

Abstract
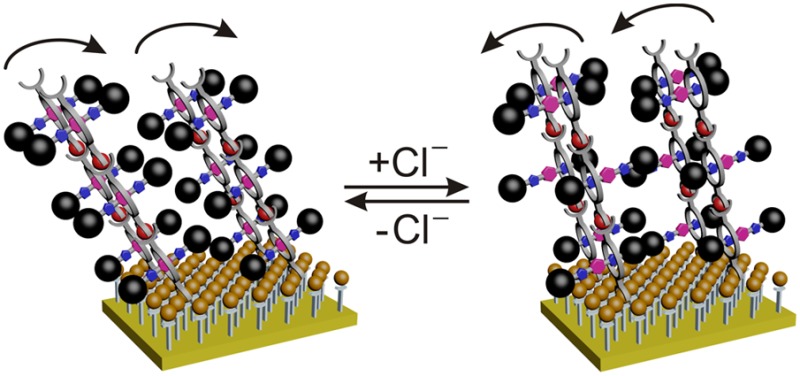
Interfaces provide the structural basis for function as, for example, encountered in nature in the membrane-embedded photosystem or in technology in solar cells. Synthetic functional multilayers of molecules cooperating in a coupled manner can be fabricated on surfaces through layer-by-layer self-assembly. Ordered arrays of stimulus-responsive rotaxanes undergoing well-controlled axle shuttling are excellent candidates for coupled mechanical motion. Such stimulus-responsive surfaces may help integrate synthetic molecular machines in larger systems exhibiting even macroscopic effects or generating mechanical work from chemical energy through cooperative action. The present work demonstrates the successful deposition of ordered mono- and multilayers of chemically switchable rotaxanes on gold surfaces. Rotaxane mono- and multilayers are shown to reversibly switch in a coupled manner between two ordered states as revealed by linear dichroism effects in angle-resolved NEXAFS spectra. Such a concerted switching process is observed only when the surfaces are well packed, while less densely packed surfaces lacking lateral order do not exhibit such effects.
Introduction
The idea of fabricating synthetic molecular machines1−5 has fascinated a whole generation of supramolecular chemists. Molecular switches,6−10 which undergo mechanical motion in response to chemical,11−14 photochemical,15,16 or electrochemical17 stimuli, represent excellent precursors for molecular machines. Especially, mechanically interlocked molecules18 such as rotaxanes have been investigated thoroughly in this respect.
In solution, molecular switches are randomly oriented, which makes the implementation of directional processes difficult. The still challenging integration of molecular switches and machines into larger ordered arrays and functional devices will help solve this problem. Inspired by nature’s use of membranes to generate order, chemists can employ soft interfaces such as Langmuir–Blodgett films19−25 or hard interfaces such as metal surfaces26−32 as alternatives to membranes to fabricate ordered arrays of synthetic molecular machines. The use of metal surfaces is particularly appealing, as the generation of self-assembled monolayers,33,34 layer-by-layer self-assembly,35,36 and the fabrication of surface-bound metal–organic frameworks (surMOFs)37−39 are well understood. Several seminal steps in this direction have been made,40,41 e.g., Leigh’s monolayers of axle-fluorinated, light-switchable [2]rotaxanes moving diiodomethane droplets uphill,42 Rapenne’s unidirectionally rotating surface-bound “piano-stool” rotors,43 Feringa’s surface-anchored overcrowded double bond motors,44 or Stoddart’s electrochemically switched cantilever-bending “molecular-muscle” [3]rotaxanes.45,46 In the latter example, the finding that a cantilever can be bent, when the rotaxanes are switched, suggests that the rotaxanes deposited on the cantilever operate in a directional way. This aspect, however, has so far not been further investigated in greater detail.
The aim of the present study is to provide evidence for a coupled motion of switchable interlocked molecules. The term coupled motion as we use it here refers to switching whole arrays of rotaxane shuttles that are aligned in a well-ordered state before switching and are again aligned in another well-ordered state after switching. For such motion, lateral order and the alignment of the deposited molecules are essential, and multilayers may enhance the achievable effects. Therefore, our study also aims to fabricate well-ordered multilayers of rotaxanes, in which a coupled motion can be observed. Here we describe novel ordered mono- and multilayers of chemically switchable amide rotaxanes on gold using either a Click chemistry approach to covalently attach them to an azide-terminated self-assembled monolayer (SAM) or a coordination-chemistry-based layer-by-layer self-assembly strategy. Evidence is provided for a preferred orientation of the rotaxanes in these layers. The rotaxane layers reversibly switch in a coupled manner between two differently ordered layer structures. The present study examines the importance of ordered arrays for coupled molecular motions in stimulus-responsive shuttle rotaxanes.
Results and Discussion
Synthesis of Macrocycles and Rotaxanes
Starting from suitably functionalized tetralactam macrocycles MCn,47 a templated one-step rotaxane synthesis has been achieved by efficiently trapping diketopiperazine 1 inside the wheels to form hydrogen-bonded pseudorotaxanes pRotn (Scheme 1; for experimental details, see the Supporting Information). The sterically demanding tritylphenyl stopper groups have been attached to the axle alkynes by a CuI-catalyzed “click” reaction with 2.6 For surface deposition, alkyne and terpyridine side chains have been introduced by Sonogashira48 and Suzuki49 cross-coupling reactions with (trimethylsilyl)acetylene and boronic acid pinacol ester 3, respectively. After trimethylsilyl (TMS) deprotection, Rot3 is ready to be clicked covalently to an azide-functionalized surface. Rot6 can be used for the deposition by metal-coordination-based layer-by-layer self-assembly. Macrocycle MC3 and singly terpyridine-substituted Rot4 serve as controls.
Scheme 1. Syntheses of the Macrocycles and Rotaxanes under Study.

Top: The axle threading reaction is templated by hydrogen-bond-mediated formation of pseudorotaxane pRotn, to which the two tritylphenyl azide stoppers are then “clicked”. Bottom: The acetylene and terpyridine side chains are attached to the iodo-substituted precursor rotaxanes by Sonogashira and Suzuki cross-coupling reactions, respectively.
Evidence for rotaxane formation comes from significant upfield shifts (Figure 1a,b) observed for the diketopiperazine protons Hd (Δδ = −1.01 ppm) and He (Δδ = −0.84 ppm), which are located inside the wheel’s cavity and experience the anisotropy of the surrounding aromatic rings. Also, the triazole proton Hf is affected by the presence of the wheel; the corresponding signal shifts to higher field by Δδ = −0.33 ppm.
Figure 1.
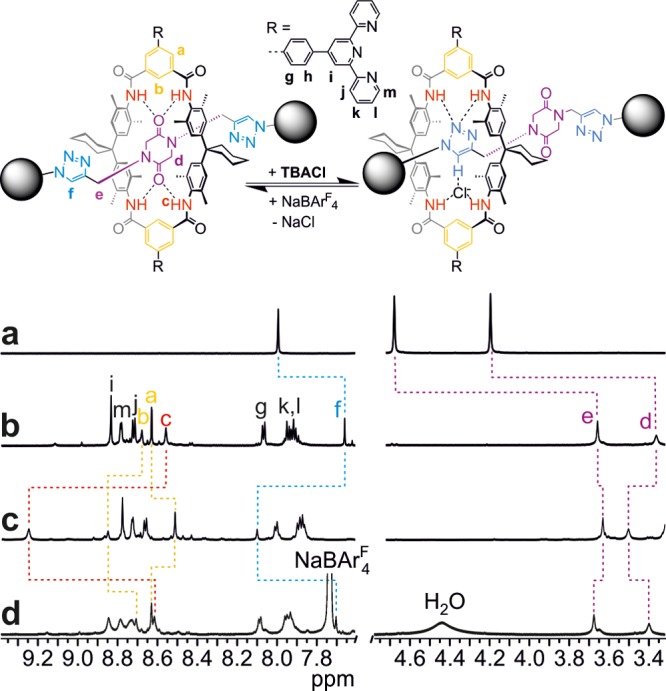
1H NMR data confirming rotaxane formation and chloride-mediated switching. Top: chloride-induced switching within rotaxane Rot6. Bottom: partial 1H NMR spectra (4 mM, CDCl3, 298 K) of (a) free, stoppered axle, (b) rotaxane Rot6 before addition of 1 equiv of tetrabutylammonium chloride ((TBA)Cl), (c) rotaxane Rot6 after addition of 1 equiv of (TBA)Cl, and (d) Rot6 after chloride removal by precipitation of NaCl with 1 equiv of sodium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (NaBArF4). Rotaxane formation is evidenced by characteristic shifts of axle signals d–f. The large signal shifts upon chloride addition and removal agree with those expected for the wheel translating from the central diketopiperazine station to one of the triazoles. The slight peak broadening and minor shift differences in spectrum d compared to spectrum b are due to the polarity change caused by the added salts.
Chemically Induced Switching of Rotaxanes in Solution
To gain insight into the switching process before transfer to the surface, a 1H NMR study of the chloride-induced axle movement of Rot6 has been performed in CDCl3 (Figure 1b–d). Clearly, chloride addition causes significant signal shifts, providing evidence for a wheel translocation from the central diketopiperazine to a triazole station, where the wheel binds mainly through C–H···Cl– interactions50,51 with the chloride hydrogen-bonded inside the wheel cavity.6,52 The downfield shifts of the signals (i) for the amide NH protons Hc (Δδ = 0.70 ppm) reflecting the change in hydrogen-bonding partner, (ii) for the triazole proton Hf (Δδ = 0.44 ppm), which moves deeper into the wheel, but at the same time interacts with the chloride anion, and (iii) for the diketopiperazine proton Hd (Δδ = 0.14 ppm), which moves somewhat away from the center of the wheel cavity, are most indicative. Also, the isophthaloyl diamide protons Ha and Hb and almost all other signals are clearly affected by the axle movement. The triazole binding motif should desymmetrize the axle as well as the wheel. As only one set of NMR signals is observed for either one, the wheel’s shuttling motion between the two triazoles and a concerted chloride flip/axle rotation both proceed fast on the NMR time scale. Removal of the chloride anion by precipitation with NaBArF yields the same spectrum as obtained prior to the switching cycle with the exception of the additional signals for the BArF anions. Consequently, the stimulus-induced structural changes in the rotaxane can be fully reversed. Analogous results have also been obtained with the diiodo-substituted precursor rotaxane Rot5 (Supporting Information, Figure S14).
Mono- and Multilayer Formation
For the deposition of a monolayer of Rot3 (Figure 2a), an azide-functionalized self-assembled monolayer (SAM) was generated by reacting a HCl-cleaned gold substrate with a 1 mM ethanol solution of bis(11-azidoundecyl) disulfide (AUD) for 24 h.53 Subsequently, the acetylene-functionalized rotaxane Rot3 was attached covalently through a click reaction catalyzed by the CuI/N-heterocyclic carbene complex Cat.54 This catalyst exhibits excellent activity in dichloromethane, a solvent that needs to be used because of the limited solubility of the rotaxane in other solvents.
Figure 2.
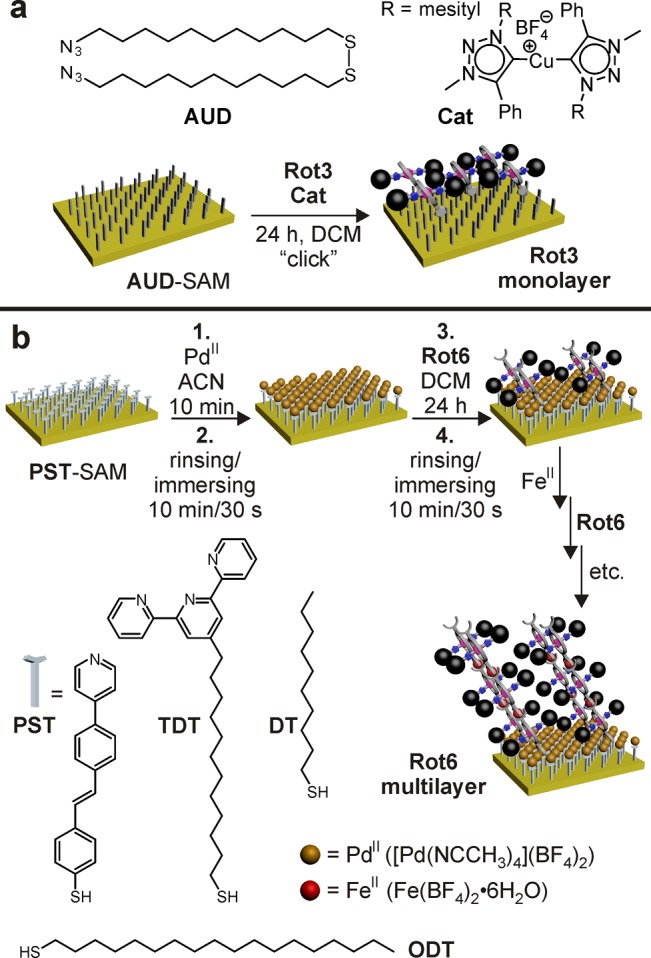
Fabrication of mono- and multilayers of switchable rotaxanes. (a) Monolayers of covalently fixed Rot3 are made by first depositing an azide-terminated SAM of AUD from ethanol solution onto a HCl-cleaned gold surface. Afterward, acetylene-substituted rotaxane Rot3 is “clicked” to the azides. (b) On a PST SAM, first PdII ions are deposited from acetonitrile and mediate the connection to the first layer of Rot6 deposited from dichloromethane solution. Subsequently, this monolayer can be converted into multilayers by an alternating deposition of FeII ions and Rot6. Similarly, a 1:3 mixed monolayer of TDT and DT can serve as a templating SAM with FeII ions connecting the next layer to it.
Figure 2b summarizes the formation of mono- and multilayers of Rot6 by coordination-based layer-by-layer self-assembly on a templating, rigid pyridine-functionalized SAM of (E)-4-(pyridin-4-yl)stilbenethiol (PST).55−57 Square-planar PdII ions connect the SAM pyridines with the rotaxane terpyridines. A 10 min metal ion deposition from a 1 mM acetonitrile solution of [Pd(NCCH3)4](BF4)2 was followed by the deposition of a monolayer of the rotaxane from a 1 mM dichloromethane solution of Rot6 which was given 24 h to assemble into an ordered monolayer array. As this Rot6 monolayer is again terminated by terpyridyl side chains, multilayers can be fabricated following this procedure repeatedly, now using FeII ions to connect the two terpyridines of adjacent rotaxanes. Alternating deposition steps with Fe(BF4)2·6H2O as the metal ion source and Rot6 thus led to a growing multilayer.
Alternatively, a mixed template SAM prepared from a 1:3 mixture of terpyridine-terminated dodecanethiol (TDT) and decanethiol (DT) (Figure 2b) can be used as the basis for multilayer growth (Supporting Information, Figures S24–S27).58 This ratio is the optimum for metal deposition, and phase segregation of the two components was ruled out earlier.59 The DT decyl chain is shorter by two carbon atoms as compared to the TDT dodecyl chain, resulting in well-ordered alkyl chains, yet flexible, unordered terpyridine groups that are exposed on top of the SAM.59 The fabrication of the TDT/DT-supported Rot6 multilayer follows the same procedure as that deposited on PST. As this SAM bears terpyridine groups as the anchors for the next layer, FeII is also used as the first metal ion layer.
To allow imaging of the deposition process, not only fully covered surfaces but also surfaces structured laterally by microcontact printing (μCP)60,61 have been produced. These surfaces have the advantage that atomic force microscopy (AFM) can be used to determine layer thicknesses by comparing the heights of the growing multilayer with the constant height of the areas passivated by a simple alkanethiol. Poly(dimethylsiloxane) stamps patterned with 10 μm wide dots spaced at 5 μm distances were inked with octadecanethiol (ODT) and then brought into contact with a freshly cleaned gold surface. Afterward, the space between the dots was backfilled with PST, followed by formation of the Rot6 multilayer as described above.
Characterization of Mono- and Multilayers
The characterization of the surface films is achieved by combining data from X-ray photoelectron spectroscopy (XPS), transmission UV/vis spectroscopy, AFM, time-of-flight secondary ion mass spectrometric (ToF-SIMS) imaging, and near-edge X-ray absorption fine structure (NEXAFS) spectroscopy. Figures 3 and 4 summarize relevant data (also see the Supporting Information).
Figure 3.
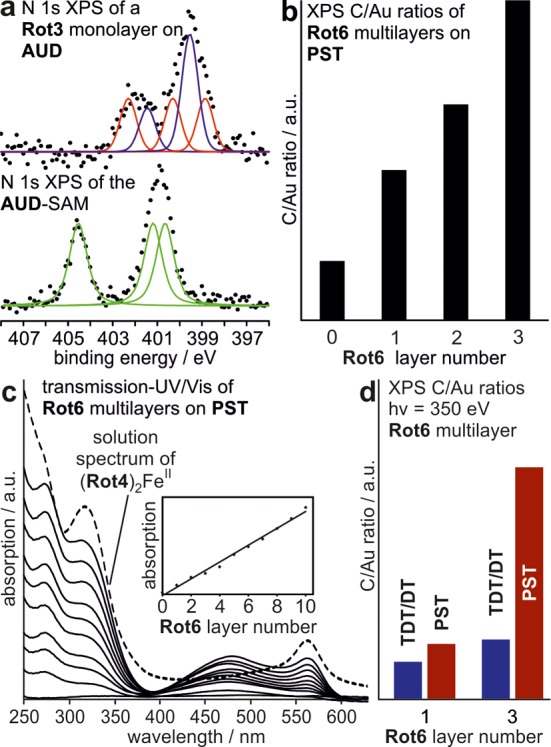
Characterization of rotaxane mono- and multilayers. (a) A comparison of the high-resolution N 1s XP spectra (excitation energy: 500 eV) of the AUD SAM before and after click deposition of Rot3 reveals conversion of the azide (green signals) to the triazole (red) superimposed by the amide and diketopiperazine N atoms of the rotaxane (blue). (b) XPS C 1s/Au 4f7/2 ratios increase with each deposited Rot6 layer. (c) Transmission UV/vis spectra of 0–10 Rot6 layers on PST/semitransparent gold. Dotted line: solution spectrum of (Rot4)2FeII. Inset: absorption at 270 nm showing a linear layer growth. (d) Relative increase of the C 1s/Au 4f7/2 ratios upon deposition of Rot6 on different templating SAMs (PST vs TDT/DT, 1:3).
Figure 4.
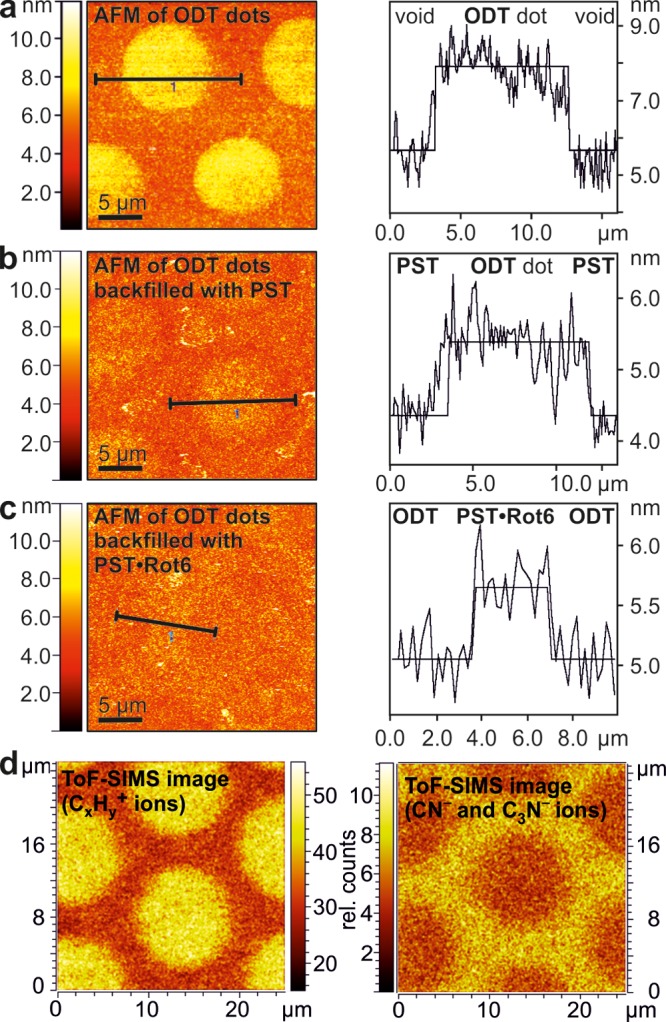
AFM images and height profiles of micropatterned surfaces functionalized (a) with ODT dots between unfilled voids, (b) with ODT dots and voids backfilled with PST, and (c) with ODT dots and voids backfilled with PST and one Rot6 layer. (d) ToF-SIMS images of the surface backfilled with PST and Rot6. Left: secondary ion intensities of CxHy+ ions indicative of ODT alkyl chains. Right: secondary ion intensities of CN– and C3N– ions indicative of the presence of pyridines.
In line with earlier results,62 the AUD SAM exhibits three signals in the high-resolution N 1s XP spectrum (Figure 3a) which can be assigned to the three azide nitrogen atoms. After the click reaction with Rot3, three signals appear shifted to those binding energies expected for the triazole nitrogen atoms. In addition, a more intense signal for the secondary amides of the macrocycle and a smaller signal for the tertiary amides of the diketopiperazine axle appear. NEXAFS spectra support this (Supporting Information, Figure S17).
Multilayer growth of Rot6 on the PST SAM can be followed by transmission UV/vis, when the multilayer is deposited on a semitransparent gold surface (Figure 3c). All bands observed in the solution spectrum of the (Rot4)2FeII control complex are also found for the rotaxanes deposited on the surface: the metal-to-ligand charge transfer (MLCT) band at ca. 560 nm, the ligand-centered band at ca. 340 nm, and the π–π* transition of the aromatic rings at ca. 270 nm. In addition, the surface plasmon band appears at around 470 nm and, as expected, shifts slightly to higher wavelengths with increasing layer thickness. A plot of the intensity at 270 nm over the rotaxane layer number clearly demonstrates a linear growth from which we conclude that the same amount of Rot6 is added in each rotaxane deposition step. No self-propagating growth is observed.57,63−66 The carbon/gold ratio extracted from the C 1s and Au 4f7/2 XPS data (Figure 3b) increases with each deposition of the rotaxane, because the number of carbon atoms in the organic layer increases and the gold signal is more attenuated at the same time caused by inelastic scattering of the photoelectrons emitted from the gold surface at the organic layers above.
A comparison of the XPS C 1s/Au 4f7/2 ratios of the Rot6 single and triple layers on PST with the same layers on TDT/DT (1:3) reveals significantly more rotaxane deposited in each layer on the PST SAM (Figure 3d). This is in marked contrast to the formation of densely packed multilayers of MC3 on TDT/DT.58 The introduction of the axle with its two bulky stopper groups increases the size of rotaxane Rot6 significantly compared to that of MC3. Thus, it does not match the grid of the terpyridine anchor points on the TDT/DT monolayer as nicely as MC3. The smaller terminal pyridines of the PST SAM offer a finer grid of coordination sites that allows Rot6 to assemble laterally more easily and to form layers of higher density.
When a micropatterned surface was examined by AFM directly after deposition of the ODT dots, a clear contrast between the ODT-passivated dots and the void space between them was observed, which translates into a layer thickness of the ODT monolayer of 1.94 nm (Figure 4a). After the space between the passivated dots is backfilled with one layer of Rot6 on PST, the contrast reverses as the rotaxane layer is now higher than the ODT layer (Figure 4b). The thickness of one rotaxane layer can be determined from Figure 4b (voids backfilled with PST alone) and Figure 4c (voids backfilled with PST and Rot6) to be ca. 1.6 nm (Supporting Information, Figure S32). This value nicely corresponds to thicknesses of 1.5–1.8 nm determined earlier for layers of macrocycle MC3.58 As the Fe–Fe distance calculated for MC3 and Rot6 oligomers is ca. 3.5 nm, the tilt angle between the rotaxane wheel and the surface can be estimated to be ca. 30°.58
The distribution of ODT and Rot6 on the micropatterned surface was also imaged by ToF-SIMS. A 25 × 25 μm2 area of the surface was scanned with 256 × 256 pixel resolution by a focused beam of Bi32+ ions using the novel “collimated burst alignment” mode for high lateral as well as high mass resolution.67 In the positive mode, the CxHy+ secondary ions characteristic for the ODT alkyl chains preferentially originate from the dot areas (Figure 4d, left). Instead, the areas covered by the PST-based multilayer of Rot6 do not contain any linear alkyl chains and produce only a very small CxHy+ ion count. Vice versa, CN– and C3N– secondary ions in the negative mode confirm the presence of pyridines and thus of Rot6 in the areas between the ODT dots (Figure 4d, right). Notably, these ions are absent in the ODT areas, indicating an excellent specificity of binding Rot6 to the PST areas.
Investigation of Chemically Induced On-Surface Switching Processes
Contact angle measurements upon repeated chloride addition and removal indicate a strong and reversible change of polarity of the rotaxane multilayers which is tentatively attributed to on-surface chloride-mediated switching (Figure 5a). When the AUD–Rot3 monolayer and a PST–Rot6 multilayer are treated with a 1 mM dichloromethane (DCM) solution of tetrabutylammonium chloride ((TBA)Cl) and then immersed for 10 min in pure dichloromethane to remove residual salt, the contact angles drop by almost 20°. This indicates a significantly higher polarity after chloride addition. Removal of the chloride with NaBArF4 (1 mM, DCM) and subsequent washing with DCM, water, and DCM again to remove salt residues bring the contact angles back to the initial values, and switching can be repeated. One might argue that the deposition of a salt is expected to increase polarity, even if it is not specifically bound and does not switch the rotaxane. Therefore, a macrocycle MC3 multilayer was used as a control, and this exhibited much smaller contact angle changes (ca. 5°). Furthermore, the contact angle changes were not affected significantly when the less lipophilic tetramethylammonium chloride ((TMA)Cl) was applied.
Figure 5.
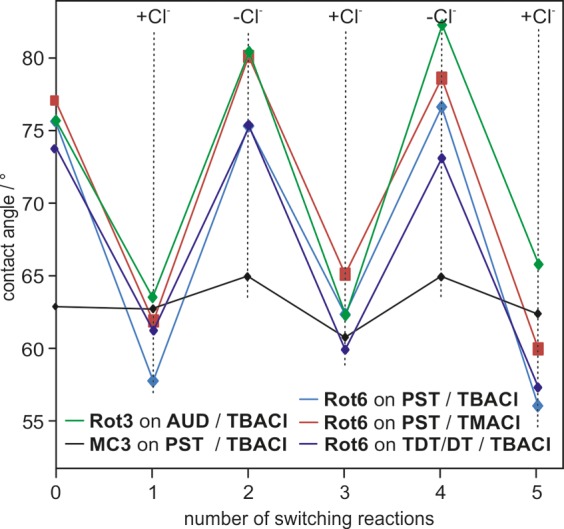
Investigation of the on-surface switching via contact angle measurements for the AUD–Rot3 monolayer and PST– and TDT/DT (1:3)–Rot6 multilayers (20 layers) over five switching steps. For comparison, an MC3 multilayer (20 layers) is examined.
These results are an indication of, but certainly not strict evidence for, on-surface rotaxane switching. Angle-resolved NEXAFS experiments provide evidence for stimulus-induced structural changes within the surface: As synchrotron light is linearly polarized, linear dichroism effects are observed when the deposited molecules have a preferred orientation.68 The transition dipole moment vectors for the excitation of core electrons into the π* (aromatic carbons) or C–H* orbitals (aliphatic carbons) line up with the electric field vector of the incident light differently at different angles, and angle-dependent intensities for the π* and C–H* resonances result. Before we discuss the NEXAFS results shown in Figures 6 and 7, let us briefly mention the following limitation: It would be very desirable to be able to analyze the NEXAFS data in much greater detail to correlate the linear dichroism effects with the details of the structures and the orientation of the rotaxanes on the surface. While this is certainly possible for simpler self-assembled monolayers, it is extremely challenging for complicated molecules such as the rotaxanes under study. Let us take the π* resonance as an example, as this is the most important one in our paper: The linear dichroism depends on the orientation of aromatic rings in the mono- and multilayers. The macrocyclic wheels are rather rigid, but contain aromatic rings in many different orientations, even when the macrocycles are fixed in a certain orientation. If one includes the axle with the trityl stoppers, additional orientations of aromatic rings come into play. Consequently, many different dichroism effects finally are superimposed, resulting in a net effect, which is typically not very large as we have experienced several times in the past for other surfaces.55,57,58 Despite this limitation, the observation of such a linear dichroism clearly indicates the rotaxanes to be ordered into arrays with preferred orientations.
Figure 6.

Evidence for on-surface switching from angle-resolved NEXAFS spectroscopy. Angle-resolved NEXAFS spectra of (a) a Rot3 monolayer on AUD, (b) a Rot6 monolayer on PST (left, (TBA)Cl; right, (TMA)Cl), and (c) a Rot6 monolayer on TDT/DT (1:3). In each case, the NEXAFS results obtained for the pristine layer, that after chloride addition, and that after chloride removal with NaBArF4 are shown (top to bottom). Red and black lines represent the NEXAFS spectra obtained at 30° and 90° angles of the incident synchrotron light beam, respectively. Difference spectra are shown in green.
Figure 7.
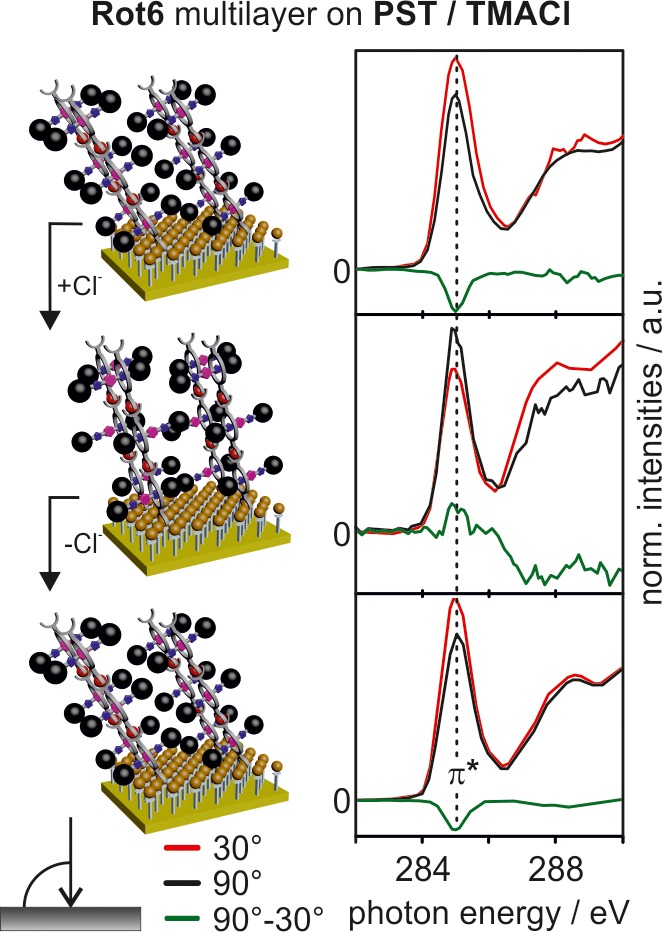
Angle-resolved NEXAFS spectra of an Rot6 triple layer. Similar to the results in Figure 7, the NEXAFS results obtained for the pristine layer, that after chloride addition, and that after chloride removal with NaBArF4 are shown (top to bottom). Red and black lines represent the NEXAFS spectra obtained at 30° and 90° angles of the incident synchrotron light beam, respectively. Difference spectra are shown in green.
The pristine monolayer of Rot3 on AUD (Figure 6a) exhibits a linear dichroism on the π* resonance—clear evidence for a preferred orientation of the rotaxane on this surface. After chloride addition, this angle dependence vanished almost completely. Consequently, either the switched rotaxanes may be disordered after switching or the dichroism effects almost exactly cancel. Chloride binding to the rotaxane thus induces a structural change, while the question of whether this is accompanied by a loss of orientation cannot clearly be answered. As neither the underlying AUD alkyl chains nor the tetrabutylammonium counterions contain π systems and thus do not affect the π* resonance, the structural change must have occurred within the rotaxane structure, and we ascribe it to the chloride-induced switching of the rotaxane. Interestingly, chloride removal with NaBArF4 leads back to angle-resolved NEXAFS spectra very similar to the initial ones. Consequently, switching the rotaxanes back into the chloride-free state yields an ordered structure similar to the initial one. The reversibility of the structural changes strongly supports rotaxane switching to be the origin of the linear dichroism changes.
The monolayer of Rot6 on PST (Figure 6b) again exhibits a linear dichroism, indicating a preferred orientation of the rotaxanes. In contrast to the Rot3 monolayer, the linear dichroism became even more pronounced upon chloride addition. Chloride removal led back to NEXAFS spectra similar to the initial ones. Clearly, this surface can be switched between two different structures, which both contain the rotaxanes in a preferred orientation. As a control, the PST–Rot6 monolayer has also been tested with the significantly larger tetrabutylammonium counterion. The results are virtually identical—indicating that the counterion is not the primary origin of the structural changes.
In marked contrast, the TDT/DT–Rot6 monolayer exhibits only negligible linear dichroism effects before and after switching (Figure 6c). This observation is interesting as this surface was found to be less densely packed (see above, Figure 3d). Even if the individual, nonordered rotaxanes deposited on the TDT/DT SAM undergo chloride-induced switching, no coupled axle movement is observed that transfers an ordered structure into another ordered structure. In turn, we conclude the reversibly switchable transition of one ordered layer structure of Rot6 on PST into a second, yet different ordered structure as a clear indication that the densely packed Rot6 layer on PST switches in a coupled manner.
Finally, the triple layer of Rot6 on PST again exhibits linear dichroism effects on the π* resonance that change with chloride addition and removal (Figure 7). The π* resonance of the initial triple layer is more intense at a 30° angle of the incident light. After chloride addition, the more intense π* resonance appears at 90° instead. Backswitching into the initial state occurs upon chloride removal. The concept of concerted switching observed for the PST–Rot6 monolayer is consequently transferable to multilayers of Rot6 on PST as well.
In principle, one might envisage another mechanism by which the transition between two ordered surfaces could be accomplished at least for those surfaces that are fabricated using the metal coordination approach: The rotaxanes on the surface might dissociate from the surface, switch, and redeposit in a different packing. This scenario can, however, be ruled out on the basis of the following arguments: (i) In those cases in which the metal ion coordination is clearly reversible (e.g., when Zn(II) is used to connect two terpyridines), no continuous layer growth is observed.58 (ii) The mono- and multilayers of macrocycles have been tested extensively, even against alkaline EDTA solutions for stability, and have not shown any degradation overnight.58 This quite high stability agrees well with our assumption of densely packed surfaces in which lateral interactions between the rotaxanes help keep them in place.
Conclusions
Amide rotaxanes that exhibit axle translocation in the wheel between two different stations in response to the presence of chloride have been synthesized and effectively deposited on gold substrates. Two approaches yielded monolayers: covalent attachment through a click reaction and a coordination-chemistry-based approach. The latter monolayer formed the basis for multilayer fabrication leading to up to 20 rotaxane layers.
Chloride-induced axle movement has successfully been transferred from the solution to the surface. Linear dichroism effects observed in angle-resolved NEXAFS experiments not only provide evidence for oriented surface-deposited rotaxanes, but clearly demonstrate switching of single and triple layers between two different layer structures. A number of control experiments and the comparison of different layers allow us to ascribe the structural changes to the rotaxane switching rather than other potential sources for structural changes.
This study represents a structural examination of ordered rotaxane multilayers on a solid support that exhibits coupled transitions between two different structures by stimulus-induced mechanical motion. In particular, the almost complete absence of linear dichroism effects in the switching experiments performed with the TDT/DT-based surfaces shows how important order is for a structurally well-defined switching between two different orientations. A dense packing of the rotaxanes under study in well-ordered layers results in limited degrees of freedom for the axles of each rotaxane through spatial constraints caused by the next neighbors. A perfectly simultaneous switching of all rotaxanes in a domain is unlikely, because the overall barrier would be expected to be very high. Therefore, we propose a nucleation/growth mechanism for the switching process: The switching of the first rotaxane is quite unfavorable, as the unswitched state packs well, whereas the switched one does not in an environment of unswitched neighbors. Therefore, a single switched unit may switch back quickly. Once the nucleus of a small ensemble of switched rotaxanes has formed, neighboring rotaxanes will switch faster, as the switched state now packs well when embedded within switched neighbors. This leads to a quicker growth of the switched domain.
A detailed understanding of the factors that govern these processes will have strong implications for the future integration of molecular machines into larger devices. Also, the conversion of chemical processes on the molecular scale into macroscopic effects may be envisaged on the basis of our study, if one, for example, considers volume changes caused by rotaxane switches tilting up in a coupled fashion.
Acknowledgments
We thank the Deutsche Forschungsgemeinschaft (Grants SCHA 893/9-1 and UN 80/8-1) and the Freie Universität Berlin for financial support. S.R. thanks the Fonds der Chemischen Industrie for a Chemiefonds Ph.D. fellowship. M.H. is grateful for financial support by the Austrian Science Fund (FWF) through the Erwin-Schrödinger fellowship program (Project J 3471-N28). S.O.K. and J.H. acknowledge the Council for Chemical Sciences of The Netherlands Organisation for Scientific Research (NWO-CW, Vici Grant 700.58.443 to J.H.). We are grateful to Dieter Treu and Dr. Thomas Gross for operating the XPS instrument at BAM 6.8. Support by Andreas Lippitz, Dr. Alexei Nefedov, and the staff at BESSY II and the HE-SGM CRG Beamline is gratefully acknowledged. We thank Dr. Stephan Hohloch and Prof. Biprajit Sarkar for providing the CuI/N-heterocyclic carbene catalyst Cat used for the click reaction on the AUD surface.
Supporting Information Available
Synthetic details, compound characterization data, multilayer fabrication procedures, and additional surface characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Coskun A.; Banaszak M.; Astumian R. D.; Stoddart J. F.; Grzybowski B. A. Chem. Soc. Rev. 2012, 41, 19–30. [DOI] [PubMed] [Google Scholar]
- Fahrenbach A. C.; Warren S. C.; Incorvati J. T.; Avestro A.-J.; Barnes J. C.; Stoddart J. F.; Grzybowski B. A. Adv. Mater. 2013, 25, 331–348. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Credi A.; Raymo F. M.; Stoddart J. F. Angew. Chem., Int. Ed. 2000, 39, 3348–3391. [DOI] [PubMed] [Google Scholar]
- Kay E. R.; Leigh D. A.; Zerbetto F. Angew. Chem., Int. Ed. 2007, 46, 72–191. [DOI] [PubMed] [Google Scholar]
- Browne W. R.; Feringa B. L. Nat. Nanotechnol. 2006, 1, 25–36. [DOI] [PubMed] [Google Scholar]
- Dzyuba E. V.; Kaufmann L.; Löw N. L.; Meyer A. K.; Winkler H. D. F.; Rissanen K.; Schalley C. A. Org. Lett. 2011, 13, 4838–4841. [DOI] [PubMed] [Google Scholar]
- Li H.; Fahrenbach A. C.; Coskun A.; Zhu Z.; Barin G.; Zhao Y.-L.; Botros Y. Y.; Sauvage J.-P.; Stoddart J. F. Angew. Chem., Int. Ed. 2011, 50, 6782–6788. [DOI] [PubMed] [Google Scholar]
- Gassensmith J. J.; Barr L.; Baumes J. M.; Paek A.; Nguyen A.; Smith B. D. Org. Lett. 2008, 10, 3343–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski B.; De Bo G.; Ward J. W.; Papmeyer M.; Kuschel S.; Aldegunde M. J.; Gramlich P. M.; Heckmann D.; Goldup S. M.; D’Souza D. M.; Fernandes A. E.; Leigh D. A. Science 2013, 339, 189–193. [DOI] [PubMed] [Google Scholar]
- Hernandez J. V.; Kay E. R.; Leigh D. A. Science 2004, 306, 1532–1537. [DOI] [PubMed] [Google Scholar]
- Carlone A.; Goldup S. M.; Lebrasseur N.; Leigh D. A.; Wilson A. J. Am. Chem. Soc. 2012, 134, 8321–8323. [DOI] [PubMed] [Google Scholar]
- Badjic J. D.; Ronconi C. M.; Stoddart J. F.; Balzani V.; Silvi S.; Credi A. J. Am. Chem. Soc. 2006, 128, 1489–1499. [DOI] [PubMed] [Google Scholar]
- Yoon H. J.; Kuwabara J.; Kim J.-H.; Mirkin C. A. Science 2010, 330, 66–99. [DOI] [PubMed] [Google Scholar]
- Yoon H. J.; Mirkin C. A. J. Am. Chem. Soc. 2008, 130, 11590–11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avellini T.; Li H.; Coskun A.; Barin G.; Trabolsi A.; Basuray A. N.; Dey S. K.; Credi A.; Silvi S.; Stoddart J. F.; Venturi M. Angew. Chem., Int. Ed. 2012, 51, 1611–1615. [DOI] [PubMed] [Google Scholar]
- Koçer A.; Walko M.; Meijberg W.; Feringa B.-L. Science 2005, 309, 755–758. [DOI] [PubMed] [Google Scholar]
- Ye T.; Kumar A. S.; Saha S.; Takami T.; Huang T. J.; Stoddart J. F.; Weiss P. S. ACS Nano 2010, 4, 3697–3701. [DOI] [PubMed] [Google Scholar]
- Molecular Catenanes, Rotaxanes and Knots: A Journey through the World of Molecular Topology; Sauvage J.-P., Dietrich-Buchecker C., Eds.; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Clemente-León M.; Credi A.; Martínez-Díaz M.-V.; Mingotaud C.; Stoddart J. F. Adv. Mater. 2006, 18, 1291–1296. [Google Scholar]
- Lee I. C.; Frank C. W.; Yamamoto T.; Tseng H.-R.; Flood A. H.; Stoddart J. F. Langmuir 2004, 20, 5809–5828. [DOI] [PubMed] [Google Scholar]
- Huang T. J.; Tseng H.-R.; Sha L.; Lu W.; Brough B.; Flood A. H.; Yu B.-D.; Celestre P. C.; Chang J. P.; Stoddart J. F.; Ho C.-M. Nano Lett. 2004, 11, 2065–2071. [Google Scholar]
- Nørgaard K.; Laursen B. W.; Nygaard S.; Kjaer K.; Tseng H.-R.; Flood A. H.; Stoddart J. F.; Bjørnholm T. Angew. Chem., Int. Ed. 2005, 44, 7035–7039. [DOI] [PubMed] [Google Scholar]
- Nørgaard K.; Jeppesen J. O.; Laursen B. W.; Simonsen J. B.; Weygand M. J.; Kjaer K.; Stoddart J. F.; Bjørnholm T. J. Phys. Chem. B 2005, 109, 1063–1066. [DOI] [PubMed] [Google Scholar]
- Jang S. S.; Jang Y. H.; Kim Y.-H.; Goddart W. A. III; Choi J. W.; Heath J. R.; Laursen B. W.; Flood A. H.; Stoddart J. F.; Nørgaard K.; Bjørnholm T. J. Am. Chem. Soc. 2005, 127, 14804–14816. [DOI] [PubMed] [Google Scholar]
- DeIonno E.; Tseng H.-R.; Harvey D. D.; Stoddart J. F.; Heath J. R. J. Phys. Chem. B 2006, 110, 7609–7612. [DOI] [PubMed] [Google Scholar]
- Lehr J.; Lang T.; Blackburn O. A.; Barendt T. A.; Faulkner S.; Davis Jason J.; Beer P. D. Chem.—Eur. J. 2013, 19, 15898–15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J. J.; Orlowski G. A.; Rahman H.; Beer P. D. Chem. Commun. 2010, 46, 54–63. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Sun Y.-L.; Song N. Acc. Chem. Res. 2014, 47, 1950–1960. [DOI] [PubMed] [Google Scholar]
- Tseng H.-R.; Wu D.; Fang N. X.; Zhang X.; Stoddart J. F. ChemPhysChem 2004, 5, 111–116. [DOI] [PubMed] [Google Scholar]
- Jang S. S.; Jang Y. H.; Kim Y.-H.; Goddard W. A. III; Flood A. H.; Laursen B. W.; Tseng H.-R.; Stoddart J. F.; Jeppesen J. O.; Choi J. W.; Steuerman D. W.; DeIonno E.; Heath J. R. J. Am. Chem. Soc. 2005, 127, 1563–1575. [DOI] [PubMed] [Google Scholar]
- Klajn R.; Fang L.; Coskun A.; Olson M. A.; Wesson P. J.; Stoddart J. F.; Grzybowski B. A. J. Am. Chem. Soc. 2009, 131, 4233–4235. [DOI] [PubMed] [Google Scholar]
- Coskun A.; Wesson P. J.; Klajn R.; Trabolsi A.; Fang L.; Olson M. A.; Dey S. K.; Grzybowski B. A.; Stoddart J. F. J. Am. Chem. Soc. 2010, 132, 4310–4320. [DOI] [PubMed] [Google Scholar]
- Ulman A. Chem. Rev. 1996, 96, 1533–1554. [DOI] [PubMed] [Google Scholar]
- Vericat C.; Vela M. E.; Benitez G.; Carro P.; Salvarezza R. C. Chem. Soc. Rev. 2010, 39, 1805–1834. [DOI] [PubMed] [Google Scholar]
- Rubinstein I.; Vaskevich A. Isr. J. Chem. 2010, 50, 333–346. [Google Scholar]
- Ariga K.; Hill J. P.; Ji Q. Chem. Phys. Phys. Chem. 2007, 9, 2319–2340. [DOI] [PubMed] [Google Scholar]
- Zacher D.; Shekhah O.; Wöll C.; Fischer R. A. Chem. Soc. Rev. 2009, 38, 1418–1429. [DOI] [PubMed] [Google Scholar]
- Zacher D.; Schmid R.; Wöll C.; Fischer R. A. Angew. Chem., Int. Ed. 2011, 50, 176–199. [DOI] [PubMed] [Google Scholar]
- Liu B.; Shekhah O.; Arslan H. K.; Liu J.; Wöll C.; Fischer R. A. Angew. Chem., Int. Ed. 2012, 51, 807–810. [DOI] [PubMed] [Google Scholar]
- Coronado E.; Gaviña P.; Tatay S. Chem. Soc. Rev. 2009, 38, 1674–1689. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Credi A.; Venturi M. ChemPhysChem 2008, 9, 202–220. [DOI] [PubMed] [Google Scholar]
- Berná J.; Leigh D. A.; Lubomska M.; Mendoza S. M.; Pérez E. M.; Rudolf P.; Teobaldi G.; Zerbetto F. Nat. Mater. 2005, 4, 704–710. [DOI] [PubMed] [Google Scholar]
- Perera U. G. E.; Ample F.; Kersell H.; Zhang Y.; Vives G.; Echeverria J.; Grisolia M.; Rapenne G.; Joachim C.; Hla S. W. Nat. Nanotechnol. 2013, 8, 46–51. [DOI] [PubMed] [Google Scholar]
- van Delden R. A.; ter Wiel M. K. J.; Pollard M. M.; Vicario J.; Koumura N.; Feringa B. L. Nature 2005, 437, 1337–1340. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Flood A. H.; Bonvallet P. A.; Vignon S. A.; Northrop B. H.; Tseng H.-R.; Jeppesen J. O.; Huang T. J.; Brough B.; Baller M.; Magonov S.; Solares S. D.; Goddard W. A.; Ho C.-M.; Stoddart J. F. J. Am. Chem. Soc. 2005, 127, 9745–9759. [DOI] [PubMed] [Google Scholar]
- Juluri B. K.; Kumar A. S.; Liu Y.; Ye T.; Yang Y.-W.; Flood A. H.; Fang L.; Stoddart J. F.; Weiss P. S.; Huang T. J. ACS Nano 2009, 3, 291–300. [DOI] [PubMed] [Google Scholar]
- Baytekin B.; Zhu S. S.; Brusilowskij B.; Illigen J.; Ranta J.; Huuskonen J.; Russo L.; Rissanen K.; Kaufmann L.; Schalley C. A. Chem.—Eur. J. 2008, 14, 10012–10028. [DOI] [PubMed] [Google Scholar]
- Pirali T.; Gatti S.; Di Brisco R.; Tacchi S.; Zaninetti R.; Brunelli E.; Massarotti A.; Sorba G.; Canonico P. L.; Moro L.; Genazzani A. A.; Tron G. C.; Billington R. A. ChemMedChem 2007, 2, 437–440. [DOI] [PubMed] [Google Scholar]
- Han F. S.; Higuchi M.; Kurth D. G. Org. Lett. 2007, 9, 559–562. [DOI] [PubMed] [Google Scholar]
- Hay B. P.; Bryantsev V. S. Chem. Commun. 2008, 2417–2428. [DOI] [PubMed] [Google Scholar]
- Castellano R. K. Curr. Org. Chem. 2004, 8, 845–865. [Google Scholar]
- Kaufmann L.; Traulsen N. L.; Springer A.; Schröder H. V.; Mäkelä T.; Rissanen K.; Schalley C. A. Org. Chem. Front. 2014, 1, 521–531. [Google Scholar]
- Heinrich T.; Traulsen C. H.-H.; Darlatt E.; Richter S.; Poppenberg J.; Traulsen N. L.; Linder I.; Lippitz A.; Dietrich P. M.; Dib B.; Unger W. E. S.; Schalley C. A. RSC Adv. 2014, 4, 17694–17702. [Google Scholar]
- Hohloch S.; Scheiffele D.; Sarkar B. Eur. J. Inorg. Chem. 2013, 3956–3965. [Google Scholar]
- Poppenberg J.; Richter S.; Darlatt E.; Traulsen C. H.-H.; Min H.; Unger W. E. S.; Schalley C. A. Surf. Sci. 2012, 606, 367–377. [Google Scholar]
- Richter S.; Poppenberg J.; Traulsen C. H.-H.; Darlatt E.; Sokolowski A.; Sattler D.; Unger W. E. S.; Schalley C. A. J. Am. Chem. Soc. 2012, 134, 6289–16297. [DOI] [PubMed] [Google Scholar]
- Richter S.; Traulsen C. H.-H.; Heinrich T.; Poppenberg J.; Leppich C.; Holzweber M.; Unger W. E. S.; Schalley C. A. J. Phys. Chem. C 2013, 117, 18980–18985. [Google Scholar]
- Poppenberg J.; Richter S.; Traulsen C. H.-H.; Darlatt E.; Baytekin B.; Heinrich T.; Deutinger P. M.; Huth K.; Unger W. E. S.; Schalley C. A. Chem. Sci. 2013, 4, 3131–3139. [Google Scholar]
- Traulsen C. H.-H.; Darlatt E.; Richter S.; Poppenberg J.; Hoof S.; Unger W. E. S.; Schalley C. A. Langmuir 2012, 28, 10755–10763. [DOI] [PubMed] [Google Scholar]
- Kaufmann T.; Ravoo B. J. Polym. Chem. 2010, 1, 371–387. [Google Scholar]
- Ruiz S. A.; Chen C. S. Soft Matter 2007, 3, 168–177. [DOI] [PubMed] [Google Scholar]
- Darlatt E.; Nefedov A.; Traulsen C. H.-H.; Poppenberg J.; Richter S.; Dietrich P. M.; Lippitz A.; Illgen R.; Kühn J.; Schalley C. A.; Wöll C.; Unger W. E. S. J. Electron Spectrosc. Relat. Phenom. 2012, 185, 621–624. [Google Scholar]
- Motiei L.; Feller M.; Evmenenko G.; Dutta P.; van der Boom M. E. Chem. Sci. 2012, 3, 66–71. [Google Scholar]
- Motiei L.; Sassi M.; Kaminker R.; Evmenenko G.; Dutta P.; Iron M. A.; van der Boom M. E. Langmuir 2011, 27, 1319–1325. [DOI] [PubMed] [Google Scholar]
- Motiei L.; Yao Y.; Choudhury J.; Yan H.; Marks T. J.; van der Boom M. E.; Facchetti A. J. Am. Chem. Soc. 2010, 132, 12528–12530. [DOI] [PubMed] [Google Scholar]
- de Ruiter G.; Lahav M.; van der Boom M. E. Acc. Chem. Res. 2014, 47, 3407–3416. [DOI] [PubMed] [Google Scholar]
- Kubicek M.; Holzlechner G.; Opitz A. K.; Larisegger S.; Hutter H.; Fleig J. Appl. Surf. Sci. 2014, 289, 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J.NEXAFS Spectroscopy; Springer: Berlin, Germany, 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.