

Abstract
Nitric oxide (NO)-releasing nanoparticles (NPs) have emerged as a wound healing enhancer and a novel antibacterial agent that can circumvent antibiotic resistance. However, the NO release from NPs over extended periods of time is still inadequate for clinical application. In this study, we developed NO-releasing poly(lactic-co-glycolic acid)-polyethylenimine (PEI) NPs (NO/PPNPs) composed of poly(lactic-co-glycolic acid) and PEI/diazeniumdiolate (PEI/NONOate) for prolonged NO release, antibacterial efficacy, and wound healing activity. Successful preparation of PEI/NONOate was confirmed by proton nuclear magnetic resonance, Fourier transform infrared spectroscopy, and ultraviolet/visible spectrophotometry. NO/PPNPs were characterized by particle size, surface charge, and NO loading. The NO/PPNPs showed a prolonged NO release profile over 6 days without any burst release. The NO/PPNPs exhibited potent bactericidal efficacy against methicillin-resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa concentration-dependently and showed the ability to bind on the surface of the bacteria. We also found that the NO released from the NO/PPNPs mediates bactericidal efficacy and is not toxic to healthy fibroblast cells. Furthermore, NO/PPNPs accelerated wound healing and epithelialization in a mouse model of a MRSA-infected wound. Therefore, our results suggest that the NO/PPNPs presented in this study could be a suitable approach for treating wounds and various skin infections.
Keywords: nitric oxide-releasing nanoparticles, PLGA, PEI, antimicrobial, wound healing
Introduction
Bacterial infections are among the major causes of infectious disease-related mortality and morbidity worldwide.1 Particularly, bacterial infections impair the process of wound healing, resulting in chronic wounds such as diabetic foot ulcers.2 Although wound infections are usually caused by multiple bacteria or fungi, the Gram-positive Staphylococcus aureus and Gram-negative Pseudomonas aeruginosa are the two most common bacteria responsible for the majority of wound infections.3–5 Notably, certain S. aureus strains have developed resistance to methicillin and the prevalence of infections caused by methicillin-resistant S. aureus (MRSA) is increasing.6 These bacteria can easily contaminate the surface of wounds and access the underlying tissue,7 thereby delaying the healing process. Owing to growing health care costs and increasing antibiotic resistance, the economic burden of the treatment of chronic wounds is rapidly growing. Considering that resistance against newly approved antibiotics develops within 2 years,8 there is an urgent need for new generations of antibiotics to fight infections.
Nitric oxide (NO) is a diatomic free radical endogenously generated by NO synthase via oxidation of the amino acid L-arginine.9 NO functions as a crucial effector and chemical messenger in diverse physiological and pathophysiological processes such as host defense, platelet aggregation, neuronal communication, regulation of vascular tone, wound healing, and immune responses.9 NO especially plays an important role as a potent endogenous antibacterial agent against a broad spectrum of bacteria in the immune response.10 NO is known to kill bacterial cells by direct or indirect oxidation through the formation of peroxynitrite (−OONO), which is the byproduct of the reaction between NO and free radical superoxide (O2*−).11 Along with antibacterial effects, NO has been recognized as a key molecule in the natural wound healing process. A previous study demonstrated that NO regulates cell proliferation, collagen formation, and wound contraction, thereby accelerating wound healing.12
Despite the beneficial effects of NO, its clinical application is hampered by its gaseous property and short half-life. Therefore, the controlled release of NO is an indispensable property in NO delivery systems. There are various NO delivery systems including nanotechnology, which has recently emerged as a new strategy for storing and releasing NO for application as an antibacterial because it provides distinct advantages when combined with antibiotics. For example, nanotechnology combined with conventional antibiotics such as vancomycin and silver ion (Ag+) exhibited greater antibacterial efficacy than the antibiotics alone.13,14 The unique feature and advantage of nanoparticles (NPs) lie in their high surface area to volume ratio, which creates chemical flexibilities and beneficial physical properties from their individual components.15 To date, several NO-releasing NPs such as silica, gold, liposomes, and dendrimers have been developed by taking advantage of nanotechnology and NO.16–22 However, these NO-releasing NPs showed an initial burst release probably due to the NO tethering only to the surface of the NPs. This was accompanied by a relatively short duration of NO release ranging from a few to up to 24 hours. Such a short duration of NO release requires frequent administration and the burst release may cause toxicity at the site of application. Therefore, NPs that release NO over a period of days in a prolonged and controlled manner without burst release would be ideal for biomedical applications.
In this study, we developed NO-releasing poly(lactic-co-glycolic acid) (PLGA)-polyethylenimine (PEI) NPs (NO/PPNPs) in a sustained manner over an extended period. We selected PLGA as the basic polymer because it is an established biocompatible and biodegradable polymer that has long been used for controlled release drug delivery.23,24 PEI was selected as the NO donor polymer. The PEI/diazeniumdiolate (PEI/NONOate) was synthesized by reacting NO with the secondary amine groups of PEI and incorporated in the matrix of PLGA NPs. The physicochemical properties of the PEI/NONOate and NO/PPNPs were characterized and their antibacterial efficacy against MRSA and P. aeruginosa were evaluated. The wound-healing activity of the NO/PPNPs was also evaluated in a mouse model of MRSA-infected wounds.
Materials and methods
Materials
PLGA (50:50 DLG 5E) was purchased from Lakeshore Biomaterials (Birmingham, AL, USA). PEI (MW 25 kDa), sodium methoxide (NaOCH3), nile red, tetrazolium dye 3-(4,5-di-methylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT), dimethylsulfoxide (DMSO) and Griess assay reagent were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). NO and nitrogen (N2) gasses were obtained from HANA gas (Gimhae, Korea). Bacto™ tryptic soy broth (TSB) and Difco™ Luria-Bertani (LB) media were purchased from BD Biosciences (San Jose, CA, USA). The LIVE/DEAD® Baclight™ bacterial viability kit (Molecular Probes) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). The Roswell Park Memorial Institute 1640 medium, trypsin, fetal bovine serum, and penicillin-streptomycin were purchased from Hyclone, Thermo Fisher Scientific Inc. Tiletamine/zolazepam (Zoletil 50®) was obtained from Virbac SA (Virbac, Carros, France) and Xylazine hydrochloride (Rompun®) was obtained from Bayer AG (Leverkusen, Germany). All other reagents and solvents were of the highest analytical grade available commercially.
Synthesis of PEI/NONOate
PEI/NONOate was synthesized as shown in Figure 1A.25 Briefly, 0.5 g of PEI was dissolved in 30 mL of a co-solvent consisting of dry tetrahydrofuran (THF):dry methanol (2:1) and NaOCH3 (1 mol equivalent with respect to the total amine sites) in dry methanol (10 mL) was added to the PEI solution. This mixture was placed in a Parr high-pressure reactor, which was flushed with 20 psi N2 gas for 15 minutes and then subsequently charged with NO gas at 80 psi for 3 days at room temperature. The NO was then vented and the reactor flushed with 20 psi N2 gas for 15 minutes. After washing the unreacted PEI with THF:methanol (2:1), the PEI/NONOate was precipitated with cold dry ether. The solvent was quickly removed by filtration and the product was washed with cold dry ether and vacuum-dried to yield the light yellow PEI/NONOate. The final product was stored at −20°C for future uses.
Figure 1.

Synthesis of PEI/NONOate, followed by NO/PPNPs fabrication.
Notes: (A) Synthesis of PEI/NONOate and (B) fabrication of NO/PNNPs.
Abbreviations: NONOate, diazeniumdiolate; NO, nitric oxide; PLGA, poly(lactic-co-glycolic acid); PEI, polyethylenimine; NPs, nanoparticles; THF, tetrahydrofuran; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; PVA, poly(vinyl alcohol); DCM, dichloromethane; NaOCH3, sodium methoxide; MeOH, methanol.
Proton nuclear magnetic resonance (1H-NMR), Fourier transform infrared spectroscopy (FTIR), and ultraviolet (UV)-visible (Vis) spectrophotometric analysis
The nuclear magnetic resonance (NMR) spectra of the PEI/NONOate were recorded in deuterium oxide (D2O) using 1H-NMR spectroscopy (500 MHz superconducting FT-NMR spectrometer, Unity Inova 500 MHz NB high resolution FT-NMR, Varian Inc, Santa Clara, CA, USA). The FTIR spectrum of the PEI/NONOate was measured using a Varian® 640 FT-IR spectrophotometer (Varian Inc) in transmittance mode with potassium bromide (KBr) plates. A total of 48 scans were taken per spectrum in the range from 4,000 to 500 cm−1 at a resolution of 4 cm−1. For the UV-Vis spectral analysis, different concentrations of the PEI/NONOate were measured in Dulbecco’s phosphate-buffered saline (DPBS) at 37°C using a UV-Vis spectrophotometer (Optizen 2120 UV, Mecasys, Republic of Korea) at predetermined time intervals. For nitrite (NO2−) analysis, Griess reagent was added to the PEI/NONOate in DPBS at 37°C and nitrite was spectrophotometrically detected by monitoring the formation of an azo dye.
NP preparation
The PPNPs and NO/PPNPs were prepared using an oil-in-water emulsification solvent evaporation method (Figure 1B). Briefly, PLGA (105 mg) was mixed with 30 mg of either PEI or PEI/NONOates for PPNPs and NO/PPNPs respectively, dissolved in 10 ml of dichloromethane and poured into a 20 mL solution of cold poly(vinyl alcohol) (1%). For fluorescence labeling, nile red (50 μg/mL in methanol) was added to the polymer solution. The solution was stirred using a high-speed homogenizer (IKA® Ultra Turrax T-10) at 14,500 rpm for 2 minutes in an ice bath, followed by probe sonication at 150 W in an ice bath for 3 minutes. The emulsion was then added to 10 mL of deionized water and stirred at 4°C for 12 hours. After the residual solvent was removed, the emulsion was centrifuged at 20,000× g at 4°C for 30 minutes and washed three times. The pellets obtained were freeze-dried and stored at −20°C for future use.
NO measurement in PEI-NONOate and NO/PPNPs
The NO content of the PEI/NONOate was measured using the Griess assay. Briefly, a known amount of the PEI/NONOate was incubated in citrate buffer (pH 4.0) at 37°C for 4 hours. After spectrophotometrically confirming the absence of a peak at 252 nm, 50 μL of the solution was mixed with 50 μL of DPBS and 100 μL of the Griess reagent. This mixture was incubated for 15 minutes at room temperature in the dark and the absorbance was measured at 540 nm using a Biorad iMark™ microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and the NO concentration was calculated with 0–100 μM sodium nitrite (NaNO2) as the standard. To determine the total amount of NO in the NO/PPNPs, the NO/PPNPs were incubated in 0.5 M sodium hydroxide (NaOH) at 37°C for 1 hour to extract PEI/NONOates by degrading PLGA polymer. The amount of NO in extracted PEI/NONOates was determined as described above.
NO release study
The NO release of PEI/NONOates and NO/PPNPs were evaluated in DPBS at 37°C. A total of 25 mg of PEI/NONOate was dispersed in 5 mL of DPBS and placed in Visking® dialysis tubing (molecular weight cut-off, 12–14 kDa), which was further immersed in 50 mL of DPBS at pH 7.4 with mild stirring at 37°C. At predetermined time intervals, 50 μL of the sample was withdrawn, mixed with DPBS (50 μL) and 100 μL Griess reagent and incubated for 15 minutes at room temperature in the dark. The NO content was analyzed using the Griess assay described above. For NO/PPNPs, 1 mg of NO/PPNPs was added to 1 mL of PBS (pH 6.0) and DPBS (pH 7.4) and incubated at 37°C with mild stirring. At predetermined time intervals, samples were centrifuged at 13,000× g for 15 minutes and the supernatants were analyzed by the Griess assay described above.
Scanning electron microscopy (SEM)
The PPNPs and NO/PPNPs were characterized using field emission SEM with an FE-SEM S4800 microscope (Hitachi Ltd., Tokyo, Japan). The NPs were placed on carbon tape and coated with platinum for 2 minutes under vacuum. The samples were then viewed with the FE-SEM at an accelerating voltage of 35 kV and the particles size (n=300) was measured using the ImageJ software (National Institutes of Health, Bethesda, MA, USA).
Particle size and zeta potential
The particle size was confirmed using a qNano size analyzer (iZON Sciences, Christchurch, New Zealand) with an air-based variable pressure module, nanopore 200 (iZON NP 200) and a calibration particle 200 nm (CP 200). Buffer containing sodium chloride (NaCl), Tris (pH 8), ethylenediaminetetraacetic acid, and triton in deionized water was used as the electrolyte to suspend the NP sample and the calibration particles. Each recorded measurement was based on 500 particles and the size was measured using the iZON control suite 2.2 software. The zeta potential was measured in water at 25°C using the Malvern zetasizer nano ZS (Malvern Instruments, Malvern, UK) with three repetitions.
Antibacterial assay
The bacterial strains used in this study were MRSA (USA300)26 and P. aeruginosa PAO1 (wild-type prototroph).27 The bacteria were inoculated on TSB agar for MRSA and LB agar for P. aeruginosa overnight at 37°C and grown to the mid-exponential phase (107 colony forming units, CFU/mL). The resulting bacterial suspension was centrifuged for 15 minutes at 8,000× g. The pellet was re-suspended in sterile DPBS and adjusted to an appropriate concentration. A total of 100 μL of the bacterial suspension (final concentration 106 CFU/mL) was incubated with 1.7 mL of either TSB or LB media for MRSA and P. aeruginosa, respectively. Then, 200 μL of the PPNPs or NO/PPNPs were added for final NP concentrations of 0.625, 1.25, 2.5, 5, and 10 mg/mL in 12-well plates. A tube containing bacteria in DPBS was used as a control. All the samples were incubated at 37°C for 24 hours in a shaking incubator, then centrifuged at 8,000× g and washed twice with 0.85% NaCl.
For the confocal study, the bacterial suspensions were stained with LIVE/DEAD® Baclight™ bacterial viability kit reagents according to the manufacturer’s protocol. The samples were observed with the FV10i Fluoview confocal microscopy to differentiate live bacteria from the dead. Bacteria stained green with Syto-9 at excitation/emission wavelengths of 539/570–620 nm were considered viable/live and those stained red with propidium iodide (PI) at excitation/emission 470/490–540 nm were considered dead. The percentage survival was analyzed in the bacterial suspensions using a flow cytometer (BD Accuri™ C6 Flow Cytometer, Ann Arbor, MI, USA). Each sample was acquired for 30 seconds at the medium flow rate (35 μL/min) with side scatter height plots threshold =10,000 to exclude debris. For bacterial viability (CFU measurement), the bacterial suspensions were diluted in DPBS from 10−1 to 10−8. A 200 μL aliquot of each dilution was plated on TSB and LB agar media for the MRSA and P. aeruginosa, respectively, and incubated at 37°C overnight. The number of colonies was enumerated, factoring in the number of viable bacteria at the time of plating.
NP adhesion to the bacteria
To evaluate the adhesion of the NPs to the bacteria, MRSA and P. aeruginosa were inoculated on coupons. The coupons were incubated for 1 hour, then immersed in culture medium containing nile red-labeled NPs (5 mg/mL) and incubated for 2 hours at 37°C with mild shaking. The coupons were washed three times with fresh medium to remove the unbound NPs and the bacteria on the coupons were detached using a bath sonicator for 15 minutes. The bacterial suspensions were washed twice with 1 mL of 0.85% NaCl by centrifugation at 8,000× g, to remove residual media components. The final bacterial suspensions were stained with Syto-9 dye and observed with a confocal microscope.
In vitro cytotoxicity study
L929 mouse fibroblasts from the Korean Cell Line Bank (KCLB, Seoul, Korea) were cultured in Roswell Park Memorial Institute 1640 medium supplemented with 10% (v/v) fetal bovine serum and antibiotics (100 IU/mL penicillin G sodium and 100 μg/mL streptomycin sulfate). The cells were maintained in an incubator supplied with 5% carbon dioxide (CO2) and a humidified air atmosphere at 37°C. Cells were seeded on a 96-well plate at 5×104 cells per well and incubated for 48 hours. The media were then replaced with fresh media containing NPs at increasing concentrations (0.625, 1.25, 2.5, and 5 mg/mL) and incubated for 24 hours. A standard MTT solution in sterile PBS was added to each well and incubated for 2 hours. The MTT solution was then removed and 150 μL of DMSO was added to each well. The absorbance measured at 540 nm was proportional to the concentration of viable cells in each well. Untreated cells were used as a control. The data were expressed as mean ± standard deviation (SD) of eight replicates (n=8). The cell viability was calculated using the following equation:
| (1) |
In vivo wound-healing assay
All animal experiments were performed in accordance with the regulations of Pusan National University and Korean Legislation on animal studies. Female BALB/c mice (7–8 weeks, Samtako Bio Korea) were used as the animal model. Prior to the development of wounds on the dorsal area, the mice were anesthetized using Zoletil® 50 and Rompun® at a ratio of 5:2. The dorsal hair was shaved with an electric razor and the back skin was excised to create 5-mm diameter full-thickness wounds. Then, a suspension containing 6×108 MRSA US300 was inoculated to induce infectious wounds. After 24 hours, 5 mg of the freeze-dried PPNPs and NO/PPNPs were topically applied. Each wound was covered with sterile gauze. Untreated mice were used as a control. The gauze on the wound lesions was replaced with new gauze at the proper time. Photographs of the wounds were taken on day 0, 1, 4, and 7 to observe the gross visual wound healing as determined by the wound area not covered by the migrating epithelial cells. The ImageJ software was used to determine the wound size reduction which was calculated as follows:
| (2) |
where W0 is the wound area at initial time 0 and Wt is the wound area at time t.
Histological processing of wound area
The cross-sectional full-thickness skin specimens and deep granulation tissue were collected on the seventh day of the experiment. Full wound areas were excised, fixed in 10% formalin for 24 hours, and blocked with paraffin. Five-micron vertical sections were fixed to glass slides and stained with hematoxylin and eosin to observe morphology. The slides were examined using light microscopy and images were digitally captured without further processing.
Statistical analysis
Statistical analysis of in vivo data was performed using unpaired Student’s t-test in GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). In cases of significant deviations from t-test, nonparametric Mann–Whitney U-test was conducted to compare the distributions of two unpaired groups. P-values of <0.05 were considered statistically significant. The data are presented as the mean ± SD.
Results and discussion
Synthesis and characterization of PEI/NONOate
The PEI/NONOate was synthesized via the reaction between the secondary amine group of PEI and two moles of NO under high pressure (80 psi) to create a relatively stable adduct structure (Figure 1). The PEI was dissolved in the THF and methanol co-solvent in the presence of NaOCH3 as the exogenous cation. The positively charged NaOCH3 counteracts the negatively charged NONOate adduct to fulfill the required electroneutral conditions.28 The conjugation between PEI and NO was verified using 1H NMR (Figure 2). The 1H NMR details are as follows: (D2O, 500 MHz) δ-1.2 (t, 6H, J =10 Hz), 1.9 (t, 2H, J =10 Hz), 2.57 (s, 1H), 2.67 (s, 1H), 3.10 (s, 1H), 3.37 (s, 1H), 3.56 (q, 6H, J =25 Hz), 3.76 (t, 2H, J =10 Hz). In the 1H NMR, the proton signals of the methylene groups near the secondary amine sites of PEI at 2.7 ppm were shifted downfield by approximately 3.1 ppm in PEI/NONOates as a result of the electron withdrawing effect of the NONOate group. The infrared measurement of PEI/NONOate was performed to confirm the presence of NONOate in the polymer (Figure 3). NONOates are known to have a characteristic peak at the range of wavenumber 1,250–1,300 cm−1.29 Our PEI/NONOate exhibited the peak at 1,284 cm−1, confirming the successful synthesis of NONOate in the PEI. Moreover, the NO/PPNPs also exhibited the characteristic NO peak at 1,275 cm−1, which was not observed in the PLGA polymer and PPNPs, indicating the successful loading of PEI/NONOate in the NPs.
Figure 2.

Characterization of PEI/NONOate by 1H NMR and FTIR.
Notes: (A) 1H NMR spectra of PEI and PEI/NONOate. (B) FTIR spectra of PEI/NONOate, PLGA, PPNPs, and NO/PPNPs. The red circles in A and B refer to proton signals of methylene groups and diazeniumdiolates group.
Abbreviations: 1H NMR, proton nuclear magnetic resonance; PEI, polyethylenimine; PLGA, poly(lactic-co-glycolic acid); FTIR, Fourier transform infrared spectroscopy; PPNPs, PLGA-PEI nanoparticles; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; NO, nitric oxide; NONOate, diazeniumdiolate.
Figure 3.

Characterization of PEI/NONOate by UV-Vis spectra.
Notes: (A) UV-Vis absorption of PEI/NONOate (violet line), PEI/NONOate with Griess reagent (green line). (B) UV-Vis spectra of PEI/NONOate in different concentrations. (C) UV-Vis spectra of PEI/NONOate in DPBS at 37°C at different times.
Abbreviations: NED, N-1-naphthylethylenediamine; UV-Vis, ultraviolet-visible; PEI, polyethylenimine; DPBS, Dulbecco’s phosphate-buffered saline; NONOate, diazeniumdiolate.
To characterize the PEI/NONOate as a NO donor activity, the UV-Vis spectra were measured under various conditions (Figure 3). The characteristic absorption at 252 nm indicated the presence of NONOate (Figure 3A, violet line). The PEI/NONOate absorption measured at 252 nm decreased in a concentration-dependent manner (Figure 3B), further confirming the presence of NONOate. The NO-releasing ability of the PEI/NONOate was examined by measuring the time course of the absorbance at 252 nm (Figure 3C). The result showed a time-dependent decrease in absorbance at 252 nm, indicating that NO is released from the PEI/NONOate time-dependently. Since the NO released is converted to NO2−, we also measured NO2− using the Griess assay (Figure 3A, green line). The presence of an absorption signal at 540 nm confirms the azo dye peak, which is the product of the reaction of the NO2− (auto-oxidation of NO released from PEI/NONOate) with the Griess reagent.30
The NO in the PEI/NONOate was measured using the Griess assay following its complete release. To accelerate the release of NO, the PEI/NONOate was incubated in an acidic condition (pH 4.0 buffer) where NONOates quickly decompose. The complete NO release was confirmed spectrophotometrically at 252 nm and the amount of NO in PEI/NONOate was 1.4 μmole/mg polymer (Table 1).
Table 1.
Characterizations of NPs
| NPs | Size
|
Zeta potential (mV) | NO amount (μmole/mg NPs) | |
|---|---|---|---|---|
| SEM | qNano | |||
| PLGA | 175±15 | 184±16 | −24.3±1.7 | Not determined |
| NPs PPNPs | 166±46 | 175±35 | +30.1±0.5 | Not determined |
| NO/PPNPs | 162±19 | 179±25 | +34.9±0.9 | 0.04±0.008 |
Note: Values were presented as means ± standard deviation of three different batches of particles.
Abbreviations: NO, nitric oxide; PLGA, poly(lactic-co-glycolic acid); NPs, nanoparticles; PEI, polyethylenimine; PPNPs, PLGA-PEI nanoparticles; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; SEM, scanning electron microscopy.
Characterization of NPs
SEM images of the prepared NPs revealed a spherical morphology with uniformed particle size (Figure 4A). The average size of the PPNPs and NO/PPNPs was 166±46 nm and 162±19 nm, respectively based on SEM images. The particle size was confirmed by qNano, which determined the sizes to be 175±35 nm and 179±25 nm for the PPNPs and NO/PPNPs, respectively, with a narrow size distribution (Figure 4B). There was no noticeable difference in particle size between the PLGA NPs, PPNPs, and NO/PPNPs (Table 1), indicating that the addition of PEI or PEI/NONOate did not affect particle size or size distribution. The zeta potential measurement (Table 1) revealed that the PLGA NPs were negatively charged (−24.3±1.7 mV) while the PPNPs and NO/PPNPs were positive at +30.1±0.5 mV and +34.9±0.9 mV, respectively. The PEI or PEI/NONOate has protonated primary amines. Therefore, their orientation on the surface of the NPs can explain the positive surface charge as shown in Figure 1B. To measure NO amount of NO/PPNPs, PEI/NONOate in the NP/PPNPs was extracted by degrading the PLGA in 0.5 M NaOH solution. It is worth noting that alkaline conditions accelerate the degradation of PLGA to water (H2O) and CO2 and maintain the stability of the NONOates.31 The NO amount of NO/PPNPs was 0.04 μmole NO per mg of NPs (Table 1).
Figure 4.

Characterization of nanoparticles.
Notes: (A) SEM images of PPNPs and NO/PPNPs, bars represent 500 nm. (B) Size distribution of PPNPs and NO/PPNPs.
Abbreviations: SEM, scanning electron microscopy; PLGA, poly(lactic-co-glycolic acid); PEI, polyethylenimine; PPNPs, PLGA-PEI nanoparticles; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; NONOate, diazeniumdiolate; NO, nitric oxide.
NO release study
NO has a short half-life and therefore the ability to facilitate its controlled release is an indispensable property of an efficient NO delivery system. As shown in Figure 5, PEI/NONOate exhibited burst release of NO (~50%) in the first 30 minutes and thereafter released ~90% in 2 hours, followed by nearly 100% in 12 hours. Interestingly, the NO/PPNPs remarkably improved the NO release profile. NO was released from the NO/PPNPs in a sustained manner over 6 days without burst release. At pH 7.4, NOPP/NPs released 0.021 μmoles NO per mg NPs in the first 24 hours (~50%) and continuously released NO for over 6 days (0.0374 μmoles NO/mg NPs, ~93.2%). The difference between PEI/NONOate and NO/PPNPs in the NO release profiles can be attributed to the incorporation of PEI/NONOate into the hydrophobic PLGA NP matrix, which restricts the spontaneous degradation of NONOate group. We also performed the NO release study at pH 6.0 since wound pH can decrease during the course of healing.32 At pH 6.0, NO/PPNPs released more NO in first 24 hours (0.0256 μmoles/mg NPs, ~64%) than at pH 7.4 and continuously released NO in sustained manner for over 6 days (0.0395 μmoles NO/mg NPs, ~98.8%). The faster NO release at pH 6.0 would be due to lower stability of NONOate at acidic pHs. A number of other NO-releasing NPs such as silica,33 gold,34 and dendrimers35 all exhibit a relatively short duration (ie, ~4 to ~24 hours) of NO release with initial burst release. Therefore, the comparatively prolonged NO release of our NO/PPNPs would be beneficial for biomedical applications such as antibacterial activity, which requires sustained NO release. Since drug release from the PLGA NPs can be controlled by changing the polymer hydrophobicity and particle size,23 NO release from NO/PPNPs could be further optimized.
Figure 5.

In vitro release profile of PEI/NONOate and NO/PPNPs.
Notes: All samples were placed in DPBS at 37°C; data presented are mean ± standard deviation; n=3.
Abbreviations: PEI, polyethylenimine; PPNPs, PLGA-PEI nanoparticles; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; NONOate, diazeniumdiolate; NO, nitric oxide; PLGA, poly(lactic-co-glycolic acid); DPBS, Dulbecco’s phosphate-buffered saline; h, hour(s).
Antibacterial activity of NPs
The antibacterial activities of the PPNPs and NO/PPNPs against MRSA and P. aeruginosa were evaluated by counting the number of CFU (Figure 6A and B) and by measuring % survival rate using flow cytometry (Figure 7C and D). We first tested the antibacterial activity of the PPNPs since PEI is known to have antibacterial activity because of its polycationic nature.36 However, both bacteria were not susceptible to the PPNPs regardless of the NP concentrations, implying that the majority of the PEI in the PPNPs is located in the inner part of the PLGA NPs, leaving only a small portion on the surface of the NPs. The proposition that the PEI is encapsulated within the NPs can also be supported by the prolonged NO release profile of the NO/PPNPs and no burst release, as mentioned previously (Figure 5). In contrast, NO/PPNPs killed both bacteria in a concentration-dependent manner. A reduction of bacterial viability by >2 logs (~99% of killing) and >4 logs (~99.99% of killing) against MRSA was observed with 5 mg/mL and 10 mg/mL of NO/PPNPs, respectively (Figure 6A). A reduction of bacterial viability by ~3 logs and >3 logs (~99.99% of killing) against P. aeruginosa was observed with 5 mg/mL and 10 mg/mL of NO/PPNPs, respectively (Figure 6B). Considering that the PPNPs did not show significant antibacterial activity, this result demonstrates that the antibacterial activity of the NO/PPNPs can be attributed solely to the NO content. NO can kill P. aeruginosa by disrupting its cellular physiology. This disruption includes inactivation of critical microbial enzymes by reacting with iron-containing proteins (especially Fe-S cluster protein) to produce a range of dinitrosyl iron complexes known as DNICs, which are mainly disruptive.37 As for MRSA, reactive nitrogen intermediates, which are by-products of the reaction between NO and the free radical superoxide (O2*−),11 have been shown to modify DNA, protein, and lipid as well as act indirectly on bacteria by modifying immune responses or other host cell functions.38
Figure 6.
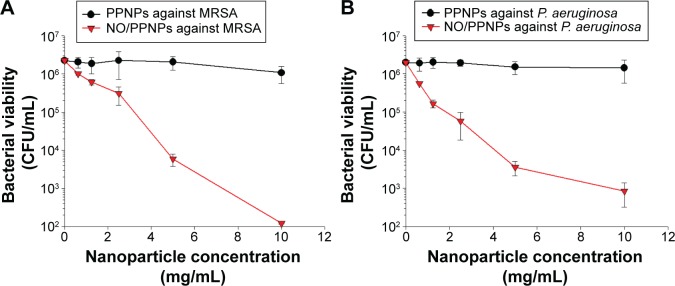
Antibacterial activity of PPNPs and NO/PPNPs against MRSA and Pseudomonas aeruginosa.
Notes: The number of CFU (A) of MRSA and (B) of P. aeruginosa. Data shown are mean ± standard deviation; n=3.
Abbreviations: PLGA, poly(lactic-co-glycolic acid); PEI, polyethylenimine; PPNPs, PLGA-PEI nanoparticles; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; MRSA, methicillin-resistant Staphylococcus aureus; CFU, colony forming units; NO, nitric oxide.
Figure 7.

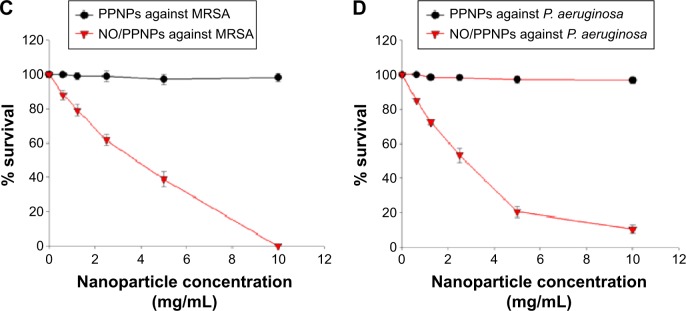
Confocal microscopy images and the percent (%) survival of MRSA (left panel) and Pseudomonas aeruginosa (right panel) after 24 hours of treatment with nanoparticles at different concentrations.
Notes: (A) PPNPs, (B) NO/PPNPs, (C) percent (%) survival against MRSA and (D) percent (%) survival against P. aeruginosa. Syto-9 fluorescence (green) indicates intact membrane of healthy bacteria, PI fluorescence (red) indicates membrane destruction and cell death. Bacterial survival at each point is presented as a percentage relative to the control group (buffer alone). Data shown are mean ± standard deviation; n=3. (A and B) Bars represent 20 μm.
Abbreviations: PLGA, poly(lactic-co-glycolic acid); PEI, polyethylenimine; PPNPs, PLGA-PEI nanoparticles; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; MRSA, methicillin-resistant Staphylococcus aureus; PI, propidium iodide; NO, nitric oxide.
The viability of the bacteria was also visualized by distinguishing live and dead bacteria using a confocal microscope and the results were quantitatively evaluated with the % survival rates by flow cytometry. The green (Syto-9) and red (PI) fluorescence represent live and dead bacteria, respectively (Figure 7). The Syto-9 stain is a cell-permeant dye that penetrates healthy bacterial cells with intact membranes and is detected primarily in the FL1 and FL2 channels of the flow cytometer, while PI only penetrates bacteria with damaged membranes. As shown in Figure 7A, despite an increase in the NP concentration, the density of the live bacteria remained the same and dead bacteria were not found. This confirms that the PPNPs do not affect the viability of MRSA and P. aeruginosa regardless of the NP concentration. However, the NO/PPNPs exhibited concentration-dependent antibacterial activity against MRSA and P. aeruginosa (Figure 7B). As the NP concentration increased, the population of live bacteria (green) decreased, while the population of dead bacteria (red) obviously increased. The result agreed with the % survival rates (Figure 7C and D).
Adhesion of NPs to bacteria
In the antibacterial study, the amount of NO released from our NO/PPNPs was approximately 0.2 μmole at the highest NP dose (10 mg/mL) over 24 hours resulting in ~0% survival for MRSA and ~10% for P. aeruginosa. Compared to other NO-releasing NPs, the NO/PPNPs killed bacteria at over ten fold lower NO concentrations. For example, Hetrick et al evaluated the bactericidal efficacy of NO-releasing silica NPs against P. aeruginosa and observed approximately 2 logs of biofilm killing (~99% killing of the bacteria) with a total NO amount of 3.2 μmoles.39 Sun et al also reported a dendrimer NO-releasing system, which showed 3 logs killing against the Gram-positive S. aureus with a total NO release of 2.47 μmoles.40 The possible mechanism by which the NO/PPNPs kill bacteria at such low NO concentrations is not fully understood. However, we hypothesized that the positive surface charge of NO/PPNPs (Table 1) facilitates the electrostatic binding of the NPs to the negatively charged bacterial surface, thereby increasing the antibacterial activity.
To test this hypothesis, we studied the binding of NPs to the bacterial surface using confocal microscopy. Negatively charged PLGA NPs were used as a complement for the positively charged NO/PPNPs. For fluorescent visualization, the NPs were labeled with nile red and incubated with the bacteria at 37°C for 2 hours. The red fluorescence represents the NPs (labeled with nile red) and green fluorescence represents bacteria stained with the Syto-9. As shown in Figure 8, the association of NO/PPNPs with MRSA and P. aeruginosa was observed. It is worth noting that the adhesion of the NPs to the bacteria was maintained even after the sonication process, which was used to detach the bacteria from the coupons for the confocal study. However, the negatively charged PLGA NPs were not found on the bacterial surface, indicating that the association between the NPs and the bacteria is an electrostatic interaction. The electrostatic adhesion of NO/PPNPs to the bacterial surface could explain why NO/PPNPs with low NO concentrations have potent antibacterial activity. NO exerts its antibacterial activity mainly on the bacterial membrane by disrupting DNA and proteins. Therefore, the penetration of NO through the bacterial membrane is a critical step in mediating its antibacterial activity. Importantly, NO has a very short half-life and a limited diffusion distance (up to 150–300 μm),31 meaning that NO from unbound NPs loses a substantial degree of activity during diffusion to the bacterial membrane. This suggests that without NP binding to bacteria, high concentrations of NO would be required to have sufficient NO on the bacterial membrane to kill bacteria. Conversely, NPs bound to bacteria can directly release NO to the bacterial membrane with minimal loss, thereby achieving sufficient antibacterial activity at low NO concentrations.
Figure 8.

Adhesion of PLGA NPs, PPNPs, and NO/PPNPs to bacteria.
Notes: (A) MRSA and (B) Pseudomonas aeruginosa. Nanoparticles were incubated with bacteria for 2 hours and images were obtained using a confocal microscope. Bacterial membrane (green) stained with Syto-9 and nanoparticles (red) labeled with nile red for visualization.
Abbreviations: PLGA, poly(lactic-co-glycolic acid); PEI, polyethylenimine; PPNPs, PLGA-PEI nanoparticles; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; MRSA, methicillin-resistant Staphylococcus aureus; NO, nitric oxide; NPs, nanoparticles.
Cytotoxicity study
We evaluated the cytotoxicity of the PLGA NPs, PPNPs, and NO/PPNPs against mouse fibroblast cells (L929) on account of their promising therapeutic application for infected wound healing. Mammalian fibroblasts cells provide a suitable model for cytotoxicity studies because they have a significant role in wound healing, epithelial–mesenchymal interaction, and development of the extracellular matrix.41 As illustrated in Figure 9, all NPs showed no significant cytotoxicity (>80% viability) against L929 fibroblast cells in the NPs concentrations up to 5 mg/mL, indicating that PEI exposed on the surface and NO released from NPs is not toxic to healthy fibroblasts at 5 mg/mL or less. At a concentration of 10 mg/mL, NPs exhibited weak cytotoxicity, showing 69.4%, 68.9%, and 64.8% of viability when treated with PLGA NPs, PPNPs, and NO/PPNPs, respectively. The viabilities of fibroblasts treated with PPNPs and NO/PPNPs at 10 mg/mL were not significantly different from those treated with PLGA NPs, indicating that cytotoxicity of the NPs at 10 mg/mL is not caused by NO, but by PLGA NPs per se. Since PLGA is known to be biocompatible, the cytotoxicity of PLGA NPs might be due to the high number of NPs. From the results, it appears that the NO released from the NO/PPNPs has potent antibacterial activities against MRSA and P. aeruginosa and minimal effect against healthy mammalian fibroblasts. In other studies, NO-releasing NPs showed strong cytotoxicity against L929 fibroblast cells. For example, the NO-releasing MAP3 silica NPs showed approximately 30% viability39 and NO-releasing dendrimer showed around 22% viability.40 The lower toxicity of our NO/PPNPs than other NO-releasing NPs can be attributed to the relatively lower amount of NO per mg of NPs as mentioned above.
Figure 9.

Viability (%) of L929 mouse fibroblast cells following 24-hour exposure to nanoparticles at different concentrations (n=8).
Abbreviations: PLGA, poly(lactic-co-glycolic acid); PEI, polyethylenimine; NPs, nanoparticles; PPNPs, PLGA-PEI nanoparticles; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; NO, nitric oxide.
In vivo wound healing assay
The in vivo wound healing assay was performed to investigate whether NO/PPNPs can accelerate the repair of bacteria- infected wounds. Full-thickness wounds were inflicted in mice and MRSA was inoculated in the wound for 1 day to develop infection. The wounds were treated with the NO/PPNPs once on day 1 because they release NO for over 6 days. Untreated mice were used as controls. The macroscopic appearances of the wounds and % wound area on different days are presented in Figure 10. On day 4 (3 days after the first treatment), the NO/PPNPs-treated group exhibited a signifi-cantly reduced wound area (64%, P<0.05) without any scab, while the untreated mice showed slightly increased wound area with scabs (crusts of dried blood and exudate over wound area during healing) over the wound bed. On day 7, clear epithelialization was observed in the NO/PPNPs-treated group and the wound area was further reduced to 25% (P<0.05). However, the untreated group showed a thicker scab without significant reduction in the wound area. The delayed wound healing in the untreated group could be due to bacterial infections. Therefore, the fast wound healing observed with NO/PPNPs treatment can be attributed to the bactericidal effect as well as wound healing activity of the NO.
Figure 10.

Wound healing assay in mice.
Notes: (A) Representative photographs of MRSA-infected wounds of BALB/c mice treated with or without NO/PPNPs. (B) Area reduction (%) profiles of the wounds. Values are mean ± standard deviation, n=4, *P<0.05 compared with untreated group.
Abbreviations: MRSA, methicillin-resistant Staphylococcus aureus; NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; PLGA, poly(lactic-co-glycolic acid); PEI, polyethylenimine; NO, nitric oxide.
Histological examination in the infected full-thickness wounds was also performed to compare the untreated wounds with NO/PPNPs-treated wounds. Figure 11 shows micrographs of hematoxylin and eosin staining section of normal mouse skin, untreated and treated with NO/PPNPs. Untreated group showed ulceration, edema, and abundance of mononuclear inflammatory cells with deep inflammatory infiltration getting through the dermal layer, NO/PPNPs groups showed increased numbers of fibroblast-like and decreased mononuclear inflammatory cells with healed skin structures close to normal healthy epidermis.
Figure 11.
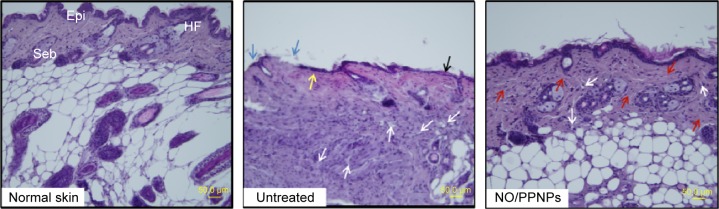
Histological sections of normal skin, untreated, and NO/PPNPs stained with hematoxylin and eosin.
Notes: Histological analysis of BALB/c mice at day 7, scale bar =50 μm. The arrows colored with black, blue, yellow, white, and red indicate edema, ulceration, early epithelialization, mononuclear inflammatory cell, and fibroblast cell, respectively.
Abbreviations: NO/PPNPs, NO-releasing PLGA-PEI nanoparticles; PLGA, poly(lactic-co-glycolic acid); PEI, polyethylenimine; NO, nitric oxide; HF, hair follicles; Epi, epidermis; Seb, sebaceous glands.
Conclusion
In this study, we successfully fabricated NO-releasing polymeric NPs by incorporating PEI/NONOate in the PLGA polymer matrix. The NO/PPNPs showed a remarkably extended NO release profile over 6 days. We found that the positive charge of the NO/PPNPs enabled their adhesion to the surface of the negatively charged MRSA and P. aeruginosa and facilitated the potent bactericidal efficacy with minimal toxicity to healthy fibroblast cells. Moreover, application of the NO/PPNPs to MRSA-infected full-thickness wounds resulted in favorable wound healing with accelerated wound size reduction. Therefore, the novel NO-releasing polymeric NPs investigated in this study could be a promising approach for enhanced wound healing and treatment of various skin infections.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2014R1A1A4A01007808) and by a grant from the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare Affairs, Republic of Korea (HI12C0529).
Footnotes
Disclosure
The authors declare that they have no conflicts of interest regarding this study.
References
- 1.Morens DM, Folkers GK, Fauci AS. The challenge of emerging and re-emerging infectious diseases. Nature. 2004;430(6996):242–249. doi: 10.1038/nature02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366(9498):1736–1743. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- 3.Chiller K, Selkin BA, Murakawa GJ. Skin microflora and bacterial infections of the skin. J Investig Dermatol Symp Proc. 2001;6(3):170–174. doi: 10.1046/j.0022-202x.2001.00043.x. [DOI] [PubMed] [Google Scholar]
- 4.Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA) Proc Natl Acad Sci U S A. 2002;99(11):7687–7692. doi: 10.1073/pnas.122108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis. 2005;41(10):1373–1406. doi: 10.1086/497143. [DOI] [PubMed] [Google Scholar]
- 6.Maranan MC, Moreira B, Boyle-Vavra S, Daum RS. Antimicrobial resistance in staphylococci: epidemiology, molecular mechanisms, and clinical relevance. Infect Dis Clin North Am. 1997;11(4):813–849. doi: 10.1016/s0891-5520(05)70392-5. [DOI] [PubMed] [Google Scholar]
- 7.Edwards R, Harding KG. Bacteria and wound healing. Curr Opin Infect Dis. 2004;17(2):91–96. doi: 10.1097/00001432-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Coates A, Hu Y. Novel approaches to developing new antibiotics for bacterial infections. Br J Pharmacol. 2007;152(8):1147–1154. doi: 10.1038/sj.bjp.0707432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scatena R, Bottoni P, Pontoglio A, Giardina B. Pharmacological modulation of nitric oxide release: new pharmacological perspectives, potential benefits and risks. Curr Med Chem. 2010;17(1):61–73. doi: 10.2174/092986710789957841. [DOI] [PubMed] [Google Scholar]
- 10.Hetrick EM, Schoenfisch MH. Reducing implant-related infections: active release strategies. Chem Soc Rev. 2006;35(9):780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 11.Burgner D, Rockett K, Kwiatkowski D. Nitric oxide and infectious diseases. Arch Dis Child. 1999;81(2):185–188. doi: 10.1136/adc.81.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Efron DT, Most D, Barbul A. Role of nitric oxide in wound healing. Curr Opin Clin Nutr Metab Care. 2000;3(3):197–204. doi: 10.1097/00075197-200005000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Gu H, Ho P, Tong E, Wang L, Xu B. Presenting vancomycin on nanoparticles to enhance antimicrobial activities. Nano letters. 2003;3(9):1261–1263. [Google Scholar]
- 14.Sambhy V, MacBride MM, Peterson BR, Sen A. Silver bromide nanoparticle/polymer composites: dual action tunable antimicrobial materials. J Am Chem Soc. 2006;128(30):9798–9808. doi: 10.1021/ja061442z. [DOI] [PubMed] [Google Scholar]
- 15.Whitesides GM. Nanoscience, nanotechnology, and chemistry. Small. 2005;1(2):172–179. doi: 10.1002/smll.200400130. [DOI] [PubMed] [Google Scholar]
- 16.Lu Y, Slomberg DL, Sun B, Schoenfisch MH. Shape-and Nitric Oxide Flux-Dependent Bactericidal Activity of Nitric Oxide-Releasing Silica Nanorods. Small. 2013;9(12):2189–2198. doi: 10.1002/smll.201201798. [DOI] [PubMed] [Google Scholar]
- 17.Slomberg DL, Lu Y, Broadnax AD, Hunter RA, Carpenter AW, Schoenfisch MH. Role of size and shape on biofilm eradication for nitric oxide-releasing silica nanoparticles. ACS Appl Mater Interfaces. 2013;5(19):9322–9329. doi: 10.1021/am402618w. [DOI] [PubMed] [Google Scholar]
- 18.Lu Y, Slomberg DL, Shah A, Schoenfisch MH. Nitric oxide-releasing amphiphilic poly(amidoamine)(PAMAM) dendrimers as antibacterial agents. Biomacromolecules. 2013;14(10):3589–3598. doi: 10.1021/bm400961r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Y, Sun B, Li C, Schoenfisch MH. Structurally diverse nitric oxide-releasing poly(propylene imine) dendrimers. Chem Mater. 2011;23(18):4227–4233. doi: 10.1021/cm201628z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Y, Slomberg DL, Schoenfisch MH. Nitric oxide-releasing chitosan oligosaccharides as antibacterial agents. Biomaterials. 2014;35(5):1716–1724. doi: 10.1016/j.biomaterials.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang SL, Kee PH, Kim H, et al. Nitric oxide-loaded echogenic liposomes for nitric oxide delivery and inhibition of intimal hyperplasia. J Am Coll Cardiol. 2009;54(7):652–659. doi: 10.1016/j.jacc.2009.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nablo BJ, Rothrock AR, Schoenfisch MH. Nitric oxide-releasing sol–gels as antibacterial coatings for orthopedic implants. Biomaterials. 2005;26(8):917–924. doi: 10.1016/j.biomaterials.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 23.Choi JS, Cao J, Naeem M, et al. Size-controlled biodegradable nanoparticles: Preparation and size-dependent cellular uptake and tumor cell growth inhibition. Colloids Surf B Biointerfaces. 2014;122:545–551. doi: 10.1016/j.colsurfb.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 24.Choi JS, Seo K, Yoo JW. Recent advances in PLGA particulate systems for drug delivery. Journal of Pharmaceutical Investigation. 2012;42(3):155–163. [Google Scholar]
- 25.Zhou Z, Annich GM, Wu Y, Meyerhoff ME. Water-soluble poly (ethylenimine)-based nitric oxide donors: Preparation, characterization, and potential application in hemodialysis. Biomacromolecules. 2006;7(9):2565–2574. doi: 10.1021/bm060361s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol. 2003;41(11):5113–5120. doi: 10.1128/JCM.41.11.5113-5120.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearson JP, Pesci EC, Iglewski BH. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J Bacteriol. 1997;179(18):5756–5767. doi: 10.1128/jb.179.18.5756-5767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frost MC, Reynolds MM, Meyerhoff ME. Polymers incorporating nitric oxide releasing/generating substances for improved biocompatibility of blood-contacting medical devices. Biomaterials. 2005;26(14):1685–1693. doi: 10.1016/j.biomaterials.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Wan A, Sun Y, Li H. Characterization of folate-graft-chitosan as a scaffold for nitric oxide release. Int J Biol Macromol. 2008;43(5):415–421. doi: 10.1016/j.ijbiomac.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 30.Griess P. Bemerkungen zu der Abhandlung der HH. Weselsky und Benedikt “Ueber einige Azoverbindungen”. [Comments on the discussion of the HH Weselsky and Benedict “on some azo compounds”] Ber Dtsch Chem Ges. 1879;12(1):426–428. German. [Google Scholar]
- 31.Yoo JW, Lee JS, Lee CH. Characterization of nitric oxide-releasing microparticles for the mucosal delivery. J Biomed Mater Res A. 2010;92(4):1233–1243. doi: 10.1002/jbm.a.32434. [DOI] [PubMed] [Google Scholar]
- 32.Gethin G. The significance of surface pH in chronic wounds. Wounds UK. 2007;3(3):52. [Google Scholar]
- 33.Shin JH, Metzger SK, Schoenfisch MH. Synthesis of nitric oxide-releasing silica nanoparticles. J Am Chem Soc. 2007;129(15):4612–4619. doi: 10.1021/ja0674338. [DOI] [PubMed] [Google Scholar]
- 34.Rothrock AR, Donkers RL, Schoenfisch MH. Synthesis of nitric oxide-releasing gold nanoparticles. J Am Chem Soc. 2005;127(26):9362–9363. doi: 10.1021/ja052027u. [DOI] [PubMed] [Google Scholar]
- 35.Stasko NA, Fischer TH, Schoenfisch MH. S-nitrosothiol-modified dendrimers as nitric oxide delivery vehicles. Biomacromolecules. 2008;9(3):834–841. doi: 10.1021/bm7011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khalil H, Chen T, Riffon R, Wang R, Wang Z. Synergy between polyethylenimine and different families of antibiotics against a resistant clinical isolate of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2008;52(5):1635–1641. doi: 10.1128/AAC.01071-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su S, Panmanee W, Wilson JJ, et al. Catalase (KatA) Plays a Role in Protection against a Anaerobic Nitric Oxide in Pseudomonas aeruginosa. PloS One. 2014;9(3):e91813. doi: 10.1371/journal.pone.0091813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang FC. Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J Clin Invest. 1997;99(12):2818. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hetrick EM, Shin JH, Paul HS, Schoenfisch MH. Anti-biofilm efficacy of nitric oxide-releasing silica nanoparticles. Biomaterials. 2009;30(14):2782–2789. doi: 10.1016/j.biomaterials.2009.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun B, Slomberg DL, Chudasama SL, Lu Y, Schoenfisch MH. Nitric oxide-releasing dendrimers as antibacterial agents. Biomacromolecules. 2012;13(10):3343–3354. doi: 10.1021/bm301109c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong T, McGrath J, Navsaria H. The role of fibroblasts in tissue engineering and regeneration. Br J Dermatol. 2007;156(6):1149–1155. doi: 10.1111/j.1365-2133.2007.07914.x. [DOI] [PubMed] [Google Scholar]