The medium was removed, and cells were treated for 24 h with 100 μL of 50, 100 and 200 μg/mL nanoparticle suspensions. After the treatment, the cell culture medium was removed and followed by washing of the plates three times with PBS. Each well received 100 μL of culture medium containing 16.7% of MTS stock solution for 1 h at 37 °C in a humidified 5% CO2 incubator. The plate was centrifuged at 2000g for 10 min in NI Eppendorf 5430 with a microplate rotor to spin down the cell debris. A 80 μL amount of the supernatant was removed from each well and transferred into a new 96 multiwell plate. The absorbance of formazan was read at 490 nm on a SpectraMax M5 microplate reader (Molecular Devices Corp., Sunnyvale, CA, USA).
Cellular H2O2 and GSH Assessment
There were 1 × 104 BEAS-2B or RAW 264.7 cells in 100 μL of culture medium plated in each well of a 96 multiwell white plate (Costar, Corning, NY) overnight. The medium was removed, and cells were treated with 100 μL of 50, 100, and 200 μg/mL of each nanoparticle suspension for the indicated time period. Cellular H2O2 was assessed 4 h after addition of the particles by adding 20 μL of ROS-Glo H2O2 substrate (Promega Corp.) to each well and then left standing for 2 h at 37 °C in a humidified 5% CO2 atmosphere. A 100 μL amount of ROS-Glo detection solution was added to the resulting mixture and incubated for 20 min at room temperature. The luminescence intensity of each well was recorded on a SpectraMax M5 microplate reader. For cellular GSH assessment, the culture medium was removed following 6 h of incubation with the particles, and each well was washed three times with PBS. Following addition of 100 μL of GSH-Glo reagent (Promega Corp.) for 0.5 h at room temperature, 100 μL of Luciferin Detection Reagent was added for an additional 15 min at room temperature. The luminescence intensity of each well was recorded on a SpectraMax M5 microplate reader.
Western Blot Analysis for Heme Oxygenase-1 (HO-1) Expression49
There were 1.6 × 105 BEAS-2B or RAW 264.7 cells in 1.6 mL of culture medium seeded into the wells of six-well plates (Costar, Corning, NY). After overnight growth, cells were treated with 1.6 mL of a 50 μg/mL nanoparticle suspension for 6 h. Cells were washed with PBS three times and harvested by scraping. Cell pellets were resuspended in cell lysis buffer containing Triton X-100 and protease inhibitors. The lysates were sonicated briefly and centrifuged, and the protein content in the supernatant was measured by the Bradford method. A 30 μg amount of total protein from each sample was electrophoresed by 10% SDS-PAGE and transferred to a PVDF membrane. After blocking, membranes were incubated with antihuman/mouse HO-1 monoclonal antibody (1:500) (ENZO Life Sciences, Plymouth Meeting, PA, USA). Membranes were overlaid with biotinylated secondary antibody (1:1000) before addition of HRP-conjugated avidin–biotin complex (1:10 000). Proteins were detected using ECL reagent according to the manufacture’s instruction.
IL-8 or TNF-α Cytokine Quantification by ELISA6
There were 1 × 104 BEAS-2B or RAW 264.7 cells in 100 μL culture medium plated overnight in each well of a 96 multiwell black plate (Costar, Corning, NY). The medium was removed, and cells were treated with each of the nanoparticle suspensions at 50, 100, and 200 μg/mL for 6 h. Plates were centrifuged at 2000g for 10 min in an Eppendorf 5430 microcentrifuge with microplate rotor to spin down the cell debris and nanoparticles. A 50 μL amount of the supernatant was removed from each well for measurement of IL-8 activity in BEAS-2B cells or TNF-α activity in RAW 264.7 cells using an OptEIA (BD Biosciences, CA) ELISA kit according to the manufacturer’s instructions. Briefly, a 96-well plate was coated with 50 μL of monoclonal anti-IL-8 or anti-TNF-α antibody for 2 h. After removal of the unbound antibody, a standard cytokine dilution series or 50 μL of each supernatant were pipetted into the precoated wells for antigen capture. After 2 h, the unbound growth factor was removed and each well in the plate was washed with a buffer five times and an enzyme-linked secondary polyclonal antibody added. Following washing, a substrate solution was added at 1:250 dilution for 30 min to allow color development. After termination of the reaction, the colorimetric intensity was measured at 450 nm on a SpectraMax M5 microplate reader.
Use of a Multiparametric Automated Assay To Access Toxic Oxidative Stress
The rationale for using this assay, which quantitatively assesses cellular oxidative stress parameters, has been previously described.50 This assay was carried out in the Molecular Shared Screening Resource laboratory in the California NanoSystems Institute, where cellular seeding of the plates, preparation of the nanoparticle working solutions, and their addition to the tissue culture plates are carried out with automated liquid handling, including a Multidrop (Thermo-Fischer, Waltham, MA), Precision 2000 (Biotek Instruments, Winooski, VT), and Hydra 96 (Robbins Scientific, Golden Valley, MN).23 Five thousand cells in 50 μL of tissue culture medium were plated into each well of a 384 multiwell plate (Greiner Bio-One, Monroe, NC), followed by overnight growth at 37 °C in a humidified 5% CO2 incubator. The medium in each well was aspirated, and 25 μL of a nanoparticle dilution series was added to quadruplicate wells to deliver doses of 0.4, 0.8, 1.6, 3.2, 6.3, 12.5, 25, 100, and 200 μg/mL. ZnO nanoparticles, as a positive control, were prepared to treat the cells at the same concentrations as for the Co3O4, PdO, and PdO/Co3O4 nanoparticles. Three cocktails of fluorescent dye mixtures were prepared by mixing the dyes with compatible wavelengths in BEGM or complete DMEM. The first cocktail contained Hoechst 33342 (1 μM), Fluo-4 (5 μM), and propidium iodide (5 μM); the second cocktail contained Hoechst 33342 (1 μM) and MitoSox Red (5 μM), and the third contained Hoechst 33342 (1 μM) and JC-1 (5 μM). The utility of these dyes, their excitation/emission wavelengths, and response profiling have been described previously.23,25,50 Each well received 2.5 μL of one of the dye mixtures for 30 min, with the plates being kept under dark conditions. Epifluorescence readings at 10× magnification were obtained hourly for the first 6 h and again at the 24 h mark using an Image-Xpress Micro high-content screening system (Molecular Devices, Sunnyvale, CA) equipped with a laser autofocus. DAPI, FITC, and TRITC filter/dichroic combinations were used to image Hoechst 33342 (blue), Fluo-4/JC-1 (green), and PI/MitoSox Red (red), respectively. Images were processed using MetaXpress software (Molecular Devices, Sunnyvale, CA). The total number of nuclei was counted in the Hoechst/DAPI channel using the following settings: The minimum width was 3 μm (about 3 pixels), the approximate maximum width was 10 μm (about 7 pixels), and the threshold intensity was 100 gray levels above background. For the FITC and TRITC channels, the approximate minimum width was 5 μm (about 6 pixels) and the approximate maximum width was 30 μm (about 22 pixels). The thresholds were set at 250 and 500 gray levels above background, respectively. The percentage of cells positive for each response parameter was calculated using MetaXpress software on the basis of the total number of Hoechst-positive cells showing increased fluorescence intensity above a defined threshold. These toxicity data were further normalized via strictly standard mean difference (SSMD) to quantify the cell responses induced by the nanoparticles.51−53 SSMD measures the magnitude of the differences between each set of quadruplicate measurements and the control population (cell population that was not exposed to ENMs) standardized by their variances with the following definition
where μ and σ denote the mean and standard deviation of the sample quadruplicate or the control population (identified by the subscripts). |SSMD| ≥ 3 indicates a significant difference between the nanoparticle-induced cell response to control. Given that the mean difference is normally distributed, a |SSMD| of 3 indicates that the probability that the sample population is different from the control population is >99%.
ICP-OES Analysis To Determine Metal Dissolution in Cell Culture Medium and Abundance of Cellular Uptake
Pd and Co dissolution for 0%, 0.6%, 2.4%, 3.7%, 4.7%, and 8.9% PdO-doped Co3O4 nanoparticles incubated in cell culture medium was determined by ICP-OES.25 A 20 μL amount of 5 mg/mL nanoparticles was mixed with 480 μL of culture medium for 24 h at 37 °C, with gentle shaking. The resulting solution was centrifuged at 20 000 rpm for 30 min, and 500 μL of the supernatant was digested by 3 mL of concentrated nitric acid at 90 °C for 3 h. The digested solution was dried by evaporation at 120 °C, and 8 mL of 5% nitric acid was added for ICP-OES measurement.
Cellular uptake of 0%, 0.6%, 2.4%, 3.7%, 4.7%, and 8.9% PdO-doped Co3O4 nanoparticles in BEAS-2B and RAW 264.7 cells was determined by ICP-OES. There were 8 × 104 BEAS-2B cells or RAW 264.7 cells in 800 μL of culture medium seeded in each well of six-well plates overnight growth in an incubator. Cells were treated with 800 μL of 50 μg/mL nanoparticles for 6 h. After treatment, cells were gently washed three times with PBS and harvested by 500 μL of 0.05% trypsin. The cell suspension was digested by 3 mL of concentrated nitric acid at 90 °C for 3 h. The well-digested solution was dried by evaporation at 120 °C, and 8 mL of 5% nitric acid was added for ICP-OES measurement.
Cellular Transmission Electron Microscopy
Cellular uptake of nondoped and PdO-doped Co3O4 nanoparticles was evaluated using TEM.23 There were 1 × 106 BEAS-2B or RAW 264.7 cells in 10 mL culture medium seeded in a 10 cm Petri dish (Costar, Corning, NY) overnight growth at 37 °C in a humidified 5% CO2 atmosphere. Cells were treated with 50 μg/mL nanoparticles for 6 h. After treatment, cells were gently washed three times with PBS and fixed in 5 mL of 2% glutaraldehyde in 0.1 M phosphate-buffered saline (PBS) for 2 h. Cells were scratched from the plate and collected for postfixation in 1% OsO4 in PBS. After fixation for 1 h, cells were dehydrated in a graded ethanol series, treated with propylene oxide, and embedded in Epon. There were 50–70 nm thick sections sliced using a Reichert-Jung Ultracut E ultramicrotome and captured on Formvar-coated copper grids. The sections were stained with uranyl acetate and Reynolds lead citrate and examined on a JEOL 100 CX transmission electron microscope at 80 kV in the UCLA BRI Electron Microscopy Core.
Assessment of Acute Toxicological Responses in the Mouse Lung by Oropharyngeal Aspiration
Eight week old male C57 BL/6 mice were purchased from Charles River Laboratories (Hollister, CA). Nanoparticle oropharyngeal aspiration was conducted using our previously published approach, with minor modifications.25 Briefly, under ketamine/xylazine (100/10 mg/kg) anesthesia, the animals were held vertically and the tongue was gently pulled out of the mouth by forceps. Nanoparticles (20 μg suspended in 50 μL of PBS containing 0.6 mg/mL mouse serum albumin and 10 μg/mL DPPC) were placed at the back of the tongue while the nose is closed. As a negative control, 50 μL of PBS containing 0.6 mg/mL mouse serum albumin and 10 μg/mL DPPC was used to treat the mice. The nose and tongue were released after at least two breaths. Animal necropsy was performed 40 h after oropharyngeal aspiration. Bronchoalveolar lavage (BAL) was performed by cannulating the trachea and gently lavaging the lung 3 times with 1 mL of sterile PBS. BAL cells were adhered onto microscopic slides for differential cell count, while BAL fluid was stored at −80 °C for assessing cytokine and chemokine levels. BAL differential cell counts were performed as described by us.25,30 IL-6 and Lix levels in the BAL fluid were analyzed using ELISA kits (BD Biosciences, San Diego, CA) according to the manufacturer’s instructions. Histological lung sections were stained with hematoxylin/eosin to visualize the inflammatory cells.
Statistical Analysis
All data were expressed as mean ± SD. All values were obtained from at least three independent experiments. Statistical significance was evaluated using two-tailed heteroscedastic Student’s t-tests according to the TTEST function in Microsoft Excel. The significant difference between groups was considered statistically significant when the p value was lower than 0.05.
Results
Synthesis and Characterization of PdO-Doped Co3O4 NPs
A series of PdO-doped Co3O4 ultrafine crystalline NPs was synthesized using FSP.54 In order to design a nanoparticle library with an increased population density of heterojunctions, Pd acetylacetonate (which is sparingly soluble in xylene) was used to increase the Pd/Co ratio before spraying. The precursor solution was heated to 50 °C for 10 min before spraying. Energy-dispersive X-ray spectroscopy (EDX) was used to confirm the presence of 0.6%, 2.4%, 3.7%, 4.7%, and 8.9% PdO (mass percentages) in Co3O4 (Figure S1, Supporting Information). TEM demonstrated that all the Co3O4 NPs assumed a spherical morphology (Figure 1a) with a primary size range from 11.1 ± 2.8 to 13.3 ± 3.7 nm (Figure 1b). BET measurements were used to demonstrate that the specific surface area of the NPs ranges from 70.3 ± 3.3 to 88.2 ± 2.8 m2/g (Figure 1b). These NPs were dispersed in distilled water, resulting in hydrodynamic sizes from 122.5 ± 1.4 to 187.6 ± 15 nm, as determined by DLS (Figure 1b). Zeta potential measurement indicated all the NPs dispersed in this medium had positive surface charges ranging from +17.2 ± 0.8 to +25.8 ± 0.7 mV (Figure 1b). In summary, the characterization data revealed a doped library with homologous particle size, surface area, hydrodynamic size, and surface charge.
Figure 1.

Physicochemical properties of nondoped and PdO-doped Co3O4 nanoparticles. (a) TEM images of the nondoped as well as the PdO-doped Co3O4 nanoparticles. (b) Primary sizes, surface area, hydrodynamic sizes, and zeta potentials of the nanoparticles in water. Particle size was determined for ∼200 particles in the perpendicular direction.
PdO Doping of Co3O4 Nanoparticles Results in Formation of Heterojunctions
Formation of PdO/Co3O4 heterojunctions was demonstrated by a variety of physicochemical methods. High-resolution TEM of 8.9% PdO/Co3O4 NPs showed highly crystalline NPs with Co3O4 lattice fringes of 0.462 nm, orientated in the (111) direction, while the corresponding lattice distance for PdO nanoparticles was 0.258 nm in the (002) direction (Figure 2a). These lattice distances were compared with the X-ray diffraction (XRD) patterns (Figure 2b). This demonstrated a lattice distance peak 0.464 nm for Co3O4, which is in close agreement with the HRTEM results. The particles were found to be lattice-to-lattice coupled, although the heteroparticles had different dspacing orientations. The XRD of 8.9% PdO/Co3O4 NPs shows the 50% and 100% peak positions (pdf no. 01-088-2434) of PdO located at 33.56° (002) and 34.14° (011) 2θ, respectively (Figure 2b). The narrow differences (0.58° 2θ) between these two positions yielded a broad PdO signature at 33.9° 2θ. Additionally, the characteristic peak at 42.34° 2θ in the (110) orientation is also assigned to PdO, providing sufficient evidence for a heteromixture.55 The evidence that the XRD results are characteristic of a heteromixture is as follows: (1) XRD data of nondoped or PdO-doped Co3O4 showed no peak shifts, which is characteristic of surface functionalization rather than PdO incorporation into the Co3O4 lattice; (2) the ionic radius of Pd2+ (1.0 Å) is much larger than the ionic radius of Co2+ (0.72 Å) and Co3+ (0.73 Å), making incorporation of the larger Pd2+ ion into the lattice of the smaller Co2+/Co3+ unlikely;56 (3) the primary and crystallite sizes of nondoped or PdO-doped Co3O4 nanoparticles were similar.
Figure 2.

Determination of PdO/Co3O4 heterojunctions on PdO-doped Co3O4 nanoparticle surfaces. (a) HRTEM of 8.9% PdO-doped Co3O4 nanoparticles. A heterojunction is clearly shown between the single-crystalline PdO particle, orientated in the (002) direction, and a single-crystalline Co3O4 particle, orientated in the (111) direction. (b) XRD spectra of 8.9% PdO-doped Co3O4 nanoparticles. Nondoped Co3O4 and 8.9% PdO-doped Co3O4 have the same Co3O4 XRD peaks, and no peak shift is observed. The (002), (110), and (011) peaks at 33.56°, 34.14°, and 42.34° 2θ indicate the presence of PdO. (c) EFTEM of 8.9% PdO-doped Co3O4 nanoparticles: (left) Co mapping and (right) Pd mapping. PdO is distributed in the Co3O4 matrix. (d) High-resolution XPS spectra of 8.9% PdO-doped Co3O4 nanoparticles. Pd 3d3/2 and Pd 3d5/2 peaks at binding energies of 341.9 and 336.7 eV are attributed to PdO. Both peaks gradually increase with increasing PdO concentration.
Collectively, these findings indicate that the particles are a mixture of two components (PdO and Co3O4), giving rise to a nanoscale heterojunction. To verify the distribution of PdO on the Co3O4 NPs, elemental maps were obtained using a postcolumn energy filter. The representative Co and Pd elemental maps for 8.9% PdO/Co3O4 NPs in energy-filtered transmission electron microscopy (EFTEM) are presented in Figure 2c. The X-ray photoelectron spectroscopy (XPS) of these particles showed Pd 3d3/2 and Pd 3d5/2 signals at binding energies of 341.9 and 336.7 eV, respectively (Figure 2d). This can be attributed to the PdO phase.55,57 These signals gradually increased with increased PdO content from 0.6% to 8.9% (Figure S2, Supporting Information). This demonstrates the increased population density of PdO on the surface. These results suggest clear evidence of heterojunction formation between the functionalized particles and the parent matrix.
Increase of the Heterojunction Density Leads to Adjusting of Ef Levels and Band Bending in PdO-Doped Co3O4 NPs
PdO doping of oxide nanoparticles is frequently used by industry to create heterojunctions that can be used for applications such as catalysts and energy storage.55,57 On the basis of the work function (Φ), the Ef energies values of PdO and Co3O4 have previously been reported as −7.9 and −6.1 eV, respectively.45,58 This arrangement would facilitate electron transfer from the higher energy Co3O4 to the lower energy PdO. Using ultraviolet photoelectron spectroscopy (UPS), we obtained Φ values for nondoped and PdO-doped Co3O4 NPs. This demonstrated Φ values of 6.03, 6.11, 6.12, 6.24, 6.49, and 6.57 eV for nondoped particles and particles doped with 0.6%, 2.4%, 3.7%, 4.7%, and 8.9% PdO, respectively. Since Φ represents the difference between the vacuum (0 eV) and Ef energy levels, the Ef values of nondoped and PdO-doped Co3O4 could be calculated. This yielded Ef values of −6.03, −6.11, −6.12, −6.24, −6.49, and −6.57 eV, respectively, for the same nondoped and doped particle series (Figure 3a). The trend toward lower Ef levels with incremental levels of PdO doping suggests electron transfers proceeding from Co3O4 to PdO. The alignment of Ef energy levels across the heterojunctions is also expected to increase hole dominance in Co3O4 (a p-type semiconductor) and possibly a change in band-gap energy. The equation Φ = χ + BB +μ (where χ, BB, and μ represent electron affinities, band bending, and electrochemical potential, respectively) can be used to measure the shift in the energy band position or BB (Figure 3b).29 We used UPS and UV–vis spectra to determine the Ec, Ev, and Ef levels of the nondoped and doped particles (Figure 3c), allowing us to calculate the χ and μ values. Figure 3c summarizes the energy band information, including Ec, Ev, and Ef, with incremental levels of PdO doping. The degree of BB was calculated as 0.08, 0.09, 0.21, 0.46, and 0.54 eV for 0.6%, 2.4%, 3.7%, 4.7%, and 8.9% PdO-doped particles, respectively (Figure 3d). The sharp increase in the BB at higher levels of PdO doping (Figure 3d) suggests formation of bigger space charge layers across the heterojunctions. The Ec values derived for nondoped Co3O4 and 0.6%, 2.4%, 3.7%, 4.7%, and 8.9% PdO-doped particles were −4.23, −4.18, −4.22, −4.33, −4.53, and −4.69 eV, respectively. Interestingly, these Ec values overlap with BRP (from −4.12 to −4.84 eV), which could allow nondoped and doped particles to accept electrons from cellular redox couples (e.g., NADPH and cytochrome c), as previously shown by us.25−27,59 In fact, we previously used this energy overlap to predict 5 metal oxides among 24 could lead to generation of cellular oxidative stress and acute pulmonary inflammation.25,27,59 It is possible that the electron capture by the Ec could be facilitated through recombination with the incremental population of holes in the doped particles, thereby speeding up oxidation of cellular redox couples and facilitating the onset of redox disequilibrium and oxidative stress.
Figure 3.

Electronic properties of nondoped and PdO-doped Co3O4 nanoparticles. (a) Ef levels of nondoped Co3O4 as well as 0.6%, 2.4%, 3.7%, 4.7%, and 8.9% PdO-doped Co3O4 nanoparticles. Ef levels were determined using UPS analysis. These energy levels declined progressively with incremental PdO doping. (b) Band bending (BB) values in relation to work function, electron affinity, and electrochemical potential. BB was determined by Φ = χ + BB + μ, with Φ, χ, BB, and μ representing work function, electron affinity, BB, and electrochemical potential, respectively. (c) Distribution of Ec, Ef, and Ev of nondoped Co3O4 as well as PdO-doped Co3O4 nanoparticles. Please notice that all Ec values show overlap with the BRP, which ranges from −4.12 to −4.84 eV. This energy overlap has previously been shown to lead to electron transfer from cellular redox couples to nanoparticles such as Co3O4. (d) BB varies in accordance with PdO content. Band bending values of nondoped and PdO-doped NPs were determined based on the equation in b. BB values increase in accordance with PdO concentration.
Heterojunction-Facilitated Oxygen Radical Generation and GSH Depletion under Abiotic Conditions
Electron transfer across the PdO/Co3O4 heterojunctions leads to accumulation of free holes and free electrons at the Co3O4 and PdO interfaces, respectively. This could lead to generation of reactive oxygen species (ROS), by the ability of either h+ to react with H2O to form hydroxyl radical (HO•) or the e– to react with O2 to form superoxide radical (O2•–).16 The fluorescent dye 2′,7′-dichlorofluorescein (DCF) is a useful reagent to assess abiotic ROS generation by nanoparticle surfaces.46 Use of DCF demonstrated a progressive increase in fluorescence (at 520 nm) with incremental (0–8.9%) PdO doping (Figure 4a). However, pure PdO NPs particles did not have an effect on fluorescence, confirming that introduction of heterojunctions is responsible for increased ROS generation. Because DCF does not identify the radical type, additional dyes were used to assess HO• or O2•– generation. Abiotic HO• generation can be assessed by 3′-(p-aminophenyl) fluorescein (APF), which emits fluorescence at 515 nm, while O2•– generation can be assessed by 3-bis(2-methoxy-4-nitro-5-sulfophehyl)-2H-tetrazolium-5-carboxanilide) (XTT), which has an absorbance maximum at 465 nm.47,60 Use of these dyes demonstrated that while PdO-doped Co3O4 NPs could induce APF fluorescence that is progressive with incremental doping (Figure 4b), there was no comparable increase in XTT absorbance (Figure 4c). Xanthine/xanthine oxidase did, however, induce XTT absorbance. Moreover, pure PdO NPs (Figure S3, Supporting Information) did not induce fluorescence or absorbance of any dye, suggesting that HO• generation takes place at PdO/Co3O4 heterojunctions.
Figure 4.

Abiotic assessment of total ROS, hydroxyl, and superoxide radical generation and GSH oxidation by pure PdO, nondoped, and PdO-doped Co3O4 nanoparticles. (a) DCF fluorescence: 29 μmol/L DCF was incubated with nanoparticles at 200 μg/mL for 6 h. Fluorescence emission spectra were collected at 500–600 nm with excitation at 490 nm. PdO functionalization significantly enhanced DCF fluorescence compared to nondoped Co3O4 NPs, suggesting increased total ROS generation. (b) APF fluorescence: 8 μmol/L APF was incubated with 200 μg/mL nanoparticles for 6 h and the fluorescence emission spectra collected at 480–600 nm using an excitation wavelength of 455 nm. PdO functionalization significantly enhanced APF fluorescence compared to nondoped Co3O4 NPs, suggesting increased HO• generation. (c) XTT absorbance: 80 μmol/L XTT was incubated with 200 μg/mL nanoparticles for 6 h. Xanthine/xanthine oxidase (X/XO) was used as a positive control. Absorbance spectra were collected at 410–550 nm. (d) GSH oxidation by nanoparticles: 62.5 μmol/L GSH was incubated with 200 μg/mL nanoparticles for 6 h, and the GSH level was determined by the luminescence-based GSH-Glo kit. (*) p < 0.05 compared with control.
Hydroxyl radicals are potentially hazardous to cells because of their ability to oxidize a range of biomolecules, including the glutathione (GSH), which plays a critical role in maintaining cellular redox homeostasis through its antioxidant effects. GSH oxidation by hydroxyl radicals (and other ROS) converts GSH to GSSG. The decrease in GSH levels can be quantitatively assessed by an abiotic GSH-Glo assay, which is based on conversion of a luciferin derivative to luciferin by glutathione S-transferase (GST) in the presence of GSH. The data showed a progressive decline in GSH levels with incremental PdO doping levels, while pure PdO did not affect GSH levels (Figure 4d).
Cytotoxicity, Cellular H2O2 Generation, and GSH Depletion by the PdO/Co3O4 Library
The ability of PdO/Co3O4 NPs to engage in hydroxyl radical generation under abiotic conditions suggests that these nanoparticles could generate oxidative stress under biological conditions. If inhaled, redox-active MOx can generate acute pulmonary inflammation.49,61 Cellular studies were performed in human bronchial epithelial (BEAS-2B) and mouse macrophages cell lines (RAW 264.7), and all nanoparticles were well dispersed in BEGM (for BEAS-2B cells) or DMEM (for RAW 264.7 cells) (Table 1). After exposure to a wide dose range (0.4–200 μg/mL) of nondoped and PdO-doped NPs, cell viability measurement by the MTS assay showed that while Co3O4 NPs decreased the viability of BEAS-2B (Figure 5a) and RAW 264.7 cells (Supporting Information Figure S4a), incremental PdO doping resulted in more toxicity that was significant at concentrations of 50, 100, and 200 μg/mL. In contrast, pure PdO did not decrease any cell viability. Further cellular H2O2 production was studied in these two cell lines by exposure to 50, 100, and 200 μg/mL of nondoped and PdO-doped NPs for 6 h. H2O2 production was assessed by the luminescence-based ROS-Glo assay. The results demonstrated that increased H2O2 release was dose dependent and proportional to the amount of PdO doping in both BEAS-2B (Figure 5b) and RAW 264.7 cells (Figure S4b, Supporting Information). In contrast, pure PdO did not induce any luminescence activity. In summary, there appears to be a consistent relationship between abiotic hydroxyl radical generation and cellular ROS production with increased heterojunction density on Co3O4 NPs.
Table 1. Hydrodynamic Size of Nanoparticles in DMEM and BEGM Cell Culture Medium.
| hydrodynamic
size (nm) |
||
|---|---|---|
| nanoparticles | DMEM | BEGM (BSA) |
| PdO | 291.4 ± 3.0 | 295 ± 5.7 |
| Co3O4 | 249.8 ± 14.0 | 251.0 ± 2.7 |
| 0.6% PdO | 231.4 ± 10.7 | 264.3 ± 11.9 |
| 2.4% PdO | 227.8 ± 9.6 | 237.7 ± 4.8 |
| 3.7% PdO | 233.6 ± 5.5 | 261.8 ± 8.1 |
| 4.7% PdO | 225.0 ± 1.9 | 261.6 ± 12.3 |
| 8.9% PdO | 249.5 ± 4.3 | 256.7 ± 3.8 |
Figure 5.

Cytotoxicity, cellular H2O2 generation, and GSH depletion in BEAS-2B cells. (a) Cytotoxicity by MTS assay. Cells were treated with 0.4–200 μg/mL of nondoped and PdO-doped Co3O4 nanoparticles as well as pure PdO nanoparticles for 24 h. Untreated cells were used as a negative control. (b) Cellular H2O2 generation determined by the luminescence-based ROS-Glo assay. Cells were treated with 50, 100, and 200 μg/mL nondoped and PdO-doped Co3O4 nanoparticles as well as pure PdO nanoaprticles for 6 h. Luminescence activity was expressed as fold increase above untreated cells (relative value = 1). (c) Cellular GSH depletion determined by the luminescence-based GSH-Glo assay. Cellular exposure to the nanoparticles was similar as in b. GSH levels were expressed as a percentage of the luminescence intensity compared to control cells (100%). (*) p < 0.05, compared to control. Results similar to a, b, and c were obtained in RAW 264.7 cells (Figure S4a, S4b, and S4c, Supporting Information).
Cellular ROS production can trigger a series of incremental oxidative stress responses, with the first tier characterized by Nrf2-mediated antioxidant enzyme expression and increased synthesis of GSH.6,49,61 This response could restore the redox homeostasis, but if ROS production increases until it overwhelms the antioxidant defense, incremental levels of oxidative stress lead to GSH depletion as well as cellular toxicity. A luminescence-based GSH-Glo assay was used to monitor cellular GSH levels in BEAS-2B and RAW 264.7 cells. Exposure to nondoped and PdO-doped Co3O4 NPs at 50, 100, and 200 μg/mL for 6 h could induce a significant and dose-dependent reduction of luminescence activity, proportional to the level of PdO doping in BEAS-2B (Figure 5c) as well as RAW 264.7 cells (Figure S4c, Supporting Information). In contrast, treatment with pure PdO NPs did not induce GSH depletion. These data demonstrate that an increased population density of heterojunctions results in increased cellular ROS production and incremental levels of oxidative stress. While this is in agreement with the abiotic data, it is important to point out that cellular ROS production could take place as a result of hydroxyl radical generation by the particles surface as well as disruption of cellular redox homeostasis and involvement of mitochondria.
Heterojunctions Exacerbate the Hierarchical Oxidative Stress Response in Cells
The hierarchical oxidative stress response is characterized by an antioxidant defense response (Tier 1), the initiation of inflammation (Tier 2), and mitochondrial-mediated cytotoxicity (Tier 3) (Figure 6a).61 According to the hierarchical oxidative stress paradigm, the lowest levels of oxidative stress (Tier 1) are associated with induction of antioxidant and detoxification enzymes that are collectively known as phase II enzymes. HO-1 is a typical phase II enzyme and plays a critical role in cytoprotection against ROS.6,61 Western blotting to assess heme oxygenase-1 (HO-1) expression (Tier 1) demonstrated increased HO-1 abundance in cells exposed to 50 μg/mL nondoped or PdO-doped Co3O4 NPs in BEAS-2B (Figure 6b) as well as RAW 264.7 cells (Figure S5a, Supporting Information). Importantly, the level of HO-1 expression was proportional to the level of PdO doping, while levels of β-actin did not change.
Figure 6.

Assessment of hierarchical oxidative stress responses in BEAS-2B cells. (a) Tiers of the hierarchical oxidative stress paradigm. These tiers include antioxidant defense (Tier 1), inflammation (Tier 2), and cytotoxicity (Tier 3). Phase II enzyme induction (such as HO-1 expression), cytokine and chemokine generation, and mitochondrion-mediated cell death are markers for Tier 1, Tier 2, and Tier 3, respectively. (b) Western blot analysis of HO-1 expression. Cells were treated with 50 μg/mL nondoped and PdO-doped Co3O4 nanoparticles for 6 h. Untreated cells were used as negative control. The intensity of HO-1 was normalized to β-actin using Image J software. (c) ELISA to assess IL-8 production in cells treated with nanoparticles at 50, 100, and 200 μg/mL for 6 h. (*) p < 0.05 compared to control (Student’s two-tailed t-test). (d) Heat map to compare the toxic oxidative stress response using a multiparameter automated screening assay. Heat maps were established using SSMD statistical analysis to evaluate the suprathreshold responses by automated epifluorescence microscopy. Response parameters included measurement of mitochondrial O2•– generation (MitoSox Red), mitochondrial membrane depolarization (JC-1), intracellular calcium flux (Fluo-4), and cell membrane permeability (PI). Cells were treated with a wide dose range of nanoparticles (0.4, 0.8, 1.6, 3.2, 6.3, 12.5, 25, 50, 100, 200 μg/mL). Epifluorescence images were collected hourly for the first 6 h and then again at 24 h.
Failure to restore redox equilibrium in Tier 1 could lead to activation of the Jun kinase (JNK) and NF-κB pro-inflammatory cascades, which are involved in the transcriptional activation of cytokine, chemokine, and adhesion gene promoters.61,62 In our previous studies of the metal oxide nanoparticle band gap,6,25 multiple pro-inflammatory cytokines (IL-8, IL-6, IL-1a, MCP-1, TNF-α MIP-1a, MIP-1b, MIP-2) were looked at and demonstrate the pro-inflammatory effects. We chose the robust IL-8 and TNF-α responses to investigate BEAS-2B and RAW 264.7 cell lines, respectively, in this article. In the present study, an ELISA to detect IL-8 release in the cellular supernatant of BEAS-2B cells demonstrated a dose-dependent and incremental increase in cytokine production with incremental levels of PdO doping (Figure 6c). Similar effects on TNF-α release were seen in RAW 264.7 cells (Figure S5b, Supporting Information). However, PdO NPs did not induce increased cytokine production in either cell type. Escalation of oxidative stress response to Tier 3 could trigger apoptosis through mitochondrial membrane depolarization (MMD) and release of pro-apoptotic factors. This response can be assessed by a multiparametric assay that contemporaneously assesses MMD together with superoxide generation, intracellular calcium flux, and increased membrane permeability as a result of cell death (Figures 6d and S5c, Supporting Information). These responses can be tracked by epifluorescence microscopy hourly for 6 h and again after 24 h. The multiparameter assay is conducted using fluorescent dyes, JC-1 (MMD), MitoSox Red (O2•–), Fluo-4 (Ca2+i), and propidium iodide (PI). Full details are provided in the Experimental Section. Use of nondoped and PdO-doped Co3O4 nanoparticles at 0.4–200 μg/mL to assess multiparameter responses in BEAS-2B (Figure 6d) and RAW 264.7 cells (Figure S5c, Supporting Information) allowed us to construct heat maps, which rank the intensity of each response in relationship to nondoped or untreated controls. The red pixels in the heat map indicate significant toxicity, while green pixels indicate the absence of a toxicological response. The data in Figures 6d and S5c, Supporting Information, demonstrate a progressive increase in multiparameter responses in BEAS-2B cells and RAW 264.7 cells with incremental PdO doping. In contrast, PdO NPs did not exert any effect. All considered, the above data indicate that the heterojunction density plays a role in the escalation of oxidative stress to Tiers 2 and 3. The increase in mitochondrial superoxide production could be due to hydroxyl radical generation on the particle surface as well as a disruption of cellular redox homeostasis, e.g., oxidation of redox couples.25,27,59
Cellular Uptake and Dissolution of Nondoped and PdO-Doped Co3O4 NPs
Since metal ion shedding can lead to metal oxide toxicity (e.g., ZnO6,30,43 and CuO25), the dissolution of nondoped and PdO-doped Co3O4 NPs in cell culture medium was investigated. NPs in culture medium (200 μg/mL) were incubated at 37 °C for 24 h, and the supernatants were collected after centrifugation for acid treatment. Following the quantitative assessment of elemental Co and Pd content by ICP-OES, it was clear that the nanoparticles had low rates of dissolution in BEGM (Figure 7a) and DEME media (Figure S6a, Supporting Information). At concentrations similar to the elemental content in the supernatant, Co2+ or Pd2+ ions did not affect cell viability (Figures 7b and S6b, Supporting Information). These results rule out cellular toxicity through NP dissolution.
Figure 7.

Metal dissolution and cellular uptake in BEAS-2B cells. (a) Metal dissolution in BEGM containing 2 mg/mL BSA: 200 μg/mL nondoped or PdO-doped Co3O4 nanoparticles were incubated at 37 °C for 24 h. Supernatants were collected and elemental Co or Pd concentrations determined by ICP-OES. (b) Cell viability assessment of BEAS-2B cells exposed to a low concentration of Co2+ or Pd2+ ion in BEGM medium. Cells were treated with 0.1, 0.2, and 0.4 μg/mL of Co2+ or Pd2+ for 24 h in BEGM medium, and untreated cells were used as negative control. Cell viability was assessed by the MTS assay. (c) ICP-OES analysis for cellular Co and Pd content in BEAS-2B cells. Cells were treated with 50 μg/mL nanoparticles for 6 h, and untreated cells were used as a control. After PBS washing, cells were harvested using 0.05% trypsin and the cell suspension digested with concentrated nitric acid at 90 °C for 3 h. The well-digested solution was diluted for ICP-OES measurement. (d) TEM images of BEAS-2B cells treated with 50 μg/mL nondoped and 8.9% PdO-doped Co3O4 nanoparticles for 6 h.
Besides metal dissolution, cellular uptake is another parameter to consider in terms of differential cytotoxicity. Cellular uptake of nondoped or doped NPs in BEAS-2B and RAW 264.7 cells was investigated by determining the cellular Co content by ICP-OES as well as TEM. After cellular exposure to 50 μg/mL nondoped or PdO-doped Co3O4 NPs for 6 h, ICP-OES analysis demonstrated the presence of 0.18–0.20 μg Co/μg protein in BEAS-2B cells (Figure 7c) or 0.12–0.15 μg Co/μg protein RAW 264.7 cells (Figure S6c, Supporting Information). Since the cellular uptake of particles was fairly uniform, this could not explain the differential toxicological effects. TEM analysis confirmed that nondoped and 8.9% PdO-doped Co3O4 NPs were taken up in approximately similar quantities in BEAS-2B (Figure 7d) as well as RAW 264.7 cells (Figure S6d, Supporting Information). These data indicate that generation of oxidant injury in accordance with doped levels and density of heterojunctions play a major role in the cytotoxicity of PdO/Co3O4 nanoparticles.
Differential Pro-Inflammatory Effects in the Lung by Co3O4 Nanoparticles with Different Levels of PdO Doping
We previously demonstrated that, based on band-gap characteristics and overlap with BRP, predictions can be made about the pro-inflammatory effects of metal oxide NPs in the lung.25 Similarly, differential cytokine production (Tier 2 response) in cells suggested that doping could accentuate tissue inflammation, e.g., in the lung. Oropharyngeal aspiration was used to compare the in vitro results to in vivo outcome in C57 BL/6 mice.6−8,25 Mice were instilled with 20 μg of NPs and sacrificed 40 h later. Bronchoalveolar lavage (BAL) fluid was collected to perform cell counts and measure cytokine and chemokine levels. We observed that incremental levels of PdO doping induced a statistically significant increase in neutrophil cell counts (Figure 8a), IL-6 (Figure 8b), and LIX (a lipopolysaccharide-induced CXC chemokine and murine homologue of IL-8) (Figure 8c) levels. These are characteristic markers for acute lung inflammation, which could be confirmed by hematoxylin and eosin (H&E) staining of lung sections. This demonstrated that incremental PdO doping is accompanied by increased inflammatory infiltrates around small pulmonary airways (Figure 8d). These acute inflammatory changes are in good agreement with the in vitro toxicological effects.
Figure 8.

Acute pulmonary inflammation in C57BL/6 mice. Animals received 20 μg of each of the NPs by oropharyngeal aspiration, followed by sacrifice after 40 h. ZnO was used as a positive control. Bronchoalveolar lavage (BAL) fluid was collected for the performance of differential cell counts as well as measuring cytokine and chemokine levels. Histological lung sections were stained with H&E. (a) Neutrophil counts in the BALF. (b) IL-6 and (c) Lix levels in the BALF. (d) H&E staining of lung sections. (*) p < 0.05.
Discussion
In the present study, we demonstrate through PdO doping of Co3O4 nanoparticles that creation of heterojunctions can be used to adjust band-gap and Fermi energy levels in measured quantities, thereby allowing us to study the relationship of semiconductor properties to cellular redox regulation and the potential to induce oxidative stress in vitro and in vivo. Through the use of FSP we were able to synthesize a Co3O4 nanoparticle library in which the gradual increase in the PdO content (up to 8.9%) and heterojunction density allowed a progressive increase in electron transfer from Co3O4 to PdO to align Ef energy levels. This alignment was accompanied by free hole accumulation at the Co3O4 interface and production of hydroxyl radicals. Interestingly, there was no concomitant superoxide generation, which could reflect the hole dominance in Co3O4, a p-type semiconductor. Although the electron flux across the heterojunctions induced upward Ec and Ev bending in Co3O4, the Ec levels of all the particles showed energy overlap with the biological redox potential (BRP). We previously demonstrated that this allows electron capture from the cellular redox couples that make up the BRP.25 Excessive oxidation of these redox couples can derange cellular redox homeostasis and lead to ROS production. The induction of cellular oxidative stress is manifested as increased heme oxygenase-1 (HO-1) expression, mitochondrial superoxide generation, TNF-α and IL-8 production, and, at the highest tier of oxidative stress, a decrease in mitochondrial membrane potential and cytotoxicity. These biological effects manifested in the lungs of mice as a progressive rise in IL-6 and Lix cytokine levels and acute inflammatory infiltrates with incremental levels of PdO doping. The in vitro and in vivo toxicity profiles of Co3O4 nanoparticles have been investigated by other researchers.63,64 Papis et al. reported that 45 nm Co3O4 nanoparticles have very low Co ion dissolution (0.1–0.2% Co ion release in M199 and MEM culture media) and show significant cellular uptake and ROS generation in ECV-304 and HepG2 cells. This leads to ∼60% and ∼45% cell death in ECV-304 and HepG2 cell lines, respectively, at a Co dose of 73 μg/mL (∼100 μg/mL of Co3O4). (63)Cho et al. reported that 18 nm Co3O4 nanoparticles release 0.08% Co ion in artificial interstitial fluid (pH = 7.4) and can induce acute inflammatory responses, including increased MCP-1/CCL2 and MIP-2/CXCL cytokine/chemokine levels in the lung of Wistar rats 24 h after instillation. All these in vitro and in vivo results are consistent with our results and demonstrate Co3O4 nanoparticles are potentially hazardous nanomaterials.64 All considered, generation of a combinatorial PdO/Co3O4 nanoparticle library with incremental heterojunction density allowed us to demonstrate that a combination of Ev, Ec, and Ef levels play a role in the generation of oxidant injury and inflammation by this p-type semiconductor.
PdO doping of Co3O4 nanoparticles is able to facilitate formation of PdO/Co3O4 heterojunctions. It is known that the electrons can flow (i) from a transition metal oxide (TMOx) to a noble metal oxide (NMOx) when Ef TMOx > Ef NMOx (ΦTMOx < ΦNMOx) or from a NMOx to a TMOx when Ef TMOx < Ef NMOx (ΦTMOx > ΦNMOx).29 Since it is known that the Ef of Co3O4 (−6.03 eV) is higher than PdO (−7.9 eV),45,58 electron transfer from the former to the latter will proceed until Ef levels are the same across the heterojunction. This is corroborated by the Ef energy analysis of the doped particles (Figure 3a), showing that incremental levels of doping (0.6%, 2.4%, 3.7%, 4.7%, and 8.9%) lead to a progressive deviation from the Ef (−6.03 eV) of Co3O4 while approximating the Ef of PdO (−7.9 eV). Electron transfer across this gradient creates a space charge (depletion) layer at the heterojunction, leading to a shift of the energy band edges. Energy band analysis of the PdO/Co3O4 NPs (Figure 3b and 3c) showed progressive upward band bending with increased PdO doping (Figure 3d). The magnitude of the BB effect is indicative of the charge density at the interface. Because Co3O4 is a p-type semiconductor in which hole dominance may interfere with recombination, the tendency toward increased hole formation at the heterojunction interface could explain the incremental •OH generation (Scheme 1). While theoretically the free e– on the PdO side of the interface should be available to generate O2–, we did not observe increased XTT absorbance (Figure 4c). While we do not fully understand this observation, it is possible that Co3O4 NPs can be efficient scavengers for hydrogen peroxides and superoxide radicals in neutral and/or alkaline conditions due to the large redox potential value (1.808V) of the Co3+/Co2+ couple.65
We demonstrate progressive dysregulation of the cellular redox equilibrium with incremental PdO doping and heterojunction density at the PdO/Co3O4 interface. This is corroborated by increased cellular ROS production and a progressive decline in cellular GSH levels. Depending on the level of oxidative stress, cells respond by increased antioxidant defense (e.g., HO-1 expression), increased production of cytokines and chemokines (Tier 2), or a decline in mitochondrial membrane potential, ATP production, and ultimately cellular death (Tier 3). In order to understand the breakdown of the redox homeostasis, we have to consider both the particle properties as well as BRP. Hydroxyl radical generation at the particle surface could damage a range of biomolecules (including GSH), depending on where the particles are located in the cells. For instance, damage to the mitochondrial membrane could interfere in electron transfer, with these organelles contributing to superoxide production (as demonstrated by increased MitoSox Red fluorescence). A second major impact of the particles could be disruption of the cellular redox homeostasis as a result of electron transfers from redox couples to the Co3O4 conduction band. Thus, redox couples, such as cytochrome c–Fe3+/cytochrome c–Fe2+, NADP+/NADPH, ascorbyl radical/ascorbate, Fe3+–citrate/Fe2+–citrate, Fe3+ ferritin/Fe2+ ferritin, which are involved in maintaining the BRP between −4.12 and −4.84 eV, could be oxidized and reduce their redox buffering capacity. This could lead to formation of oxidizing substances that decrease the levels of cellular antioxidants and/or increase cellular ROS production.27,59 The fact that Co3O4 is a p-type semiconductor could speed up the oxidation rate of the redox couples because of hole dominance, further accentuated by heterojunction formation in PdO-doped particles (Scheme 2).
Scheme 2. Generation of Oxidative Stress by Co3O4 as a Result of Ec Overlap with the BRP Could Be Accentuated by PdO Doping.
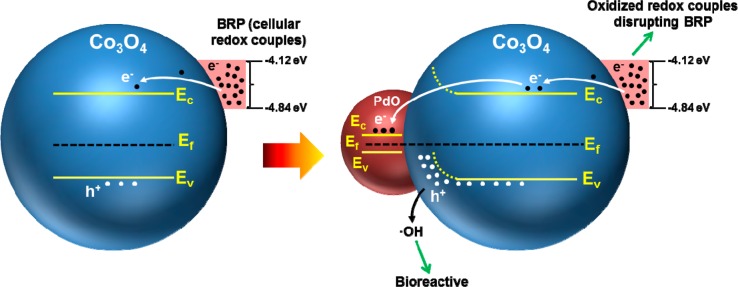
The overlap of Ec of nondoped and PdO-doped Co3O4 nanoparticles with the biological redox potential (from −4.12 to −4.84 eV) can promote electron transfer between the redox couples (that maintain the BRP) and the conduction band. PdO doping can speed up this process because the increased generation of holes could speed up the rate by which electrons transferred to the conduction band is recombined with excess holes.
A major advance in the current paper has been to demonstrate that if particle size and shape are kept constant it is possible to show for Co3O4 that Ec, Ev, and Ef contribute in an integrated fashion to biological response generation. Size and shape control helps to reduce the uncertainty about the influence these properties may have had in the analysis of the 24 commercial MOx nanoparticles from heterogeneous origin.25 All PdO/Co3O4 NPs were spherical (Figure 1a), and PdO functionlization did not change the shape (Figure 1b). Also, the crystallinity of Co3O4 NPs was retained, even at the highest level of PdO functionalization (Figure 2b). While metal dissolution is an important consideration in studying the biological effects of MOx, our ICP-OES data demonstrated very low rates (<0.1%, weight percent released metal ion compared to the whole nanoparticles) of Co and Pd shedding in cell culture medium (Figures 7a and S7a, Supporting Information). At this concentration, Co or Pd ions did not pose cytotoxic potential (Figures 7b and S6b, Supporting Information). ICP-OES and TEM analysis also confirmed that there were no differences in particle uptake in BEAS-2B and RAW 264.7 cells (Figure 7c and 7d and Figure S6c and S6d, Supporting Information). These results help to confirm that the incremental ROS production and oxidative stress in response to PdO/Co3O4 nanoparticles were due to differences in the energy properties compared to nondoped particles.
Conclusion
We investigated the role of Ec, Ev, and Ef energy in the generation of oxidative stress and inflammation by Co3O4 NPs using PdO doping. Through increasing the population density of heterojunctions on the surface of the doped NPs, alignment of the Ef levels could increase the free hole population in Co3O4, leading to a progressive increase in hydroxyl generation as well as a disruption of cellular redox equilibrium. The accompanying increase in oxidative stress generated a series of progressive cellular responses, including pro-inflammatory effects that manifested as acute inflammation in the lungs of exposed mice.
Acknowledgments
Primary support was provided by the US Public Health Service Grants, U19 ES019528 (UCLA Center for Nanobiology and Predictive Toxicology) and RO1 ES016746. The work also leveraged the infrastructure that is supported by the National Science Foundation and the Environmental Protection Agency under Cooperative Agreement Number DBI 0830117 and 1266377. S.P. amd L.M. would like to thank Prof. A. Rosenauer, Department of Solid State Physics, for the TEM imaging of the dry powder.
Supporting Information Available
Detailed information regarding high-resolution XPS, characterization of pure PdO nanoparticles, and cytotoxicity data in RAW 264.7 cell lines. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Contributions
◊ Contributed equally to this work
Supplementary Material
References
- Boro J.; Deka D.; Thakur A. J. Renewable Sustainable Energy Rev. 2012, 16, 904. [Google Scholar]
- Zhou W.; Wachs I. E.; Kiely C. J. Curr. Opin. Solid State Mater. Sci. 2012, 16, 10. [Google Scholar]
- Wetchakun K.; Samerjai T.; Tamaekong N.; Liewhiran C.; Siriwong C.; Kruefu V.; Wisitsoraat A.; Tuantranont A.; Phanichphant S. Sens. Actuators B: Chem. 2011, 160, 580. [Google Scholar]
- Puzyn T.; Rasulev B.; Gajewicz A.; Hu X.; Dasari T. P.; Michalkova A.; Hwang H.-M.; Toropov A.; Leszczynska D.; Leszczynski J. Nat. Nano 2011, 6, 175. [DOI] [PubMed] [Google Scholar]
- Antonini J. M.; Murthy G. G. K.; Rogers R. A.; Albert R.; Ulrich G. D.; Brain J. D. Toxicol. Appl. Pharmacol. 1996, 140, 188. [DOI] [PubMed] [Google Scholar]
- Xia T.; Kovochich M.; Liong M.; Mädler L.; Gilbert B.; Shi H. B.; Yeh J. I.; Zink J. I.; Nel A. E. ACS Nano 2008, 2, 2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W. S.; Duffin R.; Poland C. A.; Howie S. E. M.; MacNee W.; Bradley M.; Megson I. L.; Donaldson K. Environ. Health Perspect. 2010, 118, 1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S. L.; Duffin R.; Poland C.; Daly P.; Murphy F.; Drost E.; MacNee W.; Stone V.; Donaldson K. Environ. Health Perspect. 2009, 117, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdorster G.; Maynard A.; Donaldson K.; Castranova V.; Fitzpatrick J.; Ausman K.; Carter J.; Karn B.; Kreyling W.; Lai D.; Olin S.; Monteiro-Riviere N.; Warheit D.; Yang H.. ILSI Research Foundation/Risk Science Institute Nanomaterial Toxicity Screening Working Group, Part. Fibre Toxicol. 2005, 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushton E. K.; Jiang J.; Leonard S. S.; Eberly S.; Castranova V.; Biswas P.; Elder A.; Han X. L.; Gelein R.; Finkelstein J.; Oberdorster G. J. Toxicol. Environ. Health, Part A 2010, 73, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W. S.; Duffin R.; Thielbeer F; Bradley M.; Megson I. L.; Macnee W.; Poland C. A.; Tran C. L.; Donaldson K. Toxicol. Sci. 2012, 126, 469. [DOI] [PubMed] [Google Scholar]
- Karlsson H. L.; Gustafsson J.; Cronholm P.; Möller L. Toxicol. Lett. 2009, 188, 112. [DOI] [PubMed] [Google Scholar]
- Landsiedel R.; Ma-Hock L.; Kroll A.; Hahn D.; Schnekenburger J.; Wiench K.; Wohlleben W. Adv. Mater. 2010, 22, 2601. [DOI] [PubMed] [Google Scholar]
- Warheit D. B.; Reed K. L.; Sayes C. M. Inhalation Toxicol. 2009, 1, 61. [DOI] [PubMed] [Google Scholar]
- Sayes C.; Ivanov I. Risk Anal. 2010, 30, 1723. [DOI] [PubMed] [Google Scholar]
- George S.; Pokhrel S.; Ji Z. X.; Henderson B. L.; Xia T.; Li L. J.; Zink J. I.; Nel A. E.; Mädler L. J. Am. Chem. Soc. 2011, 133, 11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zhang W.; Niu J.; Chen Y. ACS Nano 2012, 6, 5164. [DOI] [PubMed] [Google Scholar]
- Hsiao I. L.; Huang Y.-J. Sci. Total Environ. 2011, 409, 1219. [DOI] [PubMed] [Google Scholar]
- Simon-Deckers A. l.; Loo S.; Mayne-L’hermite M.; Herlin-Boime N.; Menguy N.; Reynaud C. c.; Gouget B.; Carrière M. Environ. Sci. Technol. 2009, 43, 8423. [DOI] [PubMed] [Google Scholar]
- Cronholm P.; Karlsson H. L.; Hedberg J.; Lowe T. A.; Winnberg L.; Elihn K.; Wallinder I. O.; Möller L. Small 2013, 9, 970. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Oberdorster G.; Elder A.; Gelein R.; Mercer P.; Biswas P. Nanotoxicology 2008, 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayes C. M.; Wahi R.; Kurian P. A.; Liu Y.; West J. L.; Ausman K. D.; Warheit D. B.; Colvin V. L. Toxicol. Sci. 2006, 92, 174. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Dunphy D. R.; Jiang X.; Meng H.; Sun B.; Tarn D.; Xue M.; Wang X.; Lin S.; Ji Z.; Li R.; Garcia F. L.; Yang J.; Kirk M. L.; Xia T.; Zink J. I.; Nel A.; Brinker C. J. J. Am. Chem. Soc. 2012, 134, 15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Chen Y.; Zhang W.; Pu Z.; Jiang L.; Chen Y. Chem. Res. Toxicol. 2012, 25, 1675. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Ji Z.; Xia T.; Meng H.; Low-Kam C.; Liu R.; Pokhrel S.; Lin S.; Wang X.; Liao Y. P.; Wang M.; Li L.; Rallo R.; Damoiseaux R.; Telesca D.; Mädler L.; Cohen Y.; Zink J. I.; Nel A. E. ACS Nano 2012, 6, 4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumlee G. S.; Morman S. A.; Ziegler T. L. Rev. Mineral. Geochem. 2006, 64, 5. [Google Scholar]
- Burello E.; Worth A. P. Nanotoxicology 2011, 5, 228. [DOI] [PubMed] [Google Scholar]
- Liu R.; Zhang H. Y.; Ji Z. X.; Rallo R.; Xia T.; Chang C. H.; Nel A.; Cohen Y. Nanoscale 2013, 5, 5644. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Yates J. T. Chem. Rev. 2012, 112, 5520. [DOI] [PubMed] [Google Scholar]
- Xia T.; Zhao Y.; Sager T.; George S.; Pokhrel S.; Li N.; Schoenfeld D.; Meng H.; Lin S.; Wang X.; Wang M.; Ji Z.; Zink J. I.; Mädler L.; Castranova V.; Lin S.; Nel A. E. ACS Nano 2011, 5, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Wang D.; Han H.; Li C. Acc. Chem. Res. 2013, 46, 1900. [DOI] [PubMed] [Google Scholar]
- Mayer M. T.; Lin Y.; Yuan G.; Wang D. Acc. Chem. Res. 2013, 46, 1558. [DOI] [PubMed] [Google Scholar]
- Abdi F. F.; Han L.; Smets A. H. M.; Zeman M.; Dam B.; van de Krol R. Nat. Commun. 2013, 4, 2195. [DOI] [PubMed] [Google Scholar]
- Poizot P.; Laruelle S.; Grugeon S.; Dupont L.; Tarascon J. M. Nature 2000, 407, 496. [DOI] [PubMed] [Google Scholar]
- Svegl F.; Orel B.; Hutchins M. G.; Kalcher K. J. Electrochem. Soc. 1996, 143, 1532. [Google Scholar]
- Li W. Y.; Xu L. N.; Chen J. Adv. Funct. Mater. 2005, 15, 851. [Google Scholar]
- Yan X.; Feng D.; Heinbuch S.; Rocca J. J.; Bernstein E. R. Phys. Chem. Chem. Phys. 2010, 12, 947. [DOI] [PubMed] [Google Scholar]
- Shi J.; Zhu Y.; Zhang X.; Baeyens W. R. G.; García-Campaña A. M. TrAC Trends Anal. Chem. 2004, 23, 351. [Google Scholar]
- Minnermann M.; Grossmann H. K.; Pokhrel S.; Thiel K.; Hagelin-Weaver H.; Bäumer M.; Mädler L. Catal. Today 2013, 214, 90. [Google Scholar]
- Lang X.-Y.; Fu H.-Y.; Hou C.; Han G.-F.; Yang P.; Liu Y.-B.; Jiang Q. Nat Commun 2013, 4, 2169. [DOI] [PubMed] [Google Scholar]
- Walter M. G.; Warren E. L.; McKone J. R.; Boettcher S. W.; Mi Q. X.; Santori E. A.; Lewis N. S. Chem. Rev. 2010, 110, 6446. [DOI] [PubMed] [Google Scholar]
- Shim H.-W.; Jin Y.-H.; Seo S.-D.; Lee S.-H.; Kim D.-W. ACS Nano 2010, 5, 443. [DOI] [PubMed] [Google Scholar]
- George S.; Pokhrel S.; Xia T.; Gilbert B.; Ji Z.; Schowalter M.; Rosenauer A.; Damoiseaux R.; Bradley K. A.; Mädler L.; Nel A. E. ACS Nano 2010, 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teoh W. Y.; Amal R.; Mädler L. Nanoscale 2010, 2, 1324. [DOI] [PubMed] [Google Scholar]
- Greiner M. T.; Helander M. G.; Tang W.-M.; Wang Z.-B.; Qiu J.; Lu Z.-H. Nat. Mater. 2012, 11, 76. [DOI] [PubMed] [Google Scholar]
- Rushton E. K.; Jiang J.; Leonard S. S.; Eberly S.; Castranova V.; Biswas P.; Elder A.; Han X.; Gelein R.; Finkelstein J.; Oberdörster G. J. Toxicol. Environ. Health, Part A 2010, 73, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K.; Karasaki M.; Segawa M.; Hwang S. Y.; Tanaka T.; Ogino C.; Kondo A. MedChemComm 2010, 1, 209. [Google Scholar]
- Ji Z. X.; Jin X.; George S.; Xia T. A.; Meng H. A.; Wang X.; Suarez E.; Zhang H. Y.; Hoek E. M. V.; Godwin H.; Nel A. E.; Zink J. I. Environmental Science & Technology 2010, 44, 7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T.; Kovochich M.; Brant J.; Hotze M.; Sempf J.; Oberley T.; Sioutas C.; Yeh J. I.; Wiesner M. R.; Nel A. E. Nano Lett. 2006, 6, 1794. [DOI] [PubMed] [Google Scholar]
- Zhang H. Y.; Xia T.; Meng H.; Xue M.; George S.; Ji Z. X.; Wang X.; Liu R.; Wang M. Y.; France B.; Rallo R.; Damoiseaux R.; Cohen Y.; Bradley K. A.; Zink J. I.; Nel A. E. ACS Nano 2011, 5, 2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A.; Selfors L. M.; Forster T.; Wrobel D.; Kennedy C. J.; Shanks E.; Santoyo-Lopez J.; Dunican D. J.; Long A.; Kelleher D.; Smith Q.; Beijersbergen R. L.; Ghazal P.; Shamu C. E. Nat. Methods 2009, 6, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. D. Genomics 2007, 89, 552. [DOI] [PubMed] [Google Scholar]
- Zhang X. D. J. Biomol. Screen 2007, 12, 645. [DOI] [PubMed] [Google Scholar]
- Pokhrel S.; Nel A. E.; Mädler L. Acc. Chem. Res. 2012, 46, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L.; Peng Q.; Li Y. ChemCatChem 2011, 3, 868. [Google Scholar]
- Shannon R. D. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, A32, 751. [Google Scholar]
- Hoflund G. B.; Li Z. Appl. Surf. Sci. 2006, 253, 2830. [Google Scholar]
- Rogal J.; Reuter K.; Scheffler M. Phys. Rev. B 2004, 69. [Google Scholar]
- Burello E.; Worth A. P. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2011, 3, 298. [DOI] [PubMed] [Google Scholar]
- Brunet L. n.; Lyon D. Y.; Hotze E. M.; Alvarez P. J. J.; Wiesner M. R. Environ. Sci. Technol. 2009, 43, 4355. [DOI] [PubMed] [Google Scholar]
- Nel A.; Xia T.; Mädler L.; Li N. Science 2006, 311, 622. [DOI] [PubMed] [Google Scholar]
- McNeilly J. D.; Jimenez L. A.; Clay M. F.; MacNee W.; Howe A.; Heal M. R.; Beverland I. J.; Donaldson K. Toxicol. Lett. 2005, 158, 152. [DOI] [PubMed] [Google Scholar]
- Papis E.; Rossi F.; Raspanti M.; Dalle-Donne I.; Colombo G.; Milzani A.; Bernardini G.; Gornati R. Toxicol. Lett. 2009, 189, 253. [DOI] [PubMed] [Google Scholar]
- Cho W. S.; Duffin R.; Bradley M.; Megson I. L.; MacNee W.; Howie S. E. M.; Donaldson K. Eur. Respir. J. 2012, 39, 546. [DOI] [PubMed] [Google Scholar]
- Dong J.; Song L.; Yin J.-J.; He W.; Wu Y.; Gu N.; Zhang Y. ACS Appl. Mater. Interfaces 2014, 6, 1959. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.